

Abstract
Mitochondria are bioenergetic, biosynthetic, and signaling organelles that control various aspects of cellular and organism homeostasis. Quality control mechanisms are in place to ensure maximal mitochondrial function and metabolic homeostasis at the cellular level. Dysregulation of these pathways is a common theme in human disease. In this mini-review, we discuss how alterations of the mitochondrial network influences mitochondrial function, focusing on the molecular regulators of mitochondrial dynamics (organelle’s shape and localization). We highlight similarities and critical differences in the mitochondrial network of cancer and type 2 diabetes, which may be relevant for treatment of these diseases.
Keywords: mitochondria, fission, fusion, diabetes, cancer
Introduction
All living organisms rely on cellular and physiological mechanisms of homeostasis in order to maintain an internal environment optimal for life and function. Mitochondria are the foundation of cellular homeostasis, via their multiple roles in energy production, biosynthesis, calcium regulation and signaling, redox balance, and generation of reactive oxygen species. Not surprisingly, cells have evolved multiple mechanisms of quality control to ensure that mitochondria function at their best. These include protein import (1), folding and degradation (2), antioxidant defense mechanisms (3), mitochondrial turnover via autophagy (4), mitochondrial biogenesis (5), mitochondrial shape changes and cristae remodeling (6), and communication with the nucleus to coordinate transcriptional responses (7).
Emerging evidence indicate that mitochondrial dysfunction is associated with disparate diseases, including aging (8), neurodegenerative diseases (9), mitochondrial diseases (10), obesity (11), diabetes, and cancer. Although some controversies remain regarding whether functional or dysfunctional mitochondria are responsible for metabolic disorders, there is a resurgence of interest in understanding the mechanisms responsible for such mitochondrial alterations in disease. This review focuses on the molecular regulators of mitochondrial dynamics (organelle’s shape and localization) in cancer and metabolic pathologies.
Regulation of Mitochondrial Dynamics
Mitochondria constantly undergo shape and number changes thanks to the two opposing processes of fission and fusion (12). In turn, changes in gross mitochondrial morphology and the interconnectivity of the mitochondrial network impact on energy production (13), calcium signaling, mitochondrial DNA distribution, apoptosis, mitophagy, and segregation of mitochondria between daughter cells (6). The fine-tuning of the fusion–fission balance is crucial for cellular fitness in response to extracellular stimuli and environmental stress (14). Thus, alterations of the fission–fusion balance lead to oxidative stress, mitochondrial dysfunction, and metabolic alterations.
At the molecular level, dynamin-like GTPases orchestrate mitochondria shape changes. The fission protein dynamin-related protein 1 (DRP1) assembles into ring-like structures to constrict mitochondrial membranes in a GTP-dependent manner (6). DRP1 is recruited to mitochondria by fission protein 1 (FIS1), mitochondrial fission factor (MFF), and the mitochondrial dynamic proteins of 49 (MiD49) and 51 kDa (MiD51). On the other hand, the fusogenic proteins mitofusin 1 and 2 (MFN1/2) are located in the outer mitochondrial membrane, and tether two mitochondria through homo- and hetero-typic dimerization (13). A single GTPase, optic atrophy protein 1 (OPA1), achieves fusion of the IMM.
An expanding number of degenerative disorders are associated with mutations in the genes encoding MFN2 and OPA1, including Charcot–Marie–Tooth disease type 2A and autosomal dominant optic atrophy (15). Defective mitochondrial dynamics seem to play a more general role in the molecular and cellular pathogenesis of common neurodegenerative diseases (Alzheimer’s and Parkinson’s) (14), as well as in cardiovascular disease (16), type 2 diabetes (T2D), and cancer.
Mitochondrial Dynamics in T2D
The clinical complications of T2D include dyslipidemia, hyperglycemia (17), insulin resistance, and defects in insulin secretion from pancreatic beta cells (18). A major cause of such clinical complications is the increased production of mitochondrial ROS by hyperglycemia (17, 19). A common feature of mitochondrial morphology in T2D is an increased fragmentation (Figure 1), achieved via activation/upregulation of DRP1 and/or downregulation of MFN2 levels. In turn, increased fission and fragmentation of mitochondria was linked to HG-induced overproduction of ROS (20) and insulin secretion in mouse and human islets (21). Importantly, both HG-induced ROS and insulin secretion were blocked by inhibiting DRP1-induced fission. Furthermore, impaired mitochondrial fusion has been associated with insulin resistance in skeletal muscle (22) and with glucose intolerance and enhanced hepatic gluconeogenesis in a liver-specific MFN2 knockout (KO) mice (23). Interestingly, MFN2 KO led to increased ROS production, activation of JNK and endoplasmic reticulum (ER) stress response. Studies in rat models show that MFN2 overexpression improved insulin sensitivity and reduced lipid intermediates in muscle (24) and liver (25). At the molecular level, liver expression of MFN2 was associated with increased expression of the insulin receptor and the glucose transporter GLUT2, and activation of the PI3K/AKT2 pathway.
Figure 1.
