


Abstract
Alternative first exons diversify the transcriptomes of eukaryotes by producing variants of the 5′ Untranslated Regions (5′UTRs) and N-terminal coding sequences. Accurate transcriptome-wide detection of alternative first exons typically requires specialized experimental approaches that are designed to identify the 5′ ends of transcripts. We developed a computational pipeline SEASTAR that identifies first exons from RNA-seq data alone then quantifies and compares alternative first exon usage across multiple biological conditions. The exons inferred by SEASTAR coincide with transcription start sites identified directly by CAGE experiments and bear epigenetic hallmarks of active promoters. To determine if differential usage of alternative first exons can yield insights into the mechanism controlling gene expression, we applied SEASTAR to an RNA-seq dataset that tracked the reprogramming of mouse fibroblasts into induced pluripotent stem cells. We observed dynamic temporal changes in the usage of alternative first exons, along with correlated changes in transcription factor expression. Using a combined sequence motif and gene set enrichment analysis we identified N-Myc as a regulator of alternative first exon usage in the pluripotent state. Our results demonstrate that SEASTAR can leverage the available RNA-seq data to gain insights into the control of gene expression and alternative transcript variation in eukaryotic transcriptomes.
INTRODUCTION
Alternative transcription initiation is a major mechanism for diversifying the human transcriptome and generating tissue specific mRNA variants (1,2). Transcription initiations at alternative promoters follow two patterns (Figure 1A): initiations from distant promoters produce alternative first exons (AFE), while promoters located in close proximity typically transcribe the same exon from alternative tandem transcription start sites (TSS) producing tandem 5′UTRs. Alternative transcription initiation may change open reading frames (ORFs) of transcripts and result in novel proteins (3) or alternative protein isoforms with different N-terminal peptide sequences (4). It may also produce mRNA isoforms that code for the same protein product but with distinct 5′UTR sequences, that differ in their mRNA stability or translational efficiency (5,6). Alternative transcription initiation has been associated with transcriptome variation during development and cell differentiation (7–11).
Figure 1.

The SEASTAR pipeline for the computational identification and quantitative analysis of first exons using RNA-seq data alone. (A) Alternative transcription start sites (TSSs) can appear in two forms: alternative first exons (AFEs) and alternative tandem TSSs. (B) The reference guided transcript assembly: the reference annotation based transcript (RABT) assembly method is used to assemble novel transcripts using RNA-seq reads guided by the existing transcriptome annotation. (C) The generation of non-redundant first exons (FEs): transcripts from all samples are merged to generate a non-redundant set of FEs. (D) The quantitation of exon and splice junction coverage: reads mapped to each FE and its downstream splice junction are counted as the coverage for each FE. (E) The identification of bona fide FEs: five methods are designed and compared. The logistic regression model (highlighted in bold) is selected as the method of choice in SEASTAR due to its superior performance. (F) The detection of differential AFE usage: the percent-spliced-in (PSI) value for each AFE in each sample is calculated using the read counts and effective lengths of all AFEs within the gene. The rMATS statistical test is used to determine whether the AFE has significant differential usage between two samples or two groups of samples.
Conventionally, identification of transcription start sites and alternative first exons on a genomic scale requires specialized experimental methods, such as CAGE and 5′-RACE, that capture the true 5′ ends of polymerase II transcripts (12,13). These approaches involve elaborate experimental procedures and are not routinely used in the characterization of cellular transcriptomes. By contrast, RNA-seq data for diverse organisms, tissues, and cell types are straightforward to produce and are abundant in public repositories. Consequently, there is significant interest in utilizing RNA-seq data to identify AFEs and quantify their expression. In principle, AFEs can be identified and analyzed from RNA-seq data either by mapping sequenced reads to the existing transcriptome annotation or by carrying out de-novo transcriptome assembly (14–16). However, methods based on existing annotations alone cannot detect novel AFEs, while methods based on de-novo assembly may have false positives due to a variety of technical issues related to read coverage and bias in read distribution (17). Additionally, the discovery of AFEs should be coupled with quantitative analysis to determine differential AFE usage in specific biological conditions or cellular states. Therefore, a specialized and streamlined tool is needed for comprehensive discovery and quantitative analysis of AFEs using RNA-seq data.
Here, we describe a computational pipeline SEASTAR (Systematic Evaluation of Alternative Transcription Start Sites in RNA), designed to identify alternative first exons and alternative tandem TSSs and quantify their expression levels. SEASTAR uses a logistic regression method to reliably identify first exons (FEs), including novel exons that are not present in the current transcriptome annotation. Rigorous statistical comparison is then applied to quantify and compare AFE usage across distinct biological conditions. By benchmarking SEASTAR against a ‘gold standard’ dataset from CAGE experiments we show that it accurately predicts the positions of FEs. The FEs identified by SEASTAR carry RNA POL2 signals as well as epigenetic marks specific to active promoters, including enrichment for H3K4 trimethylation (H3K4me3) and H3K27 acetylation (H3K27ac) and depletion of H3K36 trimethylation (H3K36me3). Finally, we illustrate the utility of SEASTAR in investigating the regulation of gene expression and AFE usage by applying it to a time-course RNA-seq dataset during the reprogramming of mouse embryonic fibroblasts (MEFs) into induced pluripotent stem cells (iPSCs).
MATERIALS AND METHODS
Reconstructing first exons and quantifying their usage
The SEASTAR pipeline is composed of multiple steps (Figure 1). The first step is to reconstruct all putative first exons (FEs) by transcript assembly from the RNA-seq data (Figure 1B). The assembly is guided by the existing transcriptome annotation so users need to choose a transcriptome annotation database (e.g. Homo sapiens.Ensembl.GRCh37.72 or Mus musculus.Ensembl.NCBIM38.72), in addition to providing the aligned RNA-seq data (e.g. BAM files). We adopt the Reference Annotation Based Transcript (RABT) assembly method (18) implemented by Cufflinks (version 2.2.0, downloaded from http://cufflinks.cbcb.umd.edu/) to assemble transcript isoforms for each sample (Figure 1B). Then we merge the RABT annotation files (e.g. GTF files) of all samples to produce a complete annotation of putative FEs in the entire dataset. Overlapping putative FEs that share the same downstream 5′ splice sites are merged into the longest exon in the annotation to generate a non-redundant set of first exons (Figure 1C). We use the term FEs to represent non-redundant first exons in the remaining part of the paper. An optional choice is provided to merge only the FEs whose TSSs are within a certain user-defined distance (e.g. <100 bp), as a criterion used in the processing of CAGE data (19,20). Following the filtering criteria in CAGE analysis (19,20), putative FEs overlapping with internal exons of other annotated transcripts are discarded from downstream analyses, as such FEs are often artifacts due to recapping instead of transcription (19).
