

Abstract
Tripartite motif 31(TRIM31) is a new member of the E3 ubiquitin ligase family, which plays a role in many biological processes. It has been indicated that TRIM31 is strongly correlated with tumorigenesis. However, the impact of TRIM31 in colorectal cancer (CRC) and its underlying mechanisms are largely unknown. In this study, we found an increase in TRIM31 expression in CRC cells and a positive correlation between TRIM31 and CRC malignancy. Applying genetic interventions and cellular function tests, our results showed that TRIM31 acted as an epithelial-mesenchymal transition (EMT) inducer to promote CRC invasion and metastasis. Mechanical exploration revealed that TRIM31 mediated the upregulation of inflammatory cytokines TNF, IL-1β, and IL-6 through the NF-κB pathway, thus contributing to EMT in CRC progression. Collectively, we concluded that TRIM31 mediated chronic inflammation via the NF-κB pathway to promote invasion and metastasis in CRC. Therefore, we can infer that TRIM31 may be a valuable detective marker in primary CRC, and therapeutic intervention related to EMT suppression via TRIM31 can be a viable option for patients with advanced CRC.
Keywords: TRIM31, colorectal carcinoma, epithelial-mesenchymal transition, chronic inflammation, NF-κB pathway
Introduction
According to a survey conducted by the WHO in 2015, colorectal cancer (CRC) is the third most common malignancy worldwide. With the rapid development in diagnostic and treatment technologies in recent years, CRC can now be cured if detected early and treated adequately. The survival rate of CRC is ranked second among male (men) and third among female (women) among all types of cancer worldwide [1]. Although there are plenty of efficient therapies, such as surgery, neoadjuvant chemotherapy, and radiotherapy, CRC remains the third leading cause of cancer deaths, mostly due to recurrence and metastasis [2]. Advanced CRC is responsible for the high overall mortality rate. The 5-year survival rate in patients with stage IV CRC is 10%, which is a significant reduction from 90% in the patients with stage I CRC [3,4]. Therefore, screening and early intervention play a significant role in reducing mortality. Many challenges, including (discovering) new biomarkers, more effective therapies, and prevention of metastasis, remain.
Although the precise mechanism of metastasis in epithelial tumors is unclear, it has been reported that epithelial-mesenchymal transition (EMT) is associated with cancer progression and metastasis [5]. EMT is a biological and pathological process of epithelial cells changing phenotypically to mesenchymal cells, which plays a critical role in various processes such as embryogenesis, wound healing, organ fibrosis, and tumorigenesis [6]. During tumor progression, EMT is involved not only in migration and invasion, but also suppression of senescence and apoptosis, attenuation of cell cycle progression, as well as resistance to radiotherapy and chemotherapy [7]. In addition, all processes are mainly mediated by EMT-inducing transcription factors (EMT-TFs), controlling the expressions of target genes, non-coding RNAs, alternative splicing, and protein stability [7-9]. However, the specific mechanism that induces EMT in tumors remains unclear.
It is widely known that chronic inflammation is the key component of the tumor microenvironment acting on tumor progression [10] and there are many intricate cytokine networks contributing to carcinogenesis [11,12]. CRC, as a robust example, is consistent with this hypothesis. For instance, inflammatory bowel diseases (IBD), including Crohn’s disease and ulcerative colitis, are identified as independent risk factors for development of CRC [13,14]. It has been explicated that immune cells, inflammatory cytokines, as well as related signaling pathways, constitute a complex network, which induces progression in colon tumorigenesis [15]. However, the specific mechanism or molecule that can be targeted for prevention or treatment warrants further exploration.
The ubiquitin proteasome system (UPS) regulates ubiquitination for the degradation and turnover of intracellular proteins, playing a role in almost all biological processes, including transcriptional regulation, chromosomal control, tumorigenesis, and tumor suppression [16]. Ubiquitin ligase (E3) plays a fundamental role in ubiquitination by recruiting ubiquitin-conjugating enzyme (E2) and recognizing specific substrates through amino acid residues. Any aberration in UPS, especially the critical E3 ligase, may lead to clinically distinct phenotypes, including tumor progression. Tripartite motif (TRIM) 31 is a new member of the RING type E3 ubiquitin ligase family [16,17]. Recent reports have indicated that TRIM31 has diverse effects on the occurrence of tumors, such as tumor suppression in breast cancer [18] and non-small cell lung cancer [19], but tumor induction in gastric adenocarcinoma [20], hepatocellular carcinoma [21], and bladder cancer [22]. Many studies have shown that TRIM31 is highly expressed in the gastrointestinal tract [23]. Hence, it was inferred that TRIM31 might have an impact on CRC and clarification of the underlying mechanism was sought.
This study discusses the expression, biological function, and relevant mechanisms of TRIM31 in CRC. We found a high level of TRIM31 expression in CRC cells and patient samples. Through a series of cell function assays, it was found that TRIM31 acted as an EMT inducer, promoting CRC invasion and metastasis. Mechanical exploration revealed that TRIM31 mediated the upregulation of inflammatory cytokines TNF, IL-1β, and IL-6 through the NF-κB pathway, thus contributing to EMT in CRC progression. It was therefore concluded that TRIM31 regulated chronic inflammation via the NF-κB pathway to promote invasion and metastasis in CRC. TRIM31 can be deduced as a valuable detective marker in primary CRC samples, while therapeutic intervention related to EMT suppression via TRIM31 can be a viable option for patients with advanced CRC.