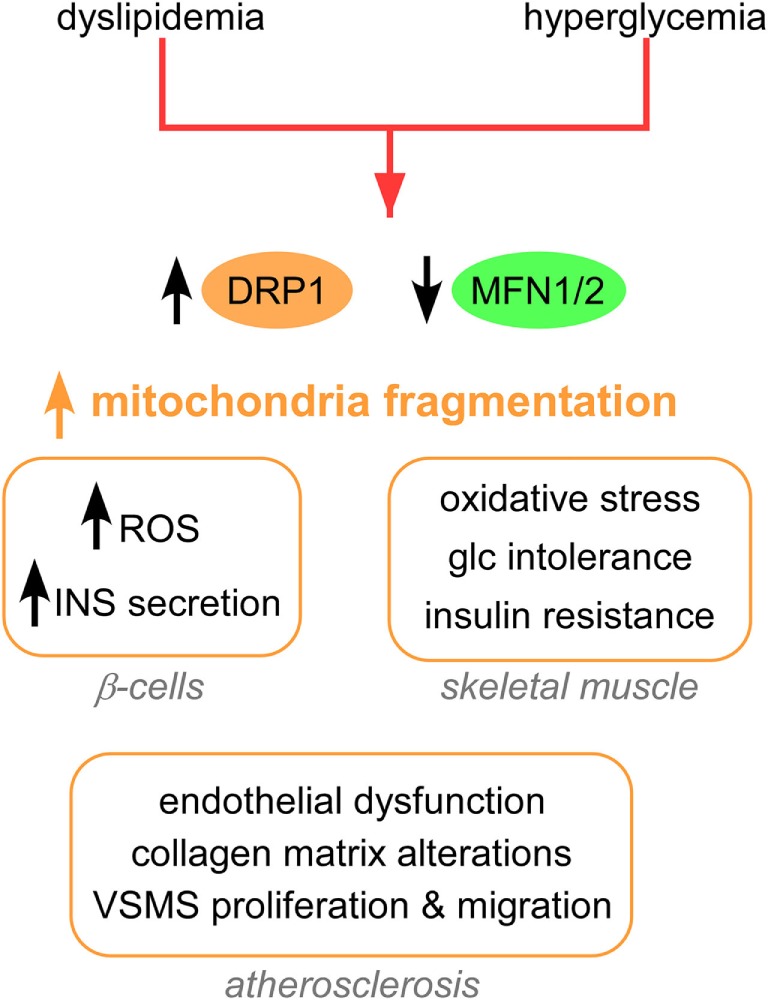
Mitochondrial shape alterations in T2D. Mitochondrial fragmentation and impaired mitochondrial trafficking are a hallmark of T2D. These changes in mitochondrial dynamics lead to pathological responses in β-cells, skeletal muscle, adipocytes, and vessels. Abbreviations: INS, insulin; Glc, glucose; T2D, type 2 diabetes.
In addition, dyslipidemia models of T2D show increased mitochondrial fission (Figure 1). Excess palmitate (PA)-induced mitochondrial fragmentation and increased mitochondrion-associated DRP1 and FIS1 in differentiated muscle cells (26). In addition, PA induced mitochondrial depolarization, lower ATP synthesis and increased oxidative stress, and reduced insulin-stimulated glucose uptake (Figure 1). Both genetic and pharmacological inhibition of DRP1 attenuated PA-induced mitochondrial fragmentation and insulin resistance. In another study, DRP1 was induced in rat islets after stimulation by free fatty acids (FFAs), and this DRP-1 upregulation was accompanied by increased pancreatic β cell apoptosis (27).
Mitochondrial fission is associated with various processes that contribute to atherosclerosis in T2D (Figure 1), including endothelial dysfunction (28), collagen matrix alteration (29), and motility and proliferation of vascular smooth muscle cells (30). From a therapeutic standpoint, silencing FIS1 or DRP1 in venous endothelial cells isolated from patients with T2D blunted HG-induced mitochondrial fission and ROS production (28). Furthermore, metformin attenuated the development of atherosclerosis in diabetic mice by reducing DRP1-mediated mitochondrial fission in an AMP-activated protein kinase (AMPK)-dependent manner (31). Mitochondrial fission induced by DRP1 also plays a critical role in the pathogenesis of microvascular [nephropathy (32), retinopathy (33), and neuropathy] and macrovascular [stroke and myocardial ischemia (34)] complications of diabetes.
In summary, we know that many of the clinical complications of T2D are associated with mitochondrial fragmentation. We also know that tipping the balance toward increased mitochondrial fragmentation in mice leads to models of T2D. Furthermore, blocking DRP1 (or increasing MFNs) ameliorated hyperglycemia, dyslipidemia, and atherosclerosis in T2D models. Less clear are the mechanisms of alterations in expression and/or activity of DRP1/MFNs. Up to date, most of the studies have shown correlation between the hallmarks of T2D and increased fragmentation of mitochondria (Table 1). However, more studies should focus on understanding the spatiotemporal regulation of DRP1 and MFN1/2 levels during the natural progression of T2D. In this context, there are a number of open questions. For example, are there alterations on the regulation of DRP1/MFNs at the transcriptional, translational, or posttranslational level? Are DRP1/MFNs regulated by insulin, glucose, FFA signaling pathways? What are the tissue- and cell-specific differences in the regulation of mitochondrial shape in T2D? Identifying such molecular pathways controlling DRP1/MFN alterations in T2D might enable therapeutic efforts in prediabetic patients to prevent full-blown settlement of the disease.
Table 1.
Mitochondrial dynamics in T2D and cancer.
