


Abstract
Exposure to many endogenous and exogenous agents can give rise to DNA alkylation, which constitutes a major type of DNA damage. Among the DNA alkylation products, alkyl phosphotriesters have relatively high frequencies of occurrence and are resistant to repair in mammalian tissues. However, little is known about how these lesions affect the efficiency and fidelity of DNA replication in cells or how the replicative bypass of these lesions is modulated by translesion synthesis DNA polymerases. In this study, we synthesized oligodeoxyribonucleotides containing four pairs (Sp and Rp) of alkyl phosphotriester lesions at a defined site, and examined how these lesions are recognized by DNA replication machinery in Escherichia coli cells. We found that the Sp diastereomer of the alkyl phosphotriester lesions could be efficiently bypassed, whereas the Rp counterparts moderately blocked DNA replication. Moreover, the Sp-methyl phosphotriester induced TT→GT and TT→GC mutations at the flanking TT dinucleotide site, and the induction of these mutations required Ada protein, which is known to remove efficiently the methyl group from the Sp-methyl phosphotriester. Together, our study provided a comprehensive understanding about the recognition of alkyl phosphotriester lesions by DNA replication machinery in cells, and revealed for the first time the Ada-dependent induction of mutations at the Sp-methyl phosphotriester site.
INTRODUCTION
DNA carries the genetic information, but it has limited chemical stability, where spontaneous hydrolysis can result in deamination of nucleobases and cleavage of N-glycosidic bonds in DNA (1). The integrity of the human genome can also be compromised upon exposure to many endogenous and exogenous agents, which may result in mutagenesis and carcinogenesis (2,3).
Alkylation is a common type of DNA damage (4), and DNA alkylation constitutes the major mechanism of action for many commonly prescribed cancer chemotherapeutic agents (5). So far, the majority of studies about DNA alkylation have focused on nucleobase modifications, which is attributed to the potential mutagenic effects of these lesions (4). Aside from forming nucleobase adducts, alkylating agents can also attack one of the non-carbon-bonded oxygen atoms of internucleotide phosphate group to yield backbone alkylation products, i.e. the alkyl phosphotriester (alkyl-PTE) lesions (6). In this vein, Me- and Et-PTEs (Figure 1) were found to account for 12–17% and 55–57% of the total alkylation in duplex DNA treated with N-methyl-N-nitrosourea and N-ethyl-N-nitrosourea, respectively (7). In addition, a very recent study showed that pyridylhydroxybutyl-PTE adducts constitute 38–55% and 34–40% of all the measured pyridine-containing DNA adducts in lung and liver tissues, respectively, of rats treated through drinking water with a tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (8). Depending on which of the two non-carbon-bound oxygen atoms on backbone phosphate is alkylated, alkyl-PTEs can form in the Sp or Rp configuration (Figure 1) (6).
Figure 1.

S p and Rp diastereomers of alkyl phosphotriester residues in DNA.
Alkyl-PTEs are known to persist in mammalian tissues. A previous study showed that, in liver tissues of mice, Et-PTE displayed longer half-life (t1/2, up to 32 days) than any nucleobase ethylation products (9). Another study by Shooter et al. (10) revealed that the t1/2 for Me-PTE was 7 days in kidney and lung tissues of C57BL mice, which is much shorter than the t1/2 values for Et-PTE in the same tissues (10–15 weeks). The poorer repair of the alkyl-PTE lesions in mammalian tissues suggest that they are more likely to be encountered by DNA replication machinery than nucleobase alkylation products.
The repair and biological consequences of alkyl-PTE lesions are under-investigated (6). In this respect, the formation of alkyl-PTEs results in neutralization of the negative charge of the phosphate backbone, which interferes with the binding of proteins to DNA, thereby resulting in an inhibition of the normal functions of many DNA-binding proteins. For instance, a single isopropyl-PTE could inhibit SF2 family of DNA helicases, including RecQ and WRN (11). It was also observed that the Sp, but not the Rp diastereomer of Me-PTE could be removed by Ada protein in Escherichia coli (12,13). In addition, in vitro biochemical studies revealed that a 50:50 mixture of Sp- and Rp-Et-PTE could inhibit primer extension mediated by T4 DNA polymerase (14). However, it remains unclear how the alkyl-PTE lesions affect DNA replication in cells.
In the present study, we employed a robust shuttle vector-based method (15,16), together with mass spectrometry, to assess how alkyl-PTE lesions with different sizes of the alkyl group (from methyl to n-butyl) and stereochemical configurations (Sp and Rp) affect the fidelity and efficiency of DNA replication in E. coli cells, and how replication across these lesions is influenced by translesion synthesis DNA polymerases and the Ada protein.
MATERIALS AND METHODS
Materials
All chemicals, unless specified, were from Sigma-Aldrich (St Louis, MO, USA) or Thermo Fisher Scientific (Pittsburg, PA, USA). Common reagents for solid-phase DNA synthesis were from Glen Research (Sterling, VA, USA) and all the unmodified oligodeoxyribonucleotides (ODNs) were from Integrated DNA Technologies (Coralville, IA, USA). 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP) was from Oakwood Products Inc. (West Columbia, SC, USA), and [γ-32P]ATP was obtained from Perkin Elmer (Piscataway, NJ, USA). All enzymes were purchased from New England Biolabs (Ipswich, MA, USA).
M13mp7(L2) plasmid, wild-type AB1157 E. coli strains, C215 E. coli strains and Ada-deficient E. coli strains were kindly provided by Prof. John M. Essigmann (17,18). Polymerase-deficient AB1157 strains [Δpol B1::spec (Pol II deficient), ΔdinB (Pol IV-deficient), ΔumuC::kan (Pol V deficient) and Δpol B1::spec ΔdinB ΔumuC::kan (Pol II, Pol IV, Pol V-triple knockout)] were generously provided by Prof. Graham C. Walker (17).