Materials and methods
Tissue specimens
Matched paired CRC tissue specimens and adjacent non-cancerous colorectal tissue specimens were obtained from the General Surgery Department at Zhongshan Hospital, Fudan University. The fresh tissues were fixed in 4% formalin and embedded in paraffin. Ethical approval for this study was obtained from the ethics committee of Zhongshan Hospital and written informed consent was obtained from each patient before the study.
Cell culture
Colorectal cancer cell lines including HT-29, SW 116, SW 620, SW 480, and embryonic kidney cell line HEK293T were used in this study, which were purchased from the Cell Bank of the Chinese Academy of Science (Shanghai, China). HT-29 cells and HEK293 cells were cultured in Dulbecco’s modified Eagle medium (DMEM), supplemented with 10% Fetal Bovine Serum (FBS) and 100 units/ml of penicillin-streptomycin, under an environment of 5% CO2 at 37°C. SW 116, SW 620, SW 480 cells were cultured in L-15 medium supplement with 10% FBS and 100 units/ml penicillin-streptomycin, under an environment of 0.038% CO2 at 37°C.
Immunohistochemical staining (IHC) and evaluation
Immunohistochemical analysis was carried out to assess the expression of TRIM31 in the paired CRC tissue specimens and adjacent non-cancerous colorectal tissue specimens. The Formalin-fixed paraffin-embedded (FFPE) specimens were sectioned at 6-μm thickness for IHC. The slides were incubated with primary antibodies TRIM31 (Proteintech, Cat# 215711-AP) at 4°C overnight. The sections were stained by incubating the slides in diaminobenzidine, followed by counterstaining with hematoxylin. All slides were analyzed using a bright field microscope.
Immunoreactivity was assessed independently by two pathologists blinded to the patient’s clinical and pathological data. All stainings were scored according to the ratio of positive staining cells and intensity of positive staining. The ratio was graded on a 1 to 4 scale (1: 0-5%; 2: 6-50%; 3: 51-75%; 4: 76-100%), and the intensity of staining was given a score between 1 and 4 (1, no staining; 2, weak staining; 3, moderate staining; 4, strong staining). A final score between 1 and 16 was given by combining the ratio and intensity score and further classified as negative (1-4), weakly positive (5-8), moderately positive (9-12), or strongly positive (>12).
TRIM31 overexpression and shRNA transfection
TRIM31 cDNA was cloned in pLenti vector and additional VSVG and Δ8.9 were used for recombinant lentivirus packaging. TRIM31 shRNA plasmid was obtained from the Institute of Biochemistry and Cell Biology, AIBS, CAS. Meanwhile, a scramble sequence was used as control. All transfections were performed with PEI reagent (Sigma-Aldrich, St. Louis, Missouri, USA).
Quantitative real-time PCR (qRT-PCR)
All RNA specimens were isolated from cultured cancer cells using Trizol (TRIzol) reagent (Invitrogen, Carlsbad, California, USA) and dissolved in diethylpyrocarbonate treated (DEPC) water. Synthesis of cDNA was performed using the Takara Reverse Transcription System Kit (Takara Biotechnology Co. Ltd., Japan). qRT-PCR reactions were performed by using the Sybr green premix kit (BioRad, Hercules, California, USA). GAPDH was used as a housekeeping gene. All reactions were reproduced in triplicate. The following primers were used in this assay: TRIM31: sense 5’-AACCTGTCACCATCGACTGTG-3’ and antisense 5’-TGATTGCGTTCTTCCTTACGG-3’; GAPDH-sense 5’-ACAACTTTGGTATCGTGGAAGG-3’ and antisense5’-GCCATCACGCCACAGTTTC-3.
Western blotting
Equal amounts of samples were separated by 10% SDS-PAGE and transferred to PVDF membranes. After blocking with 5% non-fat milk in TBST for 1 h, membranes were incubated with primary antibodies targeting TRIM31 or GAPDH overnight at 4°C, followed by incubation with HRP-conjugated secondary antibodies. Immunoreactive bands were detected with enhanced chemiluminescent HRP substrate (Millipore, Bollerica, Massachusetts, USA).
Cell proliferation assay
MTT assay was performed to evaluate cell proliferation activity. CRC cells with TRIM31 overexpression or knockdown were seeded at a density of 104 cells per well in 96-well plates. EGFP plasmid and shRNA control were used as control (?). After addition of 10 μl MTT (Sigma, Missouri, USA) to each well at 24 h intervals, cells were incubated at 37°C for 2 h. Then, 190 μl DMSO was added to each well followed by gentle vibration for 10 minutes. The proliferation status of cells was detected at 450 nm using a microplate reader. This assay was carried out in triplicate.
Transwell cell invasion assay
The invasion ability of CRC cells was determined with a Transwell chamber. 50 μl of matrigel (BD Bioscience, Franklin Lakes, New Jersey, USA) was added into the top chamber for 30 minutes at 37°C. Cells (1×104 cells/well) were resuspended in 100 μl of FBS-free medium for 24 h and placed in the upper chamber, whereas the lower chamber was filled with medium. Any non-invasive cells remaining on the upper side of the membrane were scraped off, and the cells adhering to the lower membrane were fixed with methanol and stained with crystal violet. The number was quantified and imaged under a light microscope (×200 magnification). Crystal violet staining was dissolved in 33% acetic acid and optical density was detected at 570 nm. This assay was carried out in triplicate.
Wound healing assay
The migration ability of CRC cells was determined by wound healing assay. Cells were cultured to approximately 80% confluence as a monolayer in 6-well dishes. Wounds were scratched using a new 200 μl pipette tip. After removing the detached cells with PBS, the wells were replenished by media without serum. Cells were monitored every 6 hours. Photos were taken using phase contrast microphotography (×200 magnification). This assay was carried out in triplicate.
