

Abstract
The expression of proteins during inflammatory and immune reactions is coordinated by post-transcriptional mechanisms. A particularly strong suppression of protein expression is exerted by a conserved translational silencing element (TSE) identified in the 3′ UTR of NFKBIZ mRNA, which is among the targets of the RNA-binding proteins Roquin-1/2 and MCPIP1/Regnase-1. We present evidence that in the context of the TSE MCPIP1, so far known for its endonuclease activity toward mRNAs specified by distinct stem–loop (SL) structures, also suppresses translation. Overexpression of MCPIP1 silenced translation in a TSE-dependent manner and reduced ribosome occupancy of the mRNA. Correspondingly, MCPIP1 depletion alleviated silencing and increased polysomal association of the mRNA. Translationally silenced NFKBIZ or reporter mRNAs were mostly capped, polyadenylated and ribosome associated. Furthermore, MCPIP1 silenced also cap-independent, CrPV-IRES-dependent translation. This suggests that MCPIP1 suppresses a post-initiation step. The TSE is predicted to form five SL structures. SL4 and 5 resemble target structures reported for MCPIP1 and together were sufficient for MCPIP1 binding and mRNA destabilization. Translational silencing, however, required SL1–3 in addition. Thus the NFKBIZ TSE functions as an RNA element in which sequences adjacent to the site of interaction with MCPIP1 and dispensable for accelerated mRNA degradation extend the functional repertoire of MCPIP1 to translational silencing.
INTRODUCTION
The coordinated generation, execution and termination of the response to pathogens requires tightly regulated expression of the participating proteins. Post-transcriptional mechanisms play an essential part in this regulation (1–3). Selective control of the fate of distinct mRNAs is based on the presence of sequence elements which interact with trans-acting factors that can be either regulatory RNA species or RNA-binding proteins. Transcripts encoding cytokines, transcription factors or other proteins relevant to inflammation often contain AU-rich elements (AREs) that accelerate their degradation by interacting with RNA-binding proteins. The function of the latter is targeted by signaling pathways activated by pathogen structures like bacterial lipopolysaccharides or by inflammatory mediators (4,5). In cytokine mRNAs additional elements were identified that can form stem–loop (SL) structures, including a constitutive decay element (CDE) in the TNF-α mRNA (6), and an element in the interleukin (IL-)6 mRNA that contributes to its instability (7). Each of these mRNAs represents a group of target mRNAs regulated by distinct RNA-binding proteins: Roquin-1 and 2 that interact with the CDE and related elements (8) and MCPIP1/Regnase-1 that degrades IL-6 and other mRNAs, including its own transcript, via its intrinsic RNase activity (9,10). More recent evidence suggests that these proteins control overlapping groups of target mRNAs and that their functions are interrelated (11,12).
We have identified the ZC3H12A mRNA, which encodes MCPIP1, in an approach to detect translational regulation activated by the proinflammatory cytokine IL-1 (13). The ZC3H12A mRNA belongs to a set of mRNAs which are poorly translated in unstimulated cells as indicated by their weak association with ribosomes. Cytokine stimulation strongly increases ribosome occupancy of these mRNAs (13). They include ARE-containing transcripts and their translational suppression appears to involve ARE-binding proteins K homology-type splicing regulatory protein (KSRP) (14) and tristetraprolin (15), which are targeted by the p38 MAPK pathway. A second group of transcripts, represented by NFKBIZ and ZC3H12A mRNAs, is regulated by a different mechanism that is not affected by p38 MAPK signaling, functions independently of KSRP and requires elements different from AREs (13,16). ZC3H12A encodes MCPIP1 (see above) that was identified as a protein induced by the monocyte chemoattractant protein-1 (MCP-1), hence its name MCP-induced protein (17). It is a member of a family of four paralogs (18). Its important roles in coordinating inflammatory and immune reactions as a negative feedback regulator and its implications in different disease states have been highlighted in recent reviews (3,19–21). Besides transcription factor-like activity (22), deubiquitinating function (23,24) and RNase activity have been reported for this protein, which accordingly was also named Regnase-1 (10). Both, deubiquitinating and RNase activities together can contribute to biological function of MCPIP1, as exemplified by the involvement of both activities in macrophage polarization (25). In addition to degrading mRNAs, MCPIP1 can exert RNase activity against viral RNAs (26) and pre-microRNAs (27).
The NFKBIZ mRNA encodes IκBζ, an atypical member of the IκB family of proteins that modulates transcription of genes pertinent to inflammatory and immune reactions. It is involved in the differentiation of immune cells and its deficiency causes severe derangements in the immune system (for review see (28)). Our previous analysis showed that the 3′ UTR of NFKBIZ mRNA harbors a novel translational silencing element (TSE) that is predicted to form five SL structures (13). A part of the TSE encompassing only SL4 and 5 and lacking translational silencing activity was found sufficient to destabilize reporter mRNAs. NFKBIZ and ZC3H12A mRNAs are regulated in a similar manner (ARE-independent 3′ UTR-mediated degradation and translational silencing which is overcome by IL-1 and IL-17 stimulation (13,16)). Since MCPIP1, the protein encoded by ZC3H12A, degrades its own mRNA in an autoregulatory fashion, we hypothesized that MCPIP1 not only degrades NFKBIZ mRNA as well but might also contribute to translational silencing by the NFKBIZ TSE.
We report here that MCPIP1 interacts with SL4 and 5 of the TSE. While this interaction is sufficient for mRNA destabilization, the context of SL1–3 of the TSE endows MCPIP1 with hitherto unidentified translational silencing activity.
MATERIALS AND METHODS
Cells and plasmids
HeLa cells constitutively expressing the tetracycline-controlled transactivator protein (29) were cultured as described (30). HEK-293 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal calf serum, 100 units/ml penicillin and 100 μg/ml streptomycin. Transfection with plasmids (see below) was performed by the calcium phosphate method as described (30). MCPIP1 was depleted in HeLa cells using the CRISPR/Cas9 technology. For this sgRNAs targeting the MCPIP1 gene ZC3H12A were designed (sgRNA#2, GCAGGACGCTGTGGATCTCCG; sgRNA#4, GTGAGGACAGCCACAGCCGTC) using the optimized CRISPR tool provided by the Zhang lab (http://crispr.mit.edu) and cloned into the lentiCRISPR v1 vector via BsmBI restriction sites. lentiCRISPR v1 was a gift from Feng Zhang (Addgene, now available as lentiCRISPR v2 #52961) (31) and provides stable expression of sgRNA and Cas9 nuclease from one vector. Lentiviral particles were produced by calcium phosphate transfection of HEK-293T cells with lentiCRISPR v1, pVSVG and psPAX2 (Addgene # 12260, a gift from Didier Trono). HeLa cells were transduced with lentiviral particles at low multiplicity of infection via spinfection at 300 × g for 90 min in the presence of 5 μg/ml polybrene. After centrifugation, the medium was replaced by fresh medium. Starting 48 h after transduction, cells were cultured in the presence of 2 μg/ml puromycin for 3 days to select for infected cells. The depletion of MCPIP1 was verified by immunoblotting.
The human NFKBIZ TSE sequence (nt 2273–2425; numbering according to NM_031419) and fragments thereof were amplified by reverse transcription-polymerase chain reaction (RT-PCR) using total RNA from HeLa cells. PCR products were inserted into the BglII site 3′ of the β-globin coding region in ptetBBB (32) or into newly created BglII sites downstream of the green fluorescence protein (GFP) coding sequence that was introduced into pUHD10–3 (29) to obtain pUHD10-GFP, or downstream of the firefly luciferase coding sequence in pMir-Report (Ambion). Fragments of human NFKBIZ mRNA were reverse-transcribed from HeLa cell RNA and cloned into pcDNA3.1 to express the full length mRNA (nt 1–3885) with a newly generated in-frame chimeric BamHI/BglII site that allowed specific quantitation by quantitative RT-PCR (RT-qPCR) using the following primers: sense 5′-GGAGAACGAACAGCCAGTGC, antisense 5′-CTCCAAGCTAATGGAGAGATCC. For translation driven by the internal ribosome entry site (IRES) of Cricket Paralysis Virus (CrPV) the sequence of CrPV-IRES was PCR-amplified using the plasmid pFR_CrPV_xb (a gift from Phil Sharp (Addgene plasmid # 11509)) and introduced upstream of the firefly luciferase coding sequence in pMIR-Report. A hairpin-encoding sequence (33) was inserted upstream of the CrPV IRES. For HCV-IRES-driven translation DNA corresponding to the IRES sequence (nt 30–385, NC_004102) was purchased (IDT) and inserted in frame upstream of the firefly coding sequence (34) into pMIR-Report. In the IRES-containing plasmids the firefly luciferase start codon was mutated to AAG. Renilla luciferase was expressed using the plasmid phRL-TK (Promega). The plasmid expressing GFP-MCPIP1 has been described previously (16). The expression plasmid for strep-tagged MCPIP1 (strep-MCPIP1) was obtained via insertion of the human MCPIP1 coding sequence (nt 146–1957; numbering according to NM_025079.2) in frame downstream of the Twin-Strep-tag sequence into pEXPR-IBA105 (IBA). Point mutations and deletions of strep-MCPIP1 expression plasmid and of reporter plasmids were generated via the QuikChange site directed-mutagenesis procedure (Stratagene). Primer sequences are available upon request.
