


Abstract
Left-handed Z-DNA is an extraordinary conformation of DNA, which can form by special sequences under specific biological, chemical or physical conditions. Human ADAR1, prototypic Z-DNA binding protein (ZBP), binds to Z-DNA with high affinity. Utilizing single-molecule FRET assays for Z-DNA forming sequences embedded in a long inactive DNA, we measure thermodynamic populations of ADAR1-bound DNA conformations in both GC and TG repeat sequences. Based on a statistical physics model, we determined quantitatively the affinities of ADAR1 to both Z-form and B-form of these sequences. We also reported what pathways it takes to induce the B–Z transition in those sequences. Due to the high junction energy, an intermediate B* state has to accumulate prior to the B–Z transition. Our study showing the stable B* state supports the active picture for the protein-induced B–Z transition that occurs under a physiological setting.
INTRODUCTION
Non-conventional DNA conformations, often induced upon binding by proteins or chemicals, are thought to play critical roles in biological phenomena (1–7). Several non-B conformations such as G-quadruplex and Z-DNA are known to form in a sequence-specific manner and the biophysical principle behind those structures have been intensively investigated via a broad range of methods including single-molecule techniques (8–14).
Among many non-standard forms of DNA, Z-DNA has intrigued structural biologists and biochemists alike due to its extraordinary left-handed conformation (2,3,15,16). Z-DNA has also attracted a great deal of attention due to its biological relevance in regulating transcription (17–19). In a model, Z-DNA is thought to form in the promoter region of the open chromatin structure induced by a chromatin remodeler and to maintain the open structure stably via binding by proteins such as ADAR1 or Nrf2 in order to support the downstream transcription event (18,19). On the other hand, as a consequence of transcription, Z-DNA is induced in vivo by negative supercoiling generated from movement of a RNA polymerase (20–22). It has been shown physically that the unwinding torsional stress by negative supercoiling is a key factor to induce Z-DNA formation (10,23,24). Among other non-canonical DNA structures (25,26), it may serve as a buffer for torsional stress, which regulates dynamically the effective degree of negative supercoiling of DNA in a tension-sensitive manner (10).
Besides chirality, Z-DNA and B-DNA differ by their helical pitch, Z-DNA being more elongated (15). Its stacking distance between neighboring base pairs is aZ = 0.38 nm with a helical periodicity of ∼12 bp per turn. Z-DNA is formed in a sequence-specific manner: an alternating purine-pyrimidine sequence is required for Z-DNA formation. The most common Z-DNA forming sequences are dinucleotide repeats of G and C or G and T: (GC)n or (TG)n (27). The free energy cost εZ for switching to Z-DNA is ∼0.55 kBT/bp (0.33 kcal/mol/bp) for (GC)n repeats and ∼1.1 kBT/bp (0.67 kcal/mol/bp) for (TG)n repeats (24,28) as measured by 2D gel assay. Furthermore, there is a domain wall energy EJ ≈ 8–9 kBT (∼5 kcal/mol) at each boundary between B-DNA and Z-DNA due to broken stacking at the junction.
Proteins have been found to adsorb on Z-DNA and help stabilizing the Z-DNA conformation. ADAR1 (Adenosine Deaminase Acting on RNA I), prototypic Z-DNA binding protein (ZBP), has two domains Zα and Zβ identified to bind to Z-DNA (29,30). Using a band-shift assay, Herbert et al. (31) observed that the Zα domain of human ADAR1 (hZαADAR1) can bind to various Z-DNA forming sequences such as GC and TG repeats. They also found that hZαADAR1 is sensitive to the density of negative supercoiling. From this study, it is speculated that hZαADAR1 may interact in vivo with non-canonical forms of DNA induced by, and thus absorbing, local negative supercoiling. It is further demonstrated that the logarithm (base 2) of hZαADAR1 concentration needed for switching to Z-DNA is about 2 times larger for a Z-DNA forming sequence, (TG)3, than for (GC)3, most efficient Z-DNA forming sequence. This finding is compatible with a factor of two difference in εZ mentioned above. It has been suggested in Ref. (31) that the binding site for a Zα motif is no larger than 6 bp in length and that each Zα domain can flip 6 bp of bound DNA into the Z-DNA conformation. According to the crystal structure (33,36), hZαADAR1 binds on five bases on one strand, which gives the maximum protein density on DNA in the region of compact binding. On the other hand, considering that the protein binding has a structure specific nature, a slot for binding of one protein is expected to be as long as n0 = 6 bp, which can be also considered as the size of flipping segment of DNA. Below we consider both cases (n0 = 5 and 6) for our modelling. As far as main conclusions are concerned, it is not important whether n0 is 5 or 6.
Although several studies on interaction of Z-DNA with ZBP were undertaken (32,33,37), most of the studies have been dedicated to a short Z-DNA forming sequences in a short DNA oligomer which does not include flanking B-DNA parts, which exist in in vivo conditions. Whether proteins such as hZαADAR1 can passively or actively induce Z-DNA in nature is not yet established. The molecular configuration considered below mimics the in vivo situation that a core DNA is always embedded in and sandwiched by other ordinary sequences and comprises two B/Z junctions in the converted state. Hereafter, we distinguish core sequence or core from core molecule or core fragment: the former indicates the sequence to be studied and the latter indicates the hybridized molecule (with the sequence as well as flanking sequences and overhangs), which can be ligated to a long PCR fragment.
Then, one related questions is whether negative supercoiling is really needed prior to any binding. It is likely that a spontaneous conformational fluctuation to Z-DNA is too rare to efficiently trigger ZBP adsorption if ZBP only binds to pre-formed Z-DNA, especially when the conversion involves the domain wall energy, EJ. (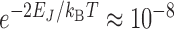 ). A simple estimate suggests that for the ZBP concentrations investigated (1–10 μM) the typical reaction time could hardly be smaller than one hour (see SI for detailed estimate). For isolated short repeats, the time scale of switching between B-state and Z-state is expected to be much smaller than our estimate and it is even shorter for the Ni2 +-mediated transitions reported in (11). The passive mechanism seems to apply for these experiments (11).
). A simple estimate suggests that for the ZBP concentrations investigated (1–10 μM) the typical reaction time could hardly be smaller than one hour (see SI for detailed estimate). For isolated short repeats, the time scale of switching between B-state and Z-state is expected to be much smaller than our estimate and it is even shorter for the Ni2 +-mediated transitions reported in (11). The passive mechanism seems to apply for these experiments (11).