Annexin V/PI assay
1×105 μl cells were harvested and resuspended in 200 μl binding buffer with 5 μl Annexin V for 15 minutes in the dark. Later, cells were resuspended in binding buffer with 5 μl PI and analyzed under flow cytometry on the 488-nm laser to evaluate for apoptosis and necrosis. This assay was carried out in triplicate.
Luciferase assay
The cells were transfected using LipofectAMINE-2000 (Invitrogen, Carlsbad, California, USA) according to the manufacturer’s instructions (protocol). Relative luciferase activity was detected with dual luciferase kit (Promega E1906) and calculated as the ratio of luciferase/renilla activity.
Statistical analysis
All relevant statistical data were analyzed by SPSS 21.0 (IBM, Chicago, Illinois, USA). Categorical variables were evaluated by the chi-squared test, and continuous variables were analyzed by the student’s t-test. Survival rates were analyzed by the Kalpan-Meier method. P values less than 0.05 were considered statistically significant.
Results
TRIM31 expression was upregulated in CRC cell linesand CRC patient samples
The expression of TRIM31 showed that TRIM31 was highly expressed in the colon (Figure 1A). We examined the expressions of TRIM31 in four CRC lines (SW480, SW 620, SW116, and HT-29) and non-CRC 293FT cells by western blot and qRT-PCR. TRIM31 was presented at a significantly higher level in all four CRC cell lines, especially in HT-29 and SW 620 (Figure 1B and 1C). For further confirmation, TRIM31 mRNA levels were compared between CRC and corresponding noncancerous tissues by qRT-PCR. As shown in Figure 1D, CRC tissues exhibited remarkably elevated TRIM31 expression compared to adjacent normal tissues.
Figure 1.
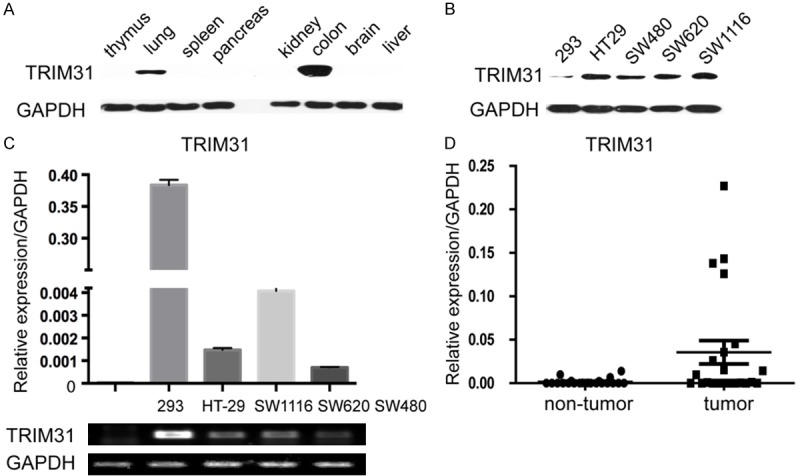
TRIM31 is highly expressed in CRC cell lines and CRC tissues. A. The Western blot analysis of TRIM 31 expression in different tissues of mice. B. Western blot analysis of TRIM 31 expression in four different CRC cell lines and 293 cell lines. Whole cell lysates (50 ug) were analyzed by Western blot and visualized by anti-TRIM31 antibody. C. Quantitative RT-PCR and semi-quantitative PCR analysis of TRIM 31 expression in four CRC cell lines, compared with 293 cell lines. Columns, mean of two independent experiments; bars, SD (n=3). (P<0.05, Student’s t test). D. Quantitative RT-PCR analysis of TRIM31 expression in 18 pairs of colorectal tumor and its corresponding normal tissues (P<0.05, Student’s t test). GAPDH was used as an internal control.
TRIM31 promotes migration and invasion capacity in CRC cells
Since TRIM31 is highly expressed in CRC patient samples and various CRC cell lines, further experiments were performed to explore its function during CRC tumorigenesis. Based on previous findings, SW480 and SW1116 had relatively low expression of TRIM31 (Figure 1B and 1C). In order to construct TRIM31 overexpression stable cell lines, TRIM31 expression plasmids were transfected into these two cell lines (Figure 2A). Meanwhile, Lentivirus-shRNA was used to infect SW620 and HT-29 cell lines to construct TRIM31 knockdown stable cell lines, in which TRIM31 expression was reduced significantly in both cell lines (Figure 3A). After carrying out a series of functional analyses involving proliferation, migration, invasion, apoptosis, and necrosis, it was confirmed that exogenous expression of TRIM31 in SW480 and SW116 cells reinforced both migration and invasion abilities (Figure 2C and 2D), but had no visible effect on proliferation, apoptosis or necrosis (Figure 2B and 2E). Results from the TRIM31 knockdown model were in line with prior findings, in which knockdown of TRIM31 attenuated cell migration and invasion (Figure 3C and 3D), while imposing minimal influence on proliferation, apoptosis, and necrosis (Figure 3B and 3E). Altogether, these results suggested that TRIM31 enhanced tumor migration and invasion of CRC.
Figure 2.