| Disease | Regulatory event | Molecular pathway | Cell function | Reference |
|---|---|---|---|---|
| T2D | DRP1 enrichment in calcified human carotid arteries | DRP1 controls matrix mineralization, cytoskeletal rearrangement, mitochondrial dysfunction, and reduced type 1 collagen secretion and alkaline phosphatase activity | Extracellular matrix changes in cardiovascular complications | (29) |
| FFA | DRP1 leads to cytC release, caspase-3 activation, and generation of ROS | Apoptosis | (27) | |
| Hyperglycemia | ROCK1 phosphorylates DRP1 | Nepropathy | (32) | |
| PA | Fragmentation was associated with increased oxidative stress, mitochondrial depolarization, loss of ATP production, and reduced insulin-stimulated glucose uptake | Insulin stimulated glucose uptake in skeletal muscle | (26) | |
| FIS1 and DRP1 increased in T2D patients | DRP1 induced ROS, and nitric oxide synthase activation | Endothelial dysfunction | (28) | |
| Hyperglycemia | HG leads to DRP1-mediated fragmentation and ROS | Cellular respiration | (20) | |
| Inflammatory signaling (TNF-α) | TNF-α induced MiR-106b which led to MFN2 downregulation | Insulin resistance | (23) | |
| Insulin | Unknown | Unknown | (30) | |
| Dyslipidemia | MFN2 prevents accumulation of lipid intermediates, including diacylglycerol and ceramides | Insulin resistance in skeletal muscle | (24) | |
| Dyslipidemia | MFN2 promotes the insulin signaling pathway (INSR/IRS2/GLUT2PI3K/AKT) | Insulin resistance in liver | (25) | |
| Hyperglycemia | MFN2 deficiency impaired insulin signaling in muscle and liver, induced ER stress, ROS production, and JNK activation | Insulin and glucose homeostasis | (23) | |
| Cancer | Oncogenic MAPK signaling | RasG12V or BRAFV600E activate ERK1/2, which then phosphorylates and activates DRP1 | Mitochondria function and cell survival | (56) |
| mTOR | mTORC1/4E-BP-dependent translation of MTFP1 leads to activation and recruitment of DRP1 to mitochondria | Cell survival | (58) | |
| Nestin | Nestin binds DRP1 and enhances DRP1 recruitment | Proliferation and invasion | (59) | |
| EHD1 | EHD1 and Rabankyrin-5 interact with the retromer complex and induce VPS35-mediated removal of inactive DRP1 from mitochondrial membranes | Unknown | (60) | |
| AMPK | AMPK phosphorylates MFF, which increases DRP1 recruitment to mitochondria | Unknown | (61) | |
| SPOP loss-of-function mutants | SPOP mutations allow localization of INF2 to mitochondria, where it recruits DRP1 | Cell migration and invasion | (62) | |
| SIRT4 | SIRT4 inhibited Drp1 phosphorylation and weakened Drp1 recruitment to the mitochondrial membrane via an interaction with FIS1 | Cell migration and invasion | (63) | |
| Estradiol | Estradiol stimulates mitochondria fission by decreasing MFN1/2 levels | Cell migration and proliferation | (66) | |
| Androgen | Androgens increase DRP1 expression via the AR | Cell proliferation | (65) | |
The upstream regulators of mitochondrial shape are presented along with the molecular mechanisms at play.
SPOP, speckle-type POZ protein; FFA, free fatty acid; FIS1, fission protein 1; DRP1, dynamin-related protein 1; AMPK, AMP-activated protein kinase; MFF, mitochondrial fission factor; T2D, type 2 diabetes; ER, endoplasmic reticulum; MFN1/2, fusogenic proteins mitofusin 1 and 2; AR, androgen receptor.
Another question that warrants further investigation is whether genetic susceptibility variants of DRP1 or MFNs are associated with T2D. A recent study in type 1 diabetes patients identified genetic factors associated with kidney disease (35). We propose that a similar approach in T2D patients could address to what extent genomic alterations of the mitochondrial shape genes are associated with disease. A potential association between genomic alterations of mitochondrial shaping genes and T2D might allow for better screening of susceptibility and/or risk prediction of certain T2D complications.
Mitochondrial Dynamics in Cancer
Recent evidence indicates that mitochondrial shape, size, and localization regulate several of the hallmarks of cancer. For instance, mitochondrial shape dynamics have been linked to metabolic adaptation, cell cycle progression (36), necroptosis (19), apoptosis (37–39), autophagy (40), tumor growth, tumor cell motility (41, 42), invasiveness, and metastasis (43). The role of mitochondrial shape changes as regulators of cancer biology is reviewed in Ref. (44). Here, we will discuss recent insights into how mitochondrial dynamics are regulated in cancer.
When considering the common alterations in mitochondria shape, we find a dichotomy between tumors with enhanced mitochondrial fragmentation versus tumors with enhanced mitochondrial fusion. For instance, hepatocellular carcinoma (45), osteosarcoma (46), medulloblastoma (47), thyroid (42), colorectal (48), endometrial (49), and breast cancer (43) show increased mitochondrial fragmentation, due to upregulation of DRP1 levels and a concomitant reduction in MFN1/2 levels. On the other hand, tumors of the prostate (50), neuroblastoma (51), leukemia (52), glioblastoma (53), and lung (54) are associated with downregulation of DRP1 and increased MFN1/2 levels. What could be driving these contrasting preferences of fission versus fusion of the mitochondrial networks in cancer? Plausible explanations could lie on the genomic landscape, hormonal/growth factor context, tumor microenvironmental conditions, and therapy responses of the tumors in question.
Oncogenic and tumor suppressor signaling converge on mitochondria to reprogram cellular metabolism (55); thus, the particular genomic events driving a tumor might favor mitochondrial shape changes to meet the metabolic demands of the tumor cells. According to this hypothesis, oncogene-induced metabolic reprogramming should induce changes in mitochondrial shape. Indeed, recent studies show that oncogenic RasG12V, BRAFV600E and MAPK/ERK (56, 57), mTOR (58) Nestin (59), and the endocytic protein EDH1 (60) increase DRP1-mediated mitochondrial fission. Similarly, the energy-sensing AMPK increased recruitment of DRP1 to mitochondria via phosphorylation of the MFF and (61). Speckle-type POZ protein loss-of-function mutations commonly found in primary prostate cancer were associated with increased DRP1 activation, mitochondrial fission, and prostate cancer cell invasion (62). Recently, loss of expression of the sirtuin SIRT4 was shown to lead to increased mitochondrial fragmentation (63). The signaling events that lead to DRP1 activation downstream of genomic and epigenetic alterations are summarized in Table 1.
