

Abstract
Low-invasive in vivo solid-phase microextraction (SPME) was used to investigate the lipid profiles of muscle tissue of living fish. Briefly, mixed mode SPME fibers were inserted into the muscle for 20 min extraction, and then the fibers were desorbed in an optimal mixture of solvents. The obtained lipid profile was then compared and contrasted to that obtained with employment of ex vivo SPME and solid-liquid extraction (SLE) from fish muscle tissue belonging to the same group of fish, following a one-year storage period. Ex vivo SPME analysis of stored muscle samples revealed 10-fold decrease in the number of detected molecular features in comparison to in vivo study. Moreover, in vivo microsampling enabled the identification of different classes of bioactive lipids, including fatty acyls, not present in the lipid profile obtained through ex vivo SPME and SLE, suggesting the alterations occurring in the unbound lipid fraction of the system under study during the storage and also indicating the advantage of the in vivo extraction approach.
Introduction
Growing interest in lipidomics over the last few years has led to the discovery and identification of hundreds of novel lipids, revealing their growing importance as it pertains to their involvement in the complex biological transformations taking place in living systems1,2. Profound analysis of lipid metabolism has proven that apart from their important roles as structural components of cell membranes and energy reservoirs, lipids are also intermediates of cellular signaling pathways and modulatory ligands for membrane proteins3–5. Knowledge concerning their significance in endogenous signaling and metabolism is continuously expanding as the roles of numerous small molecular-weight bioactive lipids are unveiled by lipidomic research, however, the mechanisms of their interaction with other cellular components and their role in tissue stability, is yet to become fully clear6,7. Hence, the availability of rapid in vivo extraction methods capable of capturing short-lived biological lipids is essential to the unveiling of the highly complex and very dynamic lipid profile8.
Currently used protocols for isolation and purification of lipids from biological matrices are tedious and time-consuming. Also, traditional sampling and separation methods used to identify trace lipids are often burdened by limitations related to the characterization of the real composition of living cells9,10. For instance, employment of certain steps involved in conventional protocols, such as invasive tissue collection (e.g. biopsy), tissue homogenization, and application of organic solvents during sample preparation, may hinder discrimination between lipids present in free form versus those previously bound to cellular components in the living system, hence introducing errors in the interpretation of the cellular lipidome11–13. Moreover, the stability of the lipid profile may also be compromised during sample storage as a result of enzymatic activity (degradation and/or aggregation processes) or chemical alterations14.
Aiming to provide adequate sample handling and fast, comprehensive extraction of small molecular weight compounds from tissue, in vivo SPME was introduced as a sample preparation method that integrates sampling, extraction, and quenching of metabolites into a single step, preserving the real metabolic profile of the analyzed matrix. In vivo SPME does not disturb the biological homeostasis of cells under study due to the solvent-free and non-exhaustive nature of SPME probes15,16. Moreover, in vivo application of SPME sampling facilitates the capture of low molecular weight and unstable metabolites present in their free form at the cellular level, thus allowing for insights into the intrinsic biochemical pathways and networks of the living system8,17,18.
In the present study, we introduced low-invasive SPME fibers with biocompatible coatings to muscle tissue of living fish (in vivo SPME) for untargeted lipidomics profiling. In order to analyze the influence of sample storage conditions on the lipidome, including potential alterations to the existing forms of lipids (bound vs. unbound), results from the in vivo study were then compared to results obtained from an ex vivo SPME study of samples collected from the same group of fish following prolonged storage conditions. Fish muscle samples were stored at −80 °C for a 1 year period, then submitted to ex vivo SPME analysis. Both sets of data, obtained from in vivo and ex vivo SPME, were then compared to results obtained by SLE, performed on homogenized fish muscle samples prepared from the same samples used for ex vivo SPME analysis. Annotation of lipids was facilitated by xMSannotator software with the use of a multistep strategy19 and was based on intensity profiles, retention time, mass defect, and isotope/adduct patterns of peaks in the data. LIPID MAPS was employed as a reference database for tentative identification of annotated molecules.
Results and Discussion
In lipidomic studies, the knowledge concerning the sample handling and storage, and their effects on the composition of lipids is limited and not consistent20,21. It has been shown that 24 months tissue storage results in compositional changes and decrease of selected lipids22, whereas other studies reported increase or decrease of the specific lipid fractions during prolonged storage23,24. However, most of the previously reported data are obtained from the experiments performed with the use of exhaustive liquid extraction techniques. The primary purpose of this study was the analysis of lipid metabolites specifically detected using in vivo SPME technique and the comparison of annotated features with the results obtained for exactly the same fish samples after one-year storage period using ex vivo SPME and SLE in order to examine the influence of storage and handling conditions on the quality of lipid composition.