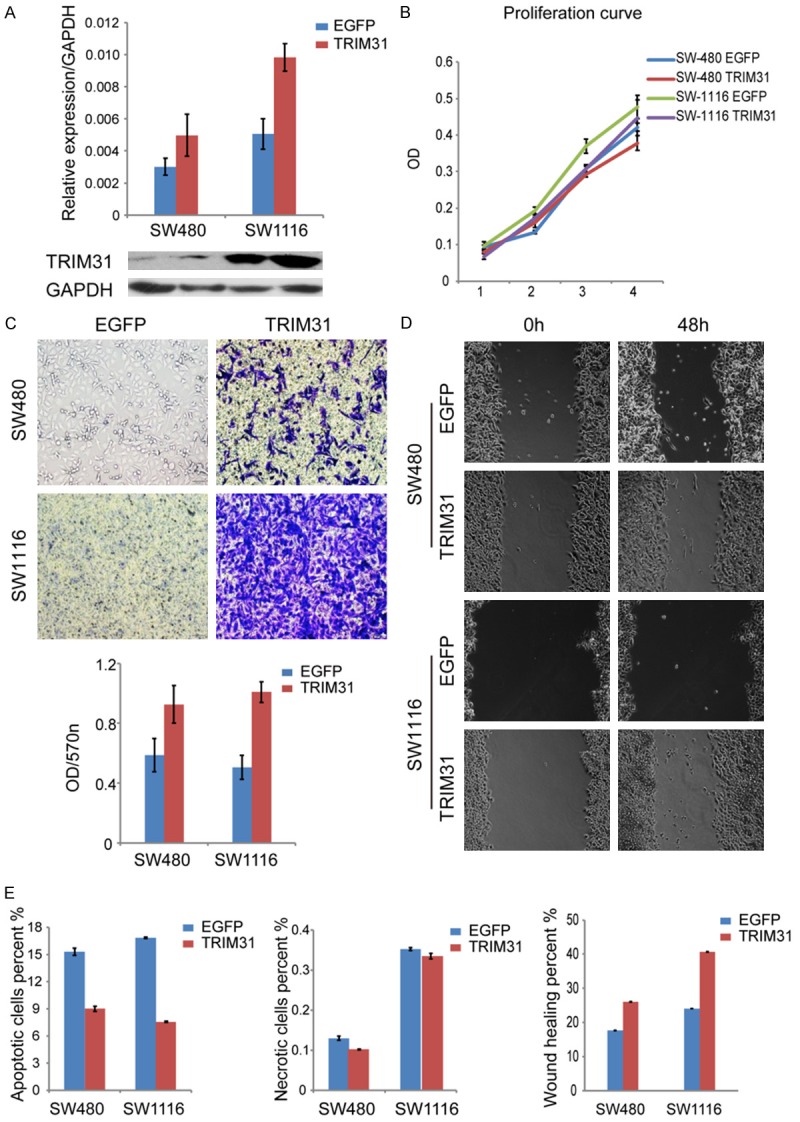
TRIM31 overexpression induced migration and invasion in CRC cells. A. TRIM31 expression plasmids were stably transfected into SW480 and SW1116 cell lines. Efficiency of overexpression in these two cell lines was evaluated by Western blotting and quantitative RT-PCR analysis (P<0.05, Student’s t test). EGFP plasmids were used for control. B. MTT assay was used to evaluate cell proliferation activity. Cells were treated with MTT and DMSO and detected at 450 nm using a microplate reader. MTT array showed no obvious difference in proliferation when TRIM31 was overexpressed in these two cell lines. (Cell density: 1×101脳104 per well). per well). (P>0.05, Student’s t test). C. Matrigel invasion assay showed that TRIM31 overexpression increased invasive capacity. The number was quantified and imaged under a light microscope (magnification: ×200). The graph below showed absorbance at 570 nm after 24 hours (P<0.05, Student’s t test). D. Wound-healing assay represented the migration ability of the cell lines. Wounds were scratched using a new 200 μl pipette tip, and cells were monitored every 6 hours. Photos were taken using phase contrast microphotography (magnification: ×200). Wound-healing assay showed that TRIM31 overexpression increased motility of CRC cells. The graph below shows mean opening distances after 48 hours (P<0.05, Student’s t test). E. Annexin V/PI assay evaluated apoptosis and necrosis in cells. Cells were treated with Annexin V and PI and analyzed under flow cytometry on the 488-nm laser. The data showed no obvious difference in apoptosis and necrosis when TRIM31 was overexpressed (P>0.05, Student’s t test). Data are shown as mean ± SD (n=3).
Figure 3.

TRIM31 knockdown inhibited migration and invasion in CRC cells. A. SW620 and HT-29 cell lines were transfected with shRNA TRIM31 (sh1 or sh2) or control shRNA (shNC). Knockdown efficiency was analyzed by Western blotting and quantitative RT-PCR analysis (P<0.05, Student’s t test). B. MTT assay showed no obvious difference in proliferation when TRIM31 was knocked down (P>0.05, Student’s t test). C. Matrigel invasion assay showed that TRIM31 knockdown decreased invasive capacity. (P<0.05, Student’s t test). D. Wound-healing assay showed that TRIM31 knockdown decreased motility of CRC cells. (P<0.05, Student’s t test). E. Annexin V/PI assay showed no obvious difference in apoptosis and necrosis when TRIM31 was knocked down (P>0.05, Student’s t test). Data are shown as mean ± SD (n=3).
TRIM31 promotes EMT via the NF-κB signaling pathway
Since EMT plays a vital role in tumorigenesis, the relationship between TRIM31 and EMT in CRC cells warranted further investigation. Biomarkers representing EMT were used to identify whether TRIM31 expression in CRC was related to EMT. Our data showed that high levels of TRIM31 induced the expression of the mesenchymal markers N-cadherin (CDH2) and Fibronectin, but inhibited the expressions of the epithelial marker E-cadherin (CDH1) and ZO1 (Figure 4A). TRIM31 knockdown cell lines had the opposite result (Figure 4B). Therefore, we inferred that TRIM31 was a critical promoter of EMT.
