

Abstract
Gamma-glutamylcyclotransferase (GGCT) was originally identified as a protein highly expressed in bladder cancer tissues by proteomic analysis, and its higher expression in a variety of cancers compared to normal tissues have been shown. Depletion of GGCT in various cancer cells results in antiproliferative effects both in vitro and in vivo; thus it is considered a promising therapeutic target. Although it has been shown that knockdown of GGCT induces cellular senescence and non-apoptotic cell death, associated with upregulation of cyclin-dependent kinase inhibitors (CDKIs) including p21WAF1/CIP1, the cellular events that follow GGCT depletion are not fully understood. Here, we show that GGCT depletion induced autophagy in MCF7 breast and PC3 prostate cancer cells. Conversely, overexpression of GGCT in NIH3T3 fibroblast under conditions of serum deprivation inhibited autophagy and increased proliferation. Simultaneous knockdown of autophagy related-protein 5, a critical effector of autophagy, along with GGCT in MCF7 and PC3 cells led to significant attenuation of the multiple cellular responses, including upregulation of CDKIs, increased numbers of senescence-associated β-galactosidase positive senescent cells, and growth inhibition. Furthermore, we show that autophagy-promoting signaling cascades including activation of the AMPK-ULK1 pathway and/or inactivation of the mTORC2-Akt pathway were triggered in GGCT-depleted cells. These results indicate that autophagy plays an important role in the growth inhibition of cancer cells caused by GGCT depletion.
Keywords: Gamma-glutamylcyclotransferase, autophagy, cyclin-dependent kinase inhibitor, p21WAF1/CIP1, cellular senescence, AMPK, ULK1, ATG5, mTORC, Akt
Introduction
Chromosome 7 open reading frame 24 was originally identified as a highly expressed protein in bladder cancer tissues by proteome analysis [1,2]; and later characterized as gamma-glutamylcyclotransferase (GGCT) [3]. GGCT levels in various cancer tissues are higher than in noncancerous tissues. A previous study involving a large cohort reported that 72% of colon cancers, 58% of uterine cervical cancers, 46% of breast cancers, and 38% of lung cancers show higher expression of GGCT protein than normal tissue, and that higher expression of GGCT protein correlates with a poor prognosis in patients with breast cancer [4]. In vitro depletion of GGCT by RNA interference inhibits proliferation in various types of cancer cell [5]. Furthermore, the anti-tumor effects of GGCT knockdown have been demonstrated in tumor-bearing mouse models using regional injection [6] and systemic administration [7] of siRNAs targeting GGCT.
In a previous study, we reported that in vitro knockdown of GGCT induces cellular senescence in multiple cell lines, due to cell type-dependent upregulation of cyclin-dependent kinase inhibitors (CDKIs) such as p21WAF1/CIP1 (p21) and/or p16INK4a (p16) [8]. Upregulation of CDKIs including p21 and p16 that cause cell cycle arrest, also mediates cellular senescence [9], which is characterized as a permanent limitation of cell division; this affects both normal cells and cancer cells [10]. However, the mechanism by which depletion of GGCT inhibits cancer cell growth and induces senescence is unclear.
Autophagy is a self-degradation system that maintains normal cellular homeostasis; it is induced in response to environmental signals such as nutrient deprivation, hormones and microbial pathogens [11]. While autophagy is basically a cytoprotective phenomenon, an imbalance in cellular metabolism may cause excessive autophagic activation and induce cell death [12]. The so-called autophagy-associated cell death is one mechanism of non-apoptotic cell death [13]. Autophagy related-protein 5 (Atg5) is an indispensable constituent of autophagosomes and plays an essential role in autophagy. Atg12 is activated via E1-like enzyme Atg7 and is covalently bound to Atg5 by E2-like enzyme Atg10, resulting in complex formation with Atg16L1. The protein complex exerts function as an E3-like enzyme for the next conjugation system, including microtubule-associated protein 1A/1B-light chain 3 (LC3) [14]. Under starvation conditions, Atg5-deficient neonatal mice lacking functional autophagy survive for much shorter periods than wild-type mice [15].
Unc-51 like autophagy activating kinase 1 (ULK1) is essential for initiation of autophagy. Once AMP-activated protein kinase (AMPK) is activated by phosphorylation at Thr172, the p-AMPK phosphorylates ULK1 directly to induce autophagy in response to cellular nutritional deficiency [16]. ULK1 harbors several phosphorylation sites, which are phosphorylated by each respective signal cascade and mediate different functions [17]. For example, activated AMPK directly phosphorylates ULK1 at Ser777 and Ser317 in response to glucose starvation, thereby initiating autophagy [18]. Under nutrient-rich conditions, ULK1 is phosphorylated at Ser757 by mTORC1, which inhibits ULK1 activation by AMPK [18]. Also, amino acids starvation activates AMPK, which induces autophagy by phosphorylating ULK1 at Ser555 [19]. In addition, activated AMPK phosphorylates Raptor, which is a component of mTORC1, thereby inhibiting mTORC1 signaling [20]. Another mTOR complex, mTORC2, increases phosphorylation of Akt [21]. The activated Akt phosphorylates Beclin1 (Atg6), and the resulting complex, which comprises Beclin1, 14-3-3 proteins, and intermediate filaments, inhibits autophagy [22].
Here, we show that depleting GGCT induces autophagy in MCF7 breast and PC3 prostate cancer cells. Conversely, overexpression of GGCT in NIH3T3 fibroblasts inhibits autophagy and increases proliferation under conditions of serum deprivation. We also demonstrate that simultaneous knockdown of Atg5 and GGCT in MCF7 and PC3 cells attenuates cellular events caused by GGCT depletion alone, including upregulation of CDKI, cellular senescence, and growth inhibition. Moreover, we show that GGCT depletion triggers the autophagy-promoting signaling cascades including activation of the AMPK-ULK1 signaling pathway and/or inactivation of the mTORC2-Akt pathway in a cell type-dependent manner.