For realization of the B–Z transition in vivo, other conversion paths must be considered, which involve some interaction (pre-complexation) between ZBPs and the DNA in B-form, and thus can be considered as active processes because there is an active role of ZBP. NMR studies using a short repeat sequence without flanking sequences (32,37) also suggests that there is an intermediate state for the B–Z transition induced by a ZBP, which is inferred from some chemical shifts attributed to the ZBP. This experiment hints at an unexpected ZBP structure in the presence of DNA prior to the B–Z transition. In the NMR experiment, we can rule out the possibility of a partial B-to-Z transition involving a B/Z junction given the short repeat sequences used.
The aim of this contribution is to elucidate the detailed pathway leading to a B–Z conversion for a core DNA sequence embedded in ‘natural’ DNA and to determine the relative stability of the states involved. The stability of the states then yields the affinity of hZαADAR1 to B-DNA and various Z-DNAs.
For the aims, we performed single-molecule Fluorescence Resonance Energy Transfer (FRET) assays and Circular Dichroism (CD) measurements. We observed the B–Z transition in those core sequences in a hZαADAR1-concentration-dependent manner. We also found that a GC repeat sequence undergoes the transition at a significantly lower hZαADAR1 concentration than a TG repeat sequence: at least two-orders-of-magnitude less hZαADAR1 is sufficient to induce the transition for the former sequence according to our results. Besides, in the B–Z transition occurring to the TG core sequence, an intermediate state was significantly populated. Though minor, the population of the intermediate state is sizable for GC repeats too.
We provide the model to rationalize these findings. The analysis of this experimental result allows us to measure the affinity of hZαADAR1 to Z-DNA (in GC and TG cores) and B-DNA. Moreover, our results including CD measurements suggest that at a low concentration of hZαADAR1, a state of hZαADAR1-bound right-handed DNA exists. Further, the adsorption of hZαADAR1 on B-form of TG repeats leads to mixed states, in which hZαADAR1-bound B- and Z-DNA coexist. The low population of the hZαADAR1-bound B-form for GC repeats strongly suggests that this intermediate form converts to Z-DNA very efficiently (quickly).
MATERIALS AND METHODS
Preparation of DNA samples and hZαADAR1
Sample preparation is described in detail elsewhere (10). We used GC-repeat and TG-repeat core fragments ((GC)11 and (TG)11), which contain 11 repeats of the respective di-nucleotide sequence, in which dyes are separated by 14 bp (Figure 1 and Table 1). The sequences of these core DNA molecules are given in Table 1. For smFRET assays, the core fragment is ligated to a long (>500 bp) PCR fragment obtained by BamHI digestion. Thus, a Z-DNA forming repeat sequence is embedded within a long, inactive, random sequence and positioned near one end of the long tether molecule. (In the ligated DNA construct, the segment outside the core to TGCC side is 6 base pairs long while the other segment to GATC side is longer than 500 bp.) hZαADAR1 was expressed and purified as described previously (33).
Figure 1.

(A) Schematic experimental arrangement. A DNA fragment that contains a Z-DNA forming core sequence ((GC)11 or (TG)11) is ligated to a biotinylated linker DNA, which is attached to NeutrAvidin (yellow), which is in turn bound on a biotinylated PEG substrate. Biotins are shown as small red circles. A donor (Cy3) and an acceptor dye (Cy5) is 14 bp apart within the core fragment. In the presence of hZαADAR1, the core sequence undergoes the protein-induced transition from B-form (black) to Z-form (blue). Both ends of the core sequence are bounded by B-form DNA (brown). Here, the core is linked to the surface via a ∼500 bp PCR fragment. (B) In the presence of ZBP such as hZαADAR1, the core sequence (gray box) undergoes the B–Z transition, which is detected via smFRET measurements. Different states of DNA (B, B* and Z) are identified via different levels of FRET, which are pictorially and schematically illustrated here by different relative sizes of dye symbols.
Table 1. Each core ((GC)11, (TG)11, and (scrambled)) is constructed by hybridizing a pair of corresponding oligonucleotides ((GC)11-1/2, (TG)11-1/2 and (scrambled)-1/2). iCy3 and iCy5 indicate internally labeled Cy3 and Cy5 dyes, respectively. 5′-Phos indicates the phosphate group at 5′ end for ligation. A core fragment is ligated to a biotin-labeled DNA fragment prepared by a PCR reaction containing biotin-dUTP, which is followed by BamHI digestion. Repeat-only DNA molecules ((TG)8 and (GC)8) as well as core DNA molecules ((TG)11 and (GC)11) used for smFRET assays were used for CD measurements. The core molecules used in this study are annotated as follows: red, core sequence; blue, cohesive overhang; black, flanking sequence.

|
Single-molecule FRET assay of the hZαADAR1-induced B–Z transition
hZαADAR1 prepared at various concentrations in an imaging buffer (50 mM Tris–HCl (pH 8.0)) containing 50 mM NaCl, and an oxygen scavenger system (2 mM Trolox, glucose oxidase (165 U/ml), catalase (2170 U/ml), and d-glucose (0.4% wt/wt)) was delivered into a chamber containing DNA constructs immobilized on a NeutrAvidin-bound PEGylated glass surface, and the sample was incubated for longer than 30 min. To prepare immobilized DNA tethers on the sample surface, we used ∼25 pM DNA. After 30–60 min incubation, we washed out unbound molecules and found there are ∼150 molecules in 50 μm × 25 μm viewfield. FRET efficiency (FE) histograms were obtained before and after introduction of hZαADAR1. Final FRET efficiency histograms were drawn with more than 5000 molecules (>50 different fields of view). After >30 min incubation, the sample appeared to reach equilibrium as FRET histograms drawn from the data acquired after different incubation times (30–120 min) were nearly identical (Supplementary Figures S5 and S6). All experiments with hZαADAR1 were carried out at room temperature. Each data set for smFRET measurement was independently acquired at least three times.
CD measurements
CD measurements were carried out with a Jasco J-815 spectrometer at room temperature. We used two different molecular designs for each type of repeat sequence. For direct comparison, we used (GC)11 and (TG)11 core molecules, which have the same scrambled flanking sequences as the molecules for smFRET. We also used (GC)8 and (TG)8 repeat-only sequences: these repeats do not have flanking B-DNA sequences, which simplifies interpretation of CD results. In CD measurements, the concentration of DNA was about 33 μM in terms of the number of base pairs. The concentrations of hZαADAR1 used were 0, 10, 30 and 50 μM. The buffer contained 10 mM Tris–HCl (pH 8.0) and 50 mM NaCl.
