

Abstract
Oncogenic K-Ras activation is a common mutational event in colorectal cancer. We previously showed that transcription factor, Krüppel-like factor 5 (KLF5), contributes to intestinal polyposis in mice with K-Ras activation. At 14 months of age, Villin-Cre/LSL-K-RasG12D mice developed small intestinal and colonic hyperplastic polyps while LSL-K-RasG12D had none. The intestinal crypts of Villin-Cre/LSL-K-RasG12D mice contained a higher number of mitotic figures and increased crypt heights compared to controls. The intestinal epithelium of Villin-Cre/LSL-K-RasG12D mice showed prolific KLF5 expression throughout and above the elongated crypts. In contrast, KLF5 expression was limited to the upper crypt region in the controls. The levels of K-Ras effectors were significantly increased in Villin-Cre/LSL-K-RasG12D as compared to controls. The Villin-Cre/LSL-K-RasG12D mice showed decreased survival upon treatment with azoxymethane (AOM) as compared to controls. Furthermore, loss of one of Klf5 alleles reduced levels of K-Ras effector proteins and prevented mortality of Villin-Cre/LSL-K-RasG12D mice upon AOM treatment. The Villin-Cre/LSL-K-RasG12D mice spontaneously develop hyperplastic intestinal polyps and display a hyper-proliferative intestinal phenotype with elongated crypts, increased numbers of mitotic figures, elevated expression of KLF5, and other pro-proliferative targets. Induction of colonic tumorigenesis with AOM is detrimental to Villin-Cre/LSL-K-RasG12D mice that is in part dependent of KLF5.
Keywords: KLF5, KRAS, intestinal hyper-proliferation, colonic tumorigenesis
Introduction
Krüppel-like factor 5 (KLF5) is a pro-proliferative transcription factor highly expressed in the crypts of the intestinal epithelium [1]. In adults, KLF5 has a crucial role in regulating the rapidly dividing transient amplifying epithelial cells of the intestine [1]. Furthermore, KLF5 expression levels are increased in transformed cells and KLF5 was shown to be a target of oncogenic HRAS and KRAS [2-4]. Oncogenic mutations in the RAS gene have been shown to occur in 50% of colon cancer [5,6] and are linked to formation of precancerous aberrant crypt foci. Previously, it was demonstrated that mice with intestine-specific over-expression of oncogenic KRAS (Villin-KRASV12G mice; expressing KRASV12G under the control of the intestine-specific villin promoter) develop intestinal adenomas by 9 months of age [7]. However, expression of oncogenic KRAS alone from its endogenous promoter in mice up to one year of age is not sufficient to drive colon carcinogenesis as accompanying secondary mutations are necessary [8,9]. We have previously shown that intestinal tumors derived from KRASV12G mice and human primary colorectal cancers with mutated KRAS contain high levels of KLF5 [2,10]. Conversely, genetic reduction of Klf5 results in a significant reduction in intestinal tumor formation in mice harboring a germline mutation in the murine Apc gene (ApcMin/+) or combined ApcMin/+ and KRASV12G mutations [10,11]. Importantly, KLF5 was shown to be both necessary and sufficient for the tumor initiating activity of β-catenin during intestinal adenoma formation in both ApcMin/+ and ApcMin/+/KRASV12G mice. In this study, we evaluated the role for KLF5 in mediating the long-term effects of oncogenic KRAS expression from the endogenous locus in the mouse intestinal epithelium.
Materials and methods
Mouse strains and maintenance
LSL-K-RasG12D (LSL; for LoxP-STOP-LoxP) and Villin-Cre mice strains were purchased from The Jackson Laboratory. LSL-K-RasG12D mice were bred into a C57BL/6 background through backcrossing for 7 generations. All animal studies were approved by Stony Brook University Institutional Animal Care and Use Committee (IACUC) and the methods were carried out in accordance with the relevant guidelines and regulations. Villin-Cre mice were crossed with LSL-K-RasG12D mice to generate double heterozygous Villin-Cre/LSL-K-RasG12D mice with intestine-specific expression of K-RasG12D from the endogenous promoter. Genotype analysis was performed as described previously [12]. Tail tips from mice were collected and processed using Red Extract-N-Amp kit according to manufacturer’s protocol (Sigma Aldrich). Allele-specific PCR analyses were performed using appropriate primers.