Figure 4.

TRIM31 overexpression induced EMT through activating the NF-κB signal pathway in CRC cells. A. Quantitative RT-PCR assay showed that TRIM31 overexpression induced EMT in CRC cells, characterized by an increase in epithelial markers such as E-cadherin, ZO1 and a decrease in mesenchymal markers such as N-cadherin and Fibronectin (P<0.05, Student’s t test). B. Quantitative RT-PCR assay showed that TRIM31 knockdown inhibited EMT reversely in CRC cells (P<0.05, Student’s t test). C. Dual luciferase assay showed that TRIM31 overexpression activated NF-κB signaling in HT-29 cells, whereas Wnt signaling was repressed (P<0.05, Student’s t test). * NFκB and IFNβ are the promoter plasmids commonly used for NF-κB signaling; Gli is the promoter plasmid commonly used for Gli signaling; CAGA and 3’TP are the promoter plasmids commonly used for TGFβ signaling; TOP/FOP is the promoter plasmid commonly used for Wnt signaling. Data are shown as mean ± SD (n=3).
To further elucidate the molecular mechanism through which excess TRIM31 induced EMT activating phenotype, we examined multiple signaling pathways, possibly being modulated. A prominent increase in the NF-κB signal pathway with overexpression of TRIM31 was observed, with a significant decrease in the Wnt pathway and no difference in Gli and TGFβ signaling (Figure 4C). In summary, the results above suggested that TRIM31 activated the NF-κB signal pathway to regulate EMT.
TRIM31 mediates upregulation of cytokines IL-1β and IL-6 to induce the NF-κB signal pathway in EMT
To ascertain the inflammatory cytokines influenced by TRIM31, we further analyzed cytokines that had been reported to induce the NF-κB pathway, among which three had significant changes. TRIM31 overexpression enhanced the secretion of cytokines TNF, IL-1β, and especially IL-6, whereas knockdown inhibited these three cytokines, especially IL-1β (Figure 5A and 5B). All these observations clarified that TRIM31 mainly mediated upregulation of cytokines TNF, IL-1β, and IL-6, which induced the NF-κB pathway and thus contributed to EMT in CRC progression.
Figure 5.
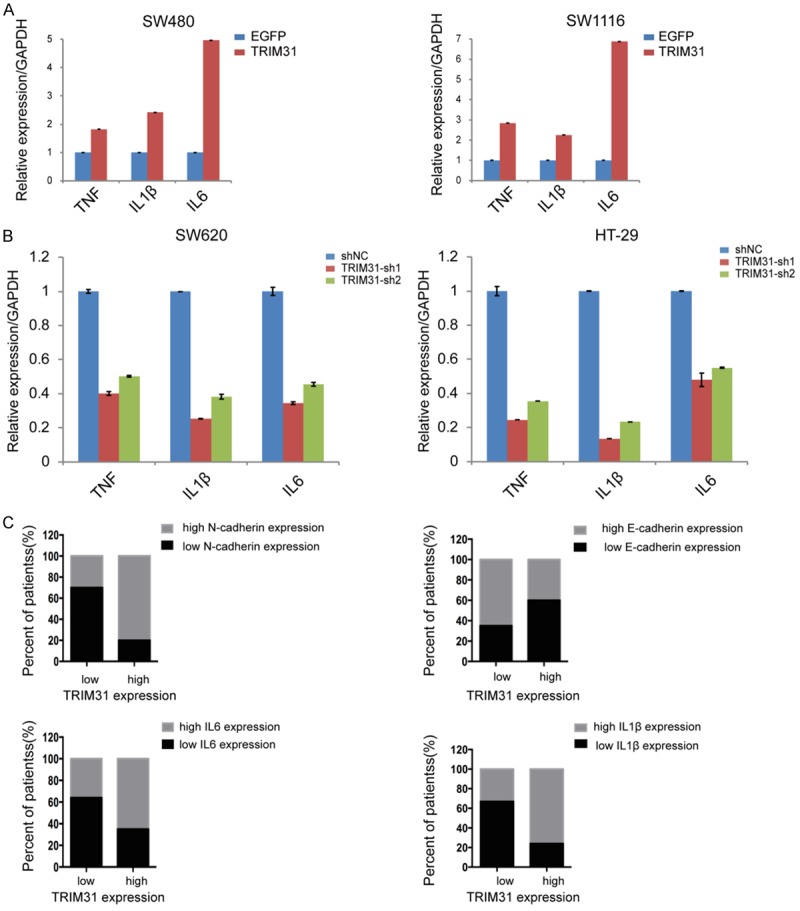
TRIM31 activated inflammatory cytokines to induce the NF-κB signal pathway in EMT. A. Quantitative RT-PCR assay showed that TRIM31 overexpression in SW480 and SW1116 cell lines significantly enhanced cytokines TNF, IL-1β, IL-6, and IL-6 (P<0.05, Student’s t test). B. Quantitative RT-PCR assay showed that TRIM31 knockdown in SW620 and HT029 cell lines inhibited the expression of three cytokines, while IL-1β had a significant decrease (P<0.05, Student’s t test). C. Quantitative RT-PCR analysis showed that the expression of TRIM31 is positively correlated with EMT and cytokines IL-1β and IL-6 activating NF-κB pathway in CRC tissues (n=87) (P<0.05, Student’s t test). Data in A and B are shown as mean ± SD (n=3).