In addition to the increasing number of oncogenes and tumor suppressors, growth factors and hormones regulate mitochondrial shape. Examples include Sonic Hedgehog (47), non-canonical Wnt ligands, pro-inflammatory cytokines, transforming growth factor-β, estradiol (64), and androgens (65). Estradiol promotes mitochondrial fragmentation through a reduction of MFN2 with parallel increase of FIS1 levels in ER+ breast cancer (66). From a translational standpoint, overexpression of MFN2 prevented estradiol-induced cell proliferation and motility (66). On the other hand, DRP1 is a transcriptional target of the androgen receptor, and androgen-stimulated DRP1 expression sensitizes prostate cancer cells to therapy-induced apoptosis (65). The possibility that other hormone-related malignancies exploit similar mechanisms of mitochondrial shape awaits further confirmation.
Tumor microenvironmental conditions exert yet another layer of regulation of mitochondrial shape. For instance, mitochondrial elongation is induced by nutrient deprivation in cancer cells (67). A hypoxic environment enhances mitochondrial fission in breast cancer (68) and glioblastoma (69). In this context, DRP1 was essential for hypoxia-stimulated cell motility. Indeed, silencing or expression of a dominant-negative mutant of DRP1 inhibited hypoxia-induced migration in both tumor cell models.
Finally, cancer cells also remodel their mitochondrial network in response to therapy. For instance, DRP1-mediated mitochondrial fragmentation is associated with cisplatin (68, 70), cytarabine and methotrexate (71), and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) (70) treatment among others. However, other therapeutic agents such as histone deacetylase inhibitors (72) produce the opposite effect, namely increased elongation of mitochondria. These opposite effects of therapy upon mitochondria morphology can be reconciled when considering the divergent signaling pathways elicited by the drugs. In the case of HDAC inhibitors, a decreased expression of FIS1 impaired DRP1 recruitment to mitochondria. These effects were independent of apoptosis induction. On the other hand, increased mitochondrial fragmentation on cisplatin and TRAIL-treated cells is coupled to apoptosis. Also worth considering, HDAC inhibitors could have additional roles in regulating mitochondrial morphology, due to non-histone-acetylating activity (acetylation of non-histone proteins, regulation of signaling kinases). A final consideration is the influence of the genomic background and tumor microenvironment on eliciting fission versus fusion upon therapy.
In summary, emerging evidence suggests that the contribution of the mitochondrial shaping genes to tumor cell biology is tumor type dependent and may reflect the genetic makeup, hormonal/growth factor context, tumor microenvironment conditions, and therapy responses of the tumor. Future efforts should aim to integrate these novel regulatory pathways and reach a comprehensive picture of the regulation of mitochondrial shape and function in cancer. Second, more emphasis should be directed toward identifying metabolic-dependent versus -independent functions of DRP1 and MFNs in cancer. For instance, which of the phenotypes associated with DRP1 activation in cancer are explained on basis of metabolism (increased glycolysis versus respiration)? Is it DRP1’s function on apoptosis (or mitochondrial localization) also important? A third area of interest for future research would be the development of anti-cancer therapies targeting mitochondrial dynamics. Encouraging fresh evidence indicates that modulating mitochondria morphology enhances anti-cancer therapies (73), particularly death receptor ligands (74–76) and antimitotic drugs (77).
Targeting Mitochondrial Dynamics
The involvement of DRP1-mediated fission in disparate diseases settings has fueled the development of pharmacological approaches to inhibit mitochondrial fission. Mitochondrial division inhibitor-1 (mdivi-1) selectively impairs the GTPase activity of DRP1, without affecting the activity of dynamin-1, MFN1/2, or OPA1 (78). The mechanism of action of mdivi-1 involves allosteric binding and stabilization of a conformational form of unassembled DRP1 that cannot polymerize. mdivi-1 treatment induces rapid mitochondrial fusion, dampens ROS production and increases ATP production. Interestingly, the original report described a second function of DRP1 in mitochondrial outer membrane polarization (MOMP). DRP1 facilitated BAX/BAK-dependent MOMP in response to C8-BID or staurosporine, independently of mitochondrial fragmentation. Thus, mdivi-1 impaired staurosporine-induced apoptosis (78). Interestingly, mdivi-1 can induce apoptosis in DRP1-KO cells (79), suggesting that mdivi-1 has off-target effects. In contrast to these initial studies in which mdivi-1 prevented apoptosis, later studies showed that mdivi-1 sensitized cells to TRAIL-dependent apoptosis (74). This potentiation of apoptosis by mdivi-1 occurred through activation of mitochondrial and ER apoptosis pathways. Thus, these controversial results suggest that mdivi-1 can act either as pro- or anti-apoptotic pharmacologic agent, depending on the cell types and apoptotic stimuli in question (80).
In T2D models, mdivi-1 prevented mitochondrial fragmentation, oxidative stress and inflammation, and improved endothelial cell function (31). Another study showed that mdivi-1 prevented HG-stimulated insulin secretion in mouse and human islets (21). Furthermore, mdivi-1 rescued palmitate-induced mitochondrial dysfunction and ROS generation, as well as insulin resistance in skeletal muscle (26). Inhibition of Drp1 with mdivi-1 improved mitochondrial function and cardiac function in a model of myocardial ischemia/reperfusion of diabetic hearts (34).
In cancer cells, DRP1 inhibition has been shown to modulate therapy sensitivity, tumor metabolism, growth, and invasiveness. For instance, mdivi-1 suppressed mitochondrial autophagy, metabolic reprogramming, cancer cell viability, and motility of breast cancer cells (81). In regards to therapy modulation, mdivi-1 potentiated TRAIL-induced apoptosis in melanoma (74, 76) and ovarian cancer models (75). Furthermore, mdivi-1 induced cell death (75) and synergized apoptotic effects of platinum agents in drug resistant ovarian tumor cells (79). However, mdivi-1 prevents apoptosis induced by cisplatin in breast cancer (68) and leukemia (52). As discussed above, these controversial results suggest that mdivi-1 can act either as pro- or anti-apoptotic agent, depending on the cell types and apoptotic stimuli in question [reviewed in Ref. (80)]. Further investigations should address the precise mechanisms dictating the differential effects of mdivi-1 on cell survival.