Azoxymethane (AOM) treatment. LSL-K-Ras G12D/Klf5flox/+, Villin-Cre/Klf5flox/+, Villin-Cre/LSL-K-Ras G12D, and Villin-Cre/LSL-K-Ras G12D/Klf5flox/+ mice were intraperitoneal injected (i.p.) with AOM (10 mg/kg), once a week for 4 consecutive weeks. Animals were observed for survival post treatment, and the small intestine and colon were removed for further analysis.
Histology and immunohistochemistry
Tissue collection and immunohistochemical analysis was performed as previously described [10]. Rabbit anti-phospho-MEK1 and anti-phospho-ERK1/2 antibodies (Cell Signaling Technology) were used at 1:100 dilution. Rabbit polyclonal antibody generated against amino acid positions 95-111 of the KLF5 protein (SDIX/Origene) were used at 1:150 dilution. Mouse monoclonal antibody anti-whole-β-catenin (Life Technologies) was used at 1:1,000 dilution. Rabbit monoclonal antibody anti-cyclin D1 (Biocare Medical) was used at 1:200 dilution. Rabbit polyclonal antibody against MKI67 (Leica Microsystems) was used at 1:400 dilution. Mouse monoclonal antibody against phosphorylated β-catenin Ser552 (Cell Signaling) was used at 1:200 dilution.
Western blot analyses
Western Blot analyses were performed as described previously [3,10]. Rabbit polyclonal antibody anti-KLF5 (SDIX/Origene) was used at 1:2,000. Rabbit anti-phospho-MEK1 and anti-phospho-ERK1/2 antibodies (Cell Signaling Technology) were used at 1:1,000 dilution. Mouse monoclonal antibody anti-β-actin (Sigma-Aldrich) was used at 1:5,000 dilution.
Statistical analysis
All statistical analysis was performed using Student’s T-test method and Mantel-Cox test with Graph Pad Prism 5 Software.
Data availability
All data generated or analyzed during this study are included in this published article.
Results
Villin-Cre mice [13] were crossed with LSL-K-RasG12D (LSL; for LoxP-STOP-LoxP) mice [14] to generate double heterozygous Villin-Cre/LSL-K-RasG12D mice with intestine-specific expression of K-RasG12D from the endogenous locus. These double heterozygous mice, along with age-matched control LSL-K-RasG12D mice, were sacrificed at 14 months of age. K-RasG12D mutation affects the architecture of the intestinal epithelium. As shown in the Figures 1A and 2A, numerous hyper-proliferative regions were observed in the small intestine and colon, respectively, of Villin-Cre/LSL-K-RasG12D, but not in the control mice (LSL-K-RasG12D). In the small intestine, the number of mitotic figures per crypt (Figure 1B) and the crypt height (Figure 1C) were significantly higher in the Villin-Cre/LSL-LRasG12D mice when compared to control. Similar findings were observed for those in the colon (Figure 2B and 2C). Additionally, the small intestine and colon of Villin-Cre/LSL-K-RasG12D mice contained many hyperplastic polyps that were absent from control mice (Figure 2D and 2E). We did not observe the development of dysplasia or neoplasia in either the small intestine or colon of Villin-Cre/LSL-K-RasG12D mice by 14 months of age. These results are consistent with previous observations indicating that endogenous K-Ras mutations alone are not sufficient to initiate carcinogenesis [8,9,15]. Previous studies using Villin-Cre/LSL-K-RasG12D mice were mainly focused on the role of