Characterization of in vivo SPME extracts obtained from muscle tissue of juvenile fish revealed that among a total number of 845 detected features with molecular masses ranging from 100 to 500 m/z (Fig. 1a), 30% were annotated as lipid species belonging to 3 different lipid categories, namely fatty acyls, sterol lipids and glycerolipids. Although SPME probes enable the extraction of compounds with molecular masses up to 1000 m/z that are present in unbound form in the tissue, the majority of the unique lipids annotated with medium to high confidence were low molecular weight fatty acyls (Table 1). Among the annotated lipids, several N-acyl amides, known as the signaling molecules of the living system, were identified in in vivo SPME samples and were not found in muscle tissue samples after the one year storage period. Moreover, in vivo SPME sampling allowed for the capture of the compound 2-decene-4,6,8-triyn-1-al, annotated as a unique fatty aldehyde, and also the compound 10 hydroxy-8E-decene-2,4,6-triynoic acid, potential end-product of fatty aldehydes conversion to fatty acids, which have been shown to play a pivotal role as a rapid energy source in developing species25,26. However, none of these compounds were present in stored fish muscle samples, suggesting the occurrence of lipidome profile alterations during storage (Table 1). Indeed, analysis of stored muscle samples (ex vivo SPME) revealed distinct differences in lipid composition, with a 10-fold decrease in the number of detected molecular features in comparison to the number of features observed for in vivo SPME samples (Fig. 1b). In addition to the observed decrease in the number of lipids detected in vivo, the appearance of a higher molecular mass lipid (above 500 m/z), a sterol conjugate - (25 R)-3α,7α-dihydroxy-5β-cholestan-27-oyl taurine, was also observed in ex vivo extracts. Comparison between in vivo and ex vivo SPME was demonstrated as an effective tool in the detection of alterations in the lipidome profile of samples over time, suggesting that unbound compounds detected during in vivo SPME of the living system may have undergone binding reactions as a consequence of morphological changes in lipid structure during sample storage.
Figure 1.
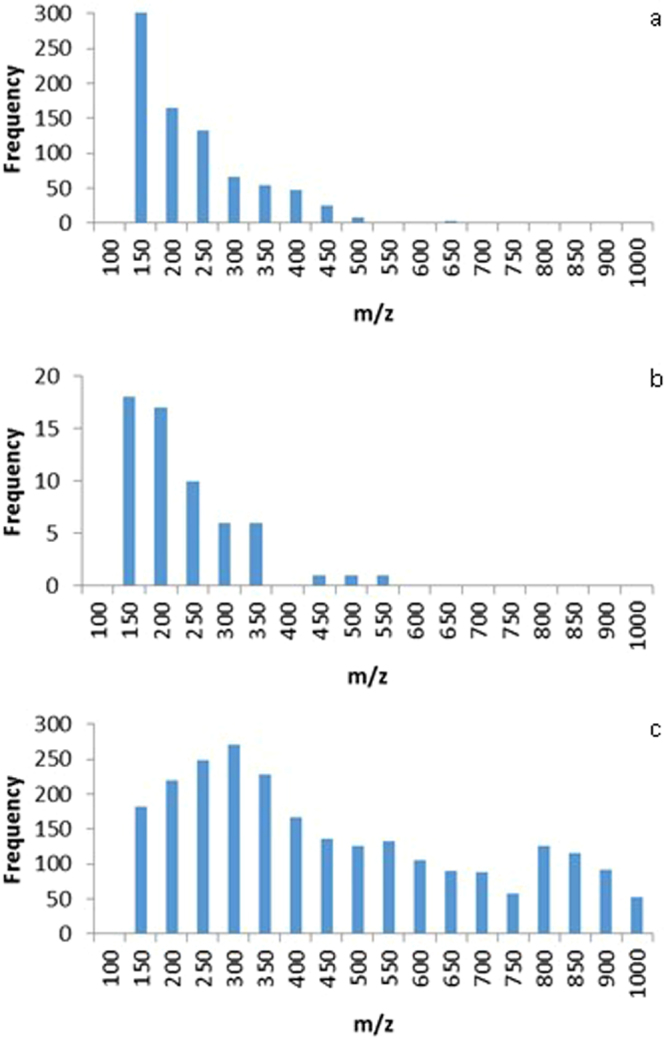
Histogram of mass distributions of detected peaks found in data obtained from (a) in vivo SPME of fish muscles in living fish; (b) ex vivo SPME analysis of non-homogenized fish tissue after a one-year storage period; and (c) solid-liquid extraction analysis of homogenized fish muscle samples after a one-year storage period.
Table 1.
Unique features with medium-to-high confidence matches annotated by LIPID MAPS.