Regarding the potential utility of mdivi-1 in the clinic, a number of questions remain open. For instance, what are the consequences of sustained in vivo inhibition of mitochondrial fission? What are the pharmacokinetics and cytotoxicity profiles for mdivi-1? Another point to consider is that mdivi-1 has poor solubility in water (80). This fact might limit the utility of mdivi-1 and might open the door for the design of new DRP1 inhibitors with improved solubility, specificity, and potency. In this regard, another pharmacological agent targets the recruitment of DRP1 to mitochondria via its interaction with FIS1. The small peptide inhibitor P110 blocks DRP1/FIS1 binding (82) and has shown promising results in neurodegenerative disease models. When tested in hepatocellular carcinoma, P110 blocked cell proliferation in vitro and in vivo (83). Future research will be needed to evaluate the utility of P100 both in T2D and cancer models.
Conclusion
Given the metabolic alterations that are a hallmark of both T2D and cancer, it is not surprising that mitochondrial alterations are a shared feature in these disparate diseases. Over the past few years, we have learnt that mitochondria are not static, solitary organelles, but they rather undergo constant changes in morphology and subcellular distribution to meet the metabolic demands of the cell. Defects in mitochondrial dynamics play a role in the molecular and cellular pathogenesis of both T2D and cancer. Now, how similar or different are these two pathologies in regards to mitochondrial dynamics? In T2D, the literature unanimously reports an increase of mitochondrial fission mediated by DRP1. In cancer, most tumors follow this same pattern of increased DRP1-mediated mitochondrial fission. However, although less frequently, tumors might display augmented mitochondrial fusion via an increase of MFN1/2 levels and/or activity. How are these differences and similarities in the mitochondrial network explained at the molecular level? Up to date, most of the studies have shown correlation between T2D and altered mitochondrial shape. More studies should focus on understanding the spatiotemporal regulation of DRP1 and MFN1/2 levels and activity during the natural progression of T2D. Likewise, there is limited information on how the genetic, epigenetic, and microenvironmental factors influence mitochondrial dynamics, or which signaling pathways integrate extracellular stimuli with mitochondrial shape in T2D. Thus, due to this limited information, is not possible to conclude if T2D and cancer utilize similar or divergent mechanisms of control of mitochondrial shape. In this regard, it would be interesting to address how metabolic pathways commonly altered both in T2D and cancer impinge on mitochondrial morphology. Examples of such pathways include PI3K/AKT and AMPK. Another question that warrants further investigation is whether other aspects of mitochondrial biology are dysregulated in these diseases. For instance, are there alterations in mitochondrial quality control, mitochondria crosstalk to other organelles, or mitochondrial localization present in both T2D and cancer?
Regarding the use of DRP1 inhibitors as anti-T2D and -cancer agents, further studies should determine long-term effects of targeting mitochondrial dynamics in vivo, and establish the pharmacokinetics and cytotoxicity profiles for mdivi-1. In addition, the involvement of potential compensatory or resistance mechanisms to mdivi-1 has not been explored yet and should be addressed in the future. An area in need of further investment is the development of selective MFN1/2 inhibitors. Despite the existence of a few DRP1 inhibitors, there is no equivalent therapeutic agent to target fusion. The fact that several tumors show increased fusion might warrant further effort in this area.
Author Contributions
MW performed literature search and review; MC conceived the project, designed the figures, and wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to apologize to those colleagues whose work could not be cited or discussed in sufficient detail due to space limitation. MC is supported by ACS IRG #16-184-56 from the American Cancer Society. The content is solely the responsibility of the author and does not necessarily represent official views of the ACS. The funders had no role in decision to publish or preparation of the manuscript.
References
- 1.Wiedemann N, Pfanner N. Mitochondrial machineries for protein import and assembly. Annu Rev Biochem (2017) 86:685–714. 10.1146/annurev-biochem-060815-014352 [DOI] [PubMed] [Google Scholar]
- 2.Jensen MB, Jasper H. Mitochondrial proteostasis in the control of aging and longevity. Cell Metab (2014) 20(2):214–25. 10.1016/j.cmet.2014.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olsen LF, Issinger OG, Guerra B. The Yin and Yang of redox regulation. Redox Rep (2013) 18(6):245–52. 10.1179/1351000213Y.0000000059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anding AL, Baehrecke EH. Cleaning house: selective autophagy of organelles. Dev Cell (2017) 41(1):10–22. 10.1016/j.devcel.2017.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ploumi C, Daskalaki I, Tavernarakis N. Mitochondrial biogenesis and clearance: a balancing act. FEBS J (2017) 284(2):183–95. 10.1111/febs.13820 [DOI] [PubMed] [Google Scholar]
- 6.Pernas L, Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol (2016) 78:505–31. 10.1146/annurev-physiol-021115-105011 [DOI] [PubMed] [Google Scholar]
- 7.Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem (2007) 76:701–22. 10.1146/annurev.biochem.76.052305.091720 [DOI] [PubMed] [Google Scholar]
