

Abstract
Aims
Hyperkalaemia risk precludes optimal renin–angiotensin–aldosterone system inhibitor use in patients with heart failure (HF), particularly those with chronic kidney disease (CKD). Patiromer is a sodium‐free, non‐absorbed potassium (K+)‐binding polymer approved for the treatment of hyperkalaemia. In PEARL‐HF, patiromer 25.2 g (fixed dose) prevented hyperkalaemia in HF patients with or without CKD initiating spironolactone. The current study evaluated the effectiveness of a lower starting dose of patiromer (16.4 g/day) followed by individualized titration in preventing hyperkalaemia and hypokalaemia when initiating spironolactone.
Methods and results
This open‐label 8‐week study enrolled 63 patients with CKD, serum K+ 4.3–5.1 mEq/L, and chronic HF, who, based on investigator opinion, should receive spironolactone. Eligible patients started spironolactone 25 mg/day and patiromer 16.8 g/day (divided into two doses), with patiromer titrated to maintain serum K+ 4.0–5.1 mEq/L. Mean (standard deviation) serum K+ was 4.78 (0.51) mEq/L at baseline; weekly values were 4.48–4.70 mEq/L during treatment. Serum K+ of 3.5–5.5 mEq/L at the end of study treatment (primary endpoint) was achieved by 57 (90.5%) patients; 53 (84.1%) had serum K+ 4.0–5.1 mEq/L. One patient (1.6%) developed hypokalaemia, and two patients (3.2%) developed hypomagnesaemia. Spironolactone was increased to 50 mg/day in all patients; 43 (68%) patients required one or more patiromer dose titration. Adverse events (AEs) occurred in 36 (57.1%) patients, with a low rate of discontinuations [four (6.3%) patients]. The most common AE was mild to moderate abdominal discomfort [four (6.3%) patients].
Conclusions
In this open‐label study, patiromer 16.8 g/day followed by individualized titration maintained serum K+ within the target range in the majority of patients with HF and CKD, all of whom were uptitrated to spironolactone 50 mg/day, patiromer was well tolerated, with a low incidence of hyperkalaemia, hypokalaemia, and hypomagnesaemia.
Keywords: Heart failure, Chronic kidney disease, Patiromer, Potassium‐binding polymer, Mineralocorticoid receptor antagonist
Introduction
Renin–angiotensin–aldosterone system inhibitors (RAASi) are guideline recommended with a Class 1 indication to reduce cardiovascular and total mortality in patients with heart failure (HF) and reduced left ventricular ejection fraction (EF).1, 2, 3, 4, 5, 6 In patients with HF with reduced EF (HFrEF) at highest risk of cardiovascular death, such as those with diabetes mellitus and/or chronic kidney disease (CKD), the risk of worsening renal function and hyperkalaemia often precludes or limits the use of optimal doses of RAASi, especially mineralocorticoid receptor antagonists (MRAs).7, 8, 9 Given that the risk of hyperkalaemia may change over time in patients with HF due to dose changes in RAASi, dietary changes, use of diuretics, and changes in renal function, it is desirable to have a titratable drug that can be used to manage serum potassium (K+) without resorting to discontinuation or dose reduction of evidence‐based HF and CKD treatments.
Patiromer is a non‐absorbed K+‐binding polymer that uses calcium rather than sodium as the cation for exchange with K+. It is approved for the treatment of hyperkalaemia in the USA and in Europe.10, 11, 12 Patiromer has been shown to reduce serum K+ in hyperkalaemic patients with CKD with or without other co‐morbidities, such as HF, diabetes mellitus, and/or hypertension.13, 14, 15 Patiromer has also been demonstrated in a randomized, double‐blind, placebo‐controlled study to prevent hyperkalaemia in normokalaemic patients with HF with or without CKD who were initiated on spironolactone.16 This study used a relatively high fixed total daily dose of patiromer (25.2 g given as a divided dose twice daily), which was associated with approximately 7% incidence of hypokalaemia (serum K+ <3.5 mEq/L).16 The present study was designed to assess a lower starting dose of patiromer followed by individualized titration based on serum K+ levels in preventing hyperkalaemia and hypokalaemia in patients with HF and CKD who were initiated on spironolactone 25 mg/day in addition to standard therapy, including one or more of the following: an angiotensin‐converting enzyme inhibitor (ACEi) and angiotensin receptor blocker (ARB) or a beta adrenergic receptor blocker (BB).
It was hypothesized that the lower starting dose followed by individually optimized dose titration would minimize the risk of hypokalaemia and still prevent hyperkalaemia, thus allowing continued usage of RAASi in these patients. The secondary aim of the study was to assess the effects of patiromer on serum K+ and the safety and tolerability of patiromer in HF patients with CKD.
Methods
This 8 week, open‐label study (registered at http://ClinicalTrials.gov, NCT01130597) was conducted at 13 centres in Georgia and Slovenia between May 2010 and September 2010. All participating sites received approval from their nationally or locally appointed Independent Ethics Committee, and all patients provided written informed consent. The study was performed in accordance with the Declaration of Helsinki, current local and national regulations, the International Conference on Harmonization, and Good Clinical Practice guidelines and fulfilled other applicable requirements governing the conduct of human clinical trials.
Patients and assessments
Eligible patients were ≥18 years old with chronic HF who, in the opinion of the investigator, should receive spironolactone therapy. Patients also had to have CKD with a screening estimated glomerular filtration rate (eGFR) of <60 mL/min/1.73 m2 based on central laboratory creatinine measurement and calculated using the Modification of Diet in Renal Disease formula.17 Serum K+ at screening and at baseline had to be 4.3–5.1 mEq/L, and patients had to be receiving at least one of the following: ACEi, ARB, or a BB. Patients were excluded if they had a history of severe gastrointestinal (GI) disorders, major GI surgery, bowel obstruction, or swallowing disorders, or if they had severe or unstable hypertensive or cardiovascular disease or severe renal disease, including history of or anticipated need for heart or kidney transplantation or dialysis during the study. Prohibited medications during the study included polymer‐based drugs, other K+ binders or phosphate binders, other K+‐sparing diuretics (including non‐study aldosterone antagonists), and K+ supplements. Investigators were instructed to maintain a constant dose of cardiac medications during the entire study. Oral diuretics (loop and thiazide) were permitted during the study if the dose was stable for at least 21 days prior to the start of the study and was expected to remain stable during the study.