Sucrose gradient fractionation and RNA isolation
Cytoplasmic lysates were prepared and centrifuged through linear sucrose gradients (10–50% sucrose) for 120 min and fractions were collected as described previously (13). A total of 0.1 volume of 3 M sodium acetate (pH 5.2) and 1 volume of isopropanol were added to each fraction for overnight precipitation at −20°C. RNA was purified using Nucleospin RNA tubes (Macherey-Nagel) following the manufacturer’s protocol including an additional rDNase digestion in solution and ethanol precipitation to minimize DNA contamination. Poly(A) mRNA was purified from total RNA with the Dynabeads® mRNA Purification Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Samples were stored at −80°C until mRNAs were quantified (see below). Results shown were confirmed in at least two independent experiments.
In vitro ribosome binding assay
Ribosomes and ribosomal subunits were purified as described by others (35). Briefly, cytoplasmic lysate of HeLa cells was layered on 2 ml sucrose cushion (0.3 M) and centrifuged for 120 min at 100 000 × g and 4°C in a TLA100.4 rotor (Beckman). Ribosomes were resuspended in buffer A (20 mM Tris–HCl, pH 7.6, 2 mM dithiothreitol (DTT), 6 mM MgCl2, 250 mM sucrose, 150 mM KCl) and adjusted to a concentration of 50–150 A260 U/ml. After addition of 1 mM puromycin, the suspension was incubated for 10 min at 0°C, then for 10 min at 37°C. The KCl concentration was increased to 0.5 M and the ribosomal subunits were resolved by centrifugation through a sucrose gradient (10–50% in buffer A containing 0.5 M KCL) for 180 min at 150 000 × g and 4°C in an SW40 rotor (Beckman). Fractions containing 40 S and 60 S ribosomal subunits as well as 80 S monosomes were collected and centrifuged for 18 h at 140 000 × g and 4°C. The pellets were resuspended in buffer B (20 mM Tris–HCl, pH 7.6, 0.1 mM ethylenediaminetetraacetic acid (EDTA), 10 mM KCl, 1 mM DTT, 1 mM MgCl2, 250 mM sucrose) and stored at −80°C.
Binding assays were performed following published procedures (36). Briefly, RNA was transcribed in vitro with the Mega-Script Kit (Ambion). A total of 2 pmol RNA were incubated with 3.5 pmol 40 S ribosomal subunits and 8 pmol of a mixture of 60 S ribosomal subunits and 80 S monosomes for 5 min at 37°C in a buffer containing 20 mM Tris, pH 7.5, 100 mM KAc, 2 mM DTT, 2.5 mM MgAc, 1 mM adenosine triphosphate (ATP), 0.4 mM GTP, 0.25 mM spermidine. Ribosomes and ribosomal subunits were separated by centrifugation through sucrose gradients, fractions collected and RNA isolated as described above.
Degradation kinetics
For monitoring the kinetics of mRNA degradation transcription was stopped by addition of actinomycin D (5 μg/ml) and total RNA was isolated at different time points thereafter, followed by RT-qPCR-based determination of mRNA amounts (see below).
mRNA detection
Northern blot analysis of β-globin mRNA was performed as previously described (30), using digoxygenin-labeled antisense probes and alkaline phosphatase-coupled anti-digoxygenin antibody (Roche). Blots were developed with the LAS 3000 imaging system (Fujifilm) using CSPD reagent (Roche). For RT-qPCR 500 ng of total RNA was reverse-transcribed using oligo(dT)18 primers (Eurofins) and RevertAid Reverse Transcriptase (Thermo Fisher Scientific). qPCRs were carried out as described (13), using TaqMan assays (Life Technologies, assay-ID Hs00230071_m1 for NFKBIZ mRNA, Hs00174131_m1 for IL6 mRNA, Hs99999905_m1 for GAPDH mRNA, Hs00262018_m1 for NFKBID mRNA, Hs00153283_m1 for NFKBIA mRNA and custom-made assays for rabbit β-globin and firefly luciferase mRNAs) or SybrGreen-based detection (Life Technologies) for mRNAs using primers as follows: GFP: sense 5′-TGCAGTGCTTCAGCCGCTAC-3′, antisense 5′-TCGCCCTCGAACTTCACCTC-3′; Renilla luciferase: sense 5′-CGTCGTGCCTCACATCGAGC-3′, antisense 5′-GCTCGAACCAAGCGGTGAGG-3′; MCPIP1: sense 5′-GGCTGGACAGAGGGAGGATT-3′, antisense 5′-TGACCCACTGAGGCAGACAG-3′; in vitro-transcribed CrPV IRES RNA: sense 5′-GCCTAATACGACTCACTATAGGGTTTAGTGAACCGTGGATC-3′, antisense 5′-GGCGGATCCTGTATCTTGAAATGTAGCAGGTAAA-3′; in vitro-transcribed TSE RNA: sense 5′-TTGGAGCCTGGCTAGCAACA-3′, antisense 5′-TTGCCAATATAAGGCAAATGGTCT-3′.
Luciferase reporter assays
Cells were co-transfected with firefly and Renilla luciferase reporter plasmids and with an expression plasmid for MCPIP1 or an empty vector as indicated. 24 h later the cells were lysed in Passive Lysis Buffer (Promega) and firefly luciferase activity was determined with a GloMax®-MultiMicroplate Multimode Reader (Promega) after mixing lysate with potassium phosphate buffer (25 mM, pH 7.8) containing 15 mM MgCl2, 1 mM ATP, 1 mM DTT, 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid and 25 μM Luciferin. Measurement of Renilla luciferase activity was described before (13). Aliquots of the cell lysates were subjected to RNA isolation and luciferase mRNA levels were quantified by RT-qPCR.
Flow cytometry
Cells were co-transfected with GFP expression plasmids together with an expression plasmid for strep-MCPIP1 or an empty vector as indicated. Twenty-four hours later the cells were trypsinized and resuspended in 1× phosphate-buffered saline (PBS) containing 1% fetal calf serum and 5 mM EDTA. Fluorescence of suspended single cells was measured in a BD Accuri C6 flow cytometer.
Immunoprecipitation
HeLa cells transfected the day before with the indicated plasmids were lysed in IP lysis buffer (20 mM β-glycerophosphate, pH 7.4, 140 mM KCl, 5 mM MgCl2, 0.25 mM DTT, 0.5% (v/v) Nonidet P-40, 0.5 mg/ml fragmin, cOmplete EDTA-free protease inhibitor cocktail (Roche)). All steps were carried out in the cold. The lysates were cleared by centrifugation for 10 min at 12 000 × g, and incubated with antibodies against GFP (monoclonal antibodies 7.1 and 13.1, Roche) or eIF4E (sc-9976, Santa Cruz, USA) or control antibodies (normal IgG, Santa Cruz) for 2 h. After addition of Protein G agarose beads (Santa Cruz) incubation was continued with constant mixing for 2 h. For pulldown of strep-tagged MCPIP1, Strep-Tactin sepharose beads (IBA) were added to the cell lysates instead. After incubation the beads were washed three times with wash buffer (20 mM β-glycerophosphate, pH 7.4, 140 mM NaCl, 5 mM MgCl2) and subjected to RNA isolation. For immunoprecipitation of capped mRNAs, 5 μg anti-m3G/m7G-cap-antibody (clone H-20, EMD Millipore) or normal IgG (Santa Cruz) was prebound to 25 μl Dynabeads protein G (Thermo Scientific) in cap-IP buffer (PBS supplemented with 0.01% Triton, 0.1 mg/ml BSA, 0.05 mg/ml poly(U) and 1 mM DTT) for 60 min at room temperature and washed twice with cap-IP wash buffer (PBS supplemented with 0.01% Triton, 1 mM DTT). After adding 2 μg total HeLa cell RNA in 200 μl cap-IP buffer the beads were incubated for 2 h at room temperature, washed four times with cap-IP wash buffer and subjected to RNA isolation. Co-purified mRNAs were quantified by RT-qPCR.
SDS-PAGE and western blot
Cell lysates were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and proteins blotted onto polyvinylidene fluoride membranes. After blocking with 5% dried milk in Tris-buffered saline containing 0.05% (v/v) Tween-20, the blots were incubated with antibodies against GFP (monoclonal antibodies 7.1 and 13.1, Roche), MCPIP1 (15D11, described in (11)), eEF2 (sc-13004-R, Santa Cruz) or GAPDH (MAB374, Millipore), followed by peroxidase-coupled secondary antibodies (Santa Cruz). Blots were developed with Immobilon Western Chemiluminescent HRP Substrate (Millipore) and chemiluminescence was detected by the LAS 3000 imaging system (Fujifilm).