RESULTS
We prepared long DNA molecules including short Z-DNA forming sequences of (GC)11 or (TG)11 which are competent to form Z-DNA with high enough hZαADAR1 concentrations. Each core DNA sequence gives high-FRET signal in the conformation of B-DNA and switches to a low-FRET state upon flipping to the conformation of Z-DNA. Our FRET experiment is hence sensitive to the conformational transition occurring to the well-defined target sequence. The fraction of molecules having the core sequence in Z-form was measured using the single molecule FRET (smFRET) technique for various protein concentrations.
FRET histograms are shown in Figure 2. The FRET efficiency (FE) values corresponding to B-DNA and Z-DNA are 0.5 and 0.15–0.2 (0.2 for TG and 0.15 for GC), respectively, as indicated in (10). For a scrambled core sequence, a FRET peak appear only at 0.5 even for a large protein concentration, which indicates that hZαADAR1 does not bind to such a random sequence stably. For GC cores, a FRET peak appears at 0.5 at low protein concentrations (<100 nM) and the peak decreases and another peak emerges at 0.15 at larger concentrations (>300 nM). For TG cores, there is no detectable change in the low-FRET range at such low concentrations (<1 μM). Intriguingly, another peak appears at FE ∼0.4 and gradually predominates over the peak at FE ∼0.5 up to [hZαADAR1] ∼1 μM. The peak of FE ∼0.4 does correspond neither to Z-DNA nor to B-DNA and hints at the existence of an intermediate state.
Figure 2.

FRET histograms for GC and TG repeats at the indicated protein (hZαADAR1) concentrations. (A) FE histograms of a scrambled sequence in the absence (bottom) and presence (top) of 50 μM hZαADAR1. (B) FE histograms of (GC)11 with various concentrations of hZαADAR1 (0, 0.1, 0.3, 0.4, 0.5 and 1 μM from bottom to top). A new peak at FE ∼0.15 corresponds to the Z-DNA state. (C) FE histograms of (TG)11 with various concentrations of hZαADAR1 (0, 0.1, 0.5, 1, 5, 10, 15 and 50 μM from bottom to top). At 50 μM hZαADAR1, the peak at FE ∼0.2 becomes dominant. The curves (blue: Z-DNA, red: B*-DNA, black: B-DNA, green: donor-only) in each panel represent multiple Gaussian fits for histogram. Two vertical dotted lines in each panel are at FE = 0 (left) and 0.5 (right), which represent donor-only and B-DNA peaks, respectively.
Figure 3 shows the fraction of each DNA state in the presence of various hZαADAR1 concentrations. The population of each state is given by the area under the corresponding peak, which was obtained by fitting the FRET histogram with a set of Gaussian functions. In the case of GC core, two FRET peaks were nearly sufficient to fit the data besides the donor-only peak at FE = 0 while three FRET peaks were necessary to fit the data for TG core. In the latter case, the population of the original high-FRET signal (FE ∼0.5) is quickly replaced by that of FE ∼0.4 in the range of [hZαADAR1] (<1 μM; the midpoint [hZαADAR1] for the transition in GC core is ∼0.3 μM) that exhibits the B–Z transition in the former case. The histograms for GC core are also fit with three Gaussian functions with a relatively small population of the intermediate state with a considerably better fitting quality.
Figure 3.
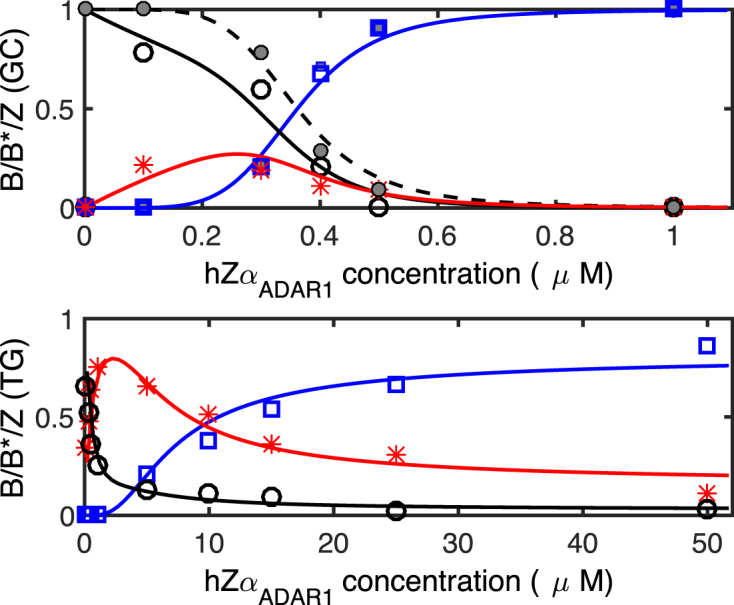
Populations of conformational states (Z: blue open square, B: black open circle, B*: red asterisk) of DNA for GC (top) and TG (bottom) repeat core sequences in the presence of indicated concentrations of hZαADAR1. Here, fits are based on the 6-binding-site model and AB and AZ were adjusted for fitting. Assuming the same B-DNA affinity for TG and GC repeats, the best fits are obtained with AZ = 16.2 kBT with a ≈ 3.7 nm (GC) and AZ = 15.8 kBT with a ≈ 3.8 nm (TG) together with AB = 9.2 kBT. From the fit, the B* state amounts to 85% of hZαADAR1 adsorbed B-DNA. The curves for each B, B* and Z state are shown in black, red, and blue (following the same convention as in Figure 2), respectively. The filled symbols (B: gray filled circle, Z: gray filled square) in the upper panel(GC) are obtained by hypothesizing no B*-state. Here, we obtained similar affinities AB = 9.2 kBT and AZ = 16.1 kBT with a = 3.8 nm.
We also identified a third state and caught a glimpse of dynamic transitions among B, Z and the third state in kinetics data. We present representative inter-converting trajectories that reveal switching events among the states in Supplementary Information (see SI). In our system, however, the time scale for protein binding and transition thereby can be very different from molecule to molecule and is very long compared with the previously reported results in (11). As we discuss later, due to the high junction energy, the transition requires a step of nucleation. Kinetics data are not conclusive to provide time scales of the transitions. We rather show that the intermediate state exists on the path of the B–Z transition as a thermodynamic state.