Patients who completed screening and satisfied the eligibility criteria returned to the clinic for baseline assessments, which included a physical examination, weight and height, resting heart rate, supine blood pressure, a 12‐lead electrocardiogram (ECG), determination of serum K+, and clinical laboratory tests, including serum chemistry, haematology, and urinalysis. Patients who continued to satisfy all eligibility criteria were enrolled in the study and were initiated on spironolactone 25 mg once daily and patiromer 16.8 g/day on Day 1. Patiromer was supplied as a powder for oral suspension, which was to be mixed with water or a low‐K+ food and consumed with meals—8.4 g with breakfast and 8.4 g with dinner.
The patiromer and spironolactone dosing algorithm is shown in Table S1 of the Supporting Information. Starting on Day 3, the patiromer dose could be adjusted up or down in increments of 8.4 g/day based on the patient's local laboratory serum K+ value in order to maintain serum K+ within the target range of 4.0–5.1 mEq/L. The only exception was if the serum K+ was <4.0 mEq/L on a visit when the patient was eligible for a spironolactone dose increase. In such cases, the patiromer dose was to be maintained. Patiromer doses could be titrated to a minimum of 0 g/day or to a maximum of 50.4 g/day. Starting on Day 7, the spironolactone dose was increased to 50 mg once daily provided that the patient's serum K+ concentration was ≤5.1 mEq/L; spironolactone dose reductions were not allowed. During the course of the study, the minimum spironolactone dose was 25 mg/day, and the maximum was 50 mg/day. Patiromer and spironolactone dose adjustments were not permitted on the same day. Patients were to discontinue both patiromer and spironolactone and were to be withdrawn from the study if their local laboratory serum K+ was <3.5 or >5.5 mEq/L at two consecutive visits despite titration of patiromer or spironolactone on the first of the two visits.
During the 8 week treatment period, serum K+ was monitored at each clinic visit, scheduled on Day 3 and weekly from Weeks 1 through 8. At each visit, 12‐lead ECG was obtained, the need for patiromer dose titration was assessed (except at the Week 8 visit), and concomitant medications and adverse events (AEs) were assessed. Vital signs, body weight, and an evaluation for spironolactone uptitration were assessed weekly from Weeks 1 through 8 (except spironolactone uptitration assessment at the Week 8 visit). Serum chemistry testing was performed at Weeks 2, 4, 6, and 8, and haematology testing was performed at Weeks 4 and 8. One week after completion of the treatment period (or following premature discontinuation of patiromer and spironolactone), patients returned to the clinic for a safety follow‐up visit.
Clinical endpoints
The primary efficacy outcome was the proportion of patients with a serum K+ 3.5–5.5 mEq/L at the end of study treatment (i.e., the final visit of the treatment period while on study drug). Secondary endpoints included (1) proportion of patients with serum K+ 3.5–5.5 mEq/L, by visit and during the entire treatment period; (2) proportion of patients with serum K+ 4.0–5.1 mEq/L, by visit and during the entire treatment period; (3) proportion of patients whose spironolactone dose could be increased to 50 mg/day; (4) mean number of patiromer titrations; (5) proportion of patients requiring patiromer uptitration or downtitration; (6) mean time to the first patiromer titration; and (7) mean dose of patiromer by visit and at the end of the study.
The safety of patiromer was assessed based on (1) AEs recorded during the study and through 7 days after the last dose of study drug; (2) clinically significant changes from baseline in clinical laboratory (haematology and serum chemistry) values, vital signs, and ECG parameters and proportion of patients who discontinued the study owing to hyperkalaemia (serum K+ >5.5 mEq/L); and (3) proportion of patients discontinuing because of hypokalaemia (serum K+ <3.5 mEq/L on two consecutive visits despite patiromer or spironolactone dose titration on the first of the two visits).
Statistical analysis
A total of 63 patients were enrolled to ensure that at least 50 patients would receive patiromer and provide efficacy data for analysis. The sample size was based on the patiromer response predicted from the PEARL‐HF study.16 For the primary efficacy endpoint, the proportion of patients with serum K+ 3.5–5.5 mEq/L at the end of the study treatment was summarized along with 95% confidence intervals (CIs). The analysis population for the primary and secondary efficacy endpoints, and for assessment of safety, was the intent‐to‐treat population (i.e., all enrolled patients). Baseline values were obtained from the last available measurement prior to the start of patiromer. For categorical secondary endpoints, the proportion of patients and 95% CIs were summarized. AEs were summarized descriptively.
Statistical analyses were performed using SAS Version 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
Baseline characteristics and disposition
A total of 63 patients [100% White, 39 (61.9%) men] were enrolled in the study, all of whom received at least one dose of patiromer and had at least one post‐baseline serum K+ assessment. The mean [standard deviation (SD)] age was 70.8 (8.5) years (Table 1). All patients had a history of HF with a mean (SD) duration of 3.9 (4.1) years, which was characterized at study entry as New York Heart Association Class II (46% of patients) or Class III (54%). Patients had a mean (SD) EF of 38.6% (9.3%); 33 (52.4%) patients had an EF <40% (Table 1). The aetiology of HF was ischaemic in 40 (63.5%) patients. Overall, 93.7% of patients had a history of hypertension, and 42.9% had diabetes. All patients had CKD; the mean (SD) eGFR was 46.2 (15.2) mL/min/1.73 m2 at baseline; 57 patients were in Stage 3a or 3b and six patients in Stage 4 or 5. The mean (SD) duration of CKD was 2.5 (3.3) years, and the most common aetiologies of CKD were hypertension only (30.2%), hypertension and diabetes (27.0%), and unknown (30.2%).