Bioinformatics analysis
Data sources
All mRNA and sequence data used in this study were acquired from the NCBI Reference Sequence Database (RefSeq) collection release 69 (37). A data set was prepared by extracting all mRNAs of NFKBIZ. The dataset included 65 mammals, 50 birds, 7 reptilian, 4 species of fish and 1 amphibian species. For a complete list of species and associated identification numbers see Supplementary Table S1.
Annotation of NFKBIZ mRNAs
mRNAs with an incompletely annotated 3′UTR were re-annotated by 3′-terminally appending the corresponding genomic sequence. The coordinates of the appended genomic sequence were defined using BLAT (38) version 3.5 with standard parameter settings. The coordinates were derived from the best hit (highest value of matches). To guarantee a minimum quality the resulting elongated mRNAs were subsequently aligned with the corresponding genomic sequences using BLAT. mRNA sequences with an alignment mean pairwise identity greater than 95% were retained.
Sequence and structure conservation analysis
A progressive multiple sequence alignment of mRNA sequences was generated using Tcoffee (39) with standard parameter settings, to localize the conserved TSE sequence. For few species with highly divergent mRNA sequence compared to mouse, the multiple alignment failed and the TSE motif was searched individually using BLASTn (40). To find motifs with a length of <20 nt a minimal word size was chosen (word size = 7) and BLASTn was run with the option task = blastn-short. A combined multiple sequence-structure alignment of the detected TSE sequences followed applying mLocarna version 1.8.1 (41) with the option local-progressive and RNAalifold (42) from the ViennaRNA package version 2.1.8–2 (43).
RESULTS
MCPIP1 interacts with the TSE of NFKBIZ mRNA
In prior studies we have obtained evidence for a translational silencing mechanism that is inactivated by the inflammatory mediators IL-1 and IL-17 and by bacterial lipopolysaccharide. It controls expression of a set of mRNAs including NFKBIZ, NFKBID and ZC3H12A (13,16). MCPIP1 was tested as a candidate protein involved in the translational silencing mechanism first by studying its interaction with these mRNAs in co-precipitation experiments (Figure 1). To minimize loss of target mRNA due to endonucleolytic activity, an RNase-deficient GFP-MCPIP1 fusion protein (GFP-MCPIP1(D141N)) (9) was expressed in HeLa cells and pulldown assays were performed with antibodies to GFP (Figure 1A). Endogenous NFKBIZ, NFKBID and ZC3H12A mRNAs were specifically co-precipitated with GFP-MCPIP1(D141N), as compared to precipitation with GFP as a control (Figure 1B). No specific enrichment was observed for mRNAs of GAPDH and NFKBIA, which encodes an IκB family member that is not regulated in the manner studied here (see Figure 1B in (13)). Translational silencing of NFKBIZ mRNA was mediated by the TSE, an RNA-element in its 3′ UTR (13). Consistently, GFP-MCPIP1(D141N) specifically co-precipitated a β-globin reporter mRNA containing the TSE in its 3′ UTR, as well as IL6 mRNA, used as a positive control (9), whereas the β-globin reporter mRNA alone was not enriched (Figure 1C). This indicates that MCPIP1 interacts with NFKBIZ mRNA through the TSE.
Figure 1.

Co-precipitation of mRNAs with MCPIP1. (A) Lysates of HeLa cells expressing GFP-tagged RNase-deficient MCPIP1 (GFP-MCPIP1(D141N)) (G-M) or GFP (G) as a control were analyzed by western blots with antibodies against MCPIP1 (upper panel). Aliquots from lysates (input) and from beads after pulldown (PD) were analyzed by antibodies against GFP (lower panel) (asterisk: heavy and light chains of anti-GFP antibodies applied for pulldown). (B and C) Total RNA was isolated from lysates and from beads after pulldown and mRNAs were quantitated by RT-qPCR. In (C) plasmids expressing β-globin mRNA without (βG) or with the NFKBIZ TSE inserted into the 3′ UTR (βG-TSE) were co-transfected. Results are expressed as fold increase in the ratio of pulldown over input for GFP-MCPIP1(D141N) as compared to GFP (set as 1) (mean ± SD for one out of three experiments with similar results; asterisk: P < 0.01 for GFP-MCPIP1(D141N) > GFP).
MCPIP1 silences TSE-containing mRNAs in part by interfering with translation
Consequences of MCPIP1 overexpression were studied in HEK-293 cells. Their endogenous MCPIP1 levels (Supplementary Figure S1A) and TSE-mediated silencing (Supplementary Figure S1B and C) were lower compared to HeLa cells, thus providing better conditions for detecting effects of increased MCPIP1 amounts. In fact, expressing low, non-toxic concentrations of strep-tagged MCPIP1 (see Supplementary Figure S1D) revealed that MCPIP1 was limiting for TSE-dependent mRNA degradation in HEK-293 cells (Supplementary Figure S1E). Western blot analysis showed that in HEK-293 cells MCPIP1 at these concentrations suppressed GFP protein translated from a TSE-containing reporter mRNA by more than 10-fold (Figure 2A) whereas the GFP-TSE mRNA was decreased only about 2-fold (Figure 2B).
Figure 2.

Increased TSE-mediated translational silencing in cells overexpressing MCPIP1. (A) HEK-293 cells were co-transfected with a GFP expression plasmid containing the TSE in its 3′ UTR (GFP-TSE) without (con) or with strep-tagged MCPIP1. Cell lysates were subjected to western blot in serial dilutions using antibodies against GFP or eEF2 as a loading control. (B) GFP-TSE mRNA was quantified by RT-qPCR (mean ± SD of quadruplicates from one out of three experiments with similar results). (C) HEK-293 cells were transfected with plasmids encoding GFP mRNAs without (GFP) or with the NFKBIZ TSE (GFP-TSE) together with empty vector (con) or strep-tagged MCPIP1 as indicated. Fluorescence intensity was determined by flow cytometry. (D) Mean fluorescence intensities (MFI,±SD of triplicates; n.s., not significant) of cells transfected with plasmids encoding GFP-TSE together with empty vector (con) or strep-tagged WT or RNase-deficient MCPIP1 (D141N). (E) Fold suppression of protein (determined as MFI) and mRNA amounts (determined by RT-qPCR and normalized to GAPDH mRNA) by the TSE or by strep-MCPIP1 expression or both was calculated by setting the values for GFP mRNA without TSE and in the absence of strep-MCPIP1 as 1 (mean ± SD, n = 5). Similar results were observed in two independent assays. (F) Cytoplasmic lysates from HEK-293 cells transfected with plasmids encoding β-globin (βG) or GFP mRNAs without or with the TSE and co-transfected with empty vector (con) or plasmids for strep-tagged WT or catalytically inactive MCPIP1 were fractionated on sucrose gradients. A typical result of adsorbance profiles at 254 nm for cells expressing empty vector (con) or strep-MCPIP1 is shown in the top panel. The amounts of mRNA in each fraction were determined by RT-qPCR and expressed in per cent of the sum detected in all fractions. Positions of ribosomal subunits, ribosomes and polysomes are indicated. The result is representative of three independent experiments.
This result was confirmed in separate experiments by flow cytometry. MCPIP1 did not affect the amount of GFP expressed by a control mRNA without TSE (Figure 2C, left panel). The presence of the TSE in the reporter mRNA significantly suppressed GFP amounts (compare black lines in Figure 2C). That suppression was markedly increased by co-expression of MCPIP1 (Figure 2C, right panel). The RNAse-deficient mutant MCPIP1(D141N) had no significant effect on protein expressed from the GFP-TSE construct (Figure 2D, presented as mean fluorescence intensity (MFI)). Quantification by RT-qPCR demonstrated much weaker suppression of GFP mRNA by MCPIP1 as compared to GFP protein (Figure 2E). This indicates that a major part of the suppressive activity of MCPIP1 is due to inhibition of translation.
Similar results, indicative of translational regulation, were obtained with firefly luciferase reporter constructs. In the HEK-293 cells the TSE exerted a mild suppressive effect on luciferase activity that was markedly increased by MCPIP1 overexpression (Supplementary Figure S2A). Note that luciferase activity was normalized to luciferase mRNA levels to document changes in activity beyond those caused by differences in mRNA amounts. Changes in luciferase mRNA amounts in our hands were minor, as compared to the marked effects of the TSE alone or in combination with MCPIP1 overexpression on GFP and β-globin mRNA levels and stability (ref. (13) and this study, Figures 2E, 3E and 4). Kinetics of luciferase mRNA and protein accumulation (Supplementary Figure S2B and C) rule out different time courses of induction as an explanation for the observed discrepancy between mRNA and protein amounts. In additional experiments (not shown) the turnover of reporter protein was not affected by MCPIP1. Taken together, the data indicate that MCPIP1 suppresses protein expression in part by interfering with translation.