TRIM31 expression is positively correlated with metastasis and poor prognosis in CRC
Though we have substantiated that TRIM31 was excessively expressed in CRC cells and pathological specimens while displaying certain effects on the function of tumor cell lines, however, the clinical feature of TRIM31 still has not been elucidated. We then tested TRIM31 mRNA expression levels in clinical specimens including all four pathological stages of CRC. We found a positive correlation in CRC progression with pathological staging (Figure 6A). In addition, we combined tissue microarray from 87 pairs of CRC specimens and corresponding normal colorectal mucosal tissues with clinical data. High expression levels of TRIM31 were found in almost half of the samples, 44 out of 87 (Table 1). The results demonstrated that TRIM31 expression had a positive correlation with CRC invasion, metastasis, and TNM staging (Figure 6A and Table 1). Kaplan-Meier survival analysis revealed CRC with high levels of TRIM31 had a shorter mean survival time (Figure 6B). The images of TRIM31 IHC staining also strengthened the positive correlation between TRIM31 and CRC malignancy (Figure 6C). Altogether, these data confirmed that TRIM31 was a significant inducer in CRC, which facilitated tumor progression in clinical patients.
Figure 6.
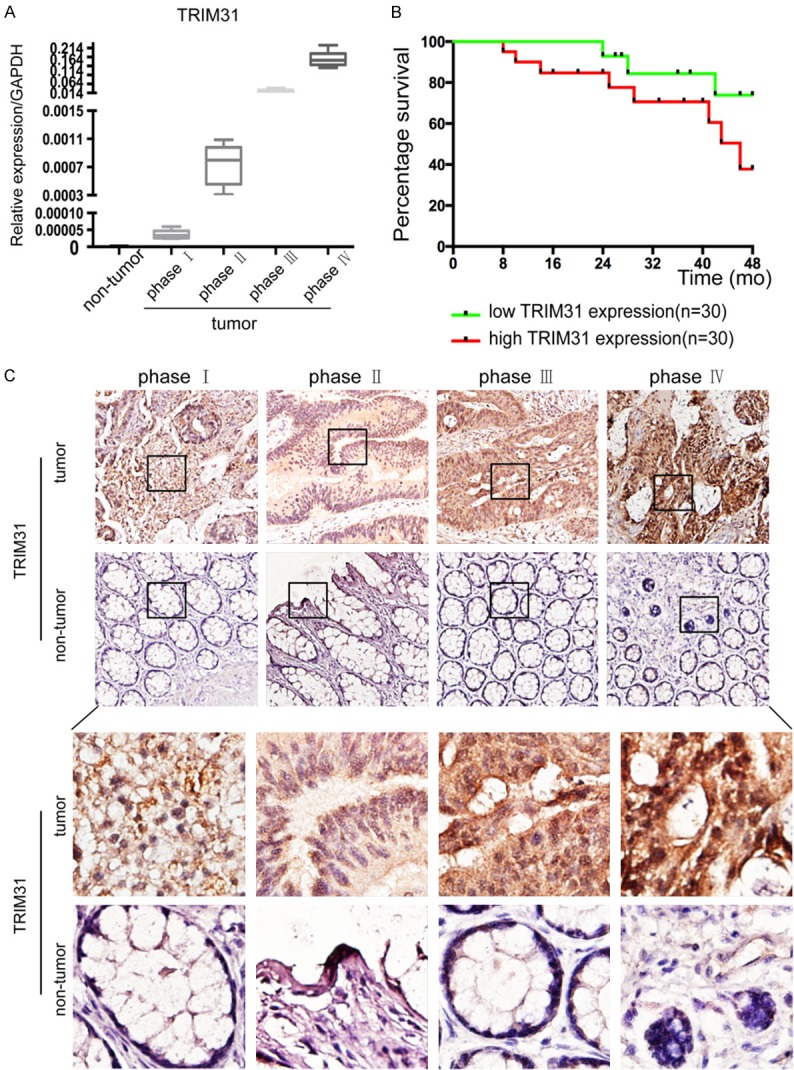
TRIM31 expression is positively correlated with clinical staging in CRC patients and is an independent indicator for poor survival. A. Quantitative RT-PCR analysis showed that TRIM31 expression increased markedly as CRC progressed (P<0.05, Student’s t test). B. Kaplan-Meier survival analysis according to TRIM31 expression in 60 CRC patients (P<0.05, log-rank test). C. The paired CRC samples and adjacent non-cancerous colorectal tissue specimens were sectioned at 6-μm thickness after being fixed in formalin and embedded in paraffin. The slides were incubated with primary anti-TRIM31 antibodies at 4°C overnight. The sections were stained by incubating the slides in diaminobenzidine, followed by counterstaining with hematoxylin. All slides were analyzed using a bright field microscope. Representative immunohistochemistry images showed the expression of TRIM31 enhanced as CRC progressed. (Previous row: original magnification ×200; next row: original magnification ×400).
Table 1.