- 8.Theurey P, Pizzo P. The aging mitochondria. Genes (Basel) (2018) 9(1):E22. 10.3390/genes9010022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arun S, Liu L, Donmez G. Mitochondrial biology and neurological diseases. Curr Neuropharmacol (2016) 14(2):143–54. 10.2174/1570159X13666150703154541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol (2017) 241(2):236–50. 10.1002/path.4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Mello AH, Costa AB, Engel JDG, Rezin GT. Mitochondrial dysfunction in obesity. Life Sci (2018) 192:26–32. 10.1016/j.lfs.2017.11.019 [DOI] [PubMed] [Google Scholar]
- 12.Otera H, Mihara K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J Biochem (2011) 149(3):241–51. 10.1093/jb/mvr002 [DOI] [PubMed] [Google Scholar]
- 13.Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Mol Cell (2016) 61(5):683–94. 10.1016/j.molcel.2016.02.022 [DOI] [PubMed] [Google Scholar]
- 14.Ranieri M, Brajkovic S, Riboldi G, Ronchi D, Rizzo F, Bresolin N, et al. Mitochondrial fusion proteins and human diseases. Neurol Res Int (2013) 2013:293893. 10.1155/2013/293893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Winger AJ, Knowlton AA. Mitochondrial dynamic changes in health and genetic diseases. Mol Biol Rep (2014) 41(11):7053–62. 10.1007/s11033-014-3663-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ong SB, Kalkhoran SB, Hernandez-Resendiz S, Samangouei P, Ong SG, Hausenloy DJ. Mitochondrial-shaping proteins in cardiac health and disease – the long and the short of it! Cardiovasc Drugs Ther (2017) 31(1):87–107. 10.1007/s10557-016-6710-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature (2001) 414(6865):813–20. 10.1038/414813a [DOI] [PubMed] [Google Scholar]
- 18.Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in beta-cell function. Nature (2001) 414(6865):788–91. 10.1038/414788a [DOI] [PubMed] [Google Scholar]
- 19.Basit F, van Oppen LM, Schockel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis (2017) 8(3):e2716. 10.1038/cddis.2017.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A (2006) 103(8):2653–8. 10.1073/pnas.0511154103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kabra UD, Pfuhlmann K, Migliorini A, Keipert S, Lamp D, Korsgren O, et al. Direct substrate delivery into mitochondrial fission-deficient pancreatic islets rescues insulin secretion. Diabetes (2017) 66(5):1247–57. 10.2337/db16-1088 [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Yang L, Gao YF, Fan ZM, Cai XY, Liu MY, et al. MicroRNA-106b induces mitochondrial dysfunction and insulin resistance in C2C12 myotubes by targeting mitofusin-2. Mol Cell Endocrinol (2013) 381(1–2):230–40. 10.1016/j.mce.2013.08.004 [DOI] [PubMed] [Google Scholar]
- 23.Sebastian D, Hernandez-Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci U S A (2012) 109(14):5523–8. 10.1073/pnas.1108220109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Wang C, Song G, Gan K, Kong D, Nie Q, et al. Mitofusion-2-mediated alleviation of insulin resistance in rats through reduction in lipid intermediate accumulation in skeletal muscle. J Biomed Sci (2013) 20:45. 10.1186/1423-0127-20-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gan KX, Wang C, Chen JH, Zhu CJ, Song GY. Mitofusin-2 ameliorates high-fat diet-induced insulin resistance in liver of rats. World J Gastroenterol (2013) 19(10):1572–81. 10.3748/wjg.v19.i10.1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol (2012) 32(2):309–19. 10.1128/MCB.05603-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng L, Men X, Zhang W, Wang H, Xu S, Fang Q, et al. Involvement of dynamin-related protein 1 in free fatty acid-induced INS-1-derived cell apoptosis. PLoS One (2012) 7(11):e49258. 10.1371/journal.pone.0049258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation (2011) 124(4):444–53. 10.1161/CIRCULATIONAHA.110.014506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rogers MA, Maldonado N, Hutcheson JD, Goettsch C, Goto S, Yamada I, et al. Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ Res (2017) 121(3):220–33. 10.1161/CIRCRESAHA.116.310293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abhijit S, Bhaskaran R, Narayanasamy A, Chakroborty A, Manickam N, Dixit M, et al. Hyperinsulinemia-induced vascular smooth muscle cell (VSMC) migration and proliferation is mediated by converging mechanisms of mitochondrial dysfunction and oxidative stress. Mol Cell Biochem (2013) 373(1–2):95–105. 10.1007/s11010-012-1478-5 [DOI] [PubMed] [Google Scholar]
- 31.Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z, et al. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes (2017) 66(1):193–205. 10.2337/db16-0915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab (2012) 15(2):186–200. 10.1016/j.cmet.2012.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong Q, Kowluru RA. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Invest Ophthalmol Vis Sci (2011) 52(12):8739–46. 10.1167/iovs.11-8045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding M, Dong Q, Liu Z, Liu Z, Qu Y, Li X, et al. Inhibition of dynamin-related protein 1 protects against myocardial ischemia-reperfusion injury in diabetic mice. Cardiovasc Diabetol (2017) 16(1):19. 10.1186/s12933-017-0501-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandholm N, Van Zuydam N, Ahlqvist E, Juliusdottir T, Deshmukh HA, Rayner NW, et al. The genetic landscape of renal complications in type 1 diabetes. J Am Soc Nephrol (2017) 28(2):557–74. 10.1681/ASN.2016020231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qian W, Choi S, Gibson GA, Watkins SC, Bakkenist CJ, Van Houten B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J Cell Sci (2012) 125(Pt 23):5745–57. 