Figure 3.

Impaired TSE-mediated translational silencing in MCPIP1-depleted cells. (A) Western blot detection of endogenous MCPIP1 in HeLa cells expressing Cas9 nuclease without or with two different sgRNAs specific for the MCPIP1-encoding ZC3H12A gene (sgZ #2 and sgZ #4), and of the two HeLa cell clones established from bulk cultures expressing sgZ #2. (B) HeLa cells analyzed in (A) were transfected with firefly luciferase plasmids without (−) or with the TSE. Relative luciferase activity was calculated as the ratio of firefly- to co-expressed Renilla luciferase-generated light units. Fold suppression (mean ± SD) by the TSE represents the ratio of activity for plasmids without TSE (set as 1) over activity for TSE-containing plasmid. (C) In HeLa cells expressing Cas9 without or with the indicated sgRNA, firefly luciferase activity generated by plasmids without (−) or with the TSE was quantified and normalized to the amounts of the corresponding mRNA. Shown is TSE-mediated fold suppression of normalized luciferase activity (mean ± SD from one out of three assays with similar results). TSE-mediated suppression of mRNA amounts was 1.9-, 1.0- and 1.6-fold in WT, sgZ #2 and sgZ #4 cells, respectively. (D) HeLa cells expressing Cas9 without (WT) or with MCPIP1-depleting sgRNAs (Mdepl) (sgZ #2, clone G4) were transfected with luciferase expression plasmids without (−) or with the TSE and—where indicated—with strep-MCPIP1 (+M). Fold suppression of firefly luciferase activity by the TSE was calculated after normalization to co-expressed Renilla luciferase as in (B). (E and F) HeLa cells lacking or expressing MCPIP1-depleting sgRNAs were transfected with plasmids for βG-TSE and GFP-TSE mRNAs. (E) Amounts of reporter and endogenous NFKBIZ mRNAs were quantitated by RT-qPCR (mean ± SD for one out of three experiments with similar results). (F) Distribution of the indicated mRNAs after sucrose-gradient fractionation of lysates from HeLa cells expressing Cas9 without (WT) or with MCPIP1 depleted (Mdepl) (sgZ #2, clone G4) was determined as described for Figure 2F).
Figure 4.

Involvement of TSE substructures in MCPIP1 binding and function. (A) Mammalian sequences matching the TSE were identified using BLAST. A combined sequence structure alignment was obtained using mLocarna followed by RNAalifold. The number of different types of base pairs for a consensus pair, i.e. compensatory mutations supporting the structure, is given by the hue, the number of incompatible pairs by the saturation of the consensus base pair. (B) Scheme of TSE and reporter mRNAs expressed and summary of results obtained for them ((13), this report and additional data, not shown). (C) HeLa cells were co-transfected with GFP-MCPIP1(D141N) and β-globin reporter constructs containing the indicated insertions. Co-pulldown of reporter mRNA with GFP-MCPIP1(D141N) was determined as in Figure 1. (D) Degradation kinetics of reporter mRNAs in HeLa cells were determined by quantification of residual mRNA at the indicated time points after blocking transcription with actinomycin D (Act. D, 5 μg/ml). (E) Degradation kinetics were determined as in (D) for HEK-293 cells expressing the indicated reporter mRNA without (con) or with strep-MCPIP1 (one out of three independent assays with similar results). (F) Sucrose gradient distribution of reporter mRNAs expressed in HeLa cells was determined as described in Figure 2F. (G) Sucrose gradient distribution of reporter mRNAs in HEK-293 cells without (con) or with co-expression of strep-MCPIP1. Results were confirmed in three independent assays.
MCPIP1 suppresses ribosome occupancy of TSE-containing mRNAs
To directly establish an effect of MCPIP1 on translation, ribosome occupancy of the GFP and the previously studied β-globin reporter mRNAs with and without TSE insertion was analyzed by sucrose gradient centrifugation (Figure 2F). Strep-MCPIP1 expression did not affect the general profile of ribosomal subunits, ribosomes and polysomes (upper panel). Both reporter mRNAs lacking the TSE were predominantly found in the polysome fractions and co-expressing strep-MCPIP1 did not significantly affect the distribution. In accordance with our previous observations using the β-globin reporter (13) the presence of the TSE shifted the distribution of both mRNAs to sub-polysomal fractions (compare open symbols for the respective reporter with and without the TSE). This shift was further increased by strep-MCPIP1 co-expression, whereas the RNase-deficient D141N mutant had no effect. Thus MCPIP1 specifically decreases ribosome occupancy of the TSE-containing mRNAs.
Depletion of MCPIP1 impairs TSE function
Expression of MCPIP1 in HeLa cells was disrupted by the CRISPR/Cas9 approach, using two different sgRNA sequences against the MCPIP1-encoding ZC3H12A gene. Cells expressing the sgRNAs and clones generated from one of the pools had undetectable levels of MCPIP1 (Figure 3A) and exhibited reduced TSE-mediated suppression of luciferase activity (Figure 3B). This was observed also after normalizing to luciferase mRNA amounts (Figure 3C), which were hardly affected by MCPIP1 depletion. This again suggests that a large part of MCPIP1 function is on the level of translation. Low suppression of luciferase activity by the TSE in MCPIP1-depleted cells could be rescued through reconstitution with strep-MCPIP1-encoding cDNA (Figure 3D). Consistent with its role in mRNA degradation, MCPIP1 depletion significantly increased the levels of the TSE-containing GFP and β-globin reporter mRNAs, which are more sensitive to this effect than the luciferase reporter, and also the level of endogenous NFKBIZ mRNA (Figure 3E). Importantly, distribution of these mRNAs was more polysomal in the MCPIP1-depleted HeLa cells as compared to the wild-type (WT) cells. Distribution of NFKBIA mRNA as a negative control was not affected (Figure 3F). These results demonstrate a role for endogenous MCPIP1 in translational silencing mediated by the NFKBIZ TSE.
Translational silencing by MCPIP1 requires an upstream element that cooperates with the MCPIP1 binding site
A combined sequence-structure alignment of mammalian TSE sequences using mLocarna (41) followed by RNAalifold (42) identified five SL structures with different evolutionary properties (Figure 4A). SL3–5 are highly conserved in mammals and were also found in birds and fish (Supplementary Figure S3A and B). The stem regions of SL4 are completely conserved in mammals, hinting at evolutionary constraints beyond maintenance of RNA secondary structure. SL3 and 5 exhibited compensatory mutations indicating a conserved role of RNA secondary structure. SL1 and 2 on the other hand appeared to be mammalian inventions. SL1 displayed some features of structural conservation throughout mammals but with considerable variation in marsupials (Supplementary Figure S3B and C). SL2 has weaker structural properties and similarly to SL1 was only detected in placental mammals, however seems to be missing in some families, e.g. Cercopithecidae (Supplementary Figure S3A).
We have reported earlier that sequences corresponding to SL4 and 5 of the NFKBIZ mRNA are sufficient for destabilization, but not for translational silencing (13). The structure-function relationship of the TSE and the involvement of MCPIP1 was addressed with a panel of TSE mutants (Figure 4B). Omission of SL 4 and 5 from the TSE resulted in complete loss of binding of GFP-MCPIP1(D141N) to the β-globin reporter mRNA, whereas omission of SL1–3 did not affect binding (Figure 4C). Of note, neither SL4 nor 5 alone were sufficient to confer interaction. Thus SL4 and 5 are necessary and sufficient for interaction between the mRNA and MCPIP1. This result correlates with functional assays in HeLa cells indicating that SL4–5 are sufficient to destabilize reporter mRNA ((13) and Figure 4D), whereas neither of the SLs alone is active (Figure 4D). Overexpressing strep-MCPIP1 in HEK-293 cells enhanced destabilization of the reporter mRNA by the TSE (Supplementary Figure S1E and Figure 4E) as well as by SL4 and 5 (Figure 4E).
Importantly, SL4 and 5 did not suffice to relocalize reporter mRNA to sub-polysomal fractions in HeLa cells (Figure 4F, upper panel). Deletion and mutation experiments indicate, however, that each of the two SL structures is essential for the ability of the TSE to decrease ribosome occupancy of the reporter mRNA (Figure 4F, lower panels). Apparently, SL4 and 5 do not function as independent units since their order and the distance between them are critical for silencing (Supplementary Figure S4). Overexpression of strep-MCPIP1 in HEK-293 cells shifted the TSE-containing reporter mRNA to sub-polysomal fractions, whereas it did not affect distribution of the β-globin-SL4–5 mRNA (Figure 4G). These results demonstrate that MCPIP1 can destabilize mRNAs without affecting their ribosome occupancy. To exert translational silencing MCPIP1 requires RNA sequences in addition to those involved in its binding.