Materials and methods
Cell culture
The MCF7, PC3, DU-145, and MDA-MB-231 cells were obtained from RIKEN BRC and cultured in DMEM supplemented with 10% FBS (HyClone, South Logan, UT) and 1% penicillin and streptomycin. The mouse embryonic fibroblast NIH3T3 cell line was purchased from American Type Culture Collection (Rockville, MD) and GGCT was overexpressed using the pCX4bsr vector as described in [2]. Control NIH3T3 cells transfected with empty vector or a GGCT expression vector were seeded in 6 cm dishes. The next day, the medium was changed for DMEM containing 2% or 10% FBS. All cells were maintained at 37°C in 5% CO2 atmosphere.
Antibodies
Antibodies specific for the following proteins were purchased: mouse monoclonal antibodies against LC3 (M186-3, MBL, Nagoya, Japan), p21 (556430, BD Biosciences, NJ), GGCT (6-1E, Cosmo Bio, Tokyo, Japan), Atg5 (M153-3, MBL), GAPDH (016-25523, Wako Pure Chemical Industries, Osaka, Japan), β-actin (013-24553, Wako), β-tubulin (T4026, Sigma-Aldrich, St. Louis, MO); and rabbit monoclonal antibodies against p16 (ab51243, Abcam, Cambridge, MA), phospho-AMPKα (Thr172, #2535, Cell Signaling Technology, Danvers, MA), phospho-ULK1 Ser555 and Ser757 (#5869 and #6888, CST), phospho-70 kDa ribosomal protein S6 kinase (p70S6K) (Thr389, #9205, CST), phospho-rapamycin insensitive companion of mTOR (Rictor) (Thr1135, #3806, CST), phospho-Akt (Ser473, #4060, CST), and the respective non-phosphorylated forms (#5832, #8054, #9476 and #4691, CST). Horse anti-mouse IgG-HRP conjugates were purchased from Vector Laboratories (PI-2000, Burlingame, CA). The HRP-linked goat anti-rabbit IgG was purchased from CST (#7074).
Western blot analysis
Cells were lysed with lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate-Na, and 0.1% SDS) supplemented with a protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan) and PhosSTOP EASYpack (Roche Diagnostic, Indianapolis, IN). The protein concentration was measured using the BCA protein assay (Bio-Rad, Hercules, CA), according to the manufacturer’s protocol. Aliquots containing 20 μg of protein were separated by SDS-PAGE and transferred to PVDF membranes (Millipore, Billerica, MA). After blocking with 3% fat-free dry milk in PBS with 0.05% Tween-20 (PBST), or PVDF Blocking Reagent for Can Get Signal (TOYOBO, Osaka, Japan), the membranes were incubated with appropriate primary and secondary antibodies diluted in 3% BSA in PBST, or Can Get Signal Immunoreaction Enhancer Solution (TOYOBO). When using antibodies against phosphorylated proteins, membranes were blocked with Blocking one-P (Nacalai Tesque), and then incubated with primary and secondary antibodies diluted in TBS with 0.05% Tween-20. Proteins were visualized using Clarity or Super Western ECL Substrate (Bio-Rad). Chemiluminescence was detected by the ChemiDoc XRS Plus system (Bio-Rad).
Transfection of siRNA targeting GGCT, Atg5
Transient transfection was performed with Lipofectamine RNAi MAX (Invitrogen, Waltham, MA), according to the manufacturer’s protocol. Synthesized siRNAs were purchased from RNAi Co., Tokyo, or from Gene Design Inc., Osaka, Japan. The sequences of siRNA targeting GGCT or non-targeting described in [8] were used. The following Atg5 siRNA sequence was used: 5’-CGAAUUCCAACUUGCUUUA-3’ (Atg5). All siRNAs were transfected at a concentration of 10 nM. For the double knockdown experiments, 20 nM non-targeting siRNA was transfected as a control.
Detection of autophagy
Five × 104 MCF7 or PC3 cells were seeded in 6-well plates, and on next day transfected with siRNAs as described above. At 4 days post-transfection, cells were stained with CYTO-ID Green detection reagent and Hoechst 33342, using the CYTO-ID Autophagy Detection Kit (Enzo Life Science, Farmingdale, NY), according to the manufacturer’s protocol. After treatment with 10 µM of chloroquine for 18 hours, images were obtained using an Eclipse Ti confocal microscope (Nikon, Tokyo, Japan).
Cell cycle analysis
Two × 105 NIH3T3 cells stably transfected with empty vector or a GGCT expression vector were seeded in 6 cm dishes. The next day, the medium was changed for DMEM containing 2% FBS. At 3 days after seeding, the cells were washed with PBS and fixed for 1 hour at -20°C in 70% ethanol, and stained with propidium iodide (20 μg/ml) in the presence of RNase A (200 μg/ml). DNA content was analyzed using BD LSRFortessa X-20 cytometer (BD Bioscience). MCF7 cells were seeded in 6-well plates and transfected with GGCT, Atg5, or non-targeting siRNA. The cells were then washed with PBS and fixed overnight at -20°C in 70% ethanol, and stained with propidium iodide as described above. DNA content was analyzed using a FACSCalibur cytometer (BD Bioscience). At least 10,000 cells were analyzed per sample.