The existence of the third, intermediate state is further supported below. To gain insight into the origin of the intermediate FRET signal, we performed CD measurements. We tested the same core molecules bearing a 6-bp flanking sequence at each end ((GC)11 and (TG)11) as those to be ligated to a PCR fragment for smFRET assays. Here, the molecule is minus a long PCR fragment. Otherwise, CD spectra are completely dominated by the B-form PCR fragment. The CD spectra from the samples are shown in Figure 4A and B. In the absence of hZαADAR1, the DNA exhibits the well-known CD spectrum of B-DNA. At larger hZαADAR1 concentrations (>10 μM), the CD spectra exhibit sign inversion around 290 nm, which indicates structural change towards Z-DNA. Interestingly, at [hZαADAR1] = 10 μM, the CD spectrum around 290 nm appears flat (ellipticity ∼ 0) for (TG)11. This near flatness of CD spectrum should result from linear superposition of two spectra with opposite helicities (right and left). The DNA with scrambled flanking sequences, however, contain regular B-DNA even after the transition is completed in the repeat sequence. Thus it is not straight forward to attribute the change in CD spectra directly to the (equal) population of the intermediate state and Z-state because of right-handed flanking sequences. To avoid unnecessary complication (and to gain clear insight into the helicity of the intermediate state), we also perform CD measurements for repeat-only sequences without flanking B-DNA sequences. Figure 4C and D displays CD spectra of (TG)8 as well as (GC)8 for various hZαADAR1 concentrations. The overall behavior of CD spectra from the repeat-only molecules are similar to those from the cores except that the latter need somewhat more hZαADAR1 to induce the transition. From FRET measurements with [hZαADAR1] >10 μM, two FRET states, left-handed Z-DNA and a state exhibiting the intermediate FRET efficiency, coexist at the hZαADAR1 concentration. Thus, the net ellipticity measured in CD should be given by the sum of the ellipticities by the two states, the intermediate state and Z-state. From the negligible ellipticity of (TG)8 with 10 μM hZαADAR1, the helicity of the intermediate state should be opposite to that of Z-DNA.
Figure 4.

CD spectra of (A) (GC)11, (B) (TG)11, (C) (GC)8 and (D) (TG)8 at various concentrations of hZαADAR1. In the absence of hZαADAR1 (black solid), the DNA exhibits a CD spectrum for typical B-DNA. With increasing concentrations of hZαADAR1 (red dashed: 10 μM, blue dotted: 30 μM, green dot-dashed: 50 μM), CD spectra continuously change from B-DNA to Z-DNA. At 10 μM hZαADAR1, the ellipticity of (TG)8 (as well as (TG)11 core) around 290 nm becomes close to zero, indicating near cancellation of the (negative) ellipticity of Z-DNA with the (positive) ellipticity of the intermediate DNA state (named B*-DNA). (GC)11 and (TG)11 have flanking scrambled sequences in both termini and thus have two B–Z junctions after the B–Z transition in the core sequence while the two repeat-only sequences, (GC)8 and (TG)8, have no B–Z junction after the B–Z transition.
Then, can the intermediate state be just B-DNA? Is the different FRET level just due to non-specific protein adsorption on B-DNA? This can be ruled out because smFRET measurements on DNA molecules of scrambled core sequence do not show such an intermediate level of FRET signal.
It appears that hZαADAR1 can sense chemical and fine structural differences of various DNA sequences and interact with their B-form configurations discriminately. GC- or TG-repeat DNAs may be softer and more deformable upon binding by hZαADAR1 towards the left-handed DNA as shown in our smFRET measurements (Figure 2) in contrast to the case of a B-DNA with random scrambled sequence, in which a steric clash prevents the protein from binding to an ordinary right-handed helix as pointed out previously (34). The change in FRET is induced by sequence-specific binding of hZαADAR1 and likely originates from structural rearrangement (however small or large) of DNA upon hZαADAR1 binding. Complexation of DNA molecules by chiral recognition is also theoretically discussed in (35). Can the intermediate state indicate the presence of a very short Z-DNA domain (one 6-bp or, at most, two (12-bp) sites of Z-form)? If this is the case, the transition between the states of FE ∼0.4 and 0.2 should be more continuous: in other words, the peak FE should change continuously from 0.5 all the way to 0.2, which is not the case here. Furthermore, from the model calculation, such a short patch of Z-form is energetically too costly to be feasible. Therefore, we name the intermediate state B*-form as it is the hZαADAR1-bound (B-DNA-like) right-handed conformation.
At this stage, it is yet unclear whether these findings reflect a difference in ZBP affinity to various sequences or manifest a difference in the number of bound ZBP needed for the B–Z conversion. To answer this question, we develop a model below.
Model
Based on the structure analysis (36), we assume that a protein occupies a segment of 6 bp in length. It is also comparable with the size of DNA which can be flipped to Z-form by hZαADAR1 binding (36). In the core molecule, the 22-bp switchable sequence allows at least three proteins to fit in tandem along the backbone of DNA. For each 6-bp slot, two hZαADAR1 proteins can bind to DNA alongside, in opposite to each other (Figure 1B). Hence there are six binding sites on each core DNA molecule (6-binding-site model). We also consider a more compact binding such that two proteins can bind to every 5-bp of DNA. This model allows as many as 10 proteins to fit in the core DNA (10-binding-site model).
The entropic part of the chemical potential for hZαADAR1 adsorption to DNA is μc = kBTln (ca3) where a and c stand for the size and concentration of hZαADAR1, respectively. By definition a3 reflects the bulk volume spanned by hZαADAR1. Here, we determine the value of a from the best fit to our statistical physics model. A typical value of μc at 1 μM is μc = −10.3 kBT (below, values for energy are all presented in unit of kBT).
We assume three different binding modes of hZαADAR1: on B-form, on de novo Z-form, and on pre-existing Z-form. The binding affinities to B-form and Z-form are denoted as AB and AZ, respectively. Upon a first adsorption by hZαADAR1, the state of DNA can be either B-form or (de novo) Z-form. Besides the junction energy cost of 2EJ for a newly formed Z-DNA domain, the free energy per base pair of Z-form is larger than that of B-form by εZ. If a protein converts n0(= 6) bp to Z-form, the additional energy cost is n0εZ. The second binding to the same site by another protein, however, does not involve this additional energy cost.
When a protein binds to free DNA, which is either B- or Z-form, the corresponding free energy cost would be −AB and −AZ + n0εZ, respectively. When two proteins fully occupy two binding sites of a 6-bp DNA segment, which is either B- or Z-form, the corresponding free energy cost would be −2AB and −2AZ + n0εZ, respectively. Thus the apparent affinity 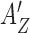 for the first protein is effectively reduced to
for the first protein is effectively reduced to 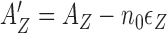 . Besides the ‘local’ free energy cost, a junction energy EJ applies for each B–Z junction. If both ends of the core sequence are flanked by B-DNA, Z-DNA formation is accompanied by two B–Z junctions with the domain wall energy of 2EJ.