Translational silencing by MCPIP1 involves its RNase domain
The post-transcriptional function of MCPIP1 so far has been ascribed to its RNA-degrading activity that is exerted by its PIN-like RNase domain (9). We asked if the role of MCPIP1 in translational control has similar or different structural requirements. Different MCPIP1 mutants were expressed in HEK-293 cells (Figure 5A) and their effects on expression of a luciferase-TSE mRNA were compared to that of WT MCPIP1 (Figure 5B). Suppressive activity of MCPIP1 was abolished by the D141N mutation that impairs both the RNase and deubiquitinating activities (RNase/DUBdef, (9,23)), and by the D225/226A mutation that selectively impairs RNase activity while leaving deubiquitinating activity intact (RNasedef, (44)). The C157A mutation that selectively impairs the latter (DUBdef, (44)) did not significantly change the TSE-mediated suppressive effect on luciferase expression. Disturbance of the recently reported head-to-tail dimerization of PIN domains that is essential for mRNA degradation (45) by the D278R mutation (Dimdef) also abolished translational silencing. Pulldown data showed that this mutant was still able to bind its target mRNA efficiently (Figure 5C). This indicates that an intact RNase activity is required for the function of MCPIP1 in translational control studied here. Silencing activity of MCPIP1 was also strongly impaired by deleting the zinc finger motif (nt 305–325, ΔZF) (Figure 5B) which is involved in RNA binding (26,27 and Figure 5C), whereas deletion of a proline-rich region in the C-terminal part (nt 458–536, ΔPRR) that is involved in oligomerization of MCPIP1 (26,27) had only a partial effect.
Figure 5.

Domains of MCPIP1 involved in translational silencing. (A) Empty vector (con) or plasmids encoding WT and different mutants of strep-MCPIP1 (see scheme) were transfected into HEK-293 cells together with firefly luciferase plasmid without (−) or with the TSE inserted. Equal expression of MCPIP1 and mutants was controlled by western blot. (B) Firefly luciferase activity was normalized to the corresponding mRNA amount quantified by RT-qPCR. Results are expressed as fold suppression with the value for firefly luciferase activity per mRNA in the absence of the TSE and co-transfected MCPIP1 set as 1 (mean ± SD for one out of three assays with similar results; asterisk, P < 0.01 for WT versus mutant; n.s., not significant) (mutants: RNase/DUBdef, RNase and deubiquitination deficient; RNasedef, RNase deficient; DUBdef, deubiquitination deficient; Dimdef, dimerization deficient; ΔZF, zinc finger deleted; ΔPRR, proline-rich region deleted). (C) Binding of the indicated MCPIP1 mutants to the reporter mRNA was determined as in Figure 1, using StrepTactin beads for pulldown.
Evidence for inhibition of translational elongation by the NFKBIZ TSE
Antibodies against the cap structure pulled down β-globin mRNA without and with the TSE from purified total HeLa cell RNA to similar extents (Figure 6A), showing that the TSE-containing mRNA is capped. Furthermore, in pulldown experiments with HeLa cell lysates antibodies against eIF4E strongly enriched β-globin mRNAs without and with the TSE as well as the endogenous NFKBIZ and GAPDH mRNAs (Supplementary Figure S5A). Northern blot analysis (Figure 6B) confirmed that insertion of the TSE into the β-globin reporter causes a redistribution from preferentially polysomal to preferentially sub-polysomal fractions. Importantly, both, the sub-polysomal and polysomal TSE reporter mRNAs run at the same position, markedly above the β-globin-TSE RNA lacking a poly(A)-tail. This was also observed for purified poly(A)+ RNA in separate experiments (Supplementary Figure S5B). Purification of poly(A)+ RNA recovered most of the β-globin mRNA irrespective of the presence of the TSE. Specifically, recovery for the subpolysomal and polysomal fractions was 48 and 93% for β-globin mRNA and 91 and 83% for β-globin-TSE mRNA, whereas about 3% of total RNA was recovered as expected for efficient depletion of non-adenylated RNA. The results indicate that the silenced mRNAs still contain long poly(A)-tails.
Figure 6.

Ribosome association of TSE-containing mRNAs. (A) Total RNA from HeLa cells transfected with plasmids expressing β-globin mRNA without (βG) or with the TSE (βG-TSE) was subjected to pulldowns with antibodies recognizing the cap structure or control antibodies. In vitro-transcribed luciferase RNA was used as a non-capped control RNA (cap(−)). Results (mean ± SD, n = 3) are expressed as fold increase in the ratio of pulldown over input for immunoprecipitation with specific as compared to control antibodies; n.s., not significantly enriched by cap-specific antibodies. (B) HeLa cells were transfected with plasmids encoding β-globin mRNA without (βG) or with the TSE (βG-TSE). RNA was isolated from pooled subpolysomal (SP) or polysomal (P) fractions after sucrose density centrifugation and analyzed by northern blot with a β-globin antisense probe. In vitro-transcribed β-globin-TSE RNA without poly(A)-tail (i.t. βG-TSE) was loaded as a size marker. The positions of βG-TSE mRNA (*), βG mRNA (**) and18 S rRNA are indicated, ethidium bromide staining of 28 S rRNA is shown for comparison of RNA amounts loaded. (C) Polysome profiles of the indicated mRNAs were determined for HeLa cells as in Figure 2F, grey line: absorbance at 254 nm. Top panel: endogenous NFBKIZ and βG-TSE reporter mRNA, middle panel: plasmid-derived NFKBIZ mRNA (pNFKBIZ), bottom panel: endogenous IL6 mRNA. For gradients in the top and bottom panels resolution was increased by collecting 0.5 ml portions for fractions 2–5 which are labeled _.1 and _.2 to ease comparison with profiles in other figures. (D) HeLa cells were transfected with plasmids expressing β-globin-TSE mRNA (βG-TSE) and GFP-tagged 60 S ribosomal protein RPL10a or GFP as a negative control. Two days later, endogenous NFKBIZ and β-globin-TSE mRNAs were quantified in the lysate and eluted from the beads after pulldown using GFP-specific antibodies. Results are expressed as fold increase (mean ± SD) of mRNA-binding to GFP-RPL10a over binding to GFP. (E) HeLa cells were left untreated (con) or incubated with harringtonine (10 μM) for 10 min. Polysome profiles of endogenous NFKBIZ and GAPDH mRNAs were obtained as in (C). (F) In vitro-transcribed CrPV IRES RNA or TSE RNA was incubated for 5 min without ( ) or with a mixture of ribosomal subunits and 80 S ribosomes (
) or with a mixture of ribosomal subunits and 80 S ribosomes ( ) purified from HeLa cells (35), followed by centrifugation through sucrose gradients (see ‘Materials and Methods’ section). Fractions were collected as in (C) and RNAs quantified by RT-pPCR, grey line: absorbance at 254 nm. The results were reproduced in two independent assays.
) purified from HeLa cells (35), followed by centrifugation through sucrose gradients (see ‘Materials and Methods’ section). Fractions were collected as in (C) and RNAs quantified by RT-pPCR, grey line: absorbance at 254 nm. The results were reproduced in two independent assays.
Extended fractionation of the sub-polysomal part of the gradients revealed that the fractions containing most of the NFKBIZ mRNA overlapped with the 80 S peak fractions, suggesting that most mRNAs were still bound by at least one ribosome (Figure 6C, upper panel). The distribution of the β-globin-TSE mRNA also overlapped with the 80 S peak, though somewhat less pronounced compared to the NFKBIZ mRNA. Therefore NFKBIZ mRNA was expressed from a plasmid containing its complete cDNA. Its distribution confirmed the overlap with the 80 S peak observed for the endogenous mRNA (Figure 6C, central panel). Interestingly, unlike NFKBIZ mRNA the AU-rich IL6 mRNA was detected also in the 40 S peak and lighter fractions (Figure 6C, lower panel), which is consistent with impaired initiation of AU-rich mRNAs (46–48). To confirm that NFKBIZ and TSE reporter mRNAs indeed are bound to a complete ribosome, we expressed the 60 S subunit protein RPL10a fused to GFP (49). As expected, this protein associated with polysomes in an EDTA-sensitive manner (Supplementary Figure S6) and enriched NFKBIZ and β-globin-TSE mRNAs in pulldown experiments (Figure 6D), confirming ribosome association.
Harringtonine, which binds to the A-site cleft of ribosomes (50), was applied to block ribosomes at the start codon and pausing sites without impeding elongation of translating ribosomes (51). The run-off of ribosomes led to the disappearance of polysomal GAPDH mRNA (Figure 6E). Small amounts of polysomal NFKBIZ mRNA that are regularly detected under control conditions were lost from the polysomal fractions upon harringtonine as well. This indicates that not all ribosomes are stalled.