Clinical pathologic correlation analysis of TRIM31 expression in different stages of human colorectal carcinoma
| Feature | Trim31 | χ2 | P values | |
|---|---|---|---|---|
|
| ||||
| Low | High | |||
| All cases | 43 | 44 | ||
| Gender | 0.3118 | 0.577 | ||
| Male | 26 | 24 | ||
| Female | 17 | 20 | ||
| Age | 0.0975 | 0.755 | ||
| >65 | 23 | 25 | ||
| ≤65 | 20 | 19 | ||
| Intravascular cancer embolus | 7.380 | 0.007 | ||
| Present | 8 | 13 | ||
| Absent | 35 | 31 | ||
| Perineuronal invasion | 4.261 | 0.039 | ||
| Present | 8 | 17 | ||
| Absent | 35 | 27 | ||
| Tumor size (cm) | 5.678 | 0.017 | ||
| >4 | 10 | 21 | ||
| ≤4 | 33 | 23 | ||
| T stage | 14.541 | 0.002 | ||
| T1 | 5 | 0 | ||
| T2 | 15 | 5 | ||
| T3 | 9 | 12 | ||
| T4 | 14 | 27 | ||
| N stage | 14.242 | 0.001 | ||
| N0 | 21 | 6 | ||
| N1 | 12 | 14 | ||
| N2 | 10 | 24 | ||
| M stage | 10.694 | 0.001 | ||
| M0 | 41 | 30 | ||
| M1 | 2 | 14 | ||
| Tumor stage | 25.513 | 0.000 | ||
| I | 8 | 0 | ||
| II | 17 | 5 | ||
| III | 16 | 25 | ||
| IV | 2 | 14 | ||
Notes: Clinical pathologic correlation analysis of TRIM31 expression in different stages of human colorectal carcinoma (P<0.05, Pearson’s chi-square test). The median expression level was used as the cutoff.
Discussion
As metastasis is the chief culprit for high mortality rates in CRC, it is critical to find new biomarkers or therapeutic targets for CRC progression. In our study, we revealed that TRIM31 regulated chronic inflammation via the NF-κB pathway to promote EMT in CRC. This finding poses TRIM31 as a potentially significant detective marker or therapeutic target, which may be applied to prevention and treatment of advanced CRC.
Since E3 enzymes perform a critical role in ubiquitination and is associated with protein degradation, interactions, and protein localization, any aberrancies may lead to abnormal cell processes and perhaps result in oncogenesis [16,24]. Therefore, E3 ligase is considered to be an efficient target for anticancer agents such as nutlins, an MDM2 antagonist, which can enhance p53 function and induce p53-related apoptosis, ultimately inhibiting the malignancies of different cancers [25,26]. Similarly, TRIM31, as an E3 enzyme, may be a potential biomarker or an efficient target of CRC. Our results confirmed that TRIM31 was upregulated in CRC cells and clinical samples.
Epithelial cells undergoing EMT detach from the primary site and have the potential to disseminate hematologically, which promotes tumor invasion and metastasis [5]. By analyzing the plasticity biomarkers of EMT, our results were consistent with the hypothesis that TRIM31 promoted CRC invasion and metastasis by inducing EMT.
Recent studies have determined that stress associated with tumorigenesis, such as inflammation, hypoxia, and oncogenesis, activates EMT by triggering numerous signaling pathways known to play critical roles in both embryogenesis and tumor development, including Wnt, TGF-β, and the NF-κB pathway [27]. TRIM31 belongs to TRIM subfamily V, along with TRIM 8 and TRIM40, which have been confirmed to be associated with inflammation-associated tumorigenesis in specific signaling pathways [17]. To clarify the specific signaling pathways and the underlying inflammatory cytokines involved in CRC controlled by TRIM31, we screened several relevant signaling pathways and found overactivation of the NF-κB signal pathway. In this process, there was also an attenuation in the Wnt signal pathway, of which the related mechanism has not been clarified. Previous findings showed that there was close interaction between the NF-κB and Wnt pathways, occurring during EMT mediation by E-cadherin and Fibronectin [12,28]. Hence, further studies are needed to clarify the potential roles of Wnt in the development of EMT.
The NF-κB pathway is a complex signaling pathway that regulates oncogenesis by mediating the expression of various target genes involved in tumor maintenance and progression [29]. It has been reported that inflammatory cytokines, such as TNFα, IL-1β, and IL-6 are the main inducers involved in the NF-κB pathway and have an effect on EMT in cancer progression [9,30]. Further experiments confirmed our hypothesis that TRIM31 induced TNF and IL-1β as well as IL-6 upregulation and had a markedly positive correlation with EMT. As a result, we concluded that TRIM31 mediated upregulation of chronic inflammation induced by the NF-κB pathway in EMT and promoted invasion and metastasis of CRC cells. The retrospective analysis of clinical data firmly supports the effect of TRIM31 in CRC.
Although more systemic chemotherapy and targeted drugs have been widely available in clinical practice, their effectiveness is often accompanied by side effects and drug resistance. Thus, new therapies are constantly being sought. Our results demonstrate that excessive TRIM31 expression in CRC mediates EMT via activation of the NF-κB pathway triggered by inflammatory cytokines, promoting CRC invasion and metastasis. Therefore, we can infer that TRIM31 may be a valuable detective marker or a regulatory target for EMT progression in CRC. Further studies are warranted to explore the detailed mechanism of TRIM31 in advanced CRC and EMT-related pathogenesis.
Acknowledgements
We would like to thank Prof. Zou (Institute of Biochemistry and Cell Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences) for providing valuable suggestions during our research. We are also grateful for access to instruments and technical support provided by the Zou Lab.
Disclosure of conflict of interest
None.