| Compound | Category | Precursor m/z | Precursor adducts | RT (min) | Sample | Confidence match | ||
|---|---|---|---|---|---|---|---|---|
| in vivo SPME | ex vivo SPME | SLE | ||||||
| 2-decene-4,6,8-triyn-1-al | Fatty Acyls | 143.04856 145.04431 144.05173 |
M + H M + 3 H M + 2 H |
0.80 | ✓ | high | ||
| N-eicosanoyl-ethanolamine | Fatty Acyls | 356.35235 | M + H | 8.10 | ✓ | medium | ||
| N-(11Z-eicosaenoyl)-ethanolamine | Fatty Acyls | 354.33658 | M + H | 8.45 | ✓ | medium | ||
| 4-[(5-aminopentyl)(hydroxy)amino]−4-oxobutanoic acid | Fatty Acyls | 219.13410 | M + H | 0.93 | ✓ | medium | ||
| N-octadecanoyl-valine | Fatty Acyls | 384.34715 | M + H | 8.97 | ✓ | medium | ||
| N-octadecanoyl-proline | Fatty Acyls | 382.33164 | M + H | 8.60 | ✓ | medium | ||
| 3-oxobutanamide | Fatty Acyls | 102.05523 103.05861 |
M + H M + 2 H |
0.90 | ✓ | ✓ | medium, high* | |
| hexadecanamide | Fatty Acyls | 256.26374 | M + H | 8.15 | ✓ | medium | ||
| pentanamide | Fatty Acyls | 102.09158 | M + H | 1.37 | ✓ | ✓ | medium | |
| Dodecanamide | Fatty Acyls | 200.20115 | M + H | 6.27 | ✓ | ✓ | medium | |
| O-(17-carboxyheptadecanoyl)carnitine | Fatty Acyls | 458.34762 | M + H | 7.62 | ✓ | medium | ||
| 3,7-dimethyl-8,11-dioxo-2E,6E,9E-dodecatrienal | Fatty Acyls | 235.13304 | M + H | 6.43 | ✓ | medium | ||
| 2-amino-hexanedioic acid | Fatty Acyls | 162.07614 | M + H | 0.93 | ✓ | medium | ||
| 2E,4E,8E,10E-dodecatetraenedioic acid | Fatty Acyls | 223.09658 | M + H | 6.10 | ✓ | medium | ||
| 7,8-diaminononanoic acid | Fatty Acyls | 189.15971 | M + H | 1.17 | ✓ | medium | ||
| 2-amino-3-oxo-hexanedioic acid | Fatty Acyls | 176.05533 | M + H | 0.90 | ✓ | medium | ||
| 12-amino-dodecanoic acid | Fatty Acyls | 216.19599 | M + H | 4.28 | ✓ | medium | ||
| 3-oxo-5S-amino-hexanoic acid OK | Fatty Acyls | 146.08116 | M + H | 0.87 | ✓ | ✓ | medium | |
| 10-hydroxy-8E-Decene-2,4,6-triynoic acid | Fatty Acyls | 177.05468 | M + H | 6.10 | ✓ | medium | ||
| 5,7,9,11,13-tetradecapentaenoic acid | Fatty Acyls | 219.13809 | M + H | 5.95 | ✓ | medium | ||
| octadecanamide | Fatty Acyls | 284.29440 286.30212 |
M + H M + 3 H |
8.85 | ✓ | ✓ | medium, high* | |
| N-(9Z,12Z-octadecadienoyl)-ethanolamine | Fatty Acyls | 324.28921 325.29257 |
M + H M + 2 H |
7.98 | ✓ | high | ||
| 3-(heptanoyloxy)−4-(trimethylazaniumyl)butanoate | Fatty Acyls | 274.20109 275.20456 |
M + H M + 2 H |
3.03 | ✓ | high | ||
| 3-{[(5Z)−3-hydroxyoct-5-enoyl]oxy}−4-(trimethylazaniumyl)butanoate | Fatty Acyls | 302.19547 303.19892 |
M + H M + 2 H |
4.19 | ✓ | high | ||
| 3-[(2E)-hex-2-enoyloxy]−4-(trimethylazaniumyl)butanoate | Fatty Acyls | 258.17035 259.17379 |
M + H M + 2 H |
5.43 | ✓ | high | ||
| 2,4,6-octatrienal | Fatty Acyls | 123.08059 124.08389 |
M + H M + 2 H |
5.34 | ✓ | high | ||
| 12-chloro-dodecanoic acid | Fatty Acyls | 235.14579 236.14932 |
M + H M + 2 H |
5.90 | ✓ | high | ||
| methyl 4-[2-(2-formyl-vinyl)−3-hydroxy-5-oxo-cyclopentyl]-butanoate | Fatty Acyls | 255.12251 256.12608 |
M + H M + 2 H |
7.40 | ✓ | high | ||
| (5Z,7E)−9,10-seco-5,7,10(19)-cholestatriene | Sterol Lipids | 369.35150 370.35492 |
M + H M + 2 H |
10.52 | ✓ | ✓ | high | |
| 26,26,26-trifluoro-25-hydroxy-27-norvitamin D3 | Sterol Lipids | 441.29831 | M + H | 10.42 | ✓ | medium | ||
| (6RS)−6,19-epidioxy-24,24-difluoro-25-hydroxy-6,19-dihydrovitamin D3 | Sterol Lipids | 469.31338 470.31429 |
M + H M + 2 H |
6.89 | ✓ | high | ||
| cholesta-5,7,8(14),22E-tetraen-3-one | Sterol Lipids | 379.30122 | M + H | 8.42 | ✓ | medium | ||
| (25 R)−3α,7α-dihydroxy-5β-cholestan-27-oyl taurine | Sterol Lipids | 542.35356 | M + H | 5.20 | ✓ | medium | ||
| 1-octadecanoyl-rac-glycerol | Glycerolipids | 359.31578 | M + H | 8.87 | ✓ | medium | ||
| 1-tetradecanoyl-glycero-3-phospho-(1-sn-glycerol) | Glycerophospholipids | 457.25547 | M + H | 9.00 | ✓ | high | ||
| 1-(4Z,7Z,10Z,13Z,16Z,19Z-docosahexaenoyl)-glycero-3-phosphoserine | Glycerophospholipids | 570.28281 572.28908 571.28463 |
M + H M + 3 H M + 2 H |
6.86 | ✓ | high | ||
| 1-(1Z-eicosenyl)−2-(4Z,7Z,10Z,13Z,16Z,19Z-docosahexaenoyl)-glycero-3-phosphoserine | Glycerophospholipids | 848.57957 | M + H | 10.47 | ✓ | high | ||
| 1-(7Z,10Z,13Z,16Z-docosatetraenoyl)-glycero-3-phosphocholine | Glycerophospholipids | 572.37175 573.37447 |
M + H M + 2 H |
7.72 | ✓ | high | ||
*For this compound, medium confidence match refers to in vivo SPME, high confidence match refers to SLE
The annotation was based on intensity profiles, retention time, mass defect, and isotope/adduct patterns of peaks. In high confidence match, non-zero xMSannotator multistage score, required adducts, N, O, P, S/C ratio check, hydrogen/carbon ratio check, abundance ratio checks for isotopes, multimers and multiply charged adducts are satisfied; in medium confidence match, pathway level correlation is satisfied18.