Chemical synthesis
General synthetic procedures for alkyloxyphosphine
The thymidine alkylphosphoramidites were synthesized following previously published procedures (Scheme 1) (19,20). Compound 1 (200 mg, 0.75 mmol) was dissolved in anhydrous diethyl ether (7 ml) in an ice bath under argon atmosphere, to which solution were added the corresponding alcohol (140 μl) and triethylamine (4.0 eq., 306 mg). The solution was stirred at room temperature overnight, mixed with 8 ml petroleum ether, and filtered. The resulting precipitate was washed with ether. The filtrate and the ether solution were pooled and evaporated under reduced pressure to yield 2a–b, which were employed directly for the synthesis in the next step.
Scheme 1.
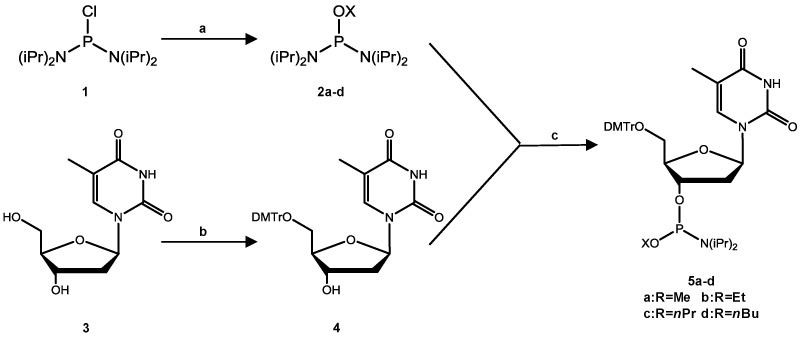
Syntheses of alkylphosphoramidite building blocks. Reagents and conditions: (A) for methyl and ethyl: ROH, triethylamine, diethyl ether, ice bath, 14 h; for n-propyl and n-butyl: ROH, triethylamine, THF, 0°C,1 h; (B) DMTr-Cl, DMAP, pyridine, room temperature, 10 h; (C) for methyl and ethyl: diisopropylamine hydrotetrazolide, CH2Cl2, room temperature, 1 h; for n-propyl and n-butyl: 0.45 M tetrazole in acetonitrile, CH2Cl2, room temperature, 5 h.
For the preparation of compounds 2c–d, compound 1 (300 mg, 1.13 mmol) in 2 ml tetrahydrofuran, the corresponding alcohol (140 μl) and triethylamine (230 μl, 1.63 mmol) were added to a round bottom flask, which was under an argon atmosphere and placed in an ice water bath. The solution was stirred at room temperature for 1 h and filtered. The filtrate was concentrated in vacuo. Pentane (10 ml) was added to the resulting solid, and the mixture was filtered. The solvent in the filtrate was removed under vacuum to yield compounds 2c–d.
General procedures for dimethoxytrityl (DMTr) protection of the 5′-hydroxyl group
Compound 4 was prepared following the previously published procedures (21). Briefly, compound 3 (100 mg, 0.41 mmol) was dissolved in anhydrous pyridine (10 ml), and to the mixture were added dimethoxytrityl chloride (1.5 eq.) and 4-dimethylaminopyridine (0.5 mol%). After stirring at room temperature for 10 h, the solution was concentrated in vacuo and the residues were purified with silica gel column chromatography by using ethyl acetate as the mobile phase to yield compound 4.
General procedures for phosphoramidite building block synthesis
To a round bottom flask, which contained a solution of compound 4 (70 mg, 0.13 mmol) in dry dichloromethane (1.5 ml), were added compound 2a–b (1 eq.) and diisopropylamine hydrotetrazolide (1 eq.), and the mixture was stirred at room temperature under argon. After 1 h, TLC analysis indicated complete conversion of the starting material. The solvent was removed in vacuo and isolated by silica gel column chromatography with a mixture of hexane, ethyl acetate, and triethylamine (49:49:2, v/v) to yield the desired products 5a–b.
For the syntheses of compounds 5c–d, compound 2c–d (1 eq.) and tetrazole in acetonitrile (0.3 ml, 0.135 mmol) were added to a round bottom flask containing a solution of compound 4 (70 mg, 0.13 mmol) in dry dichloromethane (1.5 ml), and the mixture was stirred at room temperature under argon atmosphere. The reaction was continued for 5 h, and the desired products 5c–d were purified following the aforementioned procedures for compounds 5a–b.
The reaction yields and spectroscopic characterizations of the above-synthesized products are provided in the online Supplementary Materials. The nuclear magnetic resonance (NMR) spectra for these compounds are shown in Supplementary Figures S1–S4.
ODN synthesis
The 12-mer lesion-containing ODNs, 5′-ATGGCT(X)TGCTAT-3′ (‘X’ designates the location of the alkyl-PTE), and Me-PTE-containing dimer 5′-T(Me)T-3′ were synthesized on a Beckman Oligo 1000M DNA synthesizer (Fullerton, CA) at 1 μmol scale. The corresponding phosphoramidite building block was dissolved in anhydrous acetonitrile at a concentration of 0.067 mM. Commercially available ultramild phosphoramidite building blocks were used for the incorporation of unmodified nucleotides (Glen Research Inc., Sterling, VA, USA) following the standard ODN assembly protocol. The ODNs were cleaved from the controlled pore glass support and deprotected with concentrated ammonium hydroxide at room temperature for 55 min. The solvent was removed by using a Speed-vac, and the resulting solid residues were redissolved in water and purified by high-performance liquid chromatography (HPLC).