Next, we quantify the usage of FEs by counting reads that are aligned to the exons and their downstream splice junctions (Figure 1D). We take the counts as the measure for the expression levels of FEs.
Identifying bona fide first exons
Some of the reconstructed putative FEs may reflect artifacts and noise in the RNA-seq data. We designed and tested five different methods for identifying bona fide FEs in the data, and compared them with results from CAGE to select the most reliable method as the default method in our pipeline (Figure 1E).
The first method is ‘Poisson test’. We compare the read count of an FE with the read count of its surrounding genomic region (of the same length as the FE) based on a Poisson model. The purpose is to test whether the probability of reads aligned to the FE is no more than that to its flanking genomic regions. We assume the distribution of read coverage follows the Poisson model. For each FE, we use its read count in a given sample to calculate the mean of the Poisson distribution. For its flanking regions, we take the adjacent upstream region and downstream region, each with the same length as the FE, and count the reads mapped to these regions. We compare the read counts of the FE with the two flanking regions using edgeR (v 3.4.2) (21), in which we use the Trimmed Mean of M-value (TMM) method (22) to normalize each sample and the Generalized Linear Model (GLM) method (23) to perform the likelihood ratio test. A candidate FE is called as a bona fide FE if the read count of the FE is larger than those of its upstream region and downstream region, and if the P-values of both comparisons are smaller than a given threshold.
The second method is ‘Negative Binomial test’. It uses the same strategy as the first method of Poisson test, except that we assume the distribution of read coverage on each region to follow the Negative Binomial (NB) model. We estimate the dispersion in the NB model using the Cox-Reid profile-adjusted likelihood (CR) method (23) in edgeR.
The third method is ‘Exon coverage’. We count the reads mapped to each candidate FE. A candidate FE is called as a bona fide FE if the read count is larger than a given threshold.
The fourth method is ‘Splice junction coverage’. For each FE, we count the reads mapped to its downstream splice junction as the splice junction coverage. A candidate FE is identified as a bona fide FE if the splice junction coverage is larger than a given threshold.
- The fifth method is ‘Logistic regression model’. It is a machine learning approach. The main idea is to apply a logistic regression model by combining the read coverage in methods (3) and (4). We first apply the principal component analysis (PCA) on the read coverage of methods (3) and (4) to reduce the correlation of these two measurements. Then the two principal components are used as the input of the logistic regression model. In the logistic model the combined metric is the probability that the FE is present in a given sample. All components are combined by a logistic function (Eq. 1):
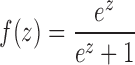
(1)
and
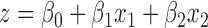 |
(2) |
where x1 and x2 are the two principal components derived from the candidate FE’s exon coverage and splice junction coverage (Eq. 2). The coefficients are determined by fitting the model on training datasets to achieve the maximum true positive rate (TPR) with the acceptable false positive rate (FPR) (e.g. 5%).
We collect the training datasets from samples with both CAGE data and RNA-seq data. On the training data, we use the function ‘glm’ in R (version 3.0.2) to regress and calculate the f(z) value by Wald statistics (implemented by the function ‘glm’). The parameters of the regression model, including the coefficients and their statistical significance, are provided in Supplementary Tables S1 and S2.
For a candidate FE, we calculate the logistic probability using the logistic function. A candidate FE with a high logistic probability is called as a bona fide FE. We select the cutoff of the logistic probability as 0.909 to control the FPR at <0.05, based on the result trained and tested by the H1-hESC nuclear data (see below).
True and false positive rate estimation for the identification of first exons
We assess the performance of the five methods above using CAGE data covering multiple cell types (see below). For each cell type, we take the TSSs detected in CAGE data as the reference positive TSSs, defined as those above a given threshold of tags per million (TPM) (e.g. TPM > 0.1) in the CAGE data of the given cell type. Reference negative TSSs are defined as those not expressed in the given cell type (e.g. TPM < 0.1) but highly expressed in any other cell type (e.g. TPM > 5) in the entire CAGE dataset. We then use the CAGE catalog of TSSs to evaluate the performance of FE identification using RNA-seq data alone. Specifically, FEs identified from RNA-seq data are classified as true positive or false positive predictions if they overlap with reference positive or negative TSSs in the CAGE data respectively. The true positive rate (TPR) is calculated as the percentage of true positive predictions among all reference positive TSSs. The false positive rate (FPR) is calculated as the percentage of false positive predictions among all reference negative TSSs. Instead of comparing the methods at specific thresholds, we use the receiver operating characteristic (ROC) curve to achieve a fair and systematic comparison. To obtain the ROC curve for each method, we set a sliding threshold over the full range of test statistics, and calculate and plot the corresponding TPR and FPR values at different thresholds for calling FEs. The area under the curve (AUC) is calculated to quantify the performance of each method.
Detecting differential usage of AFEs between biological conditions
If a gene contains two or more FEs, we consider these FEs to be AFEs. The next step in the SEASTAR pipeline is to detect differential usage of AFEs between two groups of samples. This is done by testing whether the difference in the relative proportions of the multiple AFEs between the two groups is significant above a user-defined threshold (Figure 1F). A measurement of percent-spliced-in (PSI) is defined for the relative proportion of a given AFE’s inclusion in the transcripts, which is calculated as the ratio of the RNA-seq read count on the given AFE over the total RNA-seq read count on all AFEs of the same gene. We apply the rMATS statistical method (24) for testing differential AFE ratio in the SEASTAR pipeline. The rMATS model tests for differential isoform proportion between RNA-seq sample groups while accounting for variation among biological replicates. We adopt it for differential AFE analysis in this work. Specifically, rMATS is used to compare each AFE with all other AFEs in the same gene, taking into account the RNA-seq read counts and effective transcript lengths of all AFEs. We call an AFE as differentially used if the difference in the PSI values of the AFE between the two groups exceeds a given threshold c (e.g. c > 5%) and if the false discovery rate (FDR) value is smaller than a given threshold (e.g. FDR < 0.05).