Table 1.
Baseline demographic and clinical characteristics
| Parameter | Patiromer (N = 63) |
|---|---|
| Demographics | |
| Age (years), mean ± SD | 70.8 ± 8.5 |
| Male, n (%) | 39 (61.9) |
| White, n (%) | 63 (100) |
| BMI (kg/m2), mean ± SD | 28.6 ± 3.8 |
| Cardiac history and parameters | |
| HF duration (years), mean ± SD | 3.9 ± 4.1 |
| HF aetiology: ischaemic, n (%) | 40 (63.5) |
| LVEF (%), mean ± SDa | 38.6 ± 9.3 |
| Patients with LVEF of <40%, n (%) | 33 (52.4) |
| Patients with LVEF of 40–50%, n (%) | 24 (38.1) |
| Patients with LVEF of >50%, n (%) | 5 (7.9) |
| NYHA Class, n (%) II | 29 (46.0) |
| III | 34 (54.0) |
| CKD history | |
| CKD duration (years), mean ± SDa | 2.5 ± 3.3 |
| CKD aetiology, n (%)a | |
| Hypertension (only) | 19 (30.2) |
| Hypertension and diabetes | 17 (27.0) |
| Diabetes (only) | 4 (6.3) |
| Urologic or congenital | 2 (3.2) |
| Unknown | 19 (30.2) |
| eGFR (mL/min/1.73 m2), mean ± SD | 46.2 ± 15.2 |
| Patients with eGFR (mL/min/1.73m2), n (%) | |
| <15 | 1 (1.6) |
| 15–<30 | 5 (7.9) |
| 30–45 | 26 (41.3) |
| >45 | 31 (49.2) |
| ACR (mg/g), mean ± SD [n (%)] | |
| <30 mg/g | 10.8 ± 5.7 [28 (44.4)] |
| ≥30 mg/g | 749.6 ± 1341.9 [35 (55.6)] |
| Vital signs | |
| Heart rate (b.p.m.) | 70.5 ± 12.8 |
| Systolic blood pressure (mmHg) | 133.3 ± 16.6 |
| Diastolic blood pressure (mmHg) | 82.1 ± 7.6 |
| Other co‐morbid conditions | |
| History of diabetes, n (%) | 27 (42.9) |
| History of hypertension, n (%) | 59 (93.7) |
| HF medications at baseline | 63 (100) |
| Diuretics | 52 (82.5) |
| Loop | 46 (73.0) |
| Thiazide | 11 (17.5) |
| ACEi | 45 (71.4) |
| ARB | 18 (28.6) |
| Beta‐blocker | 48 (76.2) |
| ACEi only | 9 (14.3) |
| ARB only | 5 (7.9) |
| Beta‐blocker only | 1 (1.6) |
| ACEi + ARB | 1 (1.6) |
| ACEi + beta‐blocker | 35 (55.6) |
| ARB + beta‐blocker | 12 (19.0) |
| ACEi + ARB + beta‐blocker | 0 |
ACEi, angiotensin‐converting enzyme inhibitor; ACR, albumin‐to‐creatinine ratio; ARB, angiotensin receptor blocker; BMI, body mass index; b.p.m., beats per minute; CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; HF, heart failure; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association functional class; SD, standard deviation.
n = 62.
At baseline, all patients were receiving a RAASi or a BB; 45 (71.4%) patients were on an ACEi, 18 (28.6%) were on an ARB, and 48 (76.2%) were on a BB (Table 1). One (1.6%) patient was on dual therapy of ACEi and ARB, 35 (55.6%) were on dual therapy of ACEi and BB, 12 (19.0%) were on dual therapy of ARB and BB, and no patient was on triple therapy (ACEi, ARB, and BB) (Table 1). Thirty‐two (51%) patients were receiving 50% or more of the maximal daily dose of an ACEi or ARB, while 26 (41%) patients were receiving 25% or less of the maximal daily dose of an ACEi or ARB. Of the 48 patients taking beta‐blockers at baseline, 11 (23%) were receiving 50% or more of the maximal daily dose, while 13 (27%) were receiving 25% or less of the maximal daily dose. In addition, 52 (82.5%) patients were on a diuretic at baseline, and the same proportion (82.5%) was receiving diuretic therapy during the study.
Fifty‐six (88.9%) patients completed the 8 week treatment period, and seven (11.1%) prematurely discontinued treatment; the most common reason for discontinuation was AEs (four patients; 6.3%) (see Supporting Information, Figure S1 ).
Efficacy
At baseline, the mean (SD) serum K+ was 4.78 (0.51) mEq/L. Mean (SD) change in serum K+ from baseline to end of study treatment was −0.13 (0.69) mEq/L, range: −1.9 to 1.3 mEq/L. During the treatment period, weekly mean serum K+ values were 4.48–4.70 mEq/L. At the end of study treatment, 57 (90.5%; 95% CI: 80.4%, 96.4%) patients had serum K+ 3.5–5.5 mEq/L (primary endpoint), including five of six patients with eGFR <30 mL/min/1.73 m2 at baseline. Fifty‐three (84.1%; 95% CI: 72.7%, 92.1%) patients had serum K+ 4.0–5.1 mEq/L. Figure 1 shows the proportions of patients with serum K+ in the ranges of 3.5–5.5 and 4.0–5.1 mEq/L at all post‐baseline visits. The proportion ranged from 91.2% to 100% for patients with serum K+ 3.5–5.5 mEq/L and from 72.6% to 90.3% for patients with serum K+ 4.0–5.1 mEq/L (Figure 1). Serum K+ was maintained in the range of 3.5–5.5 mEq/L in 48 (76.2%; 95% CI: 63.8%, 86.0%) patients and in the range of 4.0–5.1 mEq/L in 19 (30.2%; 95% CI: 19.2%, 43.0%) patients during the entire treatment period.
Figure 1.