HPLC
The HPLC separation was performed on an Agilent 1100 system with a Kinetex XB-C18 column (4.6 × 150 mm, 5 μm in particle size and 100 Å in pore size; Phenomenex Inc., Torrance, CA, USA). For the purification of the synthesized 12-mer ODNs, the mobile phases consisted of 50 mM triethylammonium acetate (pH 7.0, Solution A) and a mixture of solution A and acetonitrile (70/30, v/v, Solution B). A gradient of 5–30% B in 5 min and 30–60% B in 65 min was employed, and the flow rate was 0.7 ml/min. The HPLC traces for the purification of the 12-mer lesion-containing ODNs are shown in Supplementary Figure S7 and the electrospray ionization-mass spectra (ESI-MS) and tandem MS (MS/MS) of these ODNs are displayed in Supplementary Figures S8–S11.
Determination of stereochemical configurations of Me-PTE-containing ODNs
The Me-PTE-containing dimer 5′-T(Me)T-3′ was first purified by HPLC using a Synergi Fusion-RP column (10 × 150 mm, 4 μm in particle size and 80 Å in pore size; Phenomenex Inc., Torrance, CA, USA). The mobile phases were water (Solution A) and methanol (Solution B). A gradient of 5–25% B in 5 min and 25–50% B in 75 min was used, and the flow rate was 1.1 ml/min. The HPLC traces are shown in Supplementary Figure S5 and the products were identified by MS and 31P-NMR. For NMR analyses, the two purified diastereomers were dissolved in 300 μl CD3OD, and triphenylphosphine was added as the reference for chemical shift calibration. The 31P-NMR spectra were recorded on a Bruker Avance Neo 400 spectrometer at 80 MHz for 31P (Supplementary Figure S5).
To determine the stereochemical configurations of the Me-PTEs in 12 mer ODNs, we digested the lesion-containing ODNs with a cocktail of four enzymes and analyzed the digestion mixture by HPLC to examine whether the two diastereomers in 12 mer ODNs exhibited the same elution order as those in the dimers. The digestion was conducted following previously published procedures (22). Briefly, nuclease P1 (0.1 U/μg DNA), phosphodiesterase 2 (0.000125 U/μg DNA), and 1 nmol of 12 mer ODNs with Sp- or Rp-Me-PTE were incubated in a 100-μl solution containing 300 mM sodium acetate (pH 5.6) and 10 mM ZnCl2. After incubation at 37°C for 24 h, alkaline phosphatase (0.1 U/μg DNA), phosphodiesterase 1 (0.00025 U/μg DNA), and 50 μl of 0.5 M Tris–HCl (pH 8.9) were added to the digestion mixture, and the mixture was incubated at 37°C for 2 h. The enzymes were subsequently removed by chloroform extraction and the solution was dried by lyophilization. The resulting solid residue was reconstituted in doubly distilled water (100 μl) and analyzed by HPLC. A Thermo Hypersil Gold C18 column (4.6 × 250 mm, 3 μl in particle size, 300 Å in pore size, Thermo Scientific Inc., Waltham, MA) was employed for the separation of the digestion mixture. A solution of 10 mM ammonium formate and methanol were used as mobile phases A and B, respectively. The gradient profile was 0% B in 42 min, 0–2% B in 1 min, 2% B for 17 min, 2–5% B in 1 min, 5–25% B in 5 min, 25–35% B in 75 min. The HPLC traces are shown in Supplementary Figure S6.
Construction of single-stranded lesion-containing and lesion-free competitor M13 genomes
The 12-mer alkyl-PTE-containing ODNs were 5′-phosphorylated and ligated with a 10-mer ODN (5′-ACTGGAAGAC-3′) in the presence of ligation buffer with T4 DNA ligase and ATP at 16°C for 8 h. The resulting 22-mer ODNs were purified by denaturing polyacrylamide gel.
The lesion-containing and lesion-free M13mp7 (L2) genomes were prepared following previously reported procedures (Supplementary Figure S12) (21,23). First, 20 pmol of M13 plasmid was digested with 40 U EcoRI-HF at room temperature for 8 h to linearize the vector. The linearized vector was mixed with 2 scaffolds, 5′-CTTCCACTCACTGAATCATGGTCATAGCTTTC-3′ and 5′-AAAACGACGGCCAGTGAATTATAGC-3′ (25 pmol), each spanning one end of the linear vector. To the mixture was subsequently added 30 pmol of 5′-phosphorylated 22-mer lesion-containing ODN or 25-mer competitor ODN (5′-GCAGGATGTCATGGCGATAAGCTAT-3′). The resulting mixture was treated with T4 DNA ligase at 16°C for 8 h, followed by incubation with T4 DNA polymerase (22.5 U) at 16°C for 2.5 h to degrade the unligated vector and excess scaffolds. The lesion-containing and lesion-free plasmids were purified using Cycle Pure Kit (Omega) and subsequently normalized against the competitor plasmid, following published procedures (21,23).
Transfection of lesion-free, lesion-containing and competitor plasmids into E. coli cells
The lesion-free or lesion-containing M13 genome was mixed with the competitor genome at a 1:1 molar ratio. The mixtures were transfected into SOS-induced, electrocompetent AB1157 E. coli strain as well as the isogenic cells deficient in Pol II, Pol IV, Pol V, or all three in combination. The lesion-free or lesion-containing M13 genome was also mixed with the competitor genome at a 4:1 molar ratio and transfected into electrocompetent Ada-proficient (C215) and Ada-deficient (C217) E. coli cells, as described elsewhere (21). The SOS induction was conducted by irradiating E. coli cells with 254 nm ultraviolet light at a dose of 45 J/m2 (17). The AB1157, C215 and C217 E. coli cells were then grown in lysogeny broth (LB) medium at 37°C for 5.5 h. The phage was recovered from the supernatant by centrifugation at 13 200 rpm for 5 min and further transfected into SCS110 cells to increase the progeny/lesion-genome ratio. The amplified phage was purified by Qiaprep Spin M13 Kit (Qiagen) to obtain the ssM13 DNA template for PCR amplification.