Detecting differential usage of tandem TSSs
Besides detecting differential usage of AFEs, the SEASTAR pipeline also tests whether alternative tandem TSSs exist within a given FE, and whether the relative proportions of tandem TSSs significantly differ between two sample groups. The DaPars ‘change point’ statistical model, originally developed for detecting tandem alternative polyadenylation sites in RNA-seq data (25), is implemented to identify tandem TSSs. All FEs identified by SEASTAR are used as candidate regions for the DaPars analysis. DaPars first detects the internal TSS within the whole FE (i.e. the ‘change point’), then splits the whole FE into two regions at the internal TSS (change point). We use rMATS to test whether the relative proportion of the two regions is significantly different between the two sample groups. This DaPars analysis is applied to all FEs of all genes.
Processing RNA-seq and CAGE data of non-strand-specific experiments
We downloaded a public dataset containing both CAGE and RNA-seq data of the KhES embryonic stem cell line. The dataset was downloaded from the DDBJ DRA database (http://trace.ddbj.nig.ac.jp/, accession number DRA000914) of the FANTOM 5 project (26). There were four libraries containing the nuclear and cytoplasmic fractions of two replicates. Each library contained both CAGE and RNA-seq data. We downloaded the raw RNA-seq reads and aligned them to the reference human genome (UCSC hg19). The RNA-seq data were not strand-specific. We used TopHat version 1.4.1 (14) as the mapping tool, allowing no more than 3 bp mismatches. The transcriptome annotation was downloaded from the Ensembl database (version 75, http://www.ensembl.org).
We compiled the reference TSS dataset from the CAGE data, which include the annotations of TSSs along with their abundance estimates (26). We chose the KhES cell line as the cell type to be studied, and used five other cell types (two iPS cell lines including iPSCs reprogrammed from human fibroblasts and human B lymphocytes, three differentiated cell lines including fibroblasts, B lymphocytes and T lymphocytes) to select the reference negative TSSs in the studied cell type (KhES). We implemented 5-fold cross validation by splitting the reference dataset into five bins and using four bins for training and the remaining one bin for testing.
Processing RNA-seq and CAGE data of strand-specific experiments
We downloaded a public dataset containing strand-specific RNA-seq data of the H1-hES cell line. The raw RNA-seq reads were collected from the ENCODE project (https://www.encodeproject.org/experiments/), including both nuclear and cytoplasmic fractions of the H1-hES cell line. We also downloaded other public datasets containing the whole-cell RNA-seq data of the GM12878, K562, A549, HepG2, HeLa-S3, foreskin fibroblast, and SK-N-SH cells from the ENCODE project. All datasets were strand-specific paired-end RNA-seq data from rRNA-depleted PolyA+ RNA with >200 nucleotides in transcript length.
We aligned the RNA-seq reads to the reference human genome (UCSC hg19) using TopHat version 1.4.1 (14) using the strand-specific parameter (library type ‘fr-firststrand’) and allowing no more than 3 bp mismatches. The transcriptome annotation was downloaded from the Ensembl database (version 72). We downloaded the CAGE TSS annotations for all of these samples sequenced by the strand-specific CAGE protocol from the ENCODE project.
Evaluating RNA POL2 enrichment and epigenetic features of identified first exons
The aligned RNA POL2 data of the GM12878 and K562 cell lines were downloaded from ENCODE. The processed histone modification data of these cell lines were downloaded from the Roadmap Epigenomics Project (http://egg2.wustl.edu/roadmap/data/). The types of histone marks included were H3K4me3, H3K27ac and H3K36me3. They were all pre-aligned and processed by the Roadmap Project following their standard protocols which only retained the uniquely mapped reads. We used the fold-change (FC) data of each signal compared with the background signal (named as ‘Input signal’) from the Roadmap project.
For visualization, we normalized the coverage by shrinking the sequencing depth of all ChIP-seq samples to match the sample with the lowest depth, by randomly sampling reads from each sample. For each FE, we extracted a genomic region around its TSS from its upstream 5000 bp to its downstream 5000 bp. For all regions, we calculated the average coverage within each ChIP-seq sample with a 20 bp window using deepTools (27) considering the orientation of transcription.
SEASTAR analysis of time-course RNA-seq data during iPSC reprogramming
We re-analyzed our recently published mouse RNA-seq data during the time-course of iPSC reprogramming (28), which can be downloaded from the NCBI Gene Expression Omnibus (accession number GSE76233). The dataset contained samples of seven time points between day 0 to day 20 during the reprogramming of MEFs into iPSCs as well as fully reprogrammed iPSC clones. We mapped the raw RNA-seq reads to the mouse genome (mm10) and the Ensembl transcriptome annotation (release 72) using TopHat (v 1.4.1). We used SEASTAR to identify differential AFE events between day 0 and other time points from day 4 through day 20 as well as the iPSC clone. We used the threshold of c = 0.05 and FDR at 5% (Figure 1F) for calling significant difference in the relative usage of AFEs.
Temporal cluster analysis of iPSC RNA-seq data
To separate the detected differential AFE events into subgroups representing distinct temporal patterns of AFE usage during iPSC reprogramming, we performed a temporal cluster analysis on the differential AFEs using hierarchical clustering. For each possible pair of the identified differential AFEs, we calculated the Pearson Correlation Coefficient (PCC) with Jackknife resampling based on the relative usage (i.e. PSI values) of AFEs across the whole time course (seven time points × three replicates). Then we used 1 – PCC as the distance metric to perform the cluster analysis with the average linkage method. With a permutation procedure, we estimated that a PCC threshold of 0.5 corresponds to an FDR of 0.048. We then cut the clustering dendrogram at the distance threshold of 0.5, and removed sub-clusters with <5 AFEs.