Assessment of cell proliferation
MCF7 and PC3 cells were transfected with the indicated siRNAs 1 day after seeding, and viability was assessed using the standard trypan blue dye exclusion method (0.4% trypan blue solution) (Wako). The viability of NIH3T3 cells was assessed using the cell count reagent SF kit (Nacalai Tesque). Relative OD values were obtained based on absorbance at 450 nm on Day 0 (set as 1).
Staining of senescence-associated β-galactosidase
Cells were seeded in 6-well plates, and transfected with siRNAs as described above. At 4 days post-transfection, cells were stained with SA-β-Gal using a senescence kit (OZ Bioscience, San Diego, CA), according to the manufacturer’s protocol. Cells were incubated overnight at 37°C with staining solution and SA-β-Gal positive cells were counted. For each evaluation, more than 400 cells were counted in at least 12 random fields.
Statistical analysis
All data were obtained from at least three independent experiments and results were expressed as the mean ± S.D. Data were compared using a two-tailed Student’s t-test and p-values < 0.05 were deemed significant. Statistical analysis was performed using Excel software.
Results
Knockdown of GGCT induces autophagy in MCF7 and PC3 cancer cells
When autophagy is induced, LC3-I is converted to LC3-II [23]. To examine whether autophagy is induced by GGCT knockdown, we investigated the LC3-II protein levels in MCF7 breast cancer cells and PC3 prostate cancer cells transfected with GGCT-targeting siRNA or non-target siRNA. We found that GGCT knockdown increased the levels of the LC3-II (Figure 1A). This increase was attenuated by simultaneous knockdown of Atg5, which is a critical mediator of autophagy induction (Figure 1A). These results indicate that the conversion into LC3-II occurs via Atg5-mediated autophagic activity. We also confirmed that autophagosomes were formed in MCF7 and PC3 cells upon GGCT knockdown, and that formation of autophagosomes could be abrogated by co-transfection with anti-Atg5 siRNA (Figure 1B), in parallel with a positive control using a well-established autophagy inducer rapamycin (Supplementary Figure 1).
Figure 1.
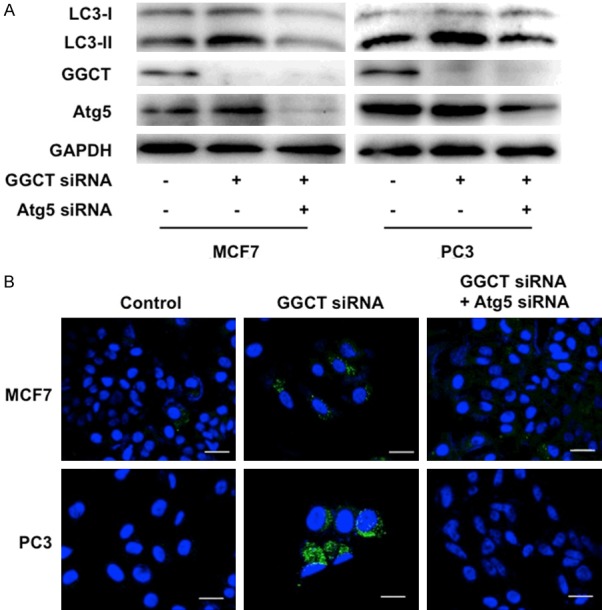
GGCT depletion induces autophagy. A: Western blot analysis of LC3, GGCT, Atg5 and GAPDH in MCF7 and PC3 cells at 4 days post-transfection with siRNA targeting GGCT and Atg5, or with a non-target control siRNA. GAPDH is shown as a loading control. B: Representative images of autophagosomes in MCF7 and PC3 cells stained by CYTO-ID Autophagy Detection Kit. Cells were stained 4 days after transfection with the indicated siRNAs. Scale bar, 20 μm.
Overexpression of GGCT prevents autophagy and promotes proliferation under conditions of serum deprivation in NIH3T3 cells
We next overexpressed GGCT in the cancer cells to address whether GGCT levels affect induction of autophagy. However, no evident phenotype was observed, suggesting that functions of GGCT would be saturated in various cancer cell lines expressing GGCT abundantly. We therefore prepared GGCT-overexpressing NIH3T3 fibroblasts and examined the effect on proliferation and autophagy under serum-deprived conditions, since it was shown that serum deprivation induces autophagy in cultured cells [23]. Western blot analysis confirmed successful overexpression of GGCT (Figure 2A). The condition with serum deprivation led to a significant increase in LC3-II levels in the control NIH3T3 cells (Figure 2B), indicating induction of autophagy. We found that overexpression of GGCT led to a marked decrease in the induction of LC3-II under the serum-deprived condition (Figure 2B). Moreover, overexpression of GGCT rescued suppressed NIH3T3 cell proliferation (Figure 2C) and promoted cell cycle progression (Figure 2D and 2E) under conditions of serum deprivation. These results clearly illustrate the autophagy-regulating and the proliferation-promoting functions of GGCT, which are consistent with our findings that knockdown of GGCT induces autophagy (Figure 1) and suppresses cell proliferation [8].
Figure 2.