. Besides the ‘local’ free energy cost, a junction energy EJ applies for each B–Z junction. If both ends of the core sequence are flanked by B-DNA, Z-DNA formation is accompanied by two B–Z junctions with the domain wall energy of 2EJ.
The total grand canonical partition function is given by 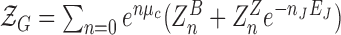 with
with 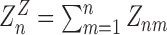 (
(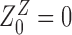 ) where Znm stands for the partition function with n adsorbed proteins among which m proteins are on Z-DNA.
) where Znm stands for the partition function with n adsorbed proteins among which m proteins are on Z-DNA. 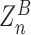 stands for the partition function of B-DNA with n adsorbed proteins. We assume that the protein adsorption on B-DNA is not correlated but its adsorption on Z-DNA is strongly correlated due to the high junction energy penalty. As we consider a short DNA molecule, we neglect formation of multiple Z-DNA domains (thus, we only consider the case of nJ = 2). In our model, the total number of hZαADAR1 molecules bound within the 22-bp-long core is up to six. We call this model 6-binding-site model. The probability to have low FRET signal is the fraction of molecule bearing Z-DNA.
stands for the partition function of B-DNA with n adsorbed proteins. We assume that the protein adsorption on B-DNA is not correlated but its adsorption on Z-DNA is strongly correlated due to the high junction energy penalty. As we consider a short DNA molecule, we neglect formation of multiple Z-DNA domains (thus, we only consider the case of nJ = 2). In our model, the total number of hZαADAR1 molecules bound within the 22-bp-long core is up to six. We call this model 6-binding-site model. The probability to have low FRET signal is the fraction of molecule bearing Z-DNA.
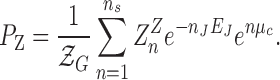 |
(1) |
The population of Z-DNA (PZ) obtained by integrating the FRET histogram in Figure 2 is fit by Eq. (1) as shown in Figure 3. We also fit the B*-state with
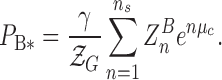 |
(2) |
For the best fit to TG data, a majority of states with n ≥ 1 (γ = 0.85 in Eq. (2)) are included for the B*-state. We will come back to the interpretation of γ <1 at the end of the discussion section. The hZαADAR1-free state or a small fraction of hZαADAR1-bound states of DNA are counted as the B-DNA state.
Our analysis suggests that hZαADAR1 also has some affinities to B-DNA as well as to Z-DNA and the latter is larger than the former by ∼7 kBT : AZ = 16.2 and 15.8 for GC and TG repeats, respectively, and AB = 9.2 (see ENDNOTE). From the 10-binding-site model, we obtain AZ = 13.4 and 13.1 for GC and TG repeats, respectively, and AB = 8.4. The molecule size a from the best fit is a ≈ 5.8 nm. The detailed description of the 10-binding-site model is given in SI. Although the values obtained for the affinities are slightly shifted from those of the 6-binding-site model, their difference is almost unchanged. Both models confirm that hZαADAR1 predominantly adsorbs to Z-DNA and also adsorbs to B-form. The difference of affinities of hZαADAR1 to GC and TG sequences in Z-form does not appear to be dramatic. This comforts the view that protein affinities are not sequence sensitive as long as they are alternating purine-pyrimidine sequences (31). The larger protein concentration for switching TG repeats to Z-form should be attributed to the higher free energy of Z-DNA in the sequence.
DISCUSSION
In the presence of hZαADAR1, the TG repeat sequence exhibits a thermodynamic distribution of states markedly different from that of the GC repeat sequence. The TG repeat sequence requires a nearly two-orders-of-magnitude higher protein concentration than the GC counterpart to induce the same level of Z-state. Moreover, we identified an intermediate state adopted by the TG repeat sequence that interacts with hZαADAR1, which we call B*-DNA as it is likely to have a similar elongation (seen from smFRET data, the peak at FE ∼0.4 for B*-DNA is close to the peak corresponding to B-DNA at FE ∼0.5) and a right-handed helicity (seen from CD data).
From the analysis of Figure 3, we find that in the case of GC core, Z-DNA forms when a few hZαADAR1 bind to B-DNA. In contrast, for TG core, the intermediate state should be further populated by hZαADAR1 and the transition to Z-DNA occurs only at higher concentrations (more than a few μM). Despite the high protein affinity to Z-DNA, the Z-DNA state is only metastable in certain circumstances due to the domain wall energy penalty and free energy cost of the Z-DNA state. If a single protein is adsorbed, the energy gain by protein binding is not large enough to stabilize Z-DNA for both (GC)n and (TG)n. The free energy cost for Z-DNA formation with a single hZαADAR1 bound, EZ = −AZ + 2EJ + n0εZ, are ∼5.2 and ∼8.9 for (GC)n and (TG)n, respectively (up to similar entropy for placing a ZBP along the target sequence). The DNA structure is thus expected to remain close to B-DNA (note that AB ∼9.2). At a larger protein concentration, more proteins would adsorb. With 2 proteins at the same slot of DNA, it can be seen that 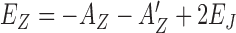 are −10.9 and −6.9 for (GC)n and (TG)n, respectively. On the other hand, the energy for the B-DNA state with 2 proteins bound is 2AB = −18.4 and this state is more stable than the Z-DNA state. With 4 proteins over two binding slots of DNA,
are −10.9 and −6.9 for (GC)n and (TG)n, respectively. On the other hand, the energy for the B-DNA state with 2 proteins bound is 2AB = −18.4 and this state is more stable than the Z-DNA state. With 4 proteins over two binding slots of DNA, 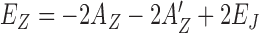 are −39.8 for (GC)n, which is lower than the energy of the B-DNA with 4 hZαADAR1 molecules bound (−4AB = −36.8) while EZ = −31.8 for (TG)n is higher than the same B-DNA energy. This suggests that four hZαADAR1 adsorbates may trigger a conversion of B-DNA to Z-DNA for (GC)n core DNA, but not for (TG)n: hZαADAR1 will form a stable B*-DNA domain first before any possible structural transition. For (TG)n, Z-DNA becomes stable (with respect to B*-DNA) if more than 5 hZαADAR1 molecules are adsorbed on DNA, which is possible at a sufficiently large hZαADAR1 concentration. At some range of protein concentration, thus, we expect both B-form and Z-form to coexist. The midpoint concentrations of hZαADAR1 at which about 50% of DNA molecules adopt the low FRET state are ∼0.3–0.4 μM for (GC)n repeats and ∼10 μM for (TG)n repeats, respectively. The concentration-dependent chemical potential values μc of hZαADAR1 at these midpoint concentrations are ∼−11.2 to −11.5 (GC) and ∼−8.0 (TG). The conversion of the core sequence to Z-DNA is possible only if several proteins are working together, requiring some accumulation of proteins on B-DNA. For hZαADAR1 adsorption on a TG core in B-form, we require that the affinity of hZαADAR1 to B-DNA be comparable to or larger than |μc| at its midpoint concentration for the B–Z transition. Our prediction for AB ≈ 9.2 is consistent with this picture.