For expression analysis of transcription factors (TFs), we collected genes encoding DNA-binding proteins involved in transcriptional regulation from JASPAR and UniPROBE (29,30). For all TFs, we obtained their gene expression levels in the iPSC time-course data (measured with fragments per kilobase of transcript per million mapped reads or FPKM) provided in our original study (28). From the entire list we identified candidate TFs with temporal changes in gene expression during iPSC reprogramming using the following criteria: (i) average FPKM across the three replicates >5 in at least one of the seven time points; (ii) significant change in FPKM values among seven time points (tested using ANOVA with P < 0.01); (iii) at least a 2-fold change between the maximum and minimum FPKM values (averaged across three replicates) among the seven time points. We considered these TFs as candidate TFs that may potentially drive differential AFE usage during iPSC reprogramming. Following the method for clustering AFEs (see above), we also performed a temporal cluster analysis of TF expression levels with a distance threshold of 0.7, corresponding to an FDR of 0.028 by permutation test.
TF motif and enrichment analysis
We collected the known binding sites of all TFs from the R package MotifDb (v 1.12.1) containing the TF motifs from JASPAR and UniPROBE (29,30). For each motif, we scanned for its occurrences in the vicinity of each AFE (from 2000 bp upstream to 500 bp downstream of the TSS) (31). Using the R function matchPWM with a score threshold of 90%, we identified and counted motif occurrences of each TF for each AFE.
We designed an enrichment analysis to identify TFs with a high potential to drive differential AFE usage during iPSC reprogramming. We first calculated the PCC with Jackknife resampling between the FPKM values of TFs and the PSI values of AFEs across the seven time points. To calculate the enrichment score and test for the significance of TFs, we adapted the gene set enrichment analysis (GSEA) algorithm (32). For each TF, we took all significant differential AFEs as the whole set and ranked them based on their raw correlation (PCC values) with the TF (from +1 to –1). Among the whole set of AFEs, the AFEs containing the TF’s motif were marked. Then we calculated the Enrichment Score (ES) for each TF using the method described in GSEA (32). We used a Kolmogorov–Smirnov (K–S) test to test for significant enrichment of AFEs containing the TF motif towards the top or the bottom of the ranked list of AFEs. We ranked all TFs based on their P-values from this enrichment analysis, followed by the Benjamini–Hochberg correction to calculate the FDRs.
RESULTS
A computational pipeline for systematic evaluation of alternative first exons and transcription start sites in RNA-seq data
We developed a multi-step computational pipeline SEASTAR to identify alternative first exons and alternative tandem TSSs and quantify their differential usage using RNA-seq data alone (Figure 1). The pipeline first reconstructs all putative FEs for each sample by reference transcriptome guided transcript assembly (Figure 1B). The transcriptome assemblies from all samples are then merged into a non-redundant transcriptome annotation (Figure 1C). This non-redundant annotation is then used to calculate the read coverage for each putative FE in each sample (Figure 1D). Next, we use the logistic regression model to identify bona fide FEs that are expressed in a given sample (Figure 1E). The logistic regression model was selected among five methods tested due to its superior performance in identifying FEs based on ‘gold standard’ reference TSS data from CAGE. Finally, the pipeline identifies differentially used AFEs and tandem TSSs by comparing their relative usage between sample groups (Figure 1F). The SEASTAR pipeline is written in Bash script and R (v 3.0.2) and is freely available for download at https://github.com/Xinglab/SEASTAR. All analyses in this paper were conducted with v0.9.4. We have modified and upgraded the package to v1.0.0 to fix compatibility issues with Mac OS X platforms.
Assessment of FE identification using reference CAGE data
CAGE is commonly considered as the ‘gold standard’ approach for mapping transcription start sites and first exons (12). To determine if it is feasible to reliably identify first exons from RNA-seq data alone, we tested five methods (Figure 1E) for detecting first exons from RNA-seq data and compared their results to the results of CAGE experiments performed on the same sample. Two of the methods, exon coverage and splice junction coverage, are simple cutoff-based methods using the number of reads mapped to the putative FEs or their downstream splice junctions. Two other methods, Poisson test and Negative Binomial test, compare the read coverage of the putative FEs to the surrounding genomic regions assuming Poisson and Negative Binomial distribution of the RNA-seq read counts, respectively. The last method is a logistic regression model based on principal component transformation of the exon and splice junction read coverage. We analyzed and compared the performance of these methods on multiple cell lines with both RNA-seq data and CAGE data. Both non-strand-specific and strand-specific RNA-seq data were tested.
We first used data for the KhES embryonic stem cell line from the FANTOM 5 project (26) to assess the five methods on non-strand-specific RNA-seq data. Using the criteria described in the Materials and Methods section, from the CAGE data we obtained a total of 9272 reference positive and 516 reference negative TSSs/FEs for the nuclear fraction, as well as 8552 reference positive and 400 reference negative TSSs/FEs for the cytoplasmic fraction. From the RNA-seq data, we reconstructed 79 699 and 70 961 putative FEs from the nuclear and cytoplasmic fractions, respectively. We compared the ROC curves and AUC values of the five methods (Figure 2A and B). The logistic regression model generated the AUC of 0.84 and 0.90 on the nuclear and cytoplasmic data respectively (Figure 2A and B), which were the highest among all methods. Moreover, the AUC values calculated by training and testing on the same data (without cross-validation) and by 5-fold cross-validation on the training data were comparable (with a difference of no more than 0.02). The ‘exon coverage’ method had a poor performance, with AUCs of 0.69 and 0.87 on the nuclear and cytoplasmic fractions, respectively.
Figure 2.

Performance assessment of the five methods for FE identification using the reference CAGE data. (A and B) The receiver operating characteristic (ROC) curves of the five methods on non-strand-specific RNA-seq data of the nuclear (A) and cytoplasmic fractions (B) of the KhES cell line. (C and D) The ROC curves of the five methods on strand-specific RNA-seq data of the nuclear (C) and cytoplasmic fractions (D) of the H1-hES cell line. The logistic regression model has the best performance in all cases.