10.1242/jcs.109769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu W, Jing L, Wang Q, Lin CC, Chen X, Diao J, et al. Bax-PGAM5L-Drp1 complex is required for intrinsic apoptosis execution. Oncotarget (2015) 6(30):30017–34. 10.18632/oncotarget.5013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas KJ, Jacobson MR. Defects in mitochondrial fission protein dynamin-related protein 1 are linked to apoptotic resistance and autophagy in a lung cancer model. PLoS One (2012) 7(9):e45319. 10.1371/journal.pone.0045319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tailor D, Hahm ER, Kale RK, Singh SV, Singh RP. Sodium butyrate induces DRP1-mediated mitochondrial fusion and apoptosis in human colorectal cancer cells. Mitochondrion (2014) 16:55–64. 10.1016/j.mito.2013.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo X, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy (2016) 12(6):999–1014. 10.1080/15548627.2016.1166318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Desai SP, Bhatia SN, Toner M, Irimia D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J (2013) 104(9):2077–88. 10.1016/j.bpj.2013.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferreira-da-Silva A, Valacca C, Rios E, Populo H, Soares P, Sobrinho-Simoes M, et al. Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS One (2015) 10(3):e0122308. 10.1371/journal.pone.0122308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene (2013) 32(40):4814–24. 10.1038/onc.2012.494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trotta AP, Chipuk JE. Mitochondrial dynamics as regulators of cancer biology. Cell Mol Life Sci (2017) 74(11):1999–2017. 10.1007/s00018-016-2451-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun X, Cao H, Zhan L, Yin C, Wang G, Liang P, et al. Mitochondrial fission promotes cell migration by Ca(2+)/CaMKII/ERK/FAK pathway in hepatocellular carcinoma. Liver Int (2017). 10.1111/liv.13660 [DOI] [PubMed] [Google Scholar]
- 46.Huang ST, Huang CC, Huang WL, Lin TK, Liao PL, Wang PW, et al. Tanshinone IIA induces intrinsic apoptosis in osteosarcoma cells both in vivo and in vitro associated with mitochondrial dysfunction. Sci Rep (2017) 7:40382. 10.1038/srep40382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malhotra A, Dey A, Prasad N, Kenney AM. Sonic hedgehog signaling drives mitochondrial fragmentation by suppressing mitofusins in cerebellar granule neuron precursors and medulloblastoma. Mol Cancer Res (2016) 14(1):114–24. 10.1158/1541-7786.MCR-15-0278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng X, Zhou D, Wei J, Lin J. Cell-cycle arrest at G2/M and proliferation inhibition by adenovirus-expressed mitofusin-2 gene in human colorectal cancer cell lines. Neoplasma (2013) 60(6):620–6. 10.4149/neo_2013_080 [DOI] [PubMed] [Google Scholar]
- 49.Cormio A, Musicco C, Gasparre G, Cormio G, Pesce V, Sardanelli AM, et al. Increase in proteins involved in mitochondrial fission, mitophagy, proteolysis and antioxidant response in type I endometrial cancer as an adaptive response to respiratory complex I deficiency. Biochem Biophys Res Commun (2017) 491(1):85–90. 10.1016/j.bbrc.2017.07.047 [DOI] [PubMed] [Google Scholar]
- 50.Philley JV, Kannan A, Qin W, Sauter ER, Ikebe M, Hertweck KL, et al. Complex-I alteration and enhanced mitochondrial fusion are associated with prostate cancer progression. J Cell Physiol (2016) 231(6):1364–74. 10.1002/jcp.25240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suwanjang W, Abramov AY, Charngkaew K, Govitrapong P, Chetsawang B. Melatonin prevents cytosolic calcium overload, mitochondrial damage and cell death due to toxically high doses of dexamethasone-induced oxidative stress in human neuroblastoma SH-SY5Y cells. Neurochem Int (2016) 97:34–41. 10.1016/j.neuint.2016.05.003 [DOI] [PubMed] [Google Scholar]
- 52.Han XJ, Shi SL, Wei YF, Jiang LP, Guo MY, Wu HL, et al. Involvement of mitochondrial dynamics in the antineoplastic activity of cisplatin in murine leukemia L1210 cells. Oncol Rep (2017) 38(2):985–92. 10.3892/or.2017.5765 [DOI] [PubMed] [Google Scholar]
- 53.Yin M, Lu Q, Liu X, Wang T, Liu Y, Chen L. Silencing Drp1 inhibits glioma cells proliferation and invasion by RHOA/ROCK1 pathway. Biochem Biophys Res Commun (2016) 478(2):663–8. 10.1016/j.bbrc.2016.08.003 [DOI] [PubMed] [Google Scholar]
- 54.Lou Y, Li R, Liu J, Zhang Y, Zhang X, Jin B, et al. Mitofusin-2 over-expresses and leads to dysregulation of cell cycle and cell invasion in lung adenocarcinoma. Med Oncol (2015) 32(4):132. 10.1007/s12032-015-0515-0 [DOI] [PubMed] [Google Scholar]
- 55.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 56.Serasinghe MN, Wieder SY, Renault TT, Elkholi R, Asciolla JJ, Yao JL, et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell (2015) 57(3):521–36. 10.1016/j.molcel.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell (2015) 57(3):537–51. 10.1016/j.molcel.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morita M, Prudent J, Basu K, Goyon V, Katsumura S, Hulea L, et al. mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol Cell (2017) 67(6):922–35.e5. 10.1016/j.molcel.2017.08.013 [DOI] [PubMed] [Google Scholar]
- 59.Wang J, Cai J, Huang Y, Ke Q, Wu B, Wang S, et al. Nestin regulates proliferation and invasion of gastrointestinal stromal tumor cells by altering mitochondrial dynamics. Oncogene (2016) 35(24):3139–50. 10.1038/onc.2015.370 [DOI] [PubMed] [Google Scholar]