Proportion of patients with serum K+ in the range of 3.5–5.5 mEq/L and 4.0–5.1 mEq/L by study visit. ET, end of treatment; K+, potassium.
The mean (SD) number of prescribed patiromer dose titrations during the study was 1.3 (1.1): 20 (31.7%) patients did not require any titration, 21 (33.3%) had one or more uptitrations, 8 (12.7%) had one or more downtitrations, and 14 (22.2%) had both one or more uptitrations and one or more downtitrations. The median (95% CI) time to the first patiromer dose titration was 21.0 (14.0, 36.0) days.
Over the course of the study, the mean (SD) duration of exposure to patiromer was 53.9 (8.6) days, and the mean (SD) dose was 18.7 (6.4) g/day. The mean (SD) dose of patiromer at the end of the study was 19.6 (10.4) g/day. In the majority of patients, the last prescribed dose was either 16.8 g/day (44.4%) or 25.2 g/day (17.5%) (see Supporting Information, Table S2 ). On average, serum K+ decreased by 0.34 mEq/L 1 week following an 8.4 g/day uptitration in patiromer dose and increased by 0.46 mEq/L 1 week following an 8.4 g/day downtitration of patiromer dose (Figure 2).
Figure 2.
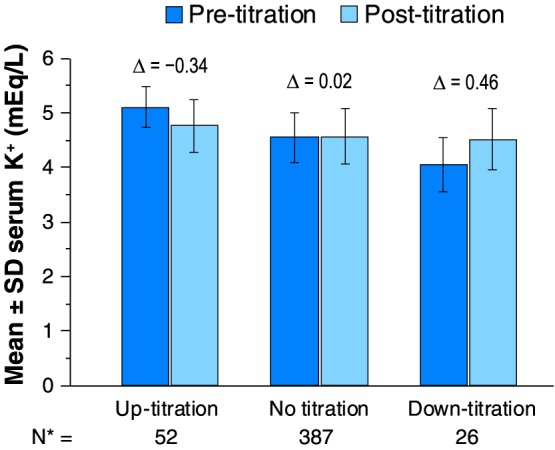
Serum K+ before and after an uptitration or downtitration. *Total number of titrations during the study. K+, potassium; SD, standard deviation.
The mean (SD) duration of exposure to spironolactone was 53.6 (8.7) days. One patient initiated treatment with spironolactone 50 mg/day (protocol deviation) and continued this dose through the completion of the study. Of the 62 patients who initiated spironolactone treatment at 25 mg/day, the MRA was successfully uptitrated to 50 mg/day in all 62 patients by Day 14 of the study. The uptitration occurred in 57 patients during the first week and in five during the second week. The median time to titration was 8 days (range: 6–16 days). The mean (SD) dose of spironolactone during the study treatment period was 45.5 (3.4) mg/day.
Patients' dosing compliance with patiromer and spironolactone was measured by examination of the drug sachets and blister packs, respectively, which were returned to the clinic at each visit. The mean (SD) dosing compliance rate was 98.7% (1.9%) for patiromer and 98.5% (5.6%) for spironolactone.
Safety
A total of 36 (57.1%) patients had at least one AE, regardless of severity, seriousness, or relatedness to patiromer (Table 2); the most common AEs (occurring in at least three patients) were worsening of renal function in eight (12.7%) patients, abdominal discomfort in four (6.3%) patients, and flatulence, headache, and hypertension, each occurring in three (4.8%) patients. In the majority of patients, the AEs were mild or moderate in intensity (83.3%) and in most patients (69.5%) were unrelated to patiromer. Mild peripheral oedema was reported in two (3.2%) patients; both cases were considered not related to patiromer by the investigator. In all eight patients with worsening of renal function, the AE was considered unrelated to patiromer by the investigator, and in three patients, the AE was considered related to spironolactone (see Supporting Information, Table S3 ). Eleven patients had patiromer‐related AEs, most commonly GI‐related (abdominal discomfort, three; flatulence, three; diarrhoea, two; abdominal pain, one). All GI‐related AEs were reported as either mild or moderate. Four (6.3%) patients had a total of six AEs that led to discontinuation of patiromer; in three patients, the AEs leading to discontinuation were serious (discussed below). The non‐serious AE leading to discontinuation was moderate diarrhoea.
Table 2.
Safety summary
| Parameter, n (%) | (N = 63) |
|---|---|
| Any adverse event | 36 (57.1) |
| Adverse eventsa occurring in ≥3 (~5%) patients | |
| Worsening of renal functiona | 8 (12.7) |
| Abdominal discomfort | 4 (6.3) |
| Flatulence | 3 (4.8) |
| Headache | 3 (4.8) |
| Hypertension | 3 (4.8) |
| Serious adverse events | 6 (9.5) |
| Patiromer‐related serious adverse events | 0 (0) |
| Adverse event leading to study discontinuation | 4 (6.3) |
Adverse events were coded to the preferred terms listed above using the Medical Dictionary for Regulatory Activities Version 12.0, except for worsening of renal function, which includes the preferred terms of renal failure acute (three patients), renal impairment (three patients), and renal failure (two patients).
During the treatment period through follow‐up, six (9.5%) patients had a total of nine serious AEs (SAEs); three patients had a single, non‐fatal SAE: acute renal failure in two patients (see Supporting Information, Table S3 ) and subcutaneous abscess in one patient. Three patients, all diabetic, experienced fatal SAEs. One patient died because of sudden cardiac death on Day 29 of the study. This patient's last measured (on Day 28) serum K+ was 4.6 mEq/L and serum magnesium was 1.7 mg/dL. Two patients died after stopping patiromer: one sudden death 5 days after completing study treatment and one sudden cardiac death 26 days after prematurely discontinuing patiromer treatment due to non‐fatal SAEs, including acute renal failure (see Supporting Information, Table S3 ). The narratives for the three patients with fatal SAEs are provided in Table S4 of the Supporting Information. None of the SAEs, including the fatal events, were considered by the study investigators as related to patiromer, and the SAEs of acute renal failure for two patients were considered to be related to spironolactone. Two of the three deaths were considered by the Safety Review Board as unlikely to be due to hypokalaemia or hyperkalaemia, and the relationship to hypokalaemia or hyperkalaemia for the one death that occurred 26 days after prematurely discontinuing patiromer treatment could not be determined.