Quantification of bypass efficiencies and mutation frequencies
A modified version of the competitive replication and adduct bypass (CRAB) assay (15,16,23,24) was employed to assess the bypass efficiencies and mutation frequencies of the alkyl-PTE lesions in E. coli cells. The sequence region of interest in the M13 template was amplified by PCR with the use of Phusion high-fidelity DNA polymerase. The primers were 5′-YCAGCTATGACCATGATTCAGTGAGTGGA-3′ and 5′-YTCGGTGCGGGCCTCTTCGCTATTAC-3′ (‘Y’ denotes the H2N(CH2)6- group conjugated to the 5′ phosphate group of the ODNs). The amplification started from annealing at 98°C for 30 s, followed by 35 cycles of amplification, each of which consisted of 10 s at 98°C, 30 s at 65°C and 15 s at 72°C, and then with a final 5-min extension at 72°C and ending at 4°C. The PCR products were purified by using Cycle Pure Kit (Omega). For determining the bypass efficiencies and mutation frequencies, 100 ng of PCR products were digested with 10 U BbsI-HF restriction endonucleases together with 10 U recombinant shrimp alkaline phosphatase (rSAP) in 10 μl CutSmart buffer (New England Biolabs) at 37°C for 25 min, followed by heating at 80°C for 10 min to deactivate the phosphatase. To the mixture were subsequently added 5 mM DTT, 1.66 pmol [γ-32P] ATP, 10 U T4 polynucleotide kinase (T4 PNK), T4 PNK buffer and water to give a total volume of 15 μl. After a 30-min incubation at 37°C, the T4 PNK was deactivated by heating the solution at 80°C for 10 min. To the resulting solution was added 10 U MluCI restriction endonuclease, and the mixture was then incubated at 37°C for 25 min. The reaction was subsequently quenched by adding 15 μl formamide gel loading buffer which contained xylene cyanol FF and bromophenol blue dyes. The mixture was resolved by using 30% native polyacrylamide gel (acrylamide:bis-acrylamide = 19:1), and the gel band intensities were determined by phosphorimager analysis on a Typhoon 9410 variable-mode imager.
The two-step restriction endonuclease digestion gave rise to a 10-mer duplex d(p*GGCMNGCTAT)/d(AATTATAGCY), where ‘M’ and ‘N’ designate the nucleobases at the dinucleotide sites initially flanking the alkyl-PTE lesions, ‘Y’ is the complementary base of ‘N’ in the opposite strand, and p* denotes the [5′-32P]-labeled phosphate. The bypass efficiency was calculated by using following equation: Bypass efficiency (%) = (lesion signal/competitor signal)/(control signal/competitor signal) × 100%.
Identification of mutagenic products by LC–MS and MS/MS
The PCR products were digested with 50 U BbsI-HF restriction endonuclease and 20 U rSAP in 250 μl CutSmart buffer at 37°C for 2 h, and the phosphatase was subsequently deactivated by heating at 80°C for 20 min. To the mixture was added 50 U MluCI and the solution was incubated at 37°C for 1 h. The solution was then extracted once with phenol/chloroform/isoamyl alcohol (25:24:1, v/v). The aqueous layer was dried, desalted by HPLC and redissolved in 20 μl water. A 10-μl aliquot was injected for LC–MS/MS analysis on an LTQ linear ion trap mass spectrometer (Thermo Electron, San Jose, CA, USA). An Agilent Zorbax SB-C18 column (0.5 × 250 mm, 5 μm in particle size) was employed, and the gradient for LC–MS/MS analysis was 5 min of 5–20% methanol followed by 35 min of 20–50% methanol in 400 mM HFIP (pH was adjusted to 7.0 with triethylamine). The mass spectrometer was set up for monitoring the fragmentation of the [M–3H]3− ions of 10-mer d(GGCMNGCTAT) and d(AATTATAGCY), with ‘M’, ‘N’, and ‘Y’ being ‘A’, ‘T’, ‘C’ or ‘G’. The fragment ions detected in MS/MS were manually assigned.
RESULTS
The objectives of this study were to assess how alkyl-PTE lesions with different stereochemical configurations and various sizes of alkyl group compromise DNA replication, and to define the respective roles of the three SOS-induced polymerases and Ada protein in bypassing and repairing these lesions in E. coli cells.
We first synthesized 12-mer ODNs harboring a site-specifically inserted alkyl-PTE lesion (Scheme 1) and characterized these ODNs by ESI-MS and MS/MS analyses (Supplementary Figures S8–S11). The ESI-MS and MS/MS results support the site of the alkyl-PTE lesion and the identity of the alkyl group being conjugated with backbone phosphate. In addition, we established the configurations of the two diastereomers of Me-PTEs based on 31P-NMR and HPLC analyses (Supplementary Figures S5 and S6). Previously, Weinfeld et al. (13,25) showed that, under the same solvent conditions, the 31P signal for the Sp diastereomer of Me-PTE appeared at a lower field than that for the Rp counterpart. We first determined the stereochemistry of the Me-PTE-containing T(Me)T based on 31P-NMR analyses, where the earlier and later eluting T(Me)T-1 and T(Me)T-2 were found to contain the Sp and Rp diastereomers of the Me-PTE, respectively. A chemical shift difference of 0.16 ppm for the two diastereomers was nearly identical to what was previously reported (26). We next digested the HPLC-purified Me-PTE-containing 12-mer ODNs with a cocktail of four enzymes, which release Me-PTE as T(Me)T. By comparing the HPLC elution properties of the T(Me)T liberated from the 12-mer ODNs with those of the standards, we demonstrated that the earlier- and later-eluting 12-mer ODNs contained the Sp- and Rp-Me-PTEs, respectively. Based on the observations that the Sp- and Rp-Me-PTE lesions in a dimer and a 12-mer ODN exhibit the same elution orders on reversed-phase HPLC columns, we assign the stereochemistry of the purified Et-, nPr- and nBu-PTEs based on their relative orders of elution from the HPLC column, i.e., the Sp diastereomer elutes earlier than the Rp diastereomer.