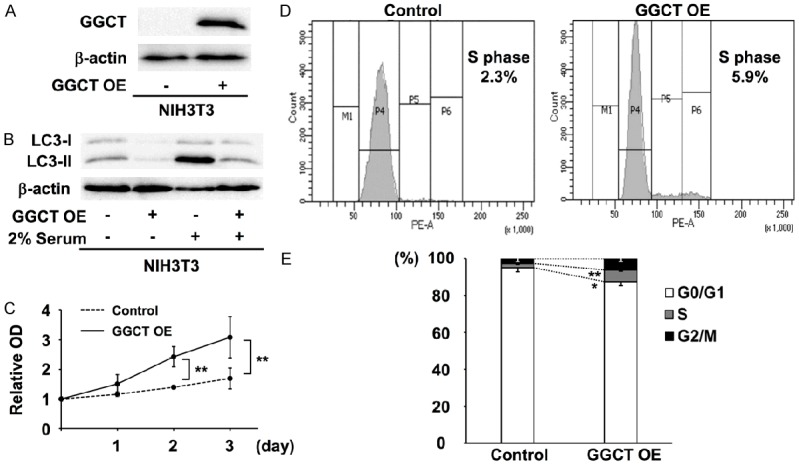
Overexpression of GGCT suppresses autophagy and promotes proliferation. (A) Western blot analysis of GGCT-overexpressing cells. (B) Western blot analyses of LC3 in control and GGCT-overexpressing NIH3T3 cells cultured in (-) 10% or (+) 2% serum-containing medium. (C) Growth curves of control and GGCT-overexpressing (OE) NIH3T3 cells cultured in 2% serum-containing medium. (D and E) Cell cycle analysis of control and GGCT-overexpressing NIH3T3 cells in DMEM supplemented with 2% FBS. Representative histograms (D) and quantified distribution (E) are shown. (*P < 0.05, **P < 0.01).
Blocking autophagy attenuates GGCT depletion-mediated CDKI upregulation and cell cycle arrest
A previous study reported that expression of p21 and/or p16 in various types of cancer cell are upregulated upon GGCT knockdown; in particular, both p21 and p16 are upregulated in MCF7 and PC3 cells [8]. We found that simultaneous knockdown of Atg5 together with GGCT in the cells attenuated upregulation of both p21 and p16 (Figure 3A). These results suggest that upregulation of these CDKIs in cancer cells requires Atg5-mediated autophagy induction. Also, depletion of GGCT from MCF7 cells induces G0/G1 phase cell cycle arrest in a p21-dependent manner [8]. Consistent with this, we found that depleting Atg5 significantly reduced G0/G1 phase population increased by GGCT knockdown, indicating significance of p21 induction via autophagy for responses to GGCT depletion (Figure 3B).
Figure 3.
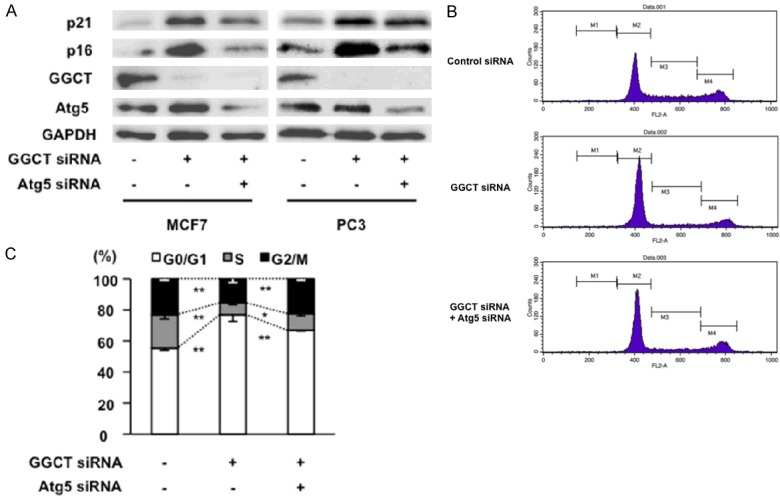
Inhibition of autophagy attenuates upregulation of CDKI and cell cycle arrest caused by GGCT knockdown. (A) Western blot analysis of p21, p16, GGCT, Atg5, and GAPDH expression in MCF7 and PC3 cells at 4 days post-transfection with siRNA targeting GGCT or with a non-target control siRNA. (B and C) Cell cycle phases in MCF7 cells were analyzed by flow cytometry at 4 days post-transfection with the indicated siRNAs. Representative histograms (B) and quantified distribution are shown (C).
Blocking autophagy rescues the cell growth inhibition caused by GGCT knockdown
Given that the induction of autophagy plays an important role in upregulating CDKIs and inducing cell cycle arrest in GGCT-depleted cells, we hypothesized that blocking autophagy would rescue the growth inhibition mediated by GGCT knockdown. Indeed, simultaneous knockdown of Atg5 and GGCT partially, but significantly rescued proliferation of MCF7 and PC3 cell lines (Figure 4). These results indicate that antiproliferative effect by depleting GGCT is, at least in part, mediated through induction of autophagy.
Figure 4.
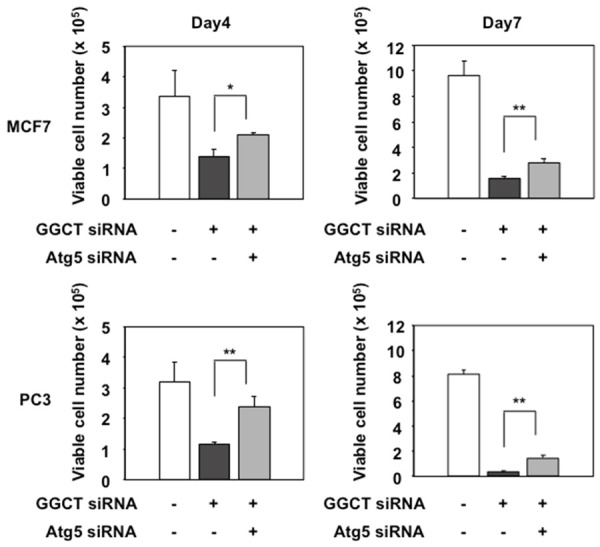
Blockade of autophagy attenuates cell growth inhibition caused by GGCT knockdown. MCF7 and PC3 cells were transfected with the indicated siRNAs. The relative number of trypan blue-negative viable cells at 4 and 7 days post-transfection is shown. (*P < 0.05, **P < 0.01).