One (1.6%) patient developed hypokalaemia (3.1 mEq/L at the Week 3 visit) based on prespecified criteria (serum K+ <3.5 mEq/L); patiromer was downtitrated, while the spironolactone dose was maintained, and at the next visit 8 days later, the serum K+ was 3.9 mEq/L. No patient discontinued the study because of hypokalaemia. A total of 15 (24%) patients developed hyperkalaemia (serum K+ >5.5 mEq/L): nine (14%) patients had an isolated event before or at the end of treatment; six (10%) patients had more than one serum K+ value >5.5 mEq/L. Serum K+ was normalized with individualized dose titration of patiromer in all but one of these patients, with the exception being the patient who was prematurely withdrawn from the study owing to hyperkalaemia. Six patients had a total of seven episodes of serum K+ >6.0 mEq/L, with the highest being 6.6 mEq/L in one patient.
Mean serum magnesium levels were slightly lower (−0.17 to −0.18 mg/dL) at all treatment visits (Week 4, Week 8, and end‐of‐treatment visits) than at baseline; however, mean levels remained within the normal range (1.8–2.4 mg/dL). At Week 8, the mean [standard error (SE)] change from baseline in serum magnesium was −0.18 (0.04) (range: −1.00 to 0.30). Eight (12.7%) patients had serum magnesium levels <1.8 mg/dL [lower limit of normal (LLN)] during the treatment period (with one having baseline level below the LLN); of these, two (3.2%) patients had levels <1.4 mg/dL (none <1.2 mg/dL). One patient with serum magnesium of 1.5 mg/dL reported moderate myalgia with onset on Day 29, which was resolved within a week after the patient received a magnesium supplement. Mean serum magnesium returned to near‐baseline values 7 days post‐treatment (change from baseline, 0.03 mg/dL). No AEs of hypomagnesaemia were reported.
There were no clinically relevant or statistically significant changes in mean serum creatinine or eGFR values at Week 8. Mean (SE) change from baseline to Week 8 was +0.04 (0.04) mg/dL (range: −0.84 to 0.97) for serum creatinine and −0.21 (1.11) mL/min/1.73 m2 (range: −16.0 to 18.0) for eGFR. At baseline, 35 (55.6%) patients had micro‐albuminuria or macro‐albuminuria [albumin‐to‐creatinine ratio (ACR) ≥30 mg/g], with a mean (SD) ACR of 750 (1342) mg/g. The mean (SE) ACR decreased from baseline to Week 8 by 291 (142) mg/g (range: −2536 to 1189; n = 30; P <0.05).
Six patients were noted to have an eGFR of <30 mL/min/1.73 m2 (range: 14–29 mL/min/1.73 m2) by central laboratory results at baseline. All of these patients were successfully uptitrated to spironolactone 50 mg/day (four at Week 1 and one at Week 2) and five were able to maintain this dose of spironolactone throughout their participation in the study. One patient was terminated early from the study because of hyperkalaemia (serum K+ 6.0 and 5.6 mEq/L at Weeks 6 and 7, respectively) while on spironolactone 50 mg/day and patiromer 8.4 g/day, and another patient was withdrawn from the study at Day 25 because of an AE of diarrhoea. From baseline to the end of treatment, there were no meaningful changes in blood pressure, heart rate, ECG intervals or findings, or physical examinations.
Discussion
Patiromer is a novel K+‐binding polymer approved to treat hyperkalaemia.10 Patiromer uses calcium, rather than sodium, as the counterion for K+ exchange.11 This avoids the potential for increases in sodium absorption and retention in patients with volume overload who may not tolerate even a small increase in sodium load, such as those with hypertension, CKD, and/or HF.18, 19 Patiromer has recently been shown to reduce serum aldosterone levels in patients with CKD and hyperkalaemia who are on RAASi.20 A previous trial, PEARL‐HF, demonstrated that, compared with placebo, a 25.2 g/day fixed dose of patiromer prevented the development of hyperkalaemia and was relatively well tolerated in patients with HF receiving standard therapy and initiating spironolactone (25–50 mg/day)16 but was associated with a 7% incidence of hypokalaemia (serum K+ <3.5 mEq/L).
The present study extends previous literature by showing that patiromer initiated at a lower starting dose of 16.8 g/day followed by individual dose titration maintained serum K+ within the target range in nearly all study patients. The study population had characteristics that may make spironolactone initiation clinically desirable but challenging: HFrEF or HF with preserved EF (HFpEF) patients with CKD and high rates of other cardiovascular risk factors, such as diabetes (43%) and hypertension (94%), all but one of whom were on one or more RAASi at baseline. Spironolactone was initiated at 25 mg/day, uptitrated to 50 mg/day to meet the target doses used in HF trials1, 4, 5; in the present trial, all participants achieved the 50 mg/day dose. At the end of the treatment period, 90.5% of patients had serum K+ in the range of 3.5–5.5 mEq/L, and 84.1% had serum K+ in the range of 4.0–5.1 mEq/L.