References
- 1.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66:271–289. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 2.El-Shami K, Oeffinger KC, Erb NL, Willis A, Bretsch JK, Pratt-Chapman ML, Cannady RS, Wong SL, Rose J, Barbour AL, Stein KD, Sharpe KB, Brooks DD, Cowens-Alvarado RL. American cancer society colorectal cancer survivorship care guidelines. CA Cancer J Clin. 2015;65:428–455. doi: 10.3322/caac.21286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brody H. Colorectal cancer. Nature. 2015;521:S1. doi: 10.1038/521S1a. [DOI] [PubMed] [Google Scholar]
- 4.Byers T, Wender RC, Jemal A, Baskies AM, Ward EE, Brawley OW. The American cancer society challenge goal to reduce US cancer mortality by 50% between 1990 and 2015: results and reflections. CA Cancer J Clin. 2016;66:359–369. doi: 10.3322/caac.21348. [DOI] [PubMed] [Google Scholar]
- 5.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 8.Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19:1438–1449. doi: 10.1038/nm.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karin M. NF-kappaB and cancer: mechanisms and targets. Mol Carcinog. 2006;45:355–361. doi: 10.1002/mc.20217. [DOI] [PubMed] [Google Scholar]
- 10.Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med. 2010;10:369–373. doi: 10.2174/156652410791316968. [DOI] [PubMed] [Google Scholar]
- 11.Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD. Complement in cancer: untangling an intricate relationship. Nat Rev Immunol. 2018;18:5–18. doi: 10.1038/nri.2017.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du Q, Geller DA. Cross-regulation between wnt and NF-κB signaling pathways. For Immunopathol Dis Therap. 2010;1:155–181. doi: 10.1615/ForumImmunDisTher.v1.i3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.So J, Tang W, Leung WK, Li M, Lo FH, Wong MTL, Sze ASF, Leung CM, Tsang SWC, Shan EHS, Chan KH, Lam BCY, Hui AJ, Chow WH, Lam TY, Lam V, Lee TW, Lo HHH, Tang CM, Wong CL, Wu JCY, Chan FKL, Sung JJY, Harbord M, Ng SC. Cancer risk in 2621 chinese patients with inflammatory bowel disease: a population-based cohort study. Inflamm Bowel Dis. 2017;23:2061–2068. doi: 10.1097/MIB.0000000000001240. [DOI] [PubMed] [Google Scholar]
- 14.Increased risk of cancer in children with inflammatory bowel disease. BMJ. 2017;358:j4285. doi: 10.1136/bmj.j4285. [DOI] [PubMed] [Google Scholar]
- 15.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138:2101–2114. e2105. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 16.Micel LN, Tentler JJ, Smith PG, Eckhardt GS. Role of ubiquitin ligases and the proteasome in oncogenesis: novel targets for anticancer therapies. J. Clin. Oncol. 2013;31:1231–1238. doi: 10.1200/JCO.2012.44.0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11:792–804. doi: 10.1038/nrc3139. [DOI] [PubMed] [Google Scholar]
- 18.Dokmanovic M, Chang BD, Fang J, Roninson IB. Retinoid-induced growth arrest of breast carcinoma cells involves co-activation of multiple growth-inhibitory genes. Cancer Biol Ther. 2002;1:24–27. doi: 10.4161/cbt.1.1.35. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Zhang Y, Zhang Y, Bai X, Peng Y, He P. TRIM31 is downregulated in non-small cell lung cancer and serves as a potential tumor suppressor. Tumour Biol. 2014;35:5747–5752. doi: 10.1007/s13277-014-1763-x. [DOI] [PubMed] [Google Scholar]
- 20.Sugiura T, Miyamoto K. Characterization of TRIM31, upregulated in gastric adenocarcinoma, as a novel RBCC protein. J Cell Biochem. 2008;105:1081–1091. doi: 10.1002/jcb.21908. [DOI] [PubMed] [Google Scholar]
- 21.Guo P, Ma X, Zhao W, Huai W, Li T, Qiu Y, Zhang Y, Han L. TRIM31 is upregulated in hepatocellular carcinoma and promotes disease progression by inducing ubiquitination of TSC1-TSC2 complex. Oncogene. 2018;37:478–488. doi: 10.1038/onc.2017.349. [DOI] [PubMed] [Google Scholar]
- 22.Zhang DQ, Zhou CK, Chen SZ, Yang Y, Shi BK. Identification of hub genes and pathways associated with bladder cancer based on co-expression network analysis. Oncol Lett. 2017;14:1115–1122. doi: 10.3892/ol.2017.6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe M, Tsukiyama T, Hatakeyama S. TRIM31 interacts with p52(Shc) and inhibits Src-induced anchorage-independent growth. Biochem Biophys Res Commun. 2009;388:422–427. doi: 10.1016/j.bbrc.2009.08.028. [DOI] [PubMed] [Google Scholar]
- 24.Buetow L, Huang DT. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat Rev Mol Cell Biol. 2016;17:626–642. doi: 10.1038/nrm.2016.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol. 2017;10:133. doi: 10.1186/s13045-017-0500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valente LJ, Aubrey BJ, Herold MJ, Kelly GL, Happo L, Scott CL, Newbold A, Johnstone RW, Huang DC, Vassilev LT, Strasser A. Therapeutic response to non-genotoxic activation of p53 by Nutlin3a is driven by PUMA-Mediated apoptosis in lymphoma cells. Cell Rep. 2016;14:1858–1866. doi: 10.1016/j.celrep.2016.01.059. [DOI] [PubMed] [Google Scholar]
- 27.Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16:488–494. doi: 10.1038/ncb2976. [DOI] [PubMed] [Google Scholar]
- 28.Ma B, Hottiger MO. Crosstalk between Wnt/β-catenin and NF-κB signaling pathway during inflammation. Front Immunol. 2016;7:378. doi: 10.3389/fimmu.2016.00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang S, Liu Z, Wang L, Zhang X. NF-kappaB signaling pathway, inflammation and colorectal cancer. Cell Mol Immunol. 2009;6:327–334. doi: 10.1038/cmi.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15:416–428. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]