We next ligated these lesion-containing ODNs into single-stranded M13mp7 plasmid and measured the bypass efficiencies and mutation frequencies of the alkyl-PTE lesions using a modified version of the competitive replication and adduct bypass assay (Figure 2 and Supplementary Figures S13-S20) (15,16). After PCR amplification and restriction digestion, the released ODNs were analyzed by LC-MS/MS and native PAGE to identify the replication products, as described elsewhere (24). As shown in Figure 2, we were able to radiolabel the 5′ terminus of either the original lesion-situated strand, i.e. d(p*GGCMNGCTAT), or its complementary strand, i.e. d(p*AATTATAGCY), by switching the orders of BbsI and MluCI digestions. Results from native PAGE analyses of the resulting radiolabeled fragments revealed that the Sp diastereomer of Me-PTE could give rise to TT→GT and TT→GC mutations at the flanking TT dinucleotide sites, whereas no mutagenic products were detectable for any other alkyl-PTE lesions. In this regard, when BbsI was added first, we could resolve [5′-32P]-labeled d(p*GGCTTGCTAT) (non-mutagenic product, 10 mer TT) from the products with TT→GT or TT→GC mutation, i.e. d(p*GGCGTGCTAT) (10 mer GT) and d(p*GGCGCGCTAT) (10 mer GC). Nonetheless, 10 mer GT and 10 mer GC could not be distinguished by native PAGE analysis (Figure 2B). To resolve these two mutagenic products, we first added MluCI to radiolabel the opposite strand, which enabled us to separate the product with TT→GT mutation, i.e. d(p*AATTATAGCA) (10 mer A), from that with TT→GC mutation, namely d(p*AATTATAGCG) (10 mer G), as shown in Figure 2C and Supplementary Figures S13–S15.
Figure 2.

Restriction enzyme digestion and radiolabeling, followed by native PAGE (30%) analysis for quantifying the bypass efficiencies and mutation frequencies of alkyl phosphotriester lesions in wild-type AB1157 E. coli cells. (A) Selective labeling of the original lesion-containing strand and its complementary strand via sequential restriction digestion. ‘SAP’ and ‘PNK’ represent shrimp alkaline phosphatase and T4 polynucleotide kinase, respectively. ‘MN’ in the sequence denotes the site where the TT dinucleotide flanking the alkyl phosphotriester lesions were initially situated. (B) Gel image showing the 13 mer (competitor genome) and 10 mer (control or lesion-containing genome) digestion products formed from the original strand, where 10 mer TT, 10 mer GC and 10 mer GT designate [5′-32P]-labeled standard ODNs 5′-GGCMNGCTAT-3′, with MN being TT, GC and GT, respectively. (C) Gel image showing 13 mer (competitor genome) and 10 mer (control or lesion-containing genome) digestion products formed from the opposite strand, where 10 mer A and 10 mer G designate the [5′-32P]-labeled standard ODNs 5′-AATTATAGCY-3′, with Y being A and G, respectively.
We further confirmed the identities of the aforementioned restriction digestion products by LC–MS/MS analysis (Supplementary Figure S16). We monitored the [M–3H]3− ions of d(GGCMNGCTAT) and d(AATTATAGCY), where ‘M’ and ‘N’ designate the nucleotides inserted at the initial TT dinucleotide flanking the alkyl-PTE site, and ‘Y’ represents the opposing nucleotide of ‘N’ in the complementary strand, respectively (Supplementary Figures S17–S20). The LC–MS/MS data showed that only Sp-Me-PTE could induce TT→GT and TT→GC mutations, which are in keeping with the results obtained from the aforementioned PAGE analyses.
Having identified the replication products, we determined the bypass efficiencies of the alkyl-PTE lesions by measuring the ratio of combined intensities of all 10-mer bands arising from the lesion-containing genome over that of the 13-mer band emanating from the lesion-free competitor genome (see Materials and Methods). The results showed that the Sp diastereomers of all four alkyl-PTEs do not appreciably impede DNA replication in E. coli cells, whereas the Rp diastereomers elicit moderate blockage effects on DNA replication (Figure 3A).
Figure 3.

Bypass efficiencies of alkyl phosphotriester lesions (A) and mutation frequencies of the Sp methyl phosphotriester lesion (B) in AB1157 E. coli strains that are proficient or deficient in SOS-induced DNA polymerases, Pol II, Pol IV, Pol V, alone or all three in combination. The data represent the means and standard deviations of results from three independent replication experiments. *, 0.01 < P < 0.05. The P values were calculated by using an unpaired two-tailed t-test and referred to comparisons with the data obtained for control cells.
We also examined the roles of the three SOS-induced polymerases (i.e. Pol II, Pol IV and Pol V) in bypassing the alkyl-PTE lesions by conducting the replication experiments using the isogenic E. coli strains that are deficient in these polymerases. The results showed that the bypass of the alkyl-PTE lesions does not require any of the three TLS polymerases (Figure 3A). Moreover, our results showed that the bypass efficiencies of these lesions were not modulated by SOS induction, either in wild-type AB1157 cells or the isogenic cells with all three SOS-induced DNA polymerases being knocked out (Figure 3A).