are −39.8 for (GC)n, which is lower than the energy of the B-DNA with 4 hZαADAR1 molecules bound (−4AB = −36.8) while EZ = −31.8 for (TG)n is higher than the same B-DNA energy. This suggests that four hZαADAR1 adsorbates may trigger a conversion of B-DNA to Z-DNA for (GC)n core DNA, but not for (TG)n: hZαADAR1 will form a stable B*-DNA domain first before any possible structural transition. For (TG)n, Z-DNA becomes stable (with respect to B*-DNA) if more than 5 hZαADAR1 molecules are adsorbed on DNA, which is possible at a sufficiently large hZαADAR1 concentration. At some range of protein concentration, thus, we expect both B-form and Z-form to coexist. The midpoint concentrations of hZαADAR1 at which about 50% of DNA molecules adopt the low FRET state are ∼0.3–0.4 μM for (GC)n repeats and ∼10 μM for (TG)n repeats, respectively. The concentration-dependent chemical potential values μc of hZαADAR1 at these midpoint concentrations are ∼−11.2 to −11.5 (GC) and ∼−8.0 (TG). The conversion of the core sequence to Z-DNA is possible only if several proteins are working together, requiring some accumulation of proteins on B-DNA. For hZαADAR1 adsorption on a TG core in B-form, we require that the affinity of hZαADAR1 to B-DNA be comparable to or larger than |μc| at its midpoint concentration for the B–Z transition. Our prediction for AB ≈ 9.2 is consistent with this picture.
From the observed midpoint concentration, we can see that the ‘quorum’ number nQ of hZαADAR1 for Z-DNA formation is nQ = 6 for TG repeats where EZ = −56.7 (note EZ = −68.7 for GC repeats). At concentrations ∼0.35 μM or less, the penalty in chemical potential (∼11.4) is larger than the energy gain by binding to B-DNA (AB = 9.2). Thus hZαADAR1 adsorption is weak unless the repeat DNA turns to Z-form. (The Z-DNA population increases very rapidly above the midpoint concentration). The stable Z-form can be formed only in GC repeat sequences as EZ <nμc (EZ and μc are negative.).
There is a clear difference in the transition behavior: Z-DNA formed in GC repeats can be stable in the natural context in which the Z-DNA-forming sequence is flanked by B-DNA regions while Z-DNA formed in TG repeats is less stable than B-DNA if n < nQ = 6. This hZαADAR1-bound B-DNA can be the candidate for the intermediate state (FE = 0.4) shown in Figure 2. For GC, switching into Z-form can be triggered by proteins directly adsorbing from bulk. For TG, however, some proteins need to be adsorbed and accumulated on B-form prior to the B–Z transition. Here we assume that the affinity of hZαADAR1 to the B-form by the GC core is the same as to the B-form by the TG core. A priori they could be slightly different without any impact on our interpretation.
We notice that the results should be interpreted with caution as they can be affected by experimental arrangements such as dye labelling. It turns out that the effect of specific experimental arrangements would not affect the main conclusions. For details, see SI.
We found that the population of the B*-state matches 85% of the (estimated) total population of the pre-complex of B-DNA and hZαADAR1 (Eq. 2). Considering that the spacing (14 bp) between donor and acceptor dyes is smaller than the size of core sequences (22 bp) (Figure 1B), we expect some fraction of adsorbed proteins not to sit between the dyes. Proteins located outside the inter-dye region make less contribution to the shift of FRET signal. This effect results in a slightly smaller value of γ = 0.85 < 1.
We also considered the 10-binding-sites model suggested by crystal structure determination (33) where the size of flipped DNA upon binding by one hZαADAR1 is only 5 bp and proteins can be packed at every 5 bp along a DNA strand matching the size of binding site (33). As far as the affinity difference is concerned, we obtained similar conclusions.
An earlier work (37) and the one following (32) mentioned an intermediate state in the path to the B–Z transition. There, it is suggested that the B-DNA turns to the Z-DNA (Zp2 state) via Bp and Zp and an estimate for protein affinity was obtained for GC, which is the easiest sequence to convert to Z-DNA. Although a similar term ‘intermediate state’ is used, the system we considered is quite different from systems considered in previous studies. In Kang et al. (37), the authors assume the 6 states, B, Bp, Bp2, Z, Zp and Zp2 and consider a single protein binding slot where up to two proteins can be adsorbed. This model would treat each 6-bp slot in the core independently. We considered a long convertible DNA segment (embedded in a B-DNA molecule) where the cooperativity plays an important role. Due to the high junction energy, a step of nucleation is required prior to conversion into Z-DNA. The B*-state we reported has to accumulate before switching the core sequence cooperatively. What we call B* may not exactly match the intermediate state mentioned in earlier literature. Dynamics is also very slow as compared to experiments in (11). This is not surprising because two systems have clear differences. Bae et al. (11) use nickel ions, which is not a common ion in a biological system and the ions help DNA to make the B–Z transition easily. Also the DNA construct used therein is terminated at the end of GC repeat while our GC or TG repeat sequence is flanked with ordinary scrambled sequence.
An interesting side issue is whether the intermediate right-handed state B* corresponds to a secondary energy minimum of the DNA core sequence itself (intrinsic) or whether it is characteristic of the protein-bound DNA system (pre-complex). In the former case, the very same state could be populated by different external stimuli. Previous experiments deal with B-to-Z conversions under negative torque or under high salt concentrations.