We reasoned that the poor performance of the ‘exon coverage’ method may be due to overlapping or adjacent antisense transcripts that cannot be distinguished due to lack of strand information. To test if strand assignment of the RNA-seq data improves the performance of FE identification, we repeated our analysis on strand-specific RNA-seq data of the H1-hES cell line from the ENCODE project. All methods except for the Poisson test had improved performance when applied to strand-specific RNA-seq data (Figure 2C and D). The logistic regression model still generated the best performance with AUCs of 0.91 and 0.95 on the nuclear and cytoplasmic fractions, respectively. Notably, the performance of the ‘exon coverage’ method improved significantly, producing results that were nearly as accurate as the logistic regression model.
The logistic regression model reliably identifies first exons across multiple cell types
To further evaluate the robustness of the logistic regression model and determine if the model parameters and performance are influenced by the type of the cells or the experimental conditions, we tested it on data from seven additional cell types profiled by the ENCODE project. These included strand-specific CAGE and whole cell RNA-seq data from the GM12878, K562, A549, HepG2, HeLa-S3, foreskin fibroblast and SK-N-SH cells. The strand-specific nuclear and cytoplasmic data from the H1-hES cell line as described above were also included in this analysis.
We tested our logistic regression model on each cell type using parameters learned from each of the other cell types. As shown in Figure 3A, the lowest AUC was 0.910 and the average AUC was 0.936. This shows that our logistic regression model was robust for different datasets, and the model parameters were not over-fitted to specific cell types and experimental conditions. The choice of the training RNA-seq dataset had little influence on the performance of the model. Based on these results, we chose to use the logistic regression parameters learned from the H1-hESC nuclear data as the default parameters in SEASTAR. We selected the cutoff of the logistic probability as 0.909 to control the FPR at <0.05, based on the ROC curve trained and tested by the H1-hESC nuclear data.
Figure 3.

Identification and features of FEs across multiple cell types. (A) The area under the curve (AUC) values of the ROC curves for nine different conditions (whole-cell data of the GM12878, K562, A549, HepG2, HeLa-S3, foreskin fibroblast and SK-N-SH samples, and both the nuclear and cytoplasmic fractions of the H1-hES cell line). To test the robustness of our logistic regression model for identifying FEs in different cell types, we tested the logistic regression model on each cell type using the parameters trained from each of the other cell types. The lowest AUC was 0.910 and the average AUC was 0.936. These results suggest that the logistic regression model was robust for different cell types, and the model parameters were not over-fitted to specific cell types. (B and C) The distributions of RNA POL2, H3K4me3, H3K27ac and H3K36me3 signals around the TSSs of different sets of FEs in the GM12878 cell line (B) and the K562 cell line (C). RNA POL2, H3K4me3 and H3K27ac are shown to be enriched around reference positive CAGE TSSs and SEASTAR FEs (known and novel). H3K36me3 is shown to be enriched downstream of reference positive CAGE TSSs and SEASTAR FEs. Reference negative CAGE TSSs and putative FEs filtered by SEASTAR show no such enrichment patterns.
SEASTAR-identified FEs bear the hallmarks of active promoters
We reasoned that if first exons identified by SEASTAR are bona fide, they should be in close proximity to epigenetic marks characteristic of active promoters, and they should exhibit the enrichment of RNA POL2 signals associated with transcription start sites. In addition, bona fide first exons should not be enriched for epigenetic modifications associated with the bodies of active genes. To test if first exons identified by SEASTAR bear the hallmarks of active promoters, we collected from ENCODE the RNA POL2 data for two cell lines, GM12878 and K562. We also collected ChIP-seq data from the Roadmap Epigenomics project on the positioning of the H3K4me3 and H3K27ac marks that are specific to active promoters, and the H3K36me3 mark which is enriched in the bodies of actively transcribed genes (33,34).
In both the GM12878 and K562 cell lines, our analysis demonstrated that the RNA POL2, H3K4me3, and H3K27ac signals were highly enriched in the regions surrounding SEASTAR-identified FEs. Both known and novel (newly discovered) SEASTAR FEs had the same pattern (Figure 3B and C). This enrichment was similar to the enrichment observed around the reference positive TSSs identified by CAGE (i.e. ‘CAGE, positive’) (Figure 3B and C). There was a characteristic dip in the epigenetic mark enrichment observed between -200 to +50bp for the H3K4me3 and H3K27ac signals, consistent with the lack of nucleosomes right at the promoters of active genes (34). The H3K36me3 signal was enriched downstream of TSSs of SEASTAR FEs, similar to the distribution pattern of H3K36me3 around the positive TSSs by CAGE (Figure 3B and C). By contrast, putative FEs filtered out by our logistic regression model as well as the reference negative TSSs by CAGE (i.e. ‘CAGE, negative’) did not show the characteristic distributions of RNA POL2 and epigenetic marks around active promoters.
We also investigated whether the promoters of SEASTAR-identified FEs overlap with CpG islands. The promoter region was defined from 2000 bp upstream to 500 bp downstream of the TSS for a given FE. The CpG island annotation was downloaded from the UCSC genome browser (hg19). We defined a promoter as overlapping with a CpG island if there is any overlapped region between them. In this analysis, we only focused on genes with two AFEs. In the GM12878 cell line, there were 2644 genes with 5288 SEASTAR-identified AFEs. Among them, 78% of upstream promoters overlapped with CpG islands, while 50% of downstream promoters overlapped with CpG islands. This trend is consistent with observations made in a previous study for upstream versus downstream promoters of the same genes (35). Similarly, we investigated the overlap of the promoters of SEASTAR-identified FEs with peak locations of histone marks from the Roadmap Epigenomics project. For H3K4me3, H3K27ac and H3K36me3, the overlapping ratios of upstream promoters were 92%, 89% and 3% respectively, while the overlapping ratios of downstream promoters were 74%, 72% and 24% respectively. It is interesting to note the opposite trend of overlapping ratios for H3K36me3 compared with H3K4me3 and H3K27ac. We reason that H3K36me3 is an indicator of transcribed regions instead of promoter regions, so downstream promoters have a higher chance of overlapping with H3K36me3.