We further determined the mutation frequencies of the Me-PTE in wild-type and polymerase-deficient AB1157 E. coli strains (Figure 3B). It turned out that the Sp-Me-PTE is moderately mutagenic, resulting in TT→GT and TT→GC mutations. Additionally, deletion of any of the three SOS-induced polymerases alone did not confer any significant alterations in mutation frequencies, though simultaneous removal of the three polymerases in SOS-induced or uninduced cells led to a slight, albeit statistically significant decrease in TT→GT mutation (Figure 3B).
The unique mutagenic property of Sp-Me-PTE parallels the previous finding that Ada protein removes the methyl group exclusively from the Sp-Me-PTE at an efficiency that is much higher than the corresponding removal of larger alkyl groups from alkyl-PTE lesions (13). Thus, we next asked whether Ada protein assumes any role in modulating the cytotoxic and mutagenic properties of the two diastereomers of Me- and Et-PTEs by conducting replication experiments with the use of isogenic E. coli cells that are proficient or deficient in Ada protein (Figure 4). Our results showed that the deletion of Ada led to a significant decline in bypass efficiency for Sp-Me-PTE, which is accompanied with the abrogation of both TT→GT and TT→GC mutations for the lesion, suggesting the involvement of Ada protein in repairing the Sp-Me-PTE and inducing mutations for this lesion. Similar as what we found for AB1157 cells, replication across Sp-Me-PTE and the two diastereomers of Et-PTE lesions are error-free in Ada-deficient C217 cells, and deletion of Ada protein did not appreciably alter the bypass efficiencies of these three lesions (Figure 4).
Figure 4.

Ada protein promotes the replicative bypass of the Sp-Me-PTE, but not the Rp-Me-PTE or the Sp-/Rp-Et-PTE lesion, and Ada is required for the mutations induced by Sp-Me-PTE. (A) Selective labelling of original lesion-containing strand and its complementary strand via sequential restriction digestion. ‘SAP’ and ‘PNK’ represent shrimp alkaline phosphatase and T4 polynucleotide kinase, respectively. (B) Gel image showing 13 mer (from the competitor genome) and 10 mer (from the control or lesion-containing genome) digestion products formed from the strand initially containing the lesion, where 10 mer TT, 10 mer GC and 10 mer GT designate [5′-32P]-labeled standard ODNs 5′-GGCMNGCTAT-3′, with MN being TT, GC and GT, respectively. (C) The bypass efficiencies of the two diastereomers of Me- and Et-PTE lesions in E. coli cells that are proficient in DNA repair or deficient in both Ada and Ogt (ΔAda, ΔOgt). The data represent the means and standard deviations of results from three independent replication experiments. ***P <0.001. The P values were calculated by using an unpaired two-tailed t-test and referred to comparisons with the data obtained for control cells.
DISCUSSION
We systematically investigated the cytotoxic and mutagenic properties of alkyl-PTE lesions in E. coli cells and our results led to several important conclusions. First, we found that the Sp and Rp diastereomers of the alkyl-PTE lesions exerted distinct effects on DNA replication in E. coli cells. The lesions in the Sp configuration displayed higher replication bypass efficiencies than the corresponding unmodified DNA, whereas those in the Rp configuration suppress DNA replication in AB1157 cells. The replication bypass efficiencies for the Sp-alkyl-PTEs, except for the methyl adduct which is removed by Ada protein, decrease with the size of alkyl group, which might be attributed to the elevated steric hindrance imposed by larger alkyl groups. In duplex DNA, the alkyl group in the Sp diastereomer is known to point perpendicularly out from the DNA double helix, whereas that of the Rp diastereomer projects into the major groove (6). Hence, the alkyl groups in the Sp configuration may assume some favorable interaction with DNA polymerases to promote translesion synthesis, where steric hindrance imposed by larger alkyl groups may diminish this enhancement effect. However, the Rp-alkyl-PTE lesions display an opposite trend, with nPr- and nBu-PTEs exhibiting higher bypass efficiencies than Me- and Et-PTEs. This result supports that the difference in stereochemical configurations of the alkyl group conjugated with the phosphate backbone can modulate the replicative bypass of these lesions. By employing a DNA template containing an equimolar mixture of Sp and Rp diastereomers of Et-PTE at a defined site, Tsujikawa et al. (14) found that the lesions could inhibit partially primer extension catalyzed by T4 DNA polymerase, but could be bypassed by E. coli DNA polymerase I. The results from that study also suggested that one of the two diastereomers was a major block to DNA replication (14), though the identity of the diastereomer could not be ascertained owing to the lack of stereochemically pure DNA substrates. It is worth noting that, due to the differential accessibilities of the two non-carbon-bound oxygen atoms on backbone phosphate to DNA alkylating agents, the Sp diastereomer is produced at a higher yield than the Rp counterpart (27). Hence, the assessment about overall biological consequences of alkyl-PTEs should also take into account the relative frequencies of occurrence of these lesions.
Second, our results demonstrated that the bypass efficiencies of the alkyl-PTE lesions were not affected by deletion of any of the three SOS-induced TLS polymerases, alone or all three in combination. Additionally, the mutation frequencies induced by Sp-Me-PTE were not influenced by the deletion of any of the three TLS polymerases alone, though the absence of all three polymerases resulted in a slight, yet statistically significant decrease in the frequency of TT→GT mutation.