Blocking autophagy inhibits cellular senescence induced by GGCT knockdown
Several recent reports demonstrated the function of autophagy as a novel effector mechanism of cellular senescence [24-28]. As knockdown of GGCT induces cellular senescence in various types of cancer cell, including breast or prostate cancer cells [8], we hypothesized that induction of autophagy would play a role in cellular senescence in GGCT-depleted cells. GGCT-depleted MCF7 and PC3 cells showed phenotypic changes characteristic of cellular senescence, i.e., flat and markedly enlarged morphology plus staining for SA-β-Gal, a specific marker of senescent cells [29]. When we blocked autophagy by simultaneously depleting Atg5 and GGCT, the flat and enlarged morphologic changes were less clear and the number of SA-β-Gal-positive senescent cells was significantly lower than that in a population of cells lacking GGCT alone (Figure 5A and 5B). These results indicate that autophagy is required for subsequent induction of cellular senescence in GGCT-depleted MCF7 and PC3 cells.
Figure 5.
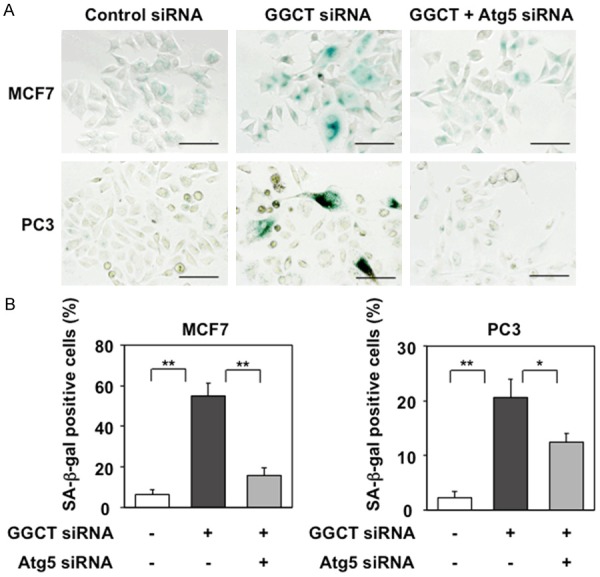
Blockade of autophagy inhibits cellular senescence induced by GGCT knockdown. A: Representative images of SA-β-Gal staining of MCF7 and PC3 cells 4 days post-transfection with the indicated siRNAs. Scale bar, 100 μm. B: The number of SA-β-Gal-positive cells was counted and the positive cell: total cell ratios are shown. (*P < 0.05, **P < 0.01).
Depleting GGCT activates the AMPK-ULK1 pathway and leads to inactivation of the mTORC2-Akt in cancer cells
Next, we analyzed signaling pathways that regulate autophagy in the GGCT-depleted cells. AMPK phosphorylates and activates ULK1, which is an essential factor for autophagy [16,18-20,30]. We found that phosphorylation of AMPKα Thr172 and ULK1 Ser555 in PC3 cells were markedly increased by GGCT knockdown (Figure 6A). We also confirmed those changes in DU-145 cells (Supplementary Figure 2). However, we observed no significant change in AMPK-ULK1 phosphorylation in GGCT-depleted MCF7 cells. We also found that the amount of p-ULK1 Ser757, which is phosphorylated by mTORC1, decreased upon GGCT depletion in MCF7 cells (Figure 6A) and MDA-MB-231 cells (Supplementary Figure 2). Phosphorylation of p70S6K, one of the major downstream targets of mTORC1 [31], significantly decreased in both GGCT-depleted MCF7 and PC3 cells (Figure 6A). These results suggest that GGCT knockdown also suppresses the mTORC1-mediated signaling pathway. We further analyzed phosphorylation of proteins within the mTORC2-Akt signaling pathway. The amount of p-Rictor Thr1135, which inhibits mTORC2 function mediating phosphorylation of Akt at Ser473 [21], increased upon GGCT depletion in MCF7 cells (Figure 6B). Consistent with this, we confirmed that expression of p-Akt Ser473, which inhibits autophagy along with Beclin1 [22], was decreased upon GGCT knockdown in both MCF7 and PC3 cells (Figure 6B). Taken together, these changes in phosphorylation status of signal mediators in response to GGCT knockdown are consistent with the observations that GGCT-depletion induces autophagy (Figure 7).
Figure 6.
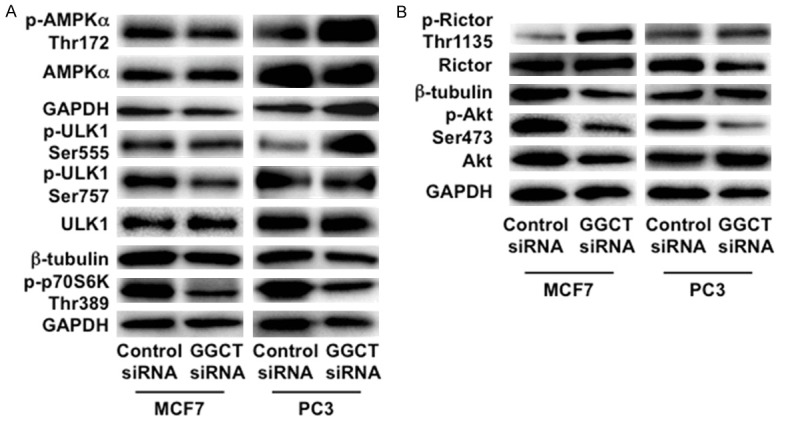
Depletion of GGCT activates AMPK-ULK1 pathway and/or leads to inactivation of the mTORC2-Akt in cancer cells. A: Western blot analyses of mediators of the AMPK-ULK1 signaling pathway (p-AMPKα Thr172, AMPKα, p-ULK1 Ser555, and ULK1) and downstream components of the mTORC1 pathway (p-ULK1 Ser757 and p-p70S6K Thr389) in MCF7 and PC3 cells 4 days post-transfection with siRNA targeting GGCT or with a non-target control siRNA. GAPDH and β-tubulin are shown as loading controls. B: Western blot analyses of p-Rictor Thr1135, Rictor, p-Akt Ser473, Akt, GAPDH and β-tubulin in MCF7 and PC3 cells 4 days post-transfection with the indicated siRNAs are shown.