In contrast, SLE method used for lipid extraction from homogenized fish muscle samples revealed the presence of many lipid compounds with higher molecular masses (up to 900 m/z) (Fig. 1c) not detected in in vivo or ex vivo SPME extracts (Table 1 and Supplementary Table 1). Owing to the employment of exhaustive extraction conditions along with sonication and the use of organic solvents, SLE enabled the release of high-abundant lipids such as sterols and glycerophospholipids from membrane and intracellular environments, and possibly of lipids belonging to aggregated structures formed during sample storage, which does not represent the unbound fraction of labile lipids detected in vivo. However, the lipid-soluble active form of vitamin D3 ((5Z,7E)-9,10-seco-5,7,10(19)-cholestatriene) detected in in vivo SPME extracts was also found in SLE samples, but not after ex vivo SPME, indicating that certain lipids found in their unbound form in living systems may undergo binding reactions as a result of storage. It should be also emphasized that in this study frozen fish muscle samples were not subjected to freeze-thaw cycles during one-year storage period and the observed differences in the lipid profile are not related to the freezing/thawing processes. Homogenization of stored tissue and application of exhaustive sample preparation method (SLE) released compounds from the aggregated structures and facilitated their detection. SLE technique has a distinct advantage in releasing lipids from their complexes with cellular components, such as proteins. Moreover, disintegration of lipid bilayer makes this method highly efficient in the analysis of high-abundant membrane lipids, such as glycerophospholipids, which are not present in the free form in the tissues and cannot be extracted with the use of SPME technique.
However, SPME, as a low-invasive technique, does not disrupt cell membrane structure and facilities the extraction of specific lipids present in unbound form in the system under study. Therefore, low-abundant and short-lived tissue components, such as signaling molecules might be captured in vivo by SPME probes, providing the insight into lipidome of the living system. Additionally, quenching of the extracted lipids during in vivo SPME approach prevents their oxidative degradation, a process commonly occurring during sample collection and handling with the use of traditional methodologies20,27. Also, possible alterations in the free fraction of lipid composition during sample storage resulting from enzymatic and non-enzymatic transformations can be monitored with the use of SPME method.
Conclusions
To summarize, results obtained from SPME of muscle tissue performed in living fish (in vivo SPME) and in muscle samples submitted to a one year storage period (ex vivo SPME) were compared and contrasted with results obtained from SLE of homogenized tissue samples, with aims of investigating the real-time profile of biological lipids and its potential disruption during sample preparation and storage. In vivo SPME sampling, as a low invasiveness microextraction method, was shown to enable the detection of unstable molecules, such as fatty acyls, known to be present in trace amounts in tissue of living organisms7. In addition, application of ex vivo SPME provided insight into alterations to the lipid composition of samples possibly stemming from the induction or suppression of chemical and biological processes throughout the storage period that result in the conversion of unbound lipids to a bound state. The findings obtained through comparisons of compounds identified in in vivo SPME extracts versus ex vivo SPME and SLE extracts sampled following a year of storage support the assertion that factors such as sample handling and storage have a profound effect on the detection of biological lipids and thus the characterization of the lipidome profile of a given system.
Materials and Methods
Materials
LC-MS grade acetonitrile (ACN), methanol, and water were purchased from Fisher Scientific (Ottawa, ON, Canada). Hexane (Hex) and acetone (Ace) were purchased from Sigma-Aldrich (Oakville, ON, Canada). Biocompatible SPME mixed mode probes (45 µm thickness, 15 mm length of coating) were provided by Supelco (Bellefonte, PA, USA). Standards used for preparation of instrumental QC samples (trans-4-(Aminomethyl)cyclohexanecarboxylic acid, Tranexemic acid, Phenylalanine-d5, Phenylalanine, Tryptophan, Progesterone) were purchased from Sigma-Aldrich (Oakville, ON, Canada).