- 60.Farmer T, Reinecke JB, Xie S, Bahl K, Naslavsky N, Caplan S. Control of mitochondrial homeostasis by endocytic regulatory proteins. J Cell Sci (2017) 130(14):2359–70. 10.1242/jcs.204537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toyama EQ, Herzig S, Courchet J, Lewis TL, Jr, Loson OC, Hellberg K, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science (2016) 351(6270):275–81. 10.1126/science.aab4138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jin X, Wang J, Gao K, Zhang P, Yao L, Tang Y, et al. Dysregulation of INF2-mediated mitochondrial fission in SPOP-mutated prostate cancer. PLoS Genet (2017) 13(4):e1006748. 10.1371/journal.pgen.1006748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu L, Dong Q, He J, Wang X, Xing J, Wang E, et al. SIRT4 inhibits malignancy progression of NSCLCs, through mitochondrial dynamics mediated by the ERK-Drp1 pathway. Oncogene (2017) 36(19):2724–36. 10.1038/onc.2016.425 [DOI] [PubMed] [Google Scholar]
- 64.Sastre-Serra J, Nadal-Serrano M, Pons DG, Roca P, Oliver J. Mitochondrial dynamics is affected by 17beta-estradiol in the MCF-7 breast cancer cell line. Effects on fusion and fission related genes. Int J Biochem Cell Biol (2012) 44(11):1901–5. 10.1016/j.biocel.2012.07.012 [DOI] [PubMed] [Google Scholar]
- 65.Choudhary V, Kaddour-Djebbar I, Lakshmikanthan V, Ghazaly T, Thangjam GS, Sreekumar A, et al. Novel role of androgens in mitochondrial fission and apoptosis. Mol Cancer Res (2011) 9(8):1067–77. 10.1158/1541-7786.MCR-10-0445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ma L, Liu Y, Geng C, Qi X, Jiang J. Estrogen receptor beta inhibits estradiol-induced proliferation and migration of MCF-7 cells through regulation of mitofusin 2. Int J Oncol (2013) 42(6):1993–2000. 10.3892/ijo.2013.1903 [DOI] [PubMed] [Google Scholar]
- 67.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A (2011) 108(25):10190–5. 10.1073/pnas.1107402108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Han XJ, Yang ZJ, Jiang LP, Wei YF, Liao MF, Qian Y, et al. Mitochondrial dynamics regulates hypoxia-induced migration and antineoplastic activity of cisplatin in breast cancer cells. Int J Oncol (2015) 46(2):691–700. 10.3892/ijo.2014.2781 [DOI] [PubMed] [Google Scholar]
- 69.Wan YY, Zhang JF, Yang ZJ, Jiang LP, Wei YF, Lai QN, et al. Involvement of Drp1 in hypoxia-induced migration of human glioblastoma U251 cells. Oncol Rep (2014) 32(2):619–26. 10.3892/or.2014.3235 [DOI] [PubMed] [Google Scholar]
- 70.Zhao C, Chen Z, Qi J, Duan S, Huang Z, Zhang C, et al. Drp1-dependent mitophagy protects against cisplatin-induced apoptosis of renal tubular epithelial cells by improving mitochondrial function. Oncotarget (2017) 8(13):20988–1000. 10.18632/oncotarget.15470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cai J, Wang J, Huang Y, Wu H, Xia T, Xiao J, et al. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell Death Dis (2016) 7(11):e2459. 10.1038/cddis.2016.370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JS, Yoon YG, Yoo SH, Jeong NY, Jeong SH, Lee SY, et al. Histone deacetylase inhibitors induce mitochondrial elongation. J Cell Physiol (2012) 227(7):2856–69. 10.1002/jcp.23027 [DOI] [PubMed] [Google Scholar]
- 73.Li G, Zhou J, Budhraja A, Hu X, Chen Y, Cheng Q, et al. Mitochondrial translocation and interaction of cofilin and Drp1 are required for erucin-induced mitochondrial fission and apoptosis. Oncotarget (2015) 6(3):1834–49. 10.18632/oncotarget.2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Akita M, Suzuki-Karasaki M, Fujiwara K, Nakagawa C, Soma M, Yoshida Y, et al. Mitochondrial division inhibitor-1 induces mitochondrial hyperfusion and sensitizes human cancer cells to TRAIL-induced apoptosis. Int J Oncol (2014) 45(5):1901–12. 10.3892/ijo.2014.2608 [DOI] [PubMed] [Google Scholar]
- 75.Wang J, Hansen K, Edwards R, Van Houten B, Qian W. Mitochondrial division inhibitor 1 (mdivi-1) enhances death receptor-mediated apoptosis in human ovarian cancer cells. Biochem Biophys Res Commun (2015) 456(1):7–12. 10.1016/j.bbrc.2014.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Suzuki-Karasaki Y, Fujiwara K, Saito K, Suzuki-Karasaki M, Ochiai T, Soma M. Distinct effects of TRAIL on the mitochondrial network in human cancer cells and normal cells: role of plasma membrane depolarization. Oncotarget (2015) 6(25):21572–88. 10.18632/oncotarget.4268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang J, Li J, Santana-Santos L, Shuda M, Sobol RW, Van Houten B, et al. A novel strategy for targeted killing of tumor cells: induction of multipolar acentrosomal mitotic spindles with a quinazolinone derivative mdivi-1. Mol Oncol (2015) 9(2):488–502. 10.1016/j.molonc.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell (2008) 14(2):193–204. 10.1016/j.devcel.2007.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Qian W, Wang J, Roginskaya V, McDermott LA, Edwards RP, Stolz DB, et al. Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget (2014) 5(12):4180–94. 10.18632/oncotarget.1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rosdah AA, Holien KJ, Delbridge LM, Dusting GJ, Lim SY. Mitochondrial fission – a drug target for cytoprotection or cytodestruction? Pharmacol Res Perspect (2016) 4(3):e00235. 10.1002/prp2.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zou P, Liu L, Zheng LD, Payne KK, Manjili MH, Idowu MO, et al. Coordinated upregulation of mitochondrial biogenesis and autophagy in breast cancer cells: the role of dynamin related protein-1 and implication for breast cancer treatment. Oxid Med Cell Longev (2016) 2016:4085727. 10.1155/2016/4085727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Qi X, Qvit N, Su YC, Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci (2013) 126(Pt 3):789–802. 10.1242/jcs.114439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhan L, Cao H, Wang G, Lyu Y, Sun X, An J, et al. Drp1-mediated mitochondrial fission promotes cell proliferation through crosstalk of p53 and NF-kappaB pathways in hepatocellular carcinoma. Oncotarget (2016) 7(40):65001–11. 10.18632/oncotarget.11339 [DOI] [PMC free article] [PubMed] [Google Scholar]