


Abstract
In type I CRISPR–Cas systems, primed adaptation of new spacers into CRISPR arrays occurs when the effector Cascade–crRNA complex recognizes imperfectly matched targets that are not subject to efficient CRISPR interference. Thus, primed adaptation allows cells to acquire additional protection against mobile genetic elements that managed to escape interference. Biochemical and biophysical studies suggested that Cascade–crRNA complexes formed on fully matching targets (subject to efficient interference) and on partially mismatched targets that promote primed adaption are structurally different. Here, we probed Escherichia coli Cascade–crRNA complexes bound to matched and mismatched DNA targets using a magnetic tweezers assay. Significant differences in complex stabilities were observed consistent with the presence of at least two distinct conformations. Surprisingly, in vivo analysis demonstrated that all mismatched targets stimulated robust primed adaptation irrespective of conformational states observed in vitro. Our results suggest that primed adaptation is a direct consequence of a reduced interference efficiency and/or rate and is not a consequence of distinct effector complex conformations on target DNA.
INTRODUCTION
Arrays of clustered regularly interspaced short palindromic repeats (CRISPR) together with CRISPR-associated (Cas) proteins constitute adaptive immune systems in prokaryotes and archaea that defend cells against invaders such as viruses, plasmids or other mobile genetic elements (1,2). CRISPR arrays contain variable spacer elements of equal length separated by repeats of identical sequence. CRISPR–Cas systems adapt to new or rapidly mutating invaders by integrating short segments of invader DNA as new CRISPR array spacers. Two most conserved Cas proteins, Cas1 and Cas2, are sufficient for spacer acquisition (3). Acquired spacers can be regarded as ‘memories’ of distinct genetic invaders. Transcripts of the CRISPR array are processed to yield short CRISPR RNAs (crRNAs). The crRNAs are part of ribonucleoprotein surveillance/effector complexes that mediate target recognition by facilitating base-pairing between the crRNA and a complementary strand of the target sequence called ‘protospacer’. Surveillance complexes of Type I CRISPR–Cas systems are large hetero-multimers, exemplified by the Type I-E surveillance complex Cascade in Escherichia coli with a stoichiometry of Cse11Cse22Cas76Cas51Cas61 (4–6). The complex first binds the protospacer-adjacent-motif (PAM)—an upstream element that is recognized by the protein component of the complex itself. It then mediates base-pairing between crRNA and the PAM proximal target base(s). Further base pairing along the target is achieved in a reversible zipper-like fashion by displacing the non-target DNA strand, resulting in a triple-strand R-loop structure (5,7). Mismatches between crRNA and DNA target represent kinetic barriers that are difficult and sometimes impossible to overcome. Particularly, PAM proximal mismatches in the so-called ‘seed region’ exhibit stronger hindrance and are thought to inhibit the R-loop nucleation (7–9). Full R-loop zipping until the PAM-distal end of the protospacer triggers a large conformational change. It mainly involves the Cse1 and Cse2 subunits and leads to a highly stable ‘locked’ state of the Cascade complex on the DNA target (10–12). Stable R-loop locking is thought to be a signal for the recruitment of the helicase-nuclease Cas3 (7), which cleaves the DNA at and around the protospacer (13–17).
In addition to mediating target recognition during interference, Cascade can promote the acquisition of new spacers from invader DNA, a response called ‘primed adaptation’ or ‘priming’ that allows to update the ‘invader memory’ of the CRISPR–Cas system (8,17,18). So far, priming has been observed for Type I-B (17–20), I-C (21), I-E (8,9,22–24) and I-F CRISPR–Cas systems (18,25). In Type I-E, priming requires all elements of the system's machinery, i.e. the surveillance complex, Cas3, and the Cas1–Cas2 adaptation complex (8,9,23). The apparent yield of priming is stimulated by the recognition of protospacers that form mismatches with the crRNA spacer or of fully matching protospacers that contain a suboptimal PAM (8,24). Spacers that are acquired in the course of primed adaptation are located in cis with such priming protospacers. In Escherichia coli, the protospacers from which new spacers are selected have almost invariably a consensus interference-proficient AAG PAM that increases the ability of CRISPR–Cas system to fight off a genetic invader. While distances between the priming site and the selected protospacer site can be substantial (tens of thousands of nucleotides), the efficiency at which new spacers are acquired drops with increasing distance from the priming site (8,18,22,23,25,26). In addition to the distance, other poorly-defined parameters such as the protospacer sequence and its local context also affect the efficiency of spacer selection (22,27); spacers from so-called ‘hot’ protospacers are selected with thousands-fold higher probability than spacers from ‘cold’ protospacers.