Approximately one‐third of the patients did not require patiromer dose uptitration to maintain their serum K+ in the range of 3.5–5.5 mEq/L. Among patients who did require patiromer dose titrations, the majority needed only one or two titrations to maintain their serum K+ levels within the target range, despite having CKD and receiving spironolactone 50 mg/day in addition to their standard therapy, including one or more RAASi. Patiromer's efficacy in preventing hyperkalaemia occurred against a background of diuretic therapy in over 80% of patients; however, a previously published analysis of OPAL‐HK data showed a similar magnitude and time course of reductions in serum K+ in CKD patients with hyperkalaemia on and not on diuretics.21
Patiromer was generally well tolerated, with only mild to moderate GI side effects in most patients and low rates of discontinuation. Furthermore, the incidence of hypokalaemia (1.6%) and serum magnesium below the LLN (12.7%) in the present study was lower compared with the rates in PEARL‐HF (7% and 24%, respectively) that used a fixed higher dose of patiromer, suggesting that patiromer initiated at a dose of 16.8 g/day with subsequent individualized dose titrations may be a more effective strategy for avoiding hypokalaemia and serum magnesium <LLN than the 25.2 g/day fixed‐dose strategy used in PEARL‐HF.16 There was a low incidence (4.8%) of renal dysfunction, and in most patients, events of hyperkalaemia were isolated, including in the six patients with serum K+ measurements ≥6.0 mEq/L; none were above 6.6 mEq/L. There were no ECG abnormalities or serious cardiovascular events associated with either hypokalaemia or hyperkalaemia.
Select patients with HF and CKD have potential to derive significant benefit from RAASi, especially MRAs,22 and MRAs have a Class Ia recommendation from the ESC to reduce the risk of HF hospitalization and death in HFrEF patients who remain symptomatic on ACEi or beta‐blocker therapy.6 In addition, spironolactone has also been shown to be effective in reducing atrial natriuretic peptide levels in HF patients on an ACEi or an ARB and in overcoming diuretic resistance in patients with HF.23, 24 However, RAASi agents are frequently not initiated or may be discontinued because of potential risk for hyperkalaemia.25, 26, 27 A prospective analysis from BIOSTAT‐CHF showed that patients with HFrEF receiving less than 50% of the recommended doses of ACEi, ARBs, and/or BBs displayed more risk of death or hospitalization for HF compared with those who receive ≥100%.28
In HFpEF or HF with mid‐range EF (HFmrEF) patients, however, no RAASi has been shown to reduce morbidity and mortality.6 Nonetheless, RAASi therapies, including MRAs, are often used in clinical practice in HFpEF and HFmrEF patients to manage coexisting conditions, such as hypertension, diabetes, and CKD and/or to provide symptom relief. In the large European Society of Cardiology Heart Failure Registry, approximately 23% of ambulatory patients with chronic HF had EF above 45%. Among these patients, 80% were prescribed an ACEi or ARB, and 41% were prescribed an MRA.27
The present study enrolled patients with HF, 47% of whom had EF >40%, and with CKD already receiving background therapy of an ACEi, ARB, and/or a BB and challenged them with a dosing regimen of spironolactone. The role of spironolactone in HFpEF patients remains uncertain,29, 30, 31 although MRAs received a Class IIb recommendation in the recently updated American Heart Association/American College of Cardiology HF guidelines to reduce hospitalization in appropriately selected patients with HFpEF.4 Nonetheless, the results of this study suggest that, in HF patients who, in the opinion of their treating physician, may benefit from spironolactone initiation and uptitration to 50 mg, patiromer was able to maintain serum K+ in the target range in the majority of patients; this was achieved with improved control of hyperkalaemia and a low incidence of hypokalaemia and hypomagnesaemia, as well as acceptable tolerability. The ability to manage or prevent hyperkalaemia may remove one obstacle to the use of MRAs in HF patients who have an indication for such therapy.
Limitations
The most important limitation in the present study is the lack of a control group. However, there was a control group in the PEARL‐HF study,16 which demonstrated a significant reduction in the incidence of hyperkalaemia in patients on patiromer vs. placebo who were initiated on spironolactone 25 mg/day with subsequent uptitration to 50 mg/day.
In addition to the inclusion of patients with HFpEF and HFmrEF, several patients in the present study who received spironolactone in an attempt to test the effectiveness of patiromer in preventing hyperkalaemia did not conform to current guidelines for the use of an MRA due to an eGFR <30 mL/min/1.73 m2. The inclusion of only six patients with an eGFR <30 mL/min/1.73 m2 in the present study was likely related to the current guidelines, which recommend initiating spironolactone in HF patients with eGFR >30 mL/min/1.73 m2.4, 6 Nonetheless, results of this study provide useful information for the development of future trials to evaluate patiromer in a broad group of patients who might be at risk for the development of hyperkalaemia if administered an ACEi or ARB and an MRA. Many of these patients, while at significantly increased risk for cardiovascular death and hospitalization for HF, are currently not treated with an MRA due to the fear of inducing hyperkalaemia.
Conclusions
Patiromer initiated at a dose of 16.8 g/day with subsequent individualized dose titrations (needed by two‐thirds of patients) maintained serum K+ within the target range in the majority of patients with HF and CKD in the present open‐label study, all of whom were uptitrated to spironolactone 50 mg/day, with a relatively low incidence of hyperkalaemia. This strategy was accomplished with a low incidence of hypokalaemia, hypomagnesaemia, and renal dysfunction. With a dose‐titration strategy, patiromer appears to be effective and safe, and our findings should allow development of further prospective, randomized controlled trials to evaluate this strategy in high‐risk patients such as those with HFrEF and eGFR <30 mL/min/1.73 m2.
Conflict of interest
Dr Bertram Pitt reports personal fees for consulting with Sanofi, Relypsa, Merck, Bayer, AstraZeneca, Boehringer Ingelheim, Forest Laboratories, scPharmaceuticals, PharmaIN, Tricida, DaVinci Therapeutics, KBP Biosciences, Stealth Peptides, and AuraSense. He has stock options with scPharmaceuticals, PharmaIN, DaVinci Therapeutics, Tricida, KBP Biosciences, and AuraSense. He serves on a data safety monitoring committee for and receives personal fees from Johnson & Johnson, Novartis, and Tenax Pharmaceuticals. He serves on a clinical events committee and receives personal fees from Juventas Therapeutics. In addition, he has a pending patent EFS ID 14916043, application number 61762661/UM‐33001/US‐1PRO for the site‐specific delivery of eplerenone to the myocardium.
Dr Bushinsky reports personal fees from Relypsa, Amgen, Sanofi/Genzyme, OPKO Health, Tricida, and Fresenius Medical Care. He also has stock options in Tricida. Dr Bushinsky reports research support from the National Institutes of Health and from the Renal Research Institute outside of the submitted work.