Third, Ada protein, which could repair the Sp-Me-PTE, is required for the TT→GT and TT→GC mutations induced by this lesion. The removal of Me-PTE by Ada protein takes time. In this vein, we also measured the bypass efficiencies and mutation frequencies of the two diastereomers of Me-PTEs at 1, 3 and 6 h following transfection, and we observed that at 1 h, the bypass efficiency of Sp-Me-PTE at 1 h is lower than that at 3 and 6 h (Supplementary Figure S21). Ada protein plays a crucial role in bacterial tolerance toward exposure to alkylating agents in a process known as adaptive response, where Ada acts both as a DNA repair protein and a transcription activator (4,28,29). In that process, the 39 kDa Ada protein undergoes proteolysis at the sole Lys-Gln linkage to yield a 19 kDa C-terminal fragment (C-Ada) and a 20 kDa N-terminal fragment (N-Ada); while Cys321 in C-Ada is a methyltransferase for the repair of O6-methylguanine and O4-methylthymine (30), Cys38 in N-Ada could remove the methyl group specifically from the Sp-Me-PTE (31). Moreover, He et al. (31) illustrated, from detailed solution (NMR) and crystal (X-ray) structure studies, that this latter methyl group transfer stimulated the binding of N-Ada with DNA through an electrostatic switch mechanism. The heightened DNA affinity led to promoter binding and transcriptional activation of several genes that are important in repairing alkylated DNA lesions including Ada, Aid, AlkA and AlkB (6). Thus, Ada protein is instrumental in protecting the genomic integrity of bacterial cells by promoting the repair of alkylated DNA lesions. Here we made an unexpected finding that, while the Ada-stimulated repair of the Sp-Me-PTE results in enhanced replicative bypass, it comes at an expense by inducing TT→GT and TT→GC mutations at the flanking nucleoside sites. Further studies are warranted for understanding how the selective interaction between Ada and Sp-Me-PTE would interfere with Watson-Crick base pairing during DNA replication and trigger specific mutations at the lesion site.
A recent genome-wide mutation analysis of 32 E. coli strains treated with N-methyl-N'-nitrosoguanidine showed that, among the 4099 identified mutations, 96.6%, 2.17% and 0.46% were G→A, T→C and T→G substitutions, respectively (32). While G→A and T→C mutations are thought to arise from methylation at the O6 of guanine and the O4 of thymine (21,33), respectively, our results suggest that T→G mutation may be attributed, in part, to the Me-PTE formed on the 3′ side of thymidine. In this context, it is worth noting that the formation frequency of the Me-PTE lesions was found to be affected by flanking sequences (34), and flanking sequences may also modulate the mutagenic properties of alkyl-PTE lesions. Thus, it will be important to investigate how the cytotoxic and mutagenic properties of the alkyl-PTE lesions are influenced by flanking sequences in the future.
In summary, our study demonstrated that the two diastereomers of alkyl-PTEs exert distinct effects on DNA replication; the Sp-alkyl-PTEs did not appreciably impede DNA replication, whereas the Rp counterparts could moderately inhibit this process. We also revealed that the mutagenic properties of the Sp-Me-PTE could be substantially modulated by Ada protein, but not by any of the three SOS-induced polymerases. Hence, our systematic shuttle-vector study on alkyl-PTE lesions offered new insights about how this group of DNA alkylation products are recognized by the E. coli DNA replication machinery. It will be important to examine how the alkyl-PTE lesions compromise DNA replication in mammalian cells in the future.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of Environmental Health Sciences [R01 ES025121]. Funding for open access charge: National Institute of Environmental Health Sciences [R01 ES025121].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993; 362:709–715. [DOI] [PubMed] [Google Scholar]
- 2. Friedberg E.C., Walker G.C., Siede W., Wood R.D.. DNA Repair and Mutagenesis. 2005; American Society for Microbiology Press. [Google Scholar]
- 3. Liu S., Wang Y.. Mass spectrometry for the assessment of the occurrence and biological consequences of DNA adducts. Chem. Soc. Rev. 2015; 44:7829–7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shrivastav N., Li D., Essigmann J.M.. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 2009; 31:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fu D., Calvo J.A., Samson L.D.. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer. 2012; 12:104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones G.D., Le Pla R.C., Farmer P.B.. Phosphotriester adducts (PTEs): DNA’s overlooked lesion. Mutagenesis. 2010; 25:3–16. [DOI] [PubMed] [Google Scholar]
- 7. Beranek D.T. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. 1990; 231:11–30. [DOI] [PubMed] [Google Scholar]
- 8. Ma B., Zarth A.T., Carlson E.S., Villalta P.W., Upadhyaya P., Stepanov I., Hecht S.S.. Identification of more than one hundred structurally unique DNA-phosphate adducts formed during rat lung carcinogenesis by the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 2018; 39:232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Den Engelse L., Menkveld G.J., De Brij R.J., Tates A.D.. Formation and stability of alkylated pyrimidines and purines (including imidazole ring-opened 7-alkylguanine) and alkylphosphotriesters in liver DNA of adult rats treated with ethylnitrosourea or dimethylnitrosamine. Carcinogenesis. 1986; 7:393–403. [DOI] [PubMed] [Google Scholar]
- 10. Shooter K.V., Slade T.A.. The stability of methyl and ethyl phosphotriesters in DNA in vivo. Chem. Biol. Interact. 1977; 19:353–361. [DOI] [PubMed] [Google Scholar]