Detecting differential AFEs and tandem TSSs
Next, we applied the rMATS statistical model to identify differentially used AFEs and tandem TSSs between the GM12878 and K562 cell lines. AFEs and tandem TSSs were considered differentially used if they had >5% change in the PSI values and rMATS FDR of <0.05. Applying these criteria we identified 2281 differential AFEs in 1340 genes. Figure 4A and B show two examples of differentially used AFEs. The RPS6KA1 gene has two alternative first exons, TSS1060 and TSS1063, that are differentially used between the two cell lines (Figure 4A). The switch detected by SEASTAR from the proximal RPS6KA1 AFE (TSS1063) in GM12878 to the distal AFE (TSS1060) in K562 was consistent with independent evidence for promoter activity from RNA POL2 ChIP-seq and CAGE experiments. Similarly, a switch from the novel distal AFE (TSS101265) in GM12878 to a proximal AFE (TSS101266) in K562 was detected in the BIN1 gene (Figure 4B). As in the case of the RPS6KA1 gene, the detected differential AFE usage was consistent with the RNA POL2 and CAGE signals at the two AFEs.
Figure 4.

Examples of differentially used AFEs and tandem TSSs between the GM12878 and K562 cell lines. (A) Differentially used AFEs in gene RPS6KA1. (B) Differentially used AFEs in gene BIN1. (C) Differentially used tandem TSSs in gene ATP6V1E2. (D) Differentially used tandem TSSs in gene SLC35D1.
We also identified 439 significant differential tandem TSSs between the two cell types. Two such examples are shown for ATP6V1E2 (Figure 4C) and SLC35D1 (Figure 4D). In both cases, the proximal TSS was active in the GM12878 cell line while the distal TSS was active in the K562 cell line. As in the examples of differential AFEs described above, the predicted switch in tandem TSS usage was consistent with the RNA POL2 ChIP-seq and CAGE signals.
SEASTAR analysis of time-course RNA-seq data during iPSC reprogramming
To determine if SEASTAR can be used to gain insights into the regulation of gene expression and AFE usage, we applied it to an RNA-seq dataset derived from the time course of reprogramming mouse embryonic fibroblasts (MEFs) into induced pluripotent stem cells (iPSCs) (28). The dataset included RNA-seq data from seven time points at days 0, 4, 7, 10, 15 and 20 as well as fully reprogrammed iPSC clones. There were three replicates at each time point. Using SEASTAR we identified differential AFEs between day 0 and other time points (day 4 through day 20 and the iPSC clones). Our analysis revealed substantial changes in AFEs at each time point compared to day 0 during reprogramming (Supplementary Table S3). To further investigate if there were distinct temporal expression patterns among these AFEs, we conducted a temporal cluster analysis of all differential AFEs in the time course (see Materials and Methods). Figure 5A shows 15 clusters of temporal expression patterns of 738 significant differential AFEs across the time course (Supplementary Table S4). The clusters showed distinct temporal expression patterns, such as graded decrease or increase in expression along the time course (clusters 1 and 2), or dramatic decrease or increase in expression from day 20 of reprogramming to the transgene-independent iPSC clones (clusters 3 and 5). 83% of these differential AFEs contain start codons in the AFEs, which may either change the N-terminal coding sequence or modulate translational efficiency via upstream AUGs or ORFs. The rest (17%) of differential AFEs do not contain start codons.
Figure 5.

Genome-wide analysis of differential AFEs and associated TFs during iPSC reprogramming. (A) Heatmap of genome-wide AFE usage levels (PSI values) during iPSC reprogramming (3 replicates for each time point). AFEs were clustered into 15 groups of distinct temporal patterns. (B) Heatmap of expression levels of 126 TFs with temporal expression changes during iPSC reprogramming. The TFs were clustered into 5 groups of distinct temporal patterns. The top 10 TFs ranked by the enrichment analysis are highlighted. (C) Heatmap of Pearson Correlation Coefficients of AFE PSI values and TF expression levels across the time course. Each row represents an AFE and each column represents a TF. The top 10 TFs ranked by the enrichment analysis are highlighted.
The temporal patterns of AFE usage suggests coordinated regulation by TFs at different stages of reprogramming. We collected a list of 308 genes encoding DNA-binding proteins involved in transcriptional regulation (29,30). From the list we identified 126 genes (Supplementary Table S5) as candidate TFs with significant temporal changes in expression during reprogramming (see Materials and Methods). Using a temporal cluster analysis of TF expression levels, we separated these 126 TFs into five distinct clusters as shown in Figure 5B (detailed data in Supplementary Table S6).
Next, we carried out an integrative analysis of AFE/TF expression as well as TF motif occurrence to identify candidate TFs that potentially drive differential AFE usage during iPSC reprogramming. We used the Jackknife PCC between AFE PSI values and TF expression levels to identify TFs whose expression levels were highly correlated with differentially used AFEs during iPSC reprogramming. We observed that many TFs correlated positively or negatively with AFE usage during the time course of reprogramming (Figure 5C). To identify TFs with potential causal roles in regulating AFE usage, we examined the occurrences of TF motifs around differential AFEs. Specifically, TFs may affect AFE usage by binding to genomic regions flanking the TSSs (36,37). The existence of a binding motif for a particular TF in proximity to a regulated AFE may indicate that the TF directly controls AFE usage. We scanned for TF motifs in the 2.5 kb genomic region (2000 bp upstream to 500 bp downstream) surrounding each TSS of AFEs and counted motif occurrences of each TF at each AFE. We then adapted the GSEA algorithm (32) to calculate the ES and significance of individual TFs for the whole set of differential AFEs, incorporating both the expression correlation between TFs and AFEs and the occurrences of TF motifs around AFEs (see Materials and Methods). Briefly, for each TF we took all differential AFEs and ranked them based on the PCC between AFE PSI values and TF expression levels across the time course, then tested for the enrichment of AFEs containing the TF’s motif towards the top or the bottom of the ranked list. This approach has the benefit of accounting for both the strength of expression correlation between TFs and AFEs, and the occurrence of TF motifs around AFEs as the potential evidence for direct regulation.