Figure 7.
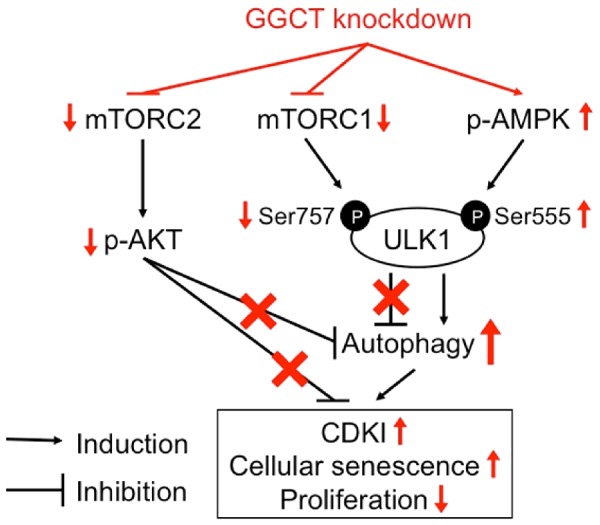
Graphical summary of the signaling cascades involved in the autophagy induction upon GGCT depletion is shown.
Discussion
It has been reported that depletion of GGCT inhibits proliferation via CDKIs induction and subsequent cellular senescence in a variety of cancer cells [8], however, the mechanisms underlying the responses are not fully understood. Here, we first demonstrate that depleting GGCT in MCF7 and PC3 cancer cells induces autophagy. Our results show that blockade of autophagy by knocking down Atg5 attenuates GGCT depletion-mediated upregulation of CDKIs, indicating a regulatory role for autophagy. Simultaneous knockdown of Atg5 attenuated upregulation of both p21 and p16 after GGCT depletion. In PC3 cells, silencing Atg5 did not weaken the upregulation of p21 but did also reduce p16. The concurrent knockdown of Atg5 and GGCT in both cell lines, however, attenuated cellular senescence and cell growth inhibition (Figure 5) induced by GGCT depletion. These findings indicate that inductions of p16 and/or p21 were shown as an indispensable factor for cellular responses (including growth inhibition and cellular senescence) to GGCT depletion in cancer cells, although the cellular events following depletion of GGCT do not deeply depend on p21 upregulation in PC3 cells [8].
Given that the inhibiting autophagy by depleting Atg5 attenuates cellular senescence induced by GGCT knockdown (Figure 5), it is conceivable that autophagy plays a role as an effector mechanism also for inducing senescence in response to GGCT depletion. Indeed, an emerging body of evidence suggests a link between autophagy and senescence; in particular, a regulatory role for autophagy in inducing senescence. For instance, a study based on a human fibroblast model revealed that oncogene-induced and oxidative stress-induced senescence could be inhibited by blocking autophagy [24,25], thereby highlighting a role for autophagy as an upstream regulator of cellular senescence induction. Anticancer drugs such as doxorubicin and camptothecin induce both autophagy and senescence, and blockade of autophagy in MCF7 and HCT116 cells reduces or delays induction of senescence [26]. In addition, a naturally occurring chemical compound, pseudolaric acid B, induces autophagy-dependent senescence in murine fibrosarcoma cells and MCF7 cells by inhibiting the Akt-mTOR pathway [27]. Li et al. recently reported that the treatment of melanoma cells harboring BRAFV600E mutation with a BRAF inhibitor, encorafenib (LGX818), blocked both the mTOR-p70S6K pathway and induction of the autophagy-dependent cellular senescence [28]. Taken together, these findings highlight the regulatory role for autophagy in senescence induction in response to therapeutic interventions against cancer cells. Our results presented herein imply that putative molecular mechanisms, as yet to be elucidated, underlying inhibition of CDKI-upregulation via autophagy blockade might be involved in autophagy-dependent senescence.
GGCT is an enzyme that plays a role in amino acid metabolism by catalyzing reactions to generate 5-oxoproline and free amino acids from gamma-glutamyl peptide in the gamma-glutamyl cycle [3]. It has been reported that a shortage of amino acids induces autophagy via AMPK activation [19], whereas sufficient amino acids activate mTORC1, which inhibits ULK1 [32]. Therefore, the mechanism underlying autophagy induction by GGCT knockdown may be explained by aberrant amino acid homeostasis. The experiments conducted herein based on overexpression demonstrated that GGCT has an autophagy-regulating function. Serum deprivation suppressed proliferation of NIH3T3 cells (data not shown) and induced autophagy (Figure 2B). In fact, augmenting GGCT function inhibited autophagy and promoted proliferation of serum-deprived NIH3T3 cells. These results suggest that enhanced GGCT function might compensate for a metabolic stress, possibly amino acid insufficiency, caused by serum insufficiency. However, particular amino acids responsible for the resulting phenotypes remain to be revealed probably by a metabolomic approach.