mutated KRAS within colonic tissue [8,9]. These reports confirm higher levels of MEK activity in Villin-Cre/LSL-K-RasG12D mice in proximal colon [8] and in colonic epithelium [9]. In our studies, we assessed the levels of K-Ras effector proteins, pMEK, pERK1/2, and cyclin D1 in normal-appearing small intestinal mucosa of the Villin-Cre/LSL-K-RasG12D and control mice (Figure 3A and 3B). We observed elevated levels of pMEK, pERK1/2, and cyclin D1 in the small intestinal epithelium in Villin-Cre/LSL-K-RasG12D as compared to control mice by western blotting and immunohistochemistry (Figure 3A and 3B, respectively). It has been previously established that KLF5 is a downstream effector of MAPK pathway [3,4,10,16,17]. Furthermore, we showed increased levels of KLF5 protein in the tissues from human colorectal cancer and in tumors developed in mice harboring KRASV12G mutation [10]. In the current study, KLF5 protein levels were elevated in the small intestinal epithelium in Villin-Cre/LSL-K-RasG12D as compared to control mice by western blotting (Figure 3A) and in the normal-appearing small intestinal mucosa from Villin-Cre/LSL-K-RasG12D mice compared to controls (data not shown). Moreover, we observed prolific KLF5 expression not only along and above the elongated crypts marking the normal intestinal epithelia but also in the Villin-Cre/LSL-K-RasG12D hyperplastic polyp sections (Figure 3Be). In contrast, KLF5 expression was limited to around the crypt-villus junction in the controls mice (Figure 3Ba). The levels of cyclin D1 (Figure 3Bd and 3Bh) and MKI67 (data not shown) were also significantly increased in Villin-Cre/LSL-K-RasG12D intestinal tissues compared to controls. The analysis of effectors of K-Ras signaling pathway in the colon showed similar results as in small intestinal epithelium with increased expression levels of pMEK and pERK1/2 in colonic tissues of Villin-Cre/LSL-K-RasG12D mice as compare to control (Figure 3Cb, 3Cc, 3Ch and 3Ci). We also observed increased levels of KLF5 and cyclin D1 in the colonic tissues of Villin-Cre/LSL-K-RasG12D in comparison to control (Figure 3Ca, 3Cd, 3Cg and 3Cj). There was no appreciable difference in the levels of protein of either total (Figure 3Cf and 3Cl) or activated β-catenin (Figure 3Ce and 3Ck) between Villin-Cre/LSL-K-RasG12D mice and controls.
Figure 1.
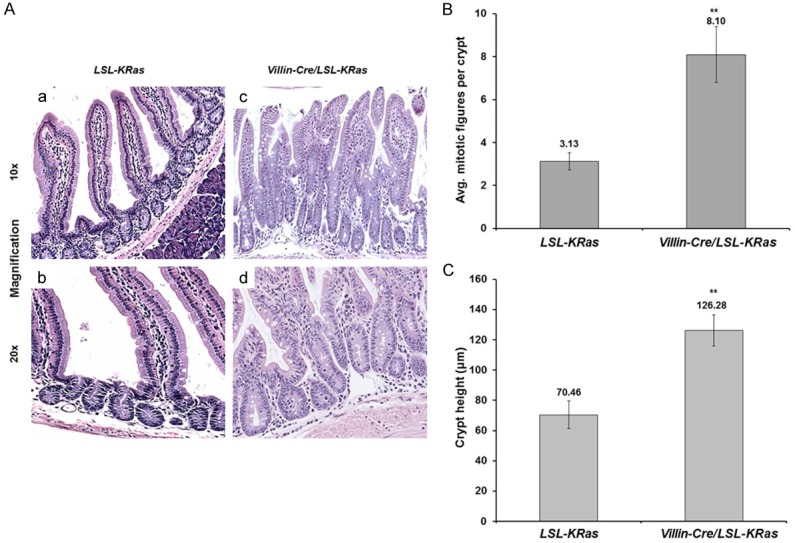
Morphological assessment of small intestines tissues of LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D mice. A. Histological staining of small intestines tissues from LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D mice. Formalin-fixed, paraffin-embedded tissue sections of small intestines were stained with hematoxylin and eosin. (a and b) represent LSL-K-RasG12D, (c and d) represent Villin-Cre/LSL-K-RasG12D mice, respectively. Images taken at 10x magnification (a and c) and 20x magnification (b and d). B. Average number of mitotic figures per crypt in LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D mice. Data represent mean ± SEM (N=3), **P<0.01. C. Crypt height (mm) in small intestinal tissues of LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D mice. Data represent mean ± SEM (N=3), **P<0.01.
Figure 2.