- 11. Suhasini A.N., Sommers J.A., Yu S., Wu Y., Xu T., Kelman Z., Kaplan D.L., Brosh R.M. Jr. DNA repair and replication fork helicases are differentially affected by alkyl phosphotriester lesion. J. Biol. Chem. 2012; 287:19188–19198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McCarthy T.V., Lindahl T.. Methyl phosphotriesters in alkylated DNA are repaired by the Ada regulatory protein of E. coli. Nucleic Acids Res. 1985; 13:2683–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weinfeld M., Drake A.F., Saunders J.K., Paterson M.C.. Stereospecific removal of methyl phosphotriesters from DNA by an Escherichia coli ada+ extract. Nucleic Acids Res. 1985; 13:7067–7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsujikawa L., Weinfield M., Reha-Krantz L.J.. Differences in replication of a DNA template containing an ethyl phosphotriester by T4 DNA polymerase and Escherichia coli DNA polymerase I. Nucleic Acids Res. 2003; 31:4965–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hong H., Cao H., Wang Y.. Formation and genotoxicity of a guanine-cytosine intrastrand cross-link lesion in vivo. Nucleic Acids Res. 2007; 35:7118–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yuan B., Cao H., Jiang Y., Hong H., Wang Y.. Efficient and accurate bypass of N2-(1-carboxyethyl)-2′-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8679–8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Neeley W.L., Delaney S., Alekseyev Y.O., Jarosz D.F., Delaney J.C., Walker G.C., Essigmann J.M.. DNA polymerase V allows bypass of toxic guanine oxidation products in vivo. J. Biol. Chem. 2007; 282:12741–12748. [DOI] [PubMed] [Google Scholar]
- 18. Rye P.T., Delaney J.C., Netirojjanakul C., Sun D.X., Liu J.Z., Essigmann J.M.. Mismatch repair proteins collaborate with methyltransferases in the repair of O6-methylguanine. DNA Repair (Amst). 2008; 7:170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dohno C., Matsuzaki K., Yamaguchi H., Shibata T., Nakatani K.. A hybridisation-dependent membrane-insertable amphiphilic DNA. Org. Biomol. Chem. 2015; 13:10117–10121. [DOI] [PubMed] [Google Scholar]
- 20. Hamamoto S., Takaku H.. New approach to the synthesis of deoxyribonucleoside phosphoramidite derivatives. Chem. Lett. 1986; 15:1401–1404. [Google Scholar]
- 21. Wang P., Amato N.J., Zhai Q., Wang Y.. Cytotoxic and mutagenic properties of O4-alkylthymidine lesions in Escherichia coli cells. Nucleic Acids Res. 2015; 43:10795–10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu Y., Wang J., Wang P., Wang Y.. Quantification of azaserine-induced carboxymethylated and methylated DNA lesions in cells by nanoflow liquid chromatography-nanoelectrospray ionization tandem mass spectrometry coupled with the stable isotope-dilution method. Anal. Chem. 2016; 88:8036–8042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Delaney J.C., Essigmann J.M.. Assays for determining lesion bypass efficiency and mutagenicity of site-specific DNA lesions in vivo. Methods Enzymol. 2006; 408:1–15. [DOI] [PubMed] [Google Scholar]
- 24. Zhai Q., Wang P., Cai Q., Wang Y.. Syntheses and characterizations of the in vivo replicative bypass and mutagenic properties of the minor-groove O2-alkylthymidine lesions. Nucleic Acids Res. 2014; 42:10529–10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Potter B.V., Connolly B.A., Eckstein F.. Synthesis and configurational analysis of a dinucleoside phosphate isotopically chiral at phosphorus. Stereochemical course of Penicillium citrum nuclease P1 reaction. Biochemistry. 1983; 22:1369–1377. [DOI] [PubMed] [Google Scholar]
- 26. Hayashi J., Hamada T., Sasaki I., Nakagawa O., Wada S., Urata H.. Synthesis of novel cationic spermine-conjugated phosphotriester oligonucleotide for improvement of cell membrane permeability. Bioorg. Med. Chem. Lett. 2015; 25:3610–3615. [DOI] [PubMed] [Google Scholar]
- 27. Weinfeld M., Drake A.F., Kuroda R., Livingston D.C.. Isolation and characterization of the diastereoisomers of a series of phosphate-ethylated dinucleoside monophosphates. Anal. Biochem. 1989; 178:93–101. [DOI] [PubMed] [Google Scholar]
- 28. Nakamura T., Tokumoto Y., Sakumi K., Koike G., Nakabeppu Y., Sekiguchi M.. Expression of the ada gene of Escherichia coli in response to alkylating agents. Identification of transcriptional regulatory elements. J. Mol. Biol. 1988; 202:483–494. [DOI] [PubMed] [Google Scholar]
- 29. Samson L., Cairns J.. A new pathway for DNA repair in Escherichia coli. Nature. 1977; 267:281–283. [DOI] [PubMed] [Google Scholar]
- 30. Samson L. The suicidal DNA repair methyltransferases of microbes. Mol. Microbiol. 1992; 6:825–831. [DOI] [PubMed] [Google Scholar]
- 31. He C., Hus J.C., Sun L.J., Zhou P., Norman D.P., Dotsch V., Wei H., Gross J.D., Lane W.S., Wagner G. et al. . A methylation-dependent electrostatic switch controls DNA repair and transcriptional activation by E. coli Ada. Mol. Cell. 2005; 20:117–129. [DOI] [PubMed] [Google Scholar]
- 32. Harper M., Lee C.J.. Genome-wide analysis of mutagenesis bias and context sensitivity of N-methyl-N'-nitro-N-nitrosoguanidine (NTG). Mutat. Res. 2012; 731:64–67. [DOI] [PubMed] [Google Scholar]
- 33. Shrivastav N., Li D., Essigmann J.M.. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 2010; 31:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma B., Zarth A.T., Carlson E.S., Villalta P.W., Upadhyaya P., Stepanov I., Hecht S.S.. Methyl DNA phosphate adduct formation in rats treated chronically with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2018; 31:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.