In vivo SPME sampling
Juvenile rainbow trout (Oncorhynchus mykiss) used in this study (13.6 ± 0.8 cm, 24.9 ± 3.8 g, n = 5) were purchased from Silver Creek Aquaculture (Erin, ON Canada). Fish were acclimatized to laboratory conditions in continuously flowing non-chlorinated water, and fed 2.0 Pt floating commercial trout ration (Martin’s Feed Mill, ON Canada). All fibers used in in vivo investigations were preconditioned in methanol/water (50/50, v/v) prior to analysis. In vivo sampling of fish muscle tissue was conducted by inserting mixed mode SPME fibers into the dorsal-epaxial muscle (near the dorsal fin) of fish initially immobilized with the use of a large foam bed. After insertion of fibers, fish were held in an aerated, covered bucket for a 20 minute period while extractions were carried out. Following extraction and the subsequent removal of fibers from the tissue, all fibers were rinsed with nanopure water to remove any excess matrix components adhered to the coating, and immersed for 90 min in 300 µL of acetonitrile/water (80/20, v/v) for desorption of lipids from the SPME coatings, with vortex agitation set at 1,000 rpm. Extract solutions were then injected to the HPLC-ESI-MS system for instrumental analysis. After completion of in vivo tissue sampling, fish were anaesthetized with 0.1% ethyl 3-amino benzoate methanesulfonate, and killed by spinal severance. Fish muscle samples were collected and immediately frozen in liquid nitrogen for further SPME sampling. All experimental protocols were in accordance with and approved by the University of Waterloo Animal Care Committee (AUPP # 14–16).
Ex vivo SPME sampling
After undergoing storage at −80 °C for one year, fish muscle tissues were submitted to analysis by ex vivo SPME. Prior to sampling, all employed mixed mode fibers were preconditioned in methanol/water (50/50, v/v). Ex vivo sampling was performed by inserting fibers into non-homogenized muscle tissue for a 20 min period. Following extraction, fibers were removed from tissue, rinsed with nanopure water, and immersed for 90 min in 300 µL of acetonitrile/water (80/20, v/v) for desorption of lipids from SPME coatings, using vortex agitation at 1,000 rpm. The obtained extract solutions were then injected to the HPLC-ESI-MS system for instrumental analysis.
Solid-liquid extraction (SLE)
Solid-liquid extractions were carried out according to the modified Mijangos method28. In brief, after performing ex vivo SPME analysis on non-homogenized tissue, 0.5 g of fish muscle was homogenized in dry ice, then placed in a tube. Following, 10 mL of Hex:Ace (50:50, v/v) was added to the sample, which was then submitted to ultrasonic assisted solid-liquid extraction (Bransonic® CPX2800H, Branson) for a 15 min period. The obtained mixture was then allowed to settle for 15 min, after which 7 mL of supernatant was collected and transferred to a new tube. The same outlined procedure was then repeated twice. All collected supernatant (21 mL) was evaporated to dryness under a nitrogen gas stream, and reconstituted in 2 mL of desorption solution used in SPME (acetonitrile/water (80/20, v/v)). The obtained extracts were then centrifuged for 5 min at 4000 rpm in tubes equipped with a 0.65 μm pore DVPP filter (Ultrafree-CL DV Centrifugal Filter, Millipore Sigma) prior to LC/MS analysis.
Liquid chromatography/mass spectrometry analysis (LC/MS)
Metabolite profiling was accomplished with the use of an LC/MS system consisted of a ThermoAccela autosampler, pumps, and an Exactive Benchtop Orbitrap System (Thermo Fisher Scientific, CA, USA). Metabolites were separated in reversed-phase, using a pentafluorophenyl column (Kinetex Phenomenex, 2.1 mm × 100 mm, 1.7 µm particle size). The flow rate was set at 300 µL/min. Mobile phase A consisted of water/formic acid (99.9/0.1, v/v), while mobile phase B consisted of acetonitrile/formic acid (99.9/0.1, v/v). The starting mobile phase conditions were 90% A from 0 to 1.0 min, followed by a linear gradient to 10% A from 1.0 to 9.0 min, and an isocratic hold at 10% A until 12.0 min. Total run time was 18 min per sample, including a 6-min re-equilibration period. Injection volume was 10 µL. Analyses were performed in positive ionization mode with a mass range of m/z 100–1000. To maintain a mass accuracy better than 5 ppm, the Exactive Benchtop Orbitrap was calibrated with a MSCAL5 standard solution (caffeine, tetrapeptide “Met-Arg-Phe-Ala”, ultramark 1621). In order to keep the instrumental conditions constant, the robustness of LC-MS method was verified via the calibration of the instrument every 24 hours during the analysis and also by using instrumental quality control samples as well as blank samples. Additionally, the developed analytical method was validated by the principal component analysis (PCA) (Supplementary Fig. 1). Score Plot showed that the pooled QCs present a stable trend within in vivo SPME and ex vivo SPME sampling. SPME fibers presented a stable performance within the extracted peaks during in vivo and ex vivo experiments (Supplementary Fig. 2).
Data processing
Raw LC/MS data sets were processed using XCMS package software for peak extraction, grouping, retention time correction, and peak filling29–31. All parameters for XCMS analysis were optimized by IPO package32. Extracted peaks were annotated with the use of the xMSannotator Integrative Scoring Algorithm19. LIPID MAPS was employed as a reference database. The annotated molecules are tentative identification, where unique features with medium to high confidence matches annotated by LIPID MAPS were selected for further discussion. The entire data processing procedure was wrapped into R-script33, which can be found in a separate section.