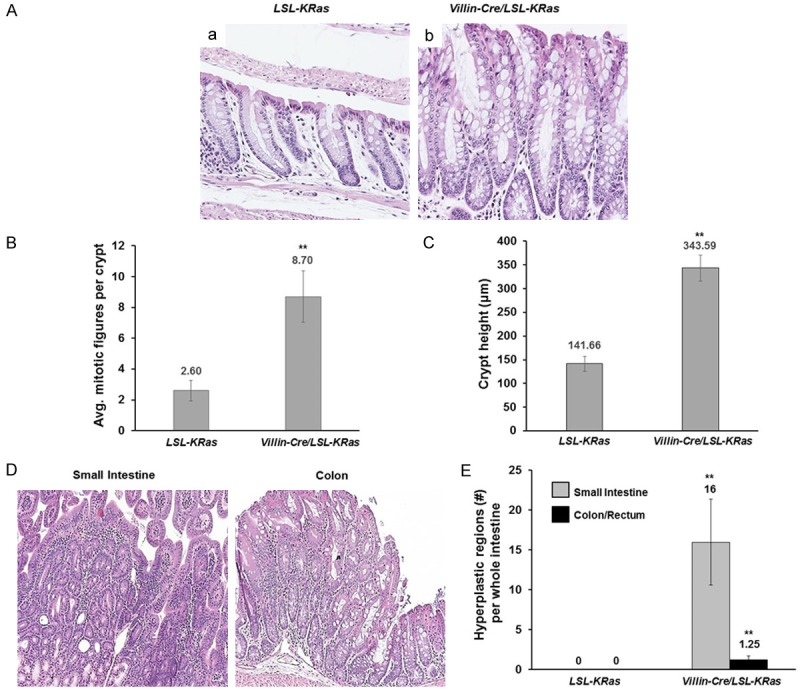
Morphological assessment of colonic tissues of LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D mice. A. Histological staining of small intestines tissues from LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D mice. Formalin-fixed, paraffin-embedded tissue sections of small intestines were stained with hematoxylin and eosin. a represents LSL-K-RasG12D, b represents Villin-Cre/LSL-K-RasG12D mice. Images taken at 10x magnification. B. Average number of mitotic figures per colonic crypt in LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D mice. Data represent mean ± SEM (N=3), **P<0.01. C. Colonic crypt height (mm) in intestinal tissues of LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D mice. Data represent mean ± SEM (N=3), **P<0.01. D. Representative images of hyperplastic regions of small intestine and colonic tissues of Villin-Cre/LSL-K-RasG12D mice. E. Quantitation of hyperplastic regions in small intestine and colon/rectum of LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D mice. Data represent mean ± SEM (N=3), **P<0.01.
Figure 3.

Upregulation of downstream targets of ERK pathway. A. Western Blot analysis of protein extract from normal appearing mucosa obtained from small intestines tissues from LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D mice. 10 ug of total protein extracts from LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D were analyzed by Western blotting using antibody against KLF5, pMEK, pERK1/2, cyclin D1 and β-actin (as a loading control). The images are shown separately as they were obtained from different gels or different parts of the same gel. B. Small intestinal tissues from LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D mice were stained with antibody against KLF5 (a, e), pMEK (b, f), pERK1/2 (c, g) and cyclin D1 (d, h). C. Colonic tissues from LSL-K-RasG12D and Villin-Cre/LSL-K-RasG12D mice were stained with antibody against KLF5 (a, g), pMEK (b, h), pERK1/2 (c, i), cyclin D1 (d, j), β-catenin Ser552 (e, k), and whole β-catenin (f, l).