Although autophagy is a cellular survival response induced by starvation, knockdown of Atg5 attenuated cell growth inhibition caused by GGCT depletion (Figure 4). These results indicate that autophagy exerts, at least in part, an inhibitory effect on proliferation of GGCT-depleted cancer cells. Excessive induction of autophagy is one mechanism of non-apoptotic cell death; presumably, sustained autophagy induced by metabolic stress leads to cell death when the turnover of protein and organelles overwhelm the capacity to synthetic them [13]. In addition, this mode of cell death, so-called autophagic cell death, occurs in cancer cells treated with some anticancer agents or drug candidates [33-37].
We found that when autophagy was induced in PC3 and DU-145 cells by GGCT depletion, levels of p-AMPKα Thr172 and p-ULK1 Ser555 increased significantly (Figure 6A), suggesting accompanying activation of the AMPK-ULK1 signaling pathway. In MCF7 and MDA-MB-231 cells, however, subtle change in phosphorylation levels at these sites on AMPKα and ULK1 were observed; rather GGCT depletion reduced p-ULK1 Ser757 (Figure 6A), an inactivated form [18]. Concordantly, GGCT depletion from both MCF7 and PC3 cells reduced the phosphorylation of p70S6K (Figure 6A), a major downstream factor of mTORC1 signaling [31]. These results suggest that the mTORC1 signaling is suppressed in GGCT-depleted cells, likely resulting in reversing inhibition of ULK1 and induction of autophagy. In addition, phosphorylated Rictor inhibits the mTORC2-Akt axis [21], which inhibits autophagy by promoting formation of a protein complex comprising Beclin1, 14-3-3, and intermediate filaments [22]. Based on our results showing that GGCT knockdown increases p-Rictor and reduced p-Akt (Figure 6B), inactivation of the mTORC2-Akt axis by increased p-Rictor may explain induction of autophagy in GGCT-depleted MCF7 cells. These findings support that GGCT depletion induces metabolic stress leading to modify various signaling pathways to promote autophagy (Figure 7).
Accumulating evidence demonstrates that cancer cell growth requires upregulation of GGCT. Our results support a novel strategy for cancer treatment based on inhibiting GGCT function. Indeed, some specific GGCT inhibitors have been studied and a lead compound developed [38,39]. Currently, however, there is no GGCT inhibitor that can be used in a clinical setting. Thus, substantial improvement in the efficacy of potential GGCT inhibitors is needed.
In summary, we provide evidence showing that GGCT knockdown induces autophagy in cancer cells, and the cellular responses to GGCT depletion including CDKI upregulation followed by induction of cellular senescence, and consequent growth inhibition are, at least in part, mediated by induction of autophagy. Consistently, GGCT depletion triggers the signaling pathways, which facilitate autophagy, including activation of AMPK-ULK1 axis and/or inactivation of the mTORC2-Akt pathway, depending on the cellular context. This study sheds new light on our understanding of mechanisms involving autophagy underlying the inhibitory effects of GGCT depletion on cancer cell growth.
Acknowledgements
This work was supported by Japan Society for the Promotion of Science grant numbers 15K10584, 15K10585, 24592384, 25460488, 16K08722, 26461436 by the Ministry of Education, Culture, Sports, Science and Technology-Supported Program for the Strategic Research Foundation at Private Universities 2015-2019, by the Takeda Science Foundation, and by a Kyoto Pharmaceutical University Fund for the Promotion of Scientific Research.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Kageyama S, Isono T, Iwaki H, Wakabayashi Y, Okada Y, Kontani K, Yoshimura K, Terai A, Arai Y, Yoshiki T. Identification by proteomic analysis of calreticulin as a marker for bladder cancer and evaluation of the diagnostic accuracy of its detection in urine. Clin Chem. 2004;50:857–66. doi: 10.1373/clinchem.2003.027425. [DOI] [PubMed] [Google Scholar]
- 2.Kageyama S, Iwaki H, Inoue H, Isono T, Yuasa T, Nogawa M, Maekawa T, Ueda M, Kajita Y, Ogawa O, Toguchida J, Yoshiki T. A novel tumor-related protein, C7orf24, identified by proteome differential display of bladder urothelial carcinoma. Proteomics Clin. 2007;1:192–9. doi: 10.1002/prca.200600468. [DOI] [PubMed] [Google Scholar]
- 3.Oakley AJ, Yamada T, Liu D, Coggan M, Clark AG, Board PG. The identification and structural characterization of C7orf24 as gamma-glutamyl cyclotransferase. An essential enzyme in the gamma-glutamyl cycle. J Biol Chem. 2008;283:22031–42. doi: 10.1074/jbc.M803623200. [DOI] [PubMed] [Google Scholar]
- 4.Gromov P, Gromova I, Friis E, Timmermans-Wielenga V, Rank F, Simon R, Sauter G, Moreira JM. Proteomic profiling of mammary carcinomas identifies C7orf24, a gamma-glutamyl cyclotransferase, as a potential cancer biomarker. J Proteome Res. 2010;9:3941–53. doi: 10.1021/pr100160u. [DOI] [PubMed] [Google Scholar]
- 5.Kageyama S, Hanada E, Ii H, Tomita K, Yoshiki T, Kawauchi A. Gamma-Glutamylcyclotransferase. A novel target molecule for cancer diagnosis and treatment. Biomed Res Int. 2015;2015:345219. doi: 10.1155/2015/345219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hama S, Arata M, Nakamura I, Kasetani T, Itakura S, Tsuchiya H, Yoshiki T, Kogure K. Prevention of tumor growth by needle-free jet injection of anti-C7orf24 siRNA. Cancer Gene Ther. 2012;19:553–7. doi: 10.1038/cgt.2012.31. [DOI] [PubMed] [Google Scholar]