R-script for data analysis
# loading the package
library(xcms)
library(BiocParallel)
library(IPO)
library(xMSannotator)
# the following code for XCMS optimation with IPO package
mzdatapath <- “data/positive/pqc/“
mzdatafiles <- list.files(mzdatapath, recursive = TRUE, full.names = TRUE)
peakpickingParameters <- getDefaultXcmsSetStartingParams(‘centWave’)
#setting levels for min_peakwidth to 10 and 20 (hence 15 is the center point)
peakpickingParameters$min_peakwidth <- c(2,10)
peakpickingParameters$max_peakwidth <- c(15,25)
#setting only one value for ppm therefore this parameter is not optimized
peakpickingParameters$ppm <- 2.5
resultPeakpicking <-
optimizeXcmsSet(files = mzdatafiles[1:4],
params = peakpickingParameters,
nSlaves = 12,
subdir = ‘rsm’)
optimizedXcmsSetObject <- resultPeakpicking$best_settings$xset
retcorGroupParameters <- getDefaultRetGroupStartingParams()
retcorGroupParameters$profStep <- 1
resultRetcorGroup <-
optimizeRetGroup(xset = optimizedXcmsSetObject,
params = retcorGroupParameters,
nSlaves = 12,
subdir = “results”)
writeRScript(resultPeakpicking$best_settings$parameters,
resultRetcorGroup$best_settings,
nSlaves = 12)
# the following code is combination the data process into one function
getopqedata <- function(path,
index = F,
xsmethod = “centWave”,
peakwidth = c(14, 25),
ppm = 2.5,
noise = 0,
snthresh = 10,
mzdiff = −0.00395,
prefilter = c(3, 100),
mzCenterFun = “wMean”,
integrate = 1,
fitgauss = FALSE,
verbose.columns = FALSE,
BPPARAM = BiocParallel::SnowParam(workers = 12),
rmethod = “obiwarp”,
plottype = “none”,
distFunc = “cor_opt”,
profStep = 1,
center = 2,
response = 1,
gapInit = 0.6176,
gapExtend = 2.4,
factorDiag = 2,
factorGap = 1,
localAlignment = 0,
gmethod = “density”,
bw = 0.25,
mzwid = 0.0021748,
minfrac = 1,
minsamp = 1,
gmax = 50,
…) {
cdffiles <- list.files(path, recursive = TRUE, full.names = TRUE)
if (index) {
cdffiles <- cdffiles[index]
}
xset <- xcms::xcmsSet(
cdffiles,
method = xsmethod,
snthresh = snthresh,
mzdiff = mzdiff,
BPPARAM = BPPARAM,
peakwidth = peakwidth,
ppm = ppm,
noise = noise,
prefilter = prefilter,
mzCenterFun = mzCenterFun,
integrate = integrate,
fitgauss = fitgauss,
verbose.columns = verbose.columns,
…
)
if (index & length(index) == 1) {
xset3 <- xset
} else{
xset <- xcms::group(
xset,
method = gmethod,
bw = bw,
mzwid = mzwid,
minfrac = minfrac,
minsamp = minsamp,
max = gmax
)
xset2 <- xcms::retcor(
xset,
method = rmethod,
plottype = plottype,
distFunc = distFunc,
profStep = profStep,
center = center,
response = response,
gapInit = gapInit,
gapExtend = gapExtend,
factorDiag = factorDiag,
factorGap = factorGap,
localAlignment = localAlignment
)
# you need group the peaks again for this corrected data
xset2 <- xcms::group(
xset2,
method = gmethod,
bw = bw,
mzwid = mzwid,
minfrac = minfrac,
minsamp = minsamp,
max = gmax
)
xset3 <- xcms::fillPeaks(xset2, BPPARAM = BPPARAM)
}
return(xset3)
}
# the following code is used to process the data
path <- “./data/lipid/exvivo/“
xset <- getopqedata(path)
path <- “./data/lipid/invivo/“
xset2 <- getopqedata(path)
path <- “./data/lipid/le/“
xset3 <- getopqedata(path)
# get the peaks group infomation for histgram
xd1 <- as.data.frame(xset@groups)
xd2 <- as.data.frame(xset2@groups)
xd3 <- as.data.frame(xset3@groups)
write.csv(xd1,‘exvivo.csv’)
write.csv(xd2,‘invivo.csv’)
write.csv(xd3,‘le.csv’)
# make annotation with one function from xMSannotator package
fanno <-
function(xset,
outloc = “./result/“,
mode = ‘pos’,
list = c(
“M + 2 H”,
“M + H + NH4”,
“M + ACN + 2 H”,
“M + 2ACN + 2 H”,
“M + H”,
“M + NH4”,
“M + Na”,
“M + ACN + H”,
“M + ACN + Na”,
“M + 2ACN + H”,
“2 M + H”,
“2 M + Na”,
“2 M + ACN + H”,
“M + 2Na-H”,
“M + H-H2O”,
“M + H-2H2O”
),
db_name = ‘HMDB’, …) {
data <- xcms::groupval(xset, ‘medret’, “into”)
adduct_weights = cbind.data.frame(Adduct = c(‘M + H’,‘M-H’),Weight = c(5,5))
mz <- xcms::groups(xset)[, 1]
time <- xcms::groups(xset)[, 4]
data <- as.data.frame(cbind(mz, time, data))
data <- unique(data)
if (mode = = ‘neg’) {
annotres <-
xMSannotator::multilevelannotation(
dataA = data,
max.mz.diff = 5,
max.rt.diff = 10,
cormethod = “pearson”,
queryadductlist = list,
mode = mode,
outloc = outloc,
db_name = db_name,
adduct_weights = adduct_weights,
num_sets = 1000,
allsteps = TRUE,
corthresh = 0.7,
NOPS_check = TRUE,
customIDs = NA,
missing.value = NA,
hclustmethod = “complete”,
deepsplit = 2,
networktype = “unsigned”,
minclustsize = 10,
module.merge.dissimilarity = 0.2,
filter.by = c(“M-H”),
biofluid.location = NA,
origin = NA,
status = “Detected and Quantified”,
boostIDs = NA,
max_isp = 5,
HMDBselect = “union”,