To address the role of KLF5 in the background of mutated KRAS in colonic tumorigenesis, we utilized established animal model for sporadic colon cancer development using azoxymethane (AOM) as a carcinogen [18]. LSL-K-RasG12D, Villin-Cre/Klf5flox/+, Villin-Cre/LSL-K-RasG12D, and Villin-Cre/LSL-K-RasG12D/Klf5flox/+ mice were injected with AOM as described in Methods (Figure 4A). We showed that Villin-Cre/LSL-K-RasG12D mice treated with AOM exhibited decreased survival as compared to the LSL-K-RasG12D, Villin-Cre/Klf5flox/+, and Villin-Cre/LSL-K-RasG12D/Klf5flox/+ mice, which did not exhibit any mortality at the end point of the experiment (Figure 4B, control mice are depicted as one lane; p value =0.0014 by Mantel-Cox test). The morphology of intestinal tissues obtained from the Villin-Cre/LSL-K-RasG12D mice showed signs of necrosis and ischemia with sporadic regions of normal mucosa (data not shown). The H&E analysis of the normal mucosa of colonic tissues shows increased height of the crypts of the Villin-Cre/LSL-K-RasG12D mice as compared to the LSL-K-RasG12D, Villin-Cre/Klf5flox/+, and Villin-Cre/LSL-K-RasG12D/Klf5flox/+ mice (Figure 4C). Our data suggest that loss of one allele of Klf5 gene in the context of K-RasG12D mutation is sufficient to neutralize oncogenic KRAS function upon carcinogen treatment. To further analyze the expression levels of components of KRAS and WNT signaling pathways we performed immunohistochemical staining of colonic tissues collected upon completion of the treatment. We show elevated levels of KLF5, pMEK, pERK1/2, and cyclin D1 in the colonic epithelium in Villin-Cre/LSL-K-RasG12D as compared to the LSL-K-RasG12D, Villin-Cre/Klf5flox/+, and Villin-Cre/LSL-K-RasG12D/Klf5flox/+ mice (Figure 5). There was no significant difference in the expression levels of total and active β-catenin between analyzed mice (Figure 5).
Figure 4.

KLF5 increases survival of Villin-Cre/LSL-K-RasG12D in AOM-induced carcinogenesis murine model. A. Design of experiment: LSL-K-RasG12D/Klf5fl/+, Villin-Cre/Klf5fl/+, Villin-Cre/LSL-K-RasG12D, and Villin-Cre/LSL-K-RasG12D/Klf5fl/+ mice were injected once a week for four weeks with AOM and tissues were collected 2 and 4 weeks after last AOM injection. B. Kaplan-Meier survival curves of LSL-K-RasG12D/Klf5fl/+ (N=4), Villin-Cre/Klf5fl/+ (N=4), Villin-Cre/LSL-K-RasG12D (N=3), and Villin-Cre/LSL-K-RasG12D/Klf5fl/+ (N=4) mice after injections with AOM, P=0.0014 using Mantel-Cox test. C. H&E staining of colonic tissues from LSL-K-RasG12D/Klf5fl/+ (a), Villin-Cre/Klf5fl/+ (b), Villin-Cre/LSL-K-RasG12D (c), and Villin-Cre/LSL-K-RasG12D/Klf5fl/+ (d) mice after AOM treatment.
Figure 5.

Immunohistochemical staining of effector of ERK and Wnt signaling pathways. Colonic tissues of LSL-K-RasG12D/Klf5fl/+, Villin-Cre/Klf5fl/+, Villin-Cre/LSL-K-RasG12D, and Villin-Cre/LSL-K-RasG12D/Klf5fl/+ mice after injections with AOM were stained for KLF5 (A, G, M, and S), pMEK (B, H, N, and T), pERK1/2 (C, I, O, and U), cyclin D1 (D, J, P, and V), β-catenin Ser552 (E, K, Q, and W), and whole β-catenin (F, L, R, and X), respectively.