Taken together, the results suggest that the majority of NFKBIZ mRNA molecules is trapped in a ribosome-associated state. This may result from an interaction between the TSE and the ribosome, either mediated by TSE-binding proteins or by direct binding. The latter hypothesis was tested by incubating in vitro-transcribed TSE RNA with purified ribosomes and ribosomal subunits (Figure 6F). As a positive control, RNA was prepared corresponding to the CrPV IRES, which assembles ribosomal subunits in the absence of additional cellular proteins (52,53). Sucrose gradient centrifugation showed a clear shift of the IRES RNA to ribosomal fractions as expected. In contrast, the TSE RNA remained in the lighter fractions despite the presence of ribosomes. This result does not support a direct TSE–ribosome interaction.
The association of polyadenylated mRNA with eIF4E and a complete ribosome would imply that the TSE inhibits translation at a step following subunit joining. This interpretation was confirmed by an independent approach exploiting the properties of viral IRESs. The CrPV IRES circumvents the dependence on host cell initiation factors by directly accommodating the 40 S and 60 S subunits in a way that starts translation at a codon in the IRES sequence ((54) and references therein). To suppress confounding cap- and initiation factor-dependent translation of mRNA from a CrPV-IRES-luciferase construct, a stable hairpin structure (−50 kcal/mol (33)) was placed close to the 5′ end of the mRNA and the luciferase start codon was mutated (Figure 7A). Luciferase expression by this construct was indeed IRES-dependent as it was decreased by more than 100-fold after mutating 2 nt in the IRES that are essential for translation (IGRmut14 in (55)) (Supplementary Figure S7A). CrPV-dependent firefly luciferase expression was suppressed by the presence of the TSE to about the same extent as cap-dependent expression (Figure 7A). Since the suppressive effect of the TSE was maintained, it appears to affect a step subsequent to 60 S joining, presumably elongation. In agreement with this result, suppression also affected translation depending on the HCV-IRES, which in addition to ribosome subunits requires the ternary complex and eIF5B to accomodate the 60 S subunit (56) (Supplementary Figure S7B and C). Importantly, overexpression of strep-MCPIP1 in HEK-293 cells increased TSE-dependent suppression of luciferase activity not only when translation was cap-dependent (Supplementary Figure S2), but also when translation was dependent on the CrPV-IRES (Figure 7B). These results indicate that in the context of the NFKBIZ TSE MCPIP1 represses translational elongation, but further experiments are needed to define the underlying molecular mechanism of this activity of MCPIP1.
Figure 7.
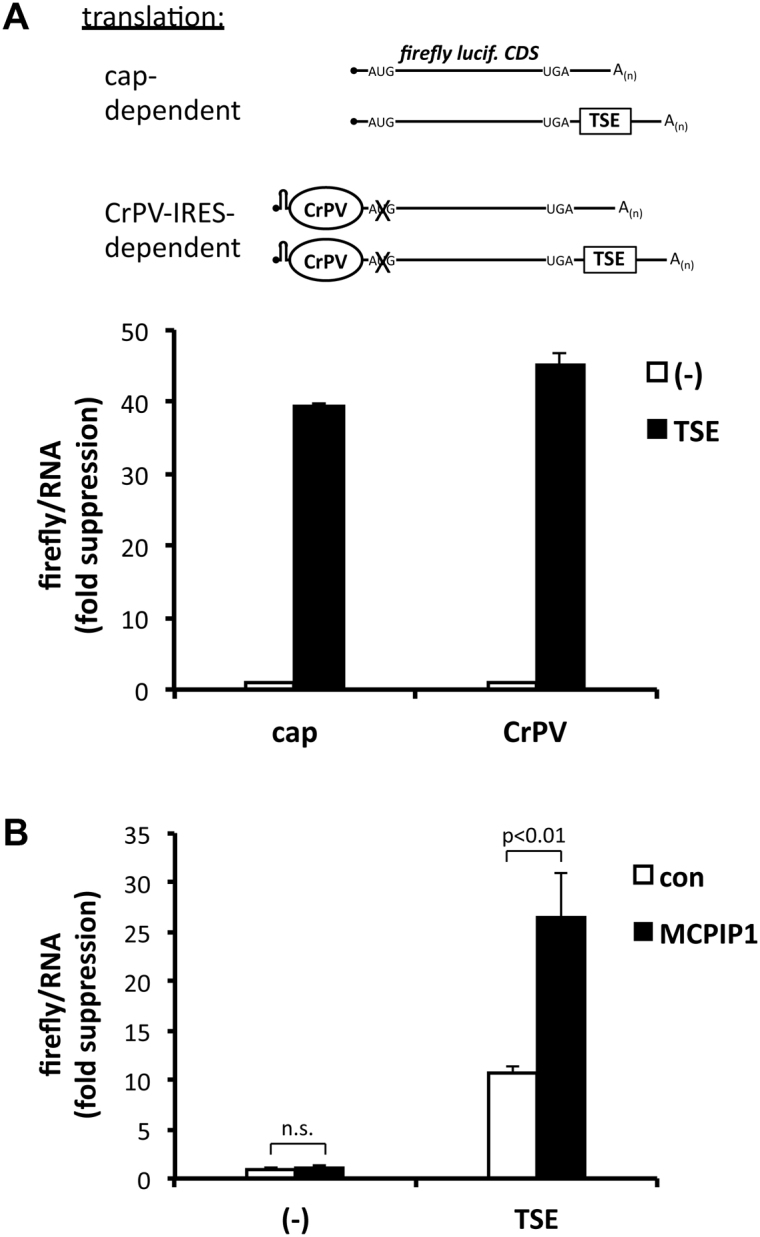
Suppressive effect of the TSE and MCPIP1 on IRES-dependent translation. (A) HeLa cells were transfected with conventional firefly luciferase plasmids without (−) or with the TSE for cap-dependent translation or with the corresponding plasmids encoding a hairpin structure followed by the CrPV IRES and a mutated luciferase start codon for IRES-dependent translation (see scheme and Supplementary Figure S6A). Luciferase activity was normalized to luciferase mRNA amounts. Results are expressed as fold inhibition by the TSE as compared to luciferase without TSE (mean ± SD for one out of five assays with similar results). (B) In HEK-293 cells co-transfected with empty vector (con) or strep-MCPIP1 expression plasmid suppression of firefly luciferase activity by the TSE was determined for CrPV-dependent translation as in (A).
DISCUSSION
Our results provide evidence for a complex architecture of an mRNA element, the NFKBIZ TSE, in which a core structure, SL4 and 5, imposes rapid degradation by providing a binding site for a protein with RNase activity, MCPIP1. Adjacent structures, SL1–3, extend the function of the core unit and its interaction partner(s) to translational silencing.
The NFKBIZ mRNA harbors a regulatory element (the TSE) that exerts strong suppression of protein expression, due to the combined effects of accelerated mRNA degradation and translational silencing (13). The similarity in regulation of the NFKBIZ and ZC3H12A mRNAs previously observed by us ((13,16) see ‘Introduction’ section) and the reported autoregulatory degradation of ZC3H12A mRNA by MCPIP1 (10) prompted us to consider the involvement of MCPIP1 in TSE function. Indeed NFKBIZ transcripts were found to belong to a group of target mRNAs shared between MCPIP1 and Roquin-1/2 (11). The results presented here indicate that MCPIP1 not only plays a role in enhancing mRNA degradation but also contributes to translational silencing mediated by the TSE.
MCPIP1 apparently exerts these functions by direct interaction with the mRNA. The TSE sequence shows high cross-species homology to enable folding into evolutionary conserved SL structures (Figure 4A). Translational silencing required the presence of the complete TSE, whereas destabilization of reporter mRNAs was elicited also by the core of the TSE alone, which contains SL4 and 5 ((13) and this report). As shown in Figure 4, this part enabled the binding and destabilizing function of MCPIP1. Furthermore, mutations in this part compromised destabilization and translational silencing. Thus by binding to SL4 and 5 MCPIP1 participates in TSE-mediated destabilization as well as translational silencing.
While there is general agreement that MCPIP1 targets SL structures, information about the interaction of MCPIP1 with target RNAs is still limited. For microRNA targets the efficiency of cleavage by MCPIP1 appears to decrease with the size of the loops (27). On the other hand, global target identification by RNA cross-linking and immunoprecipitation experiments indicate a preference for structures with a stem length of 3–7 nt and a loop size of 3 nt, predominantly of pyrimidine-purine-pyrimidine sequence (12). SL4 and SL5 of the TSE both conform to these criteria. However, the presence of either of them alone in a reporter mRNA did not suffice for efficient interaction with MCPIP1 (Figure 4C) or destabilization (Figure 4D). Perhaps MCPIP1 interacts with only one SL structure that is stabilized or preferentially folded only in the presence of the other. On the other hand, mRNAs containing either SL4 or 5 alone were successfully co-precipitated with Roquin-1, indicating that they are stably formed independently of each other (our unpublished results). Each of the SLs alone might not have sufficient affinity for productive interaction. On the other hand, we observed loss of function by changing the distance between SL4 and 5 or changing the order to SL5–4 (Supplementary Figure S4). This argues against two equivalent sub-optimal binding sites. It appears more likely that SL4 and 5 constitute a distinct functional entity.