To the best of our knowledge, none of these experiments currently hint at the existence of an intermediate state (in B–Z transition) of the DNA itself. In our salt experiments, the same (GC)11 core was converted to Z-DNA but (TG)11 core did not go to Z-DNA as shown by the fact that the FRET efficiency remains at 0.3. This result is shown in SI (Supplementary Figure S7). There are a small number of molecules for (GC)11, the FRET efficiency of which is coincidentally in the same range as that of B*-DNA. They are unlikely to be related to (salt-induced) Z-DNA because the population is insensitive to salt concentration (see Supplementary Figure S8) and the third FRET state is not observed as an intermediate state in the dynamic trajectories of the salt-induced B–Z transition (see Supplementary Figure S9). As pointed out recently in (39), the intermediary FRET signal is likely to originate from some minor misfolded states. The mechanical experiment is, however, the more direct one. As is in the current case, the torsional angle (strain) rather than the torque should be controlled to give room for the formation of B*-DNA. We however could not observe it. At present, it seems more likely that B* is characteristic of the pre-complex rather than intrinsic.
CONCLUSIONS
We provide a model for protein-induced Z-DNA formation in a more realistic context where the Z-forming DNA sequence is embedded within a long random sequence and the junction energies naturally come into play for cooperative switching. We show the stable intermediate state B* in the path of the B–Z transition and make a connection to the protein-bound B-state. Due to the high junction energy, a step of nucleation is necessary prior to the B–Z transition. The B* state we reported has to accumulate before switching the core sequence cooperatively. These nucleation and cooperativity are not addressed in the earlier literature that mention an intermediate state (32).
We successfully and quantitatively predict the affinity values of hZαADAR1 for different Z-DNA forming sequences and also for B-DNA. The formation of the B* pre-complex can be observed only from Z-DNA forming sequences not from scrambled sequences, which strongly suggests that hZαADAR1 binds to DNA in a sequence-specific manner and likely induces the B–Z transition after binding. Then the affinities to Z-DNA forming sequences are measured to be almost the same for GC and TG repeats, ∼10 kcal/mole, which is about ∼1.7 kcal/mole per ZBP-bound bp. Despite this insensitivity of the protein affinity, the paths to Z-DNA reveal a sequence-dependent behavior, mainly due to different stability of the Z-DNA state in those sequences. In GC repeats, the B* pre-complex easily decays into the complex of ZBP-Z-DNA. In TG repeats, proteins accumulate on the B-DNA (B*-state) before converting into a Z-state.
A stable Z-DNA requires six hZαADAR1 proteins to bind to one TG core while four hZαADAR1 proteins would be sufficient for a GC core. At the mid-point hZαADAR1 concentration, it is expected that the B*-state corresponds to an altered B-state by adsorption of three to five hZαADAR1 molecules.
Our results are not only compatible with recent NMR experiments (32) but also provide a (very plausible) scenario to understand what is happening in terms of the interaction of hZαADAR1 and DNA sequences. Consequently, our study showing the presence of a stable B* state supports the active picture for the protein-induced B–Z transition which occurs under a physiologically relevant mechanical context.
We further speculate that the right-handed pre-complex accumulates torsional stress within the core and thus lowers the free energy barrier to the B-to-Z-transition presumably by stabilizing the transient state in the transition. Given that chemically realistic simulations of the complex of ZBP and Z-DNA remain difficult (38), we believe that our work provides crucial insights into the protein-mediated B–Z transition of Z-DNA forming sequences.
ENDNOTE
The molecule sizes a from the best fits are a ≈ 3.7 and 3.8 nm for GC and TG, respectively, and they are somewhat larger than 6 bp in length. As mentioned before, it corresponds to the bulk size of the whole Zα domain. Hypothesizing the absence of B* in GC repeats, we obtain similar affinity AZ = 16.1 with a = 3.8 nm and γ = 0.85.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
National Research Foundation of Korea [NRF-2014R1A1A2055681 and NRF-2017R1A2B4010632 to N.-K.L., NRF-2016R1A2B4014855 to S.-C.H.]: Institute for Basic Science [IBS-R023-D1 to S.-C.H.]. Funding for open access charge: Institute for Basic Science [IBS-R023-D1].
Conflict of interest statement. None declared.
REFERENCES
- 1. Wells R.D. Non-B DNA conformations, mutagenesis and disease. Trends Biochem. Sci. 2007; 32:271–278. [DOI] [PubMed] [Google Scholar]
- 2. Rich A., Norheim A., Wang A.H.-J.. The chemistry and biology of left-handed Z-DNA. Annu. Rev. Biochem. 1984; 53:791–846. [DOI] [PubMed] [Google Scholar]
- 3. Rich A. Z-DNA: The long road to biological function. Nat. Rev. Genet. 2003; 4:566–572. [DOI] [PubMed] [Google Scholar]
- 4. Mirkin S.M., Frank-Kamenetskii M.D.. H-DNA and related structures. Annu. Rev. Biophys. Biomol. Struct. 1994; 23:541–576. [DOI] [PubMed] [Google Scholar]
- 5. Williamson J.R. G-quartet structures in telomere DNA. Annu. Rev. Biophys. Biomol. Struct. 1994; 23:703–730. [DOI] [PubMed] [Google Scholar]
- 6. Gueron M., Leroy J.-L.. The i-motif in nucleic acids. Curr. Opinion. Struct. Biol. 2000; 10:326–331. [DOI] [PubMed] [Google Scholar]
- 7. Lobachev K.L., Rattray A., Narayanan V.. Hairpin-and cruciform-mediated chromosome breakage: causes and consequences in eukaryotic cells. Front. Biosci. 2007; 12:4208–4220. [DOI] [PubMed] [Google Scholar]
- 8. Lee J.Y., Okumus B., Kim D.S., Ha T.J.. Extreme conformational diversity in human telomeric DNA. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:18938–18943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee N.-K., Park J.-S., Johner A., Obukhov S., Hyon J.-Y., Lee K.J., Hong S.-C.. Elasticity of cisplatin-bound DNA reveals the degree of cisplatin binding. Phys. Rev. Lett. 2008; 101:248101. [DOI] [PubMed] [Google Scholar]
- 10. Lee M., Kim S.H., Hong S.-C.. Minute negative superhelicity is sufficient to induce the B–Z transition in the presence of low tension. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:4985–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bae S., Kim D., Kim K.K., Kim Y.-G., Hohng S.. Intrinsic Z-DNA Is stabilized by the conformational selection mechanism of Z-DNA-binding proteins. J. Am. Chem. Soc. 2011; 133:668–671. [DOI] [PubMed] [Google Scholar]