Discussion
Our studies indicate that Villin-Cre/LSL-K-RasG12D mice spontaneously develop hyperplastic intestinal polyps at a mature age. This is in concordance with previous observations that Villin-Cre/LSL-K-RasG12D mice developed hyper-proliferative regions within small intestinal epithelium and colon [9,15,19]. This phenotype is characterized by an increase in the number of mitotic figures, an increase in crypt heights and formation of hyperplastic polyps. Other studies, using different regulatory elements to drive K-Ras expression demonstrated no alteration of small intestinal epithelium during homeostasis [20,21]. Analysis of crypt height, mitotic index and apoptosis levels and the levels of cyclin D1 and β-catenin showed no significant differences [20]. On other hand, Janssen et al demonstrated that activation of KRAS in the intestinal epithelium leads formation of intestinal tumors and distress [7]. This murine model was characterized by increased levels of phosphorylated ERK1/2 [7]. In our model, alterations to the morphology of intestinal epithelium are concurrent with the activation of pMEK and pERK signaling, and increased levels of cyclin D1 and MKI67, however these did not induce formation of dysplasia or neoplasia. A similar observation regarding activation of MAPK signaling pathway and higher mitotic index was reported by Haigis et al [9]. Their studies confirmed our observation of a hyper-proliferative state within colonic tissues upon KRAS activation [9,15]. Bennecke et al showed that activation of oncogenic K-ras induces serrated hyperplasia and is characterized by p16ink4a overexpression and induction of senescence [22]. They reported increased mitotic index and phosphorylation of ERK in the small intestinal epithelium but not in colon [22]. We showed that the activation of KRAS from the endogenous regulatory elements does not affect WNT signaling effector, β-catenin. Thus, the increased levels of KLF5 observed in Villin-Cre/LSL-K-RasG12D mice in normal appearing mucosa and in the hyperplastic polyps are the direct results of activation of the KRAS pathway. Previous reports showed accelerated tumorigenesis in mice with activation of KRAS with simultaneous loss of Apc that led to decrease of survival [20]. Additionally, the authors observed dysregulation of KRAS pathway manifested by increased levels of pMEK and sporadic nuclear staining of pERK within adenomas [20]. Using a different tumorigenesis model, we showed that Villin-Cre/LSL-K-RasG12D mice are more susceptible to the treatment with AOM in comparison to the controls mice, and they succumbed within one week after the last treatment. This suggest that the hyper-proliferative and hyper-plastic phenotype observed during the homeostasis in Villin-Cre/LSL-K-RasG12D mice in the colonic tissue is further exuberated by carcinogen treatment that lead to animal death. In contrast to our data, previous report showed that AOM-treated mice with intestinal activation of KRAS developed colonic tumors but did not die due to AOM treatment [22]. However, Bennecke and colleagues did not show any morphological changes to the colonic epithelium during homeostasis, contrary to our data. Importantly, in our model reduction in the expression levels of Klf5 by fifty percent rescues animals with KRAS activation after carcinogen treatment. The reduction in KLF5 contributes to downregulation of pMEK and pERK activity without any impact on β-catenin levels of activity.
In summary, our data indicate that KLF5 is a major mediator of KRAS pathway during sporadic carcinogenesis and that the Villin-Cre/LSL-K-RasG12D mice can serve as a model for spontaneous intestinal polyposis and inducible colon tumorigenesis driven by oncogenic K-Ras from its endogenous locus.
Acknowledgements
The authors would like to thank Asma Nusrat (University of Michigan, Department of Pathology, Ann Arbor, MI) and Kenneth R. Shroyer (Stony Brook University, Department of Pathology, Stony Brook, NY) for help with tissue analysis and Amr Ghaleb (Stony Brook University, Department of Pathology, Stony Brook, NY) for help with data analysis. This work was supported by grants from National Institutes of Health (DK052230, DK093680, and CA084197).
Disclosure of conflict of interest
None.