One reason might be that an additional protein that interacts with one of them is crucial for the binding of MCPIP1 to the other. In that respect it is of interest that these structures also interact with Roquin-1/2 ((8,12), our unpublished results), which can enhance mRNA degradation by recruiting the Ccr4–Caf1–Not deadenylase complex (8). The role of Roquin-1/2 in TSE function at present is unclear. An alternative explanation for the requirement of two SLs may be found in reports stating that also MCPIP1 functions as multimers. Binding and cleavage of pre-miRNAs was dependent on the C-terminus that harbors a proline-rich region important for oligomerization (27) and similar observations have been made for viral RNA targets (26). Most recently a structurally distinct mode of oligomerization was presented (45) according to which a head-to-tail dimerization involving the PIN domains is required for full catalytic activity. Since an MCPIP1 mutant deficient in that kind of dimerization lost silencing activity (Figure 5), it could be speculated with respect to our results on the cis-requirements that an MCPIP1 dimer binds to a corresponding target structure consisting of two SLs.
Importantly, MCPIP1 accelerated degradation of reporter mRNAs containing the complete TSE or only SL4 and 5. The presence of SL1–3 adjacent to SL4 and 5 extends the functional repertoire of MCPIP1 to translational silencing (Figure 4). The latter appears to occur at a step following 60 S subunit joining. This is suggested by (i) the ribosome-association of the TSE-containing mRNAs (Figure 6C and D) and (ii) the sensitivity of initiation-independent, CrPV-IRES-driven translation to TSE- and MCPIP1-mediated silencing (Figure 7). However, final proof for elongation as the step inhibited will require detailed molecular investigation in vitro. It is intriguing that translational silencing appears to require the RNase activity of MCPIP1. Theoretically it is possible that the conformational change caused by a mutation which impairs RNase activity also affects a function distinct from it. An independent function appears unlikely, however, in view of our concordant results of abolished silencing obtained with RNase-impairing mutations at different sites, including one interfering with the head-to-tail dimerization required for RNase activity (45). MCPIP1 may cleave the mRNA and stop translation thereby, while the fragments may persist for some time. The results in Figure 6 indicate, however, that most of the silenced mRNA is capped, polyadenylated and maintains its expected size in northern blots. We did not obtain any evidence for fragmentation in additional experiments (not shown). MCPIP1 exerts endonucleolytic activity not only against mRNAs but also against other types of RNA (26,27). Therefore it could be speculated that in the context of TSE-mediated silencing the RNase activity of MCPIP1, rather than cleaving the mRNA, is required for inactivating ribosomal or tRNA at the NFKBIZ transcript. tRNA fragments have been reported to interfere with translation at different steps, including elongation (57).
How the part of the TSE encompassing SL1–3, which has no effect on mRNA degradation or translation on its own (13), complements the function of SL4 and –5 and their interacting protein(s) to achieve translational silencing is not known. We considered the possibility that SL1–3 of the TSE function by direct interaction with the ribosome or its subunits rather than by recruiting additional regulatory factors. However, in vitro-experiments failed to show binding of the TSE to ribosomes or ribosomal subunits (Figure 6F). Concerning factors interacting with SL1–3, the hnRNP E1 was considered as a candidate member, as it has been demonstrated to participate in translational silencing of 15-lipoxygenase mRNA by inhibiting 60 S subunit joining (58). It is also part of an mRNP complex that selectively forms at distinct mRNAs and stalls translational elongation by inhibiting release of eEF1A1 from the ribosomal A site (59). In our hands, knockdown of hnRNP E1 and/or E2 did not affect TSE-mediated suppression of ribosome occupancy (not shown). Future attempts to globally identify interactors of MCPIP1 and the functional characterization of candidate proteins binding to SL1–3 will further our understanding of MCPIP1 function and TSE-mediated silencing of NFKBIZ expression.
The results of this study provide evidence that MCPIP1 takes part not only in the destabilizing effect but also in translational silencing exerted by the NFKBIZ TSE. The latter depends on sequence elements that are not essential for binding and destabilization and may be lacking in other MCPIP1 target mRNAs. Our observations therefore imply that the targets identified by transcriptome-wide screens for a given RNA-binding protein may include subsets that differ in the functional consequences imposed by the protein. Given the role of MCPIP1 in limiting expression of genes that play central roles in inflammatory and immune reactions, a comprehensive understanding of its function will include the investigation of its targets on the protein level to account for translational silencing as underlying molecular mechanism.
Supplementary Material
ACKNOWLEDGEMENTS
We thank our Bachelor students Johannes Greve and Isabel Grote for their contributions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [Ho 1116/5–2 to H.H., SPP-1935, SFB-1054 project A03, HE3359/4–1 to V.H.]; Initiative and Networking Fund of the Helmholtz Association [VH-NG738 to J.H.]; ERC-StG Grant (to V.H.); Friedrich Baur Foundation (to G.B.); Fritz Thyssen Foundation [10.16.1.021MN to V.H.]; Else Fresenius-Kröner Foundation [2015_A158 to V.H.]. Funding for open access charge: Deutsche Forschungsgemeinschaft.
Conflict of interest statement. None declared.
REFERENCES
- 1. Anderson P. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat. Rev. Immunol. 2010; 10:24–35. [DOI] [PubMed] [Google Scholar]
- 2. Piccirillo C.A., Bjur E., Topisirovic I., Sonenberg N., Larsson O.. Translational control of immune responses: from transcripts to translatomes. Nat. Immunol. 2014; 15:503–511. [DOI] [PubMed] [Google Scholar]
- 3. Jeltsch K.M., Heissmeyer V.. Regulation of T-cell signaling and autoimmunity by RNA-binding proteins. Curr. Opin. Immunol. 2016; 39:127–135. [DOI] [PubMed] [Google Scholar]
- 4. Stoecklin G., Anderson P.. Posttranscriptional mechanisms regulating the inflammatory response. Adv. Immunol. 2006; 89:1–37. [DOI] [PubMed] [Google Scholar]
- 5. Tiedje C., Holtmann H., Gaestel M.. The role of mammalian MAPK signaling in regulation of cytokine mRNA stability and translation. J. Interferon Cytokine Res. 2014; 34:220–232. [DOI] [PubMed] [Google Scholar]
- 6. Stoecklin G., Lu M., Rattenbacher B., Moroni C.. A constitutive decay element promotes tumor necrosis factor alpha mRNA degradation via an AU-rich element-independent pathway. Mol. Cell. Biol. 2003; 23:3506–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paschoud S., Dogar A.M., Kuntz C., Grisoni-Neupert B., Richman L., Kuhn L.C.. Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol. Cell. Biol. 2006; 26:8228–8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leppek K., Schott J., Reitter S., Poetz F., Hammond M.C., Stoecklin G.. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell. 2013; 153:869–881. [DOI] [PubMed] [Google Scholar]
- 9. Matsushita K., Takeuchi O., Standley D.M., Kumagai Y., Kawagoe T., Miyake T., Satoh T., Kato H., Tsujimura T., Nakamura H. et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. 2009; 458:1185–1190. [DOI] [PubMed] [Google Scholar]
- 10. Iwasaki H., Takeuchi O., Teraguchi S., Matsushita K., Uehata T., Kuniyoshi K., Satoh T., Saitoh T., Matsushita M., Standley D.M. et al. The IkappaB kinase complex regulates the stability of cytokine-encoding mRNA induced by TLR-IL-1R by controlling degradation of regnase-1. Nat. Immunol. 2011; 12:1167–1175. [DOI] [PubMed] [Google Scholar]
- 11. Jeltsch K.M., Hu D., Brenner S., Zoller J., Heinz G.A., Nagel D., Vogel K.U., Rehage N., Warth S.C., Edelmann S.L. et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote TH17 differentiation. Nat. Immunol. 2014; 15:1079–1089. [DOI] [PubMed] [Google Scholar]
- 12. Mino T., Murakawa Y., Fukao A., Vandenbon A., Wessels H.H., Ori D., Uehata T., Tartey S., Akira S., Suzuki Y. et al. Regnase-1 and roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell. 2015; 161:1058–1073. [DOI] [PubMed] [Google Scholar]
- 13. Dhamija S., Doerrie A., Winzen R., Dittrich-Breiholz O., Taghipour A., Kuehne N., Kracht M., Holtmann H.. IL-1-induced post-transcriptional mechanisms target overlapping translational silencing and destabilizing elements in IkappaBzeta mRNA. J. Biol. Chem. 2010; 285:29165–29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dhamija S., Kuehne N., Winzen R., Doerrie A., Dittrich-Breiholz O., Thakur B.K., Kracht M., Holtmann H.. Interleukin-1 activates synthesis of interleukin-6 by interfering with a KSRP-dependent translational silencing mechanism. J. Biol. Chem. 2011; 286:33279–33288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tiedje C., Ronkina N., Tehrani M., Dhamija S., Laass K., Holtmann H., Kotlyarov A., Gaestel M.. The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet. 2012; 8:e1002977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dhamija S., Winzen R., Doerrie A., Behrens G., Kuehne N., Schauerte C., Neumann E., Dittrich-Breiholz O., Kracht M., Holtmann H.. Interleukin-17 (IL-17) and IL-1 activate translation of overlapping sets of mRNAs, including that of the negative regulator of inflammation, MCPIP1. J. Biol. Chem. 2013; 288:19250–19259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou L., Azfer A., Niu J., Graham S., Choudhury M., Adamski F.M., Younce C., Binkley P.F., Kolattukudy P.E.. Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ. Res. 2006; 98:1177–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liang J., Wang J., Azfer A., Song W., Tromp G., Kolattukudy P.E., Fu M.. A novel CCCH-zinc finger protein family regulates proinflammatory activation of macrophages. J. Biol. Chem. 2008; 283:6337–6346. [DOI] [PubMed] [Google Scholar]