Following the enrichment analysis, we ranked all 126 temporally regulated TFs by their P-values for enrichment scores (Supplementary Table S7). The top 10 TFs based on the enrichment analysis are highlighted in Figure 5B and C. Among them, we found multiple TFs known to be key regulators of reprogramming including the top ranked N-Myc (Mycn) gene (with P-value of 0.00028). AFEs containing the Mycn motif were significantly enriched towards the top of the AFEs positively correlated with Mycn expression in our enrichment analysis (Figure 6A). We further investigated the expression level of Mycn (Figure 6B), as well as the average PSI values of AFEs that contain the Mycn motif and have strong positive correlation with Mycn expression (PCC > 0.5) (Figure 6C). The significant increase of Mycn expression during iPSC reprogramming (P-value = 8.9e–16, ANOVA test) was accompanied by an increase in the relative usage of these AFEs. The coordinated change in expression levels between Mycn and the differentially used AFEs containing the Mycn motif suggests that Mycn binds to and promotes the usage of these AFEs. Mycn is known to play an essential role in the maintenance of pluripotency (38). Mycn can cooperate with other TFs to reprogram adult cells into other differentiated cells (39) or into iPS cells (40). Msx2, another transcription factor identified in our enrichment analysis, is a major driver of de-differentiation in mammalian muscle cells (41). Collectively, these data imply that TFs with high scores from the enrichment analysis of differential AFEs play important roles in iPSC reprogramming and the regulation of the pluripotent state.
Figure 6.

N-Myc (Mycn) as a potential driver of differential AFEs during iPSC reprogramming. (A) The gene set enrichment analysis (GSEA) like plot for N-Myc (Mycn). Differential AFEs were ranked based on the correlation of their PSI values to Mycn expression levels during the time course. Differential AFEs containing the Mycn motif were significantly enriched towards the top of the list. (B) The expression levels of Mycn across the time course. (C) The PSI values of Mycn-associated AFEs across the time course. The PSI value of each individual AFE is drawn in gray and the red curve represents the average of all AFEs.
DISCUSSION
We report a computational pipeline SEASTAR that reliably identifies FEs and performs quantitative analyses of AFE usage using RNA-seq data alone. The enrichment of epigenetic marks specific to active promoters as well as RNA POL2 signals around the SEASTAR FEs suggests that these FEs originate at bona fide transcription start sites and are not experimental artifacts. To achieve the optimal performance we explored various methods and found that the logistic regression model, which combines the read coverage for the putative first exon and its downstream splice junction, has the best performance in identifying first exons from RNA-seq data, regardless of whether the data has strand information or not. By contrast, the exon-based method (‘Exon coverage’) has a poor performance on non-strand-specific data, but is a close second on strand-specific data. This discrepancy is likely due to the presence of antisense transcription at active promoters which may confound the identification of FEs in non-strand-specific data using only the exon count information.
We should note that all RNA-seq data used in this work are either from major consortia projects (FANTOM, ENCODE) or from our own published work, in which stringent QC criteria had been applied to ensure RNA quality prior to sequencing. Therefore, we expect that the possible degeneration of RNA-seq coverage near 5′ ends of mRNAs did not severely affect the ability of SEASTAR to identify alternative and differential AFE events in these datasets, considering that SEASTAR uses RNA-seq signals on both the AFE and its downstream splice junction. Nonetheless, this issue could be more severe for short AFEs, or for RNA-seq data of low quality and degraded RNA samples.
SEASTAR compares RNA-seq data of distinct biological conditions to identify differentially used AFEs and tandem TSSs. By analyzing the time course RNA-seq data of reprogramming MEFs into iPSCs, we demonstrated the utility of SEASTAR in studying the temporal control of gene expression and AFE usage. Furthermore, we developed an enrichment analysis method that considers the coordinated expression of TFs and AFEs as well as the occurrences of TF motifs around AFEs to identify candidate TFs that drive differential AFE usage. This enrichment analysis pinpointed Mycn as a key regulator of AFE usage during iPSC reprogramming. Collectively, SEASTAR is a comprehensive software package for the computational identification and quantitative analysis of AFEs and alternative tandem TSSs using RNA-seq data. It can be used in lieu of CAGE analysis, when suitable CAGE data is often not available or impractical to obtain due to technical challenges or limitations. Given the popularity of RNA-seq as well as the rapid accumulation of RNA-seq data in public repositories, we anticipate that SEASTAR can provide valuable and novel insights into AFE usage and regulation in diverse RNA-seq studies.
DATA AVAILABILITY
The SEASTAR pipeline is written in Bash script and R (v 3.0.2) and is freely available for download at https://github.com/Xinglab/SEASTAR.
Supplementary Material
ACKNOWLEDGEMENTS
We wish to thank Tevfik Umut Dincer, Shihao Shen, Jinkai Wang, Juw Won Park and Yang Guo for technical assistance and comments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
National Institutes of Health [R01ES024995 to Y.X., R01EY025536 to P.S.] (in part); Department of Defense Breast Cancer Research Program [W81XWH-15-1-0349 to P.S.]; National Natural Science Foundation of China [NSFC 61721003 to X.Z.]; National Basic Research Program of China [2012CB316504 to X.Z.]. Funding for open access charge: NSFC Grant 61721003.
Conflict of interest statement. None declared.