- 12. Oberstrass F.C., Fernandes L.E., Bryant Z.. Torque measurements reveal sequence-specific cooperative transitions in supercoiled DNA. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:6106–6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koirala D., Dhakal S., Ashbridge B., Sannohe Y., Rodriguez R., Sugiyama H., Balasubramanian S., Mao H.. A single-molecule platform for investigation of interactions between G-quadruplexes and small-molecule ligands. Nat. Chem. 2014; 3:782–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salerno D., Brogioli D., Cassina V., Turchi D., Beretta G.L., Seruggia D., Ziano R., Zunino F., Mantegazza F.. Magnetic tweezers measurements of the nanomechanical properties of DNA in the presence of drugs. Nucleic Acids Res. 2010; 38:7089–7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang A.H., Quigley G.J., Kolpak F.J., Crawford J.L., van Boom J.H., van der Marel G., Rich A.. Molecular structure of a left-handed double helical DNA fragment at atomic resolution. Nature. 1979; 282:680–686. [DOI] [PubMed] [Google Scholar]
- 16. Pohl F.M., Jovin T.M.. Salt-induced co-operative conformational change of a synthetic DNA: Equilibrium and kinetics studies with poly(dG-dC). J. Mol. Biol. 1972; 67:375–396. [DOI] [PubMed] [Google Scholar]
- 17. Wittig B., Wölfl S., Dorbic T., Vahrson W., Rich A.. Transcription of human c-myc in permeabilized nuclei is associated with formation of Z-DNA in three discrete regions of the gene. EMBO J. 1992; 11:4653–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu R., Liu H., Chen X., Kirby M., Brown P.O., Zhao K.. Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell. 2001; 106:309–318. [DOI] [PubMed] [Google Scholar]
- 19. Maruyama A., Mimura J., Harada N., Itoh K.. Nrf2 activation is associated with Z-DNA formation in the human HO-1 promoter. Nucleic Acids Res. 2013; 41:5223–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peck L.J., Wang J.C.. Transcriptional block caused by a negative supercoiling induced structural change in an alternating CG sequence. Cell. 1985; 40:129–137. [DOI] [PubMed] [Google Scholar]
- 21. Rahmouni A.R., Wells R.D.. Stabilization of Z DNA in vivo by localized supercoiling. Science. 1989; 246:358. [DOI] [PubMed] [Google Scholar]
- 22. Wittig B., Dorbic T., Rich A.. The level of Z-DNA in metabolically active, permeabilized mammalian cell nuclei is regulated by torsional strain. J. Cell Biol. 1989; 108:755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vologodskii A.V., Frank-Kamenetskii M.D.. Left-handed Z form in superhelical DNA: A theoretical study. J. Biomol. Struct. Dyn. 1984; 1:1325–1333. [DOI] [PubMed] [Google Scholar]
- 24. Peck L.J., Wang J.C.. Energetics of B-to-Z transition in DNA. Proc. Natl. Acad. Sci. U.S.A. 1983; 80:6206–6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Son A., Kwon A.Y., Johner A., Hong S.-C., Lee N.-K.. Underwound DNA under tension: L-DNA vs. plectoneme. EPL. 2014; 105:48002. [Google Scholar]
- 26. Kwon A.Y., Nam G.M., Johner A., Kim S., Hong S.-C, Lee N.-K.. Competition between B–Z and B-L transitions in a single DNA molecule: Computational studies. Phys. Rev. E. 2016; 93:022411. [DOI] [PubMed] [Google Scholar]
- 27. Fuertes M., Cepeda V., Aloso C., Perez J.. Molecular mechanism for the BZ transition in the example of Poly d(G-C)·d(G-C) Polymers. A Critical Review. Chem. Rev. 2006; 106:2045–2064. [DOI] [PubMed] [Google Scholar]
- 28. Ho P. The non-B-DNA structure of d(CA/TG)n does not differ from that of Z-DNA. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:9549–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Herbert A., Alfken J., Kim Y.G., Mian I.S., Nishikura K., Rich A.. A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:8421–8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schwartz T., Rould M.A., Lowenhaupt K., Herbert A., Rich A.. Crystal structure of the Zα domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science. 1999; 284:1841–1845. [DOI] [PubMed] [Google Scholar]
- 31. Herbert A., Schade M., Lowenhaupt K., Alfken J., Schwartz T., Shlyakhtenko L.S., Lyubchenko Y.L., Rich A.. The Zα domain from human ADAR1 binds to the Z-DNA conformer of many different sequences. Nucleic Acids Res. 1998; 26:3486–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee A.R., Park C.J., Cheong H.K., Ryu K.-S., Park J.-W., Kwon M.-Y., Lee J., Kim K.K., Choi B.-S., Lee J.-H.. Solution structure of the Z-DNA binding domain of PKR-like protein kinase from Carassius auratus and quantitative analyses of the intermediate complex during B–Z transition. Nucleic Acids Res. 2016; 44:2936–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ha S.C., Lowenhaupt K., Rich A., Kim Y.G., Kim K.K.. Crystal structure of a junction between B-DNA and Z-DNA reveals two extruded bases. Nature. 2005; 437:1183–1186. [DOI] [PubMed] [Google Scholar]
- 34. Schade M, Turner C.J., Kühne R., Schmieder P., Lowenhaupt K., Herbert A., Rich A., Oschkinat H.. The solution structure of the Zα domain of the human RNA editing enzyme ADAR1 reveals a prepositioned binding surface for Z-DNA. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:12465–12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nyrkova I.A., Semenov A.N.. Theory of chiral recognition in DNA condensation. Soft Matter. 2009; 5:979–989. [Google Scholar]
- 36. Schwartz T., Behlke J., Lowenhaupt K., Heinemann U., Rich A.. Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat. Struct. Biol. 2001; 8:761–765. [DOI] [PubMed] [Google Scholar]
- 37. Kang Y.-M., Bang J., Lee E.-H., Ahn H.-C., Seo Y.-J., Kim K.K., Kim Y.-G., Choi B.-S., Lee J.-H.. NMR spectroscopic elucidation of the B–Z transition of a DNA double helix induced by the Zα domain of human ADAR1. J. Am. Chem. Soc. 2009; 131:11485–11491. [DOI] [PubMed] [Google Scholar]
- 38. Wang Q., Li L., Wang X., Liu H., Yao X.. Understanding the recognition mechanisms of Zα domain of human editing enzyme ADAR1 (hZαADAR1) and various Z-DNAs from molecular dynamics simulation. J. Mol. Model. 2014; 20:2500. [DOI] [PubMed] [Google Scholar]
- 39. Ivanov I.E., Lebel P., Oberstrass F.C., Starr C.H., Parente A.C., Ierokomos A., Bryant Z.. Multimodal measurements of single-molecule dynamics using FluoRBT. Biophys. J. 2018; 114:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.