- 19. Kolattukudy P.E., Niu J.. Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circ. Res. 2012; 110:174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jura J., Skalniak L., Koj A.. Monocyte chemotactic protein-1-induced protein-1 (MCPIP1) is a novel multifunctional modulator of inflammatory reactions. Biochim. Biophys. Acta. 2012; 1823:1905–1913. [DOI] [PubMed] [Google Scholar]
- 21. Uehata T., Akira S.. mRNA degradation by the endoribonuclease Regnase-1/ZC3H12a/MCPIP-1. Biochim. Biophys. Acta. 2013; 1829:708–713. [DOI] [PubMed] [Google Scholar]
- 22. Niu J., Azfer A., Zhelyabovska O., Fatma S., Kolattukudy P.E.. Monocyte chemotactic protein (MCP)-1 promotes angiogenesis via a novel transcription factor, MCP-1-induced protein (MCPIP). J. Biol. Chem. 2008; 283:14542–14551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liang J., Saad Y., Lei T., Wang J., Qi D., Yang Q., Kolattukudy P.E., Fu M.. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J. Exp. Med. 2010; 207:2959–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Niu J., Shi Y., Xue J., Miao R., Huang S., Wang T., Wu J., Fu M., Wu Z.H.. USP10 inhibits genotoxic NF-kappaB activation by MCPIP1-facilitated deubiquitination of NEMO. EMBO J. 2013; 32:3206–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kapoor N., Niu J., Saad Y., Kumar S., Sirakova T., Becerra E., Li X., Kolattukudy P.E.. Transcription factors STAT6 and KLF4 implement macrophage polarization via the dual catalytic powers of MCPIP. J. Immunol. 2015; 194:6011–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin R.J., Chien H.L., Lin S.Y., Chang B.L., Yu H.P., Tang W.C., Lin Y.L.. MCPIP1 ribonuclease exhibits broad-spectrum antiviral effects through viral RNA binding and degradation. Nucleic Acids Res. 2013; 41:3314–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suzuki H.I., Arase M., Matsuyama H., Choi Y.L., Ueno T., Mano H., Sugimoto K., Miyazono K.. MCPIP1 ribonuclease antagonizes dicer and terminates microRNA biogenesis through precursor microRNA degradation. Mol. Cell. 2011; 44:424–436. [DOI] [PubMed] [Google Scholar]
- 28. Annemann M., Plaza-Sirvent C., Schuster M., Katsoulis-Dimitriou K., Kliche S., Schraven B., Schmitz I.. Atypical IkappaB proteins in immune cell differentiation and function. Immunol. Lett. 2016; 171:26–35. [DOI] [PubMed] [Google Scholar]
- 29. Gossen M., Bujard H.. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Winzen R., Kracht M., Ritter B., Wilhelm A., Chen C.Y., Shyu A.B., Muller M., Gaestel M., Resch K., Holtmann H.. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999; 18:4969–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelsen T.S., Heckl D., Ebert B.L., Root D.E., Doench J.G. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014; 343:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu N., Loflin P., Chen C.Y., Shyu A.B.. A broader role for AU-rich element-mediated mRNA turnover revealed by a new transcriptional pulse strategy. Nucleic Acids Res. 1998; 26:558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Babendure J.R., Babendure J.L., Ding J.H., Tsien R.Y.. Control of mammalian translation by mRNA structure near caps. RNA. 2006; 12:851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Filbin M.E., Vollmar B.S., Shi D., Gonen T., Kieft J.S.. HCV IRES manipulates the ribosome to promote the switch from translation initiation to elongation. Nat. Struct. Mol. Biol. 2013; 20:150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pestova T.V., Hellen C.U., Shatsky I.N.. Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol. Cell. Biol. 1996; 16:6859–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pestova T.V., Lomakin I.B., Hellen C.U.. Position of the CrPV IRES on the 40S subunit and factor dependence of IRES/80S ribosome assembly. EMBO Rep. 2004; 5:906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kent W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002; 12:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Notredame C., Higgins D.G., Heringa J.. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000; 302:205–217. [DOI] [PubMed] [Google Scholar]
- 40. Altschul S.F., Madden T.L., Schaffer A.A., Zhang J.H., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Will S., Reiche K., Hofacker I.L., Stadler P.F., Backofen R.. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007; 3:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bernhart S.H., Hofacker I.L., Will S., Gruber A.R., Stadler P.F.. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics. 2008; 9:474–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lorenz R., Bernhart S.H., Siederdissen C.H.Z., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithm Mol. Biol. 2011; 6:26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qi D., Huang S., Miao R., She Z.G., Quinn T., Chang Y., Liu J., Fan D., Chen Y.E., Fu M.. Monocyte chemotactic protein-induced protein 1 (MCPIP1) suppresses stress granule formation and determines apoptosis under stress. J. Biol. Chem. 2011; 286:41692–41700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yokogawa M., Tsushima T., Noda N.N., Kumeta H., Enokizono Y., Yamashita K., Standley D.M., Takeuchi O., Akira S., Inagaki F.. Structural basis for the regulation of enzymatic activity of Regnase-1 by domain-domain interactions. Sci. Rep. 2016; 6:22324–22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Piecyk M., Wax S., Beck A.R., Kedersha N., Gupta M., Maritim B., Chen S., Gueydan C., Kruys V., Streuli M. et al. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J. 2000; 19:4154–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tao X., Gao G.. Tristetraprolin recruits eukaryotic initiation factor 4E2 to repress translation of AU-rich element-containing mRNAs. Mol. Cell. Biol. 2015; 35:3921–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fu R., Olsen M.T., Webb K., Bennett E.J., Lykke-Andersen J.. Recruitment of the 4EHP-GYF2 cap-binding complex to tetraproline motifs of tristetraprolin promotes repression and degradation of mRNAs with AU-rich elements. RNA. 2016; 22:373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heiman M., Schaefer A., Gong S., Peterson J.D., Day M., Ramsey K.E., Suarez-Farinas M., Schwarz C., Stephan D.A., Surmeier D.J. et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008; 135:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gurel G., Blaha G., Moore P.B., Steitz T.A.. U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J. Mol. Biol. 2009; 389:146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ingolia N.T., Lareau L.F., Weissman J.S.. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011; 147:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pestova T.V., Hellen C.U.. Translation elongation after assembly of ribosomes on the Cricket paralysis virus internal ribosomal entry site without initiation factors or initiator tRNA. Genes Dev. 2003; 17:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wilson J.E., Pestova T.V., Hellen C.U., Sarnow P.. Initiation of protein synthesis from the A site of the ribosome. Cell. 2000; 102:511–520. [DOI] [PubMed] [Google Scholar]
- 54. Fernandez I.S., Bai X.C., Murshudov G., Scheres S.H., Ramakrishnan V.. Initiation of translation by cricket paralysis virus IRES requires its translocation in the ribosome. Cell. 2014; 157:823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wilson J.E., Powell M.J., Hoover S.E., Sarnow P.. Naturally occurring dicistronic cricket paralysis virus RNA is regulated by two internal ribosome entry sites. Mol. Cell. Biol. 2000; 20:4990–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lukavsky P.J. Structure and function of HCV IRES domains. Virus Res. 2009; 139:166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gebetsberger J., Zywicki M., Kunzi A., Polacek N.. tRNA-derived fragments target the ribosome and function as regulatory non-coding RNA in Haloferax volcanii. Archaea. 2012; 2012:260909–260919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ostareck D.H., Ostareck-Lederer A., Shatsky I.N., Hentze M.W.. Lipoxygenase mRNA silencing in erythroid differentiation: the 3′UTR regulatory complex controls 60S ribosomal subunit joining. Cell. 2001; 104:281–290. [DOI] [PubMed] [Google Scholar]
- 59. Hussey G.S., Chaudhury A., Dawson A.E., Lindner D.J., Knudsen C.R., Wilce M.C., Merrick W.C., Howe P.H.. Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Mol. Cell. 2011; 41:419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.