REFERENCES
- 1. Davuluri R.V., Suzuki Y., Sugano S., Plass C., Huang T.H.M.. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet. 2008; 24:167–177. [DOI] [PubMed] [Google Scholar]
- 2. Landry J.R., Mager D.L., Wilhelm B.T.. Complex controls: the role of alternative promoters in mammalian genomes. Trends Genet. 2003; 19:640–648. [DOI] [PubMed] [Google Scholar]
- 3. Quelle D.E., Zindy F., Ashmun R.A., Sherr C.J.. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995; 83:993–1000. [DOI] [PubMed] [Google Scholar]
- 4. Goossens S., Janssens B., Vanpoucke G., De Rycke R., van Hengel J., van Roy F.. Truncated isoform of mouse alpha T-catenin is testis-restricted in expression and function. FASEB J. 2007; 21:647–655. [DOI] [PubMed] [Google Scholar]
- 5. Rojas-Duran M.F., Gilbert W.V.. Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA. 2012; 18:2299–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang X., Hou J., Quedenau C., Chen W.. Pervasive isoform-specific translational regulation via alternative transcription start sites in mammals. Mol. Syst. Biol. 2016; 12:875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pal S., Gupta R., Kim H., Wickramasinghe P., Baubet V., Showe L.C., Dahmane N., Davuluri R.V.. Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res. 2011; 21:1260–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davis W. Jr., Schultz R.M.. Developmental change in TATA-box utilization during preimplantation mouse development. Dev. Biol. 2000; 218:275–283. [DOI] [PubMed] [Google Scholar]
- 9. Pozner A., Lotem J., Xiao C., Goldenberg D., Brenner O., Negreanu V., Levanon D., Groner Y.. Developmentally regulated promoter-switch transcriptionally controls Runx1 function during embryonic hematopoiesis. BMC Dev. Biol. 2007; 7:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rathjen P.D., Toth S., Willis A., Heath J.K., Smith A.G.. Differentiation inhibiting activity is produced in matrix-associated and diffusible forms that are generated by alternate promoter usage. Cell. 1990; 62:1105–1114. [DOI] [PubMed] [Google Scholar]
- 11. Salomonis N., Schlieve C.R., Pereira L., Wahlquist C., Colas A., Zambon A.C., Vranizan K., Spindler M.J., Pico A.R., Cline M.S. et al. Alternative splicing regulates mouse embryonic stem cell pluripotency and differentiation. PNAS. 2010; 107:10514–10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shiraki T., Kondo S., Katayama S., Waki K., Kasukawa T., Kawaji H., Kodzius R., Watahiki A., Nakamura M., Arakawa T. et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. PNAS. 2003; 100:15776–15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rapid amplification of 5′ complementary DNA ends (5′ RACE). Nat. Methods. 2005; 2:629–630. [DOI] [PubMed] [Google Scholar]
- 14. Trapnell C., Pachter L., Salzberg S.L.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011; 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Conesa A., Madrigal P., Tarazona S., Gomez-Cabrero D., Cervera A., McPherson A., Szczesniak M.W., Gaffney D.J., Elo L.L., Zhang X. et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016; 17:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roberts A., Pimentel H., Trapnell C., Pachter L.. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011; 27:2325–2329. [DOI] [PubMed] [Google Scholar]
- 19. FANTOM Consortium and the RIKEN PMI and CLST (DGT) Forrest A.R., Kawaji H., Rehli M., Baillie J.K., de Hoon M.J., Haberle V., Lassmann T.. A promoter-level mammalian expression atlas. Nature. 2014; 507:462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carninci P., Sandelin A., Lenhard B., Katayama S., Shimokawa K., Ponjavic J., Semple C.A., Taylor M.S., Engstrom P.G., Frith M.C. et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006; 38:626–635. [DOI] [PubMed] [Google Scholar]
- 21. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robinson M.D., Oshlack A.. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010; 11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCarthy D.J., Chen Y.S., Smyth G.K.. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012; 40:4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shen S., Park J.W., Lu Z.X., Lin L., Henry M.D., Wu Y.N., Zhou Q., Xing Y.. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. PNAS. 2014; 111:E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xia Z., Donehower L.A., Cooper T.A., Neilson J.R., Wheeler D.A., Wagner E.J., Li W.. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat. Commun. 2014; 5:5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fort A., Hashimoto K., Yamada D., Salimullah M., Keya C.A., Saxena A., Bonetti A., Voineagu I., Bertin N., Kratz A. et al. Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat. Genet. 2014; 46:558–566. [DOI] [PubMed] [Google Scholar]
- 27. Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T.. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cieply B., Park J.W., Nakauka-Ddamba A., Bebee T.W., Guo Y., Shang X., Lengner C.J., Xing Y., Carstens R.P.. Multiphasic and dynamic changes in alternative splicing during induction of pluripotency are coordinated by numerous RNA-binding proteins. Cell Rep. 2016; 15:247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mathelier A., Zhao X., Zhang A.W., Parcy F., Worsley-Hunt R., Arenillas D.J., Buchman S., Chen C.Y., Chou A., Ienasescu H. et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014; 42:D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hume M.A., Barrera L.A., Gisselbrecht S.S., Bulyk M.L.. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2015; 43:D117–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsankov A.M., Gu H., Akopian V., Ziller M.J., Donaghey J., Amit I., Gnirke A., Meissner A.. Transcription factor binding dynamics during human ES cell differentiation. Nature. 2015; 518:344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roadmap Epigenomics, C. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K.. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129:823–837. [DOI] [PubMed] [Google Scholar]
- 35. Wang J., Ungar L.H., Tseng H., Hannenhalli S.. MetaProm: a neural network based meta-predictor for alternative human promoter prediction. BMC Genomics. 2007; 8:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang J., Zhuang J., Iyer S., Lin X., Whitfield T.W., Greven M.C., Pierce B.G., Dong X., Kundaje A., Cheng Y. et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012; 22:1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Valen E., Sandelin A.. Genomic and chromatin signals underlying transcription start-site selection. Trends Genet. 2011; 27:475–485. [DOI] [PubMed] [Google Scholar]
- 38. Smith K.N., Singh A.M., Dalton S.. Myc represses primitive endoderm differentiation in pluripotent stem cells. Cell Stem Cell. 2010; 7:343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mizoshiri N., Kishida T., Yamamoto K., Shirai T., Terauchi R., Tsuchida S., Mori Y., Ejima A., Sato Y., Arai Y. et al. Transduction of Oct6 or Oct9 gene concomitant with Myc family gene induced osteoblast-like phenotypic conversion in normal human fibroblasts. Biochem. Biophys. Res. Commun. 2015; 467:1110–1116. [DOI] [PubMed] [Google Scholar]
- 40. Nakagawa M., Takizawa N., Narita M., Ichisaka T., Yamanaka S.. Promotion of direct reprogramming by transformation-deficient Myc. PNAS. 2010; 107:14152–14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yilmaz A., Engeler R., Constantinescu S., Kokkaliaris K.D., Dimitrakopoulos C., Schroeder T., Beerenwinkel N., Paro R.. Ectopic expression of Msx2 in mammalian myotubes recapitulates aspects of amphibian muscle dedifferentiation. Stem Cell Res. 2015; 15:542–553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The SEASTAR pipeline is written in Bash script and R (v 3.0.2) and is freely available for download at https://github.com/Xinglab/SEASTAR.