mass_defect_window = 0.01,
pathwaycheckmode = “pm”,
mass_defect_mode = mode
)
}else{
annotres <-
xMSannotator::multilevelannotation(
dataA = data,
max.mz.diff = 5,
max.rt.diff = 10,
cormethod = “pearson”,
queryadductlist = list,
mode = mode,
outloc = outloc,
db_name = db_name,
adduct_weights = adduct_weights,
num_sets = 1000,
allsteps = TRUE,
corthresh = 0.7,
NOPS_check = TRUE,
customIDs = NA,
missing.value = NA,
hclustmethod = “complete”,
deepsplit = 2,
networktype = “unsigned”,
minclustsize = 10,
module.merge.dissimilarity = 0.2,
filter.by = c(“M + H”),
biofluid.location = NA,
origin = NA,
status = “Detected and Quantified”,
boostIDs = NA,
max_isp = 5,
HMDBselect = “union”,
mass_defect_window = 0.01,
pathwaycheckmode = “pm”,
mass_defect_mode = mode
)
}
return(annotres)
}
# annotate the data
annoexvivo <- fanno(xset,outloc = ‘data/lipid/exvivo/sub/‘,db_name = ‘LipidMaps’,num_nodes = 12)
annoinvivo <- fanno(xset2,outloc = ‘data/lipid/invivo/sub/‘,db_name = ‘LipidMaps’,num_nodes = 12)
anole <- fanno(xset3,outloc = ‘data/lipid/le/sub/‘,db_name = ‘LipidMaps’,num_nodes = 12).
Electronic supplementary material
Acknowledgements
This work was supported by Environment Canada through the Environmental Damages Fund (Grant EC-129114).
Author Contributions
J.P. and A.R. designed experiments. V.B. performed in vivo SPME sampling and LC/MS analysis of these samples, and collected fish tissue samples. A.R. and M.Y. performed ex vivo SPME sampling, SLE and LC/MS analysis. M.Y. preformed data processing and compound annotation. A.R. interpreted obtained data and prepared the manuscript with help from M.Y., V.B., L.B., M.S. and J.P.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-25428-2.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hyötyläinen T, Orešič M. Systems biology strategies to study lipidomes in health and disease. Progress in Lipid Research. 2014;55:43–60. doi: 10.1016/j.plipres.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Zhao, Y.-Y., Cheng, X. & Lin, R.-C. Chapter One – Lipidomics Applications for Discovering Biomarkers of Diseases in Clinical Chemistry. International Review of Cell and Molecular Biology313 (2014). [DOI] [PubMed]
- 3.Badin PM, Langin D, Moro C. Dynamics of skeletal muscle lipid pools. Trends in Endocrinology and Metabolism. 2013;24:607–615. doi: 10.1016/j.tem.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Watt MJ, Hoy AJ, Muoio DM, Coleman RA. Distinct roles of specific fatty acids in cellular processes: implications for interpreting and reporting experiments. Am. J. Physiol. Endocrinol. Metab. 2012;302:E1–3. doi: 10.1152/ajpendo.00418.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quehenberger O, et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010;51:3299–3305. doi: 10.1194/jlr.M009449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpenter T, et al. Use of reversed phase HP liquid chromatography to assay conversion of N-acylglycines to primary fatty acid amides by peptidylglycine-α-amidating monooxygenase. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004;809:15–21. doi: 10.1016/j.jchromb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 7.Divito EB, Cascio M. Metabolism, physiology, and analyses of primary fatty acid amides. Chemical Reviews. 2013;113:7343–7353. doi: 10.1021/cr300363b. [DOI] [PubMed] [Google Scholar]
- 8.Bessonneau, V. et al. In vivo microsampling to capture the elusive exposome. Sci. Rep. 7 (2017). [DOI] [PMC free article] [PubMed]
- 9.Teo CC, et al. Advances in sample preparation and analytical techniques for lipidomics study of clinical samples. TrAC - Trends in Analytical Chemistry. 2015;66:1–18. doi: 10.1016/j.trac.2014.10.010. [DOI] [Google Scholar]
- 10.Divito, E. B., Kroniser, K. M. & Cascio, M. Multidimensional liquid chromatography coupled with tandem mass spectrometry for identification of bioactive fatty acyl derivatives. Front. Physiol. 7 (2016). [DOI] [PMC free article] [PubMed]
- 11.Liebisch G, Ekroos K, Hermansson M, Ejsing CS. Reporting of lipidomics data should be standardized. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids. 2017;1862:747–751. doi: 10.1016/j.bbalip.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen A, Rudge SA, Zhang Q, Wakelam MJ. Using lipidomics analysis to determine signalling and metabolic changes in cells. Current Opinion in Biotechnology. 2017;43:96–103. doi: 10.1016/j.copbio.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 13.Skonberg C, Artmann A, Cornett C, Hansen SH, Hansen HS. Pitfalls in the sample preparation and analysis of N-acylethanolamines. J. Lipid Res. 2010;51:3062–73. doi: 10.1194/jlr.D004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jurowski K, et al. Analytical Techniques in Lipidomics: State of the Art. Critical Reviews in Analytical Chemistry. 2017;47:418–437. doi: 10.1080/10408347.2017.1310613. [DOI] [PubMed] [Google Scholar]