Dr Kitzman reports personal fees and other from Relypsa. He also reports personal fees from AbbVie, Merck, AstraZeneca, and Bayer, as well as grants from Bayer, AstraZeneca, and Novartis.
Dr Ruschitzka reports personal fees from SJM, Servier, Zoll, AstraZeneca, Sanofi, Cardiorentis, Novartis, Amgen, BMS, Pfizer, Fresenius, Vifor, Roche, Bayer, and Abbott and grants from SJM and Novartis. He also reports serving on the steering committee meetings of clinical trials for Cardiorentis, Fresenius, and Vifor and participating at advisory board meetings for AstraZeneca, Sanofi, Amgen, BMS, Pfizer, and Roche.
Dr Metra reports consulting honoraria from Amgen, Novartis, Relypsa, and Servier.
Dr Filippatos reports committee fees from Novartis, Bayer, Servier, and Vifor.
Dr Rossignol reports personal fees (consulting) from Bayer, Novartis, Relypsa, AstraZeneca, Stealth Peptides, Fresenius, Grünenthal, Vifor Fresenius Medical Care Renal Pharma, and CTMA; lecture fees from CVRx; being a cofounder of CardioRenal.
Drs Du Mond, Garza, and Berman report employment with Relypsa, Inc., a Vifor Pharma Group Company. Dr Berman also has a patent (WO 2014/058905) pending.
Dr Lainscak reports personal fees from Relypsa and other from Relypsa and personal fees from Novartis, Relypsa, and Pfizer outside the submitted work.
Funding
This study was funded by Relypsa, Inc., a Vifor Pharma Group Company.
Supporting information
Figure S1. Patient disposition.
Table S1. Patiromer and spironolactone dosing algorithm.
Table S2. Percentage of patients on each dose level of patiromer as last prescribed.
Table S3. Summary of non‐serious and serious adverse events related to worsening renal function in eight patients.
Table S4. Narratives of deaths.
Table S5. Investigator list.
Acknowledgements
Editorial assistance was provided by Narender Dhingra, MBBS, PhD, of AlphaBioCom, LLC, and funded by Relypsa, Inc., a Vifor Pharma Group Company.
Pitt, B. , Bushinsky, D. A. , Kitzman, D. W. , Ruschitzka, F. , Metra, M. , Filippatos, G. , Rossignol, P. , Du Mond, C. , Garza, D. , Berman, L. , Lainscak, M. , and on behalf of the Patiromer‐204 Investigators (2018) Evaluation of an individualized dose titration regimen of patiromer to prevent hyperkalaemia in patients with heart failure and chronic kidney disease. ESC Heart Failure, 5: 257–266. doi: 10.1002/ehf2.12265.
Clinical trial registration: http://ClinicalTrials.gov registry identifier NCT01130597
References
- 1. Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341: 709–717. [DOI] [PubMed] [Google Scholar]
- 2. Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, Bittman R, Hurley S, Kleinman J, Gatlin M, Eplerenone Post‐Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators . Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348: 1309–1321. [DOI] [PubMed] [Google Scholar]
- 3. Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B, EMPHASIS‐HF Study Group . Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364: 11–21.21073363 [Google Scholar]
- 4. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, Drazner MH, Filippatos GS, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PN, Stevenson LW, Westlake C. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol 2017; 70: 776–803. [DOI] [PubMed] [Google Scholar]
- 5. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, Drazner MH, Filippatos G, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PE, Stevenson LW, Westlake C. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol 2016; 27: 1476–1488. [DOI] [PubMed] [Google Scholar]
- 6. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, Gonzalez‐Juanaty JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GP, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P, Authors/Task Force Members; Document Reviewers . ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016; 18: 891–975. [DOI] [PubMed] [Google Scholar]
- 7. Einhorn LM, Zhan M, Hsu VD, Walker LD, Moen MF, Seliger SL, Weir MR, Fink JC. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med 2009; 169: 1156–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jain N, Kotla S, Little BB, Weideman RA, Brilakis ES, Reilly RF, Banerjee S. Predictors of hyperkalemia and death in patients with cardiac and renal disease. Am J Cardiol 2012; 109: 1510–1513. [DOI] [PubMed] [Google Scholar]
- 9. Lainscak M, Cleland JG, Lenzen MJ, Follath F, Komajda M, Swedberg K. International variations in the treatment and co‐morbidity of left ventricular systolic dysfunction: data from the EuroHeart Failure Survey. Eur J Heart Fail 2007; 9: 292–299. [DOI] [PubMed] [Google Scholar]
- 10. Veltassa (patiromer) for oral suspension [package insert]. Relypsa, Inc. ; 2016.
- 11. Li L, Harrison SD, Cope MJ, Park C, Lee L, Salaymeh F, Madsen D, Benton WW, Berman L, Buysse J. Mechanism of action and pharmacology of patiromer, a nonabsorbed cross‐linked polymer that lowers serum potassium concentration in patients with hyperkalemia. J Cardiovasc Pharmacol Ther 2016; 21: 456–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. European Medicines Agency . Veltassa. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/004180/human_med_002141.jsp&mid=WC0b01ac058001d124 (9 January 2018).