- 7.Ran R, Liu Y, Gao H, Kuang Q, Zhang Q, Tang J, Fu H, Zhang Z, He Q. PEGylated hyaluronic acid-modified liposomal delivery system with anti-γ-glutamylcyclotransferase siRNA for drug-resistant MCF-7 breast cancer therapy. J Pharm Sci. 2014;104:476–84. doi: 10.1002/jps.24163. [DOI] [PubMed] [Google Scholar]
- 8.Matsumura K, Nakata S, Taniguchi K, Ii H, Ashihara E, Kageyama S, Kawauchi A, Yoshiki T. Depletion of γ-glutamylcyclotransferase inhibits breast cancer cell growth via cellular senescence induction mediated by CDK inhibitor upregulation. BMC Cancer. 2016;1:748. doi: 10.1186/s12885-016-2779-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang H. Molecular signaling and genetic pathways of senescence: its role in tumorigenesis and aging. J Cell Physiol. 2007;210:567–74. doi: 10.1002/jcp.20919. [DOI] [PubMed] [Google Scholar]
- 10.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–7. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mizushima N, Levine B, Cuervo A, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–41. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keller CW, Lünemann JD. Autophagy and autophagy-related proteins in CNS autoimmunity. Front Immunol. 2017;8:165. doi: 10.3389/fimmu.2017.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 16.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-Activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roach PJ. AMPK -> ULK1 -> autophagy. Mol Cell Biol. 2011;31:3082–4. doi: 10.1128/MCB.05565-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghislat G, Patron M, Rizzuto R, Knecht E. Withdrawal of essential amino acids increases autophagy by a pathway involving Ca2+/calmodulin-dependent kinase kinase-β (CaMKK-β) J Biol Chem. 2012;287:38625–36. doi: 10.1074/jbc.M112.365767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5:e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30:908–21. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338:956–9. doi: 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavaré S, Arakawa S, Shimizu S, Watt FM, Narita M. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo Y, Zou P, Zou J, Wang J, Zhou D, Liu L. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKα dependent manner. Exp Gerontol. 2011;46:860–7. doi: 10.1016/j.exger.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goehe RW, Di X, Sharma K, Henderson SC, Valerie K, Rodier F, Davalos AR, Gewirtz DA. The autophagy-senescence connection in chemotherapy: must tumor cells (self) eat before they sleep? J Pharmacol Exp Ther. 2012;343:763–78. doi: 10.1124/jpet.112.197590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qi M, Zhou H, Fan S, Li Z, Yao G, Tashiro S, Onodera S, Xia M, Ikejima T. mTOR inactivation by ROS-JNK-p53 pathway plays an essential role in psedolaric acid B induced autophagy-dependent senescence in murine fibrosarcoma L929 cells. Eur J Pharmacol. 2013;715:76–88. doi: 10.1016/j.ejphar.2013.05.051. [DOI] [PubMed] [Google Scholar]
- 28.Li Z, Jiang K, Zhu X, Lin G, Song F, Zhao Y, Piao Y, Liu J, Cheng W, Bi X, Gong P, Song Z, Meng S. Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016;370:332–44. doi: 10.1016/j.canlet.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 29.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan XY, Tian C, Wang H, Xu Y, Ren K, Zhang BY, Gao C, Shi Q, Meng G, Zhang LB, Zhao YJ, Shao QX, Dong XP. Activation of the AMPK-ULK1 pathway plays an important role in autophagy during prion infection. Sci Rep. 2015;5:14728. doi: 10.1038/srep14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J. 2012;441:1–21. doi: 10.1042/BJ20110892. [DOI] [PubMed] [Google Scholar]
- 32.Shimobayashi M, Hall MN. Multiple amino acid sensing inputs to mTORC1. Cell Res. 2016;26:7–20. doi: 10.1038/cr.2015.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2004;101:18030–5. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene. 2005;24:980–91. doi: 10.1038/sj.onc.1208095. [DOI] [PubMed] [Google Scholar]
- 35.Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell. 2008;14:90–102. doi: 10.1016/j.ccr.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nozaki R, Kono T, Bochimoto H, Watanabe T, Oketani K, Sakamaki Y, Okubo N, Nakagawa K, Takeda H. Zanthoxylum fruit extract from Japanese pepper promotes autophagic cell death in cancer cells. Oncotarget. 2016;7:70437–70446. doi: 10.18632/oncotarget.11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsieh YY, Lo HL, Yang PM. EZH2 inhibitors transcriptionally upregulate cytotoxic autophagy and cytoprotective unfolded protein response in human colorectal cancer cells. Am J Cancer Res. 2016;6:1661–80. [PMC free article] [PubMed] [Google Scholar]
- 38.Ii H, Yoshiki T, Hoshiya N, Uenishi J. Synthesis and GGCT inhibitory activity of N-Glutaryl-L-alanine analogues. Chem Pharm Bull. 2016;64:785–92. doi: 10.1248/cpb.c16-00167. [DOI] [PubMed] [Google Scholar]
- 39.Yoshiya T, Ii H, Tsuda S, Mochizuki M, Kageyama S, Yoshiki T. Design of fluorogenic probes and fluorescent-tagged inhibitors for γ-glutamyl cyclotransferasem. J Pept Sci. 2017;23:618–623. doi: 10.1002/psc.2984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.