- 15.Ouyang, G., Vuckovic, D. & Pawliszyn, J. Nondestructive sampling of living systems using in vivo solid-phase microextraction. Chem. Rev. 111 (2011). [DOI] [PubMed]
- 16.Reyes-Garcés N, Bojko B, Hein D, Pawliszyn J. Solid Phase Microextraction Devices Prepared on Plastic Support as Potential Single-Use Samplers for Bioanalytical Applications. Anal. Chem. 2015;87:9722–9730. doi: 10.1021/acs.analchem.5b01849. [DOI] [PubMed] [Google Scholar]
- 17.Birjandi AP, Mirnaghi FS, Bojko B, Wąsowicz M, Pawliszyn J. Application of solid phase microextraction for quantitation of polyunsaturated Fatty acids in biological fluids. Anal. Chem. 2014;86:12022–9. doi: 10.1021/ac502627w. [DOI] [PubMed] [Google Scholar]
- 18.Vuckovic D, Risticevic S, Pawliszyn J. In vivo solid-phase microextraction in metabolomics: Opportunities for the direct investigation of biological systems. Angewandte Chemie - International Edition. 2011;50:5618–5628. doi: 10.1002/anie.201006896. [DOI] [PubMed] [Google Scholar]
- 19.Uppal K, Walker DI, Jones DP. xMSannotator: An R Package for Network-Based Annotation of High-Resolution Metabolomics Data. Anal. Chem. 2017;89:1063–1067. doi: 10.1021/acs.analchem.6b01214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolf C, Quinn PJ. Lipidomics: Practical aspects and applications. Progress in Lipid Research. 2008;47:15–36. doi: 10.1016/j.plipres.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Zivkovic AM, et al. Effects of sample handling and storage on quantitative lipid analysis in human serum. Metabolomics. 2009;5:507–516. doi: 10.1007/s11306-009-0174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kronenberg F, Lobentanz EM, König P, Utermann G, Dieplinger H. Effect of sample storage on the measurement of lipoprotein[a], apolipoproteins B and A-IV, total and high density lipoprotein cholesterol and triglycerides. J. Lipid Res. 1994;35:1318–1328. [PubMed] [Google Scholar]
- 23.Evans K, Mitcheson J, Laker MF. Effect of storage at −70 °C on lipid, lipoprotein and apolipoprotein concentrations. Clin. Chim. Acta. 1997;258:219–229. doi: 10.1016/S0009-8981(96)06458-3. [DOI] [PubMed] [Google Scholar]
- 24.Bachorik PS, Walker R, Brownell KD, Stunkard AJ, Kwiterovich PO. Determination of high density lipoprotein-cholesterol in stored human plasma. J. Lipid Res. 1980;21:608–616. [PubMed] [Google Scholar]
- 25.Rizzo WB. Fatty aldehyde and fatty alcohol metabolism: Review and importance for epidermal structure and function. Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids. 2014;1841:377–389. doi: 10.1016/j.bbalip.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dautel, S. E. et al. Lipidomics reveals dramatic lipid compositional changes in the maturing postnatal lung. Sci. Rep. 7 (2017). [DOI] [PMC free article] [PubMed]
- 27.Niki E. Biomarkers of lipid peroxidation in clinical material. Biochimica et Biophysica Acta - General Subjects. 2014;1840:809–817. doi: 10.1016/j.bbagen.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 28.Mijangos L, Bizkarguenaga E, Prieto A, Fernández LA, Zuloaga O. Simultaneous determination of a variety of endocrine disrupting compounds in carrot, lettuce and amended soil by means of focused ultrasonic solid-liquid extraction and dispersive solid-phase extraction as simplified clean-up strategy. J. Chromatogr. A. 2015;1389:8–18. doi: 10.1016/j.chroma.2015.02.036. [DOI] [PubMed] [Google Scholar]
- 29.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 30.Tautenhahn R, Böttcher C, Neumann S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics. 2008;9:504. doi: 10.1186/1471-2105-9-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benton HP, Want EJ, Ebbels TMD. Correction of mass calibration gaps in liquid chromatography-mass spectrometry metabolomics data. Bioinformatics. 2010;26:2488–2489. doi: 10.1093/bioinformatics/btq441. [DOI] [PubMed] [Google Scholar]
- 32.Libiseller G, et al. IPO: a tool for automated optimization of XCMS parameters. BMC Bioinformatics. 2015;16:118. doi: 10.1186/s12859-015-0562-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.R Core Team. R Core Team (2017). R: A language and environment for statistical computing. R Found. Stat. Comput. Vienna, Austria. http//www.R-project.org/. R Foundation for Statistical Computing (2017).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.