References
- 1.McConnell BB, Ghaleb AM, Nandan MO, Yang VW. The diverse functions of Kruppel-like factors 4 and 5 in epithelial biology and pathobiology. Bioessays. 2007;29:549–557. doi: 10.1002/bies.20581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nandan MO, McConnell BB, Ghaleb AM, Bialkowska AB, Sheng H, Shao J, Babbin BA, Robine S, Yang VW. Kruppel-like factor 5 mediates cellular transformation during oncogenic KRAS-induced intestinal tumorigenesis. Gastroenterology. 2008;134:120–130. doi: 10.1053/j.gastro.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nandan MO, Yoon HS, Zhao W, Ouko LA, Chanchevalap S, Yang VW. Kruppel-like factor 5 mediates the transforming activity of oncogenic H-Ras. Oncogene. 2004;23:3404–3413. doi: 10.1038/sj.onc.1207397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nandan MO, Chanchevalap S, Dalton WB, Yang VW. Kruppel-like factor 5 promotes mitosis by activating the cyclin B1/Cdc2 complex during oncogenic Ras-mediated transformation. FEBS Lett. 2005;579:4757–4762. doi: 10.1016/j.febslet.2005.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011;12:594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 6.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 7.Janssen KP, el-Marjou F, Pinto D, Sastre X, Rouillard D, Fouquet C, Soussi T, Louvard D, Robine S. Targeted expression of oncogenic K-ras in intestinal epithelium causes spontaneous tumorigenesis in mice. Gastroenterology. 2002;123:492–504. doi: 10.1053/gast.2002.34786. [DOI] [PubMed] [Google Scholar]
- 8.Calcagno SR, Li S, Colon M, Kreinest PA, Thompson EA, Fields AP, Murray NR. Oncogenic K-ras promotes early carcinogenesis in the mouse proximal colon. Int J Cancer. 2008;122:2462–2470. doi: 10.1002/ijc.23383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, Settleman J, Giovannini M, Jacks T. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40:600–608. doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nandan MO, Ghaleb AM, McConnell BB, Patel NV, Robine S, Yang VW. Kruppel-like factor 5 is a crucial mediator of intestinal tumorigenesis in mice harboring combined ApcMin and KRASV12 mutations. Mol Cancer. 2010;9:63. doi: 10.1186/1476-4598-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McConnell BB, Bialkowska AB, Nandan MO, Ghaleb AM, Gordon FJ, Yang VW. Haploinsufficiency of Kruppel-like factor 5 rescues the tumor-initiating effect of the Apc(Min) mutation in the intestine. Cancer Res. 2009;69:4125–4133. doi: 10.1158/0008-5472.CAN-08-4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McConnell BB, Klapproth JM, Sasaki M, Nandan MO, Yang VW. Kruppel-like factor 5 mediates transmissible murine colonic hyperplasia caused by citrobacter rodentium infection. Gastroenterology. 2008;134:1007–1016. doi: 10.1053/j.gastro.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 14.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, Hingorani SR, Zaks T, King C, Jacobetz MA, Wang L, Bronson RT, Orkin SH, DePinho RA, Jacks T. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 15.Trobridge P, Knoblaugh S, Washington MK, Munoz NM, Tsuchiya KD, Rojas A, Song X, Ulrich CM, Sasazuki T, Shirasawa S, Grady WM. TGF-beta receptor inactivation and mutant Kras induce intestinal neoplasms in mice via a beta-catenin-independent pathway. Gastroenterology. 2009;136:1680–1688. e7. doi: 10.1053/j.gastro.2009.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong JT, Chen C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell Mol Life Sci. 2009;66:2691–2706. doi: 10.1007/s00018-009-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y, Goldstein BG, Nakagawa H, Katz JP. Kruppel-like factor 5 activates MEK/ERK signaling via EGFR in primary squamous epithelial cells. FASEB J. 2007;21:543–550. doi: 10.1096/fj.06-6694com. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg DW, Giardina C, Tanaka T. Mouse models for the study of colon carcinogenesis. Carcinogenesis. 2009;30:183–196. doi: 10.1093/carcin/bgn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng Y, Bommer GT, Zhao J, Green M, Sands E, Zhai Y, Brown K, Burberry A, Cho KR, Fearon ER. Mutant KRAS promotes hyperplasia and alters differentiation in the colon epithelium but does not expand the presumptive stem cell pool. Gastroenterology. 2011;141:1003–1013. e1–10. doi: 10.1053/j.gastro.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sansom OJ, Meniel V, Wilkins JA, Cole AM, Oien KA, Marsh V, Jamieson TJ, Guerra C, Ashton GH, Barbacid M, Clarke AR. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc Natl Acad Sci U S A. 2006;103:14122–14127. doi: 10.1073/pnas.0604130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim SH, Roth KA, Moser AR, Gordon JI. Transgenic mouse models that explore the multistep hypothesis of intestinal neoplasia. J Cell Biol. 1993;123:877–893. doi: 10.1083/jcb.123.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennecke M, Kriegl L, Bajbouj M, Retzlaff K, Robine S, Jung A, Arkan MC, Kirchner T, Greten FR. Ink4a/Arf and oncogene-induced senescence prevent tumor progression during alternative colorectal tumorigenesis. Cancer Cell. 2010;18:135–146. doi: 10.1016/j.ccr.2010.06.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.