- 13. Bakris GL, Pitt B, Weir MR, Freeman MW, Mayo MR, Garza D, Stasiv Y, Zawadski R, Beerman L, Bushinsky DA, AMETHYST‐DA Investigators . Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST‐DN randomized clinical trial. JAMA 2015; 314: 151–161. [DOI] [PubMed] [Google Scholar]
- 14. Weir MR, Bakris GL, Bushinsky DA, Mayo MR, Garza D, Stasiv Y, Wittes J, Christ‐Schmidt H, Berman L, Pitt B, OPAL‐HK Investigators . Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015; 372: 211–221. [DOI] [PubMed] [Google Scholar]
- 15. Pitt B, Bakris GL, Bushinsky DA, Garza D, Mayo MR, Stasiv Y, Christ‐Shmidt H, Berman L, Weir MR. Effect of patiromer on reducing serum potassium and preventing recurrent hyperkalaemia in patients with heart failure and chronic kidney disease on RAAS inhibitors. Eur J Heart Fail 2015; 17: 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pitt B, Anker SD, Bushinsky DA, Kitzman DW, Zannad F, Huang IZ, PEARL‐HF Investigators . Evaluation of the efficacy and safety of RLY5016, a polymeric potassium binder, in a double‐blind, placebo‐controlled study in patients with chronic heart failure (the PEARL‐HF) trial. Eur Heart J 2011; 32: 820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med 1999; 130: 461–470. [DOI] [PubMed] [Google Scholar]
- 18. Bushinsky DA, Spiegel DM, Gross C, Benton WW, Fogli J, Hill Gallant KM, Du Mond C, Block GA, Weir MR, Pitt B. Effect of patiromer on urinary ion excretion in healthy adults. Clin J Am Soc Nephrol 2016; 11: 1769–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Epstein M, Pitt B. Recent advances in pharmacological treatments of hyperkalemia: focus on patiromer. Expert Opin Pharmacother 2016; 17: 1435–1448. [DOI] [PubMed] [Google Scholar]
- 20. Weir MR, Bakris GL, Gross C, Mayo MR, Garza D, Stasiv Y, Yuan J, Berman L, Williams GH. Treatment with patiromer decreases aldosterone in patients with chronic kidney disease and hyperkalemia on renin–angiotensin system inhibitors. Kidney Int 2016; 90: 696–704. [DOI] [PubMed] [Google Scholar]
- 21. Weir MR, Mayo MR, Garza D, Arthur SA, Berman L, Bushinsky D, Wilson DJ, Epstein M. Effectiveness of patiromer in the treatment of hyperkalemia in chronic kidney disease patients with hypertension on diuretics. J Hypertens 2017; 35: S57–S63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zannad F, Alla F, Dousset B, Perez A, Pitt B, RALES Investigators . Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Circulation 2000; 102: 2700–2706. [DOI] [PubMed] [Google Scholar]
- 23. RALES Investigators . Effectiveness of spironolactone added to an angiotensin‐converting enzyme inhibitor and a loop diuretic for severe chronic congestive heart failure (the Randomized Aldactone Evaluation Study [RALES]). Am J Cardiol 1996; 78: 902–907. [DOI] [PubMed] [Google Scholar]
- 24. Hensen J, Abraham WT, Durr JA, Schrier RW. Aldosterone in congestive heart failure: analysis of determinants and role in sodium retention. Am J Nephrol 1991; 11: 441–446. [DOI] [PubMed] [Google Scholar]
- 25. Perazella MA. Drug‐induced hyperkalemia: old culprits and new offenders. Am J Med 2000; 109: 307–314. [DOI] [PubMed] [Google Scholar]
- 26. Palmer BF. Managing hyperkalemia caused by inhibitors of the renin–angiotensin–aldosterone system. N Engl J Med 2004; 351: 585–592. [DOI] [PubMed] [Google Scholar]
- 27. Maggioni AP, Anker SD, Dahlstrom U, Filippatos G, Ponikowski P, Zannad F, Amir O, Chioncel O, Leiro MC, Drozdz J, Erglis A, Fazlibegovic E, Fonseca C, Fruhwald F, Gatzov P, Goncalvesova E, Hassanein M, Hradec J, Kavoliunine A, Lainscak M, Logeart D, Merkley B, Metra M, Persson H, Seferovic P, Temizhan A, Tousoulis D, Tavazzi L, Heart Failure Association of the ESC . Are hospitalized or ambulatory patients with heart failure treated in accordance with European Society of Cardiology guidelines? Evidence from 12440 patients of the ESC Heart Failure Long‐Term Registry. Eur J Heart Failure 2013; 15: 1173–1184. [DOI] [PubMed] [Google Scholar]
- 28. Ouwerkerk W, Voors AA, Anker SD, Cleland JG, Dickstein K, Filippatos G, van der Harst P, Hillege HL, Lang CC, Ter Maaten JM, Ng LL, Ponikowski P, Samani NJ, van Veldhuisen DJ, Zannad F, Metra M, Zinderman AH. Determinants and clinical outcome of uptitration of ACE‐inhibitors and beta‐blockers in patients with heart failure: a prospective European study. Eur Heart J 2017; 38: 1883–1890. [DOI] [PubMed] [Google Scholar]
- 29. Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, Causell N, Desai AS, Diaz R, Fleg JL, Gordeev I, Harty B, Heitner JF, Kenwood CT, Lewis EF, O'Meara E, Probstfield JL, Shaburishvili T, Shah SJ, Solomon SD, Sweitzer NK, Yang S, McKinlay SM, TOPCAT Investigators . Spironolactone for heart failure with preserved ejection fraction. N Engl J Med 2014; 370: 1383–1392. [DOI] [PubMed] [Google Scholar]
- 30. Pitt B, Gheorghiade M. Geographic variation in heart failure trials: time for skepticism? Eur J Heart Fail 2014; 16: 601–602. [DOI] [PubMed] [Google Scholar]
- 31. Pfeffer MA, Claggett B, Assmann S, Boineua R, Anand IS, Clausell N, Desai AS, Diaz R, Fleg JL, Gordeev I, Heitner JF, Lewis EF, O'Meara E, Rouleau JL, Probstfield JL, Shaburishvili T, Sha SJ, Solomon SD, Sweitzer NK, McKinlay SM, Pitt B. Regional variation in patients and outcomes in the Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial. Circulation 2015; 131: 34–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Patient disposition.
Table S1. Patiromer and spironolactone dosing algorithm.
Table S2. Percentage of patients on each dose level of patiromer as last prescribed.
Table S3. Summary of non‐serious and serious adverse events related to worsening renal function in eight patients.
Table S4. Narratives of deaths.
Table S5. Investigator list.