Two main alternative mechanistic models to explain priming have been proposed. In the conformational-control model Cascade adopts a distinct conformation that supports priming compared to a conformation that supports interference. The model is based on an observation that for some target sequences that support priming but strongly attenuate interference, Cas3 recruitment is decreased, but can be restored with the help of Cas1–Cas2 (28). On such protospacers, the Cse1 subunit of Cascade adopts predominantly an open conformation in contrast to a closed conformation found on protospacers that, once recognized, promote interference (29). Thus, priming could be a consequence of a specific recognition of the open-form Cascade-target complex by Cas3 and the Cas1–Cas2 complex. In the extreme case of this model, acquisition of new spacers could occur without interference initiated at the priming site.
Within the conceptually simpler interference-based model, both interference and priming are consequences of the same process of target DNA degradation. The model is based on the observations that (i) during the short time window before their destruction, matching targets with consensus PAMs support more robust primed spacer acquisition than mismatched targets that are poorly interfered with (27) and (ii) target DNA fragments generated by Cas3 fuel priming (30). During attenuated but not completely suppressed interference, invader plasmids and phages can replicate and persist for longer periods of time inside cells despite of ongoing CRISPR interference. As a result, Cas3-generated fragments of foreign DNA, which are substrates for adaptation, will also be present for longer time, allowing spacer acquisition events to occur over longer periods. In contrast, a rapid interference reaction quickly depletes the invader DNA providing insufficient time for adaptation (31).
In this work, we aim to distinguish between the two models by systematically investigating Cascade binding, DNA cleavage, and priming on a range of target substrates with mutations in the PAM and the PAM proximal seed region. To this end, we used a combination of single-molecule magnetic tweezers experiments, bulk-biochemical in vitro characterization, and in vivo measurements of priming. All tested target variants exhibited slower Cascade binding and R-loop formation rates compared to the fully matched target with consensus PAM. PAM mutations as well as seed mutations next to the PAM, for which the Cse1 subunit of Cascade is expected to adopt an open form, were found to attenuate R-loop locking as well as cleavage of preformed, fully extended R-loops by Cas3. In contrast, seed mutations more distal from the PAM exhibited bona fide locking and cleavage expected for closed Cse1 conformation. Surprisingly, all targets supported priming independent of the locking strength. Thus, priming can occur independently of Cascade conformations on the priming protospacer. Priming rates as well as the preferred sites of spacer acquisition were indistinguishable for strongly locked mutated targets and targets with attenuated locking. These data are consistent with the interference-based model of primed adaptation that is independent of effector complex conformations at the priming site.
MATERIALS AND METHODS
DNA constructs for MT assays
Constructs containing the WT g8 protospacer and its variants were cloned into plasmid pUC19 (NEB) at the single SmaI (NEB) site by blunt end ligation. The 73 bp insert DNA carried the target sequence (PAM and protospacer variant) in its center (see Supplementary Table S1). Ligation products were transformed into NEB 5α E. coli cells. Plasmids were purified and the presence of protospacer variant sought was confirmed by sequencing. DNA constructs for magnetic tweezers experiments were obtained by amplifying a 2.2 kb fragment containing the target sequence from the corresponding plasmid (10,32). At either end of the fragment, a biotinylated and a digoxigenin-modified 0.6 kb DNA handle was ligated after digestion of the fragment and the handles with SpeI and NotI (both from NEB).
Protein purification
Cascade containing the g8 spacer crRNA was overexpressed in the E. coli strain KD418 (33) co-transformed with the plasmids pCDF-casABCDE, a derivative of pWUR400 (4) encoding the Cascade complex with an N-terminally Strep-tagged Cse2 subunit (33) and pWUR615 containing seven g8 spacers in the CRISPR array (34). The Cascade complex was purified by affinity chromatography using a Strep-trap column (4) followed by size-exclusion chromatography using a Superose 6 (GE) gel filtration column. The complex concentration was calculated from absorbance at 280 nm using an extinction coefficient of 725 000 M−1cm−1. Purified Cas3 protein was generously provided by Prof. Scott Bailey (John Hopkins University).
Magnetic tweezers experiments
Magnetic tweezers measurements were carried out using a home-built magnetic tweezers setup (35) and automated bead tracking in real time at 120 Hz (36). Forces were calibrated using the bead fluctuations along the ‘long-pendulum’ direction (37). DNA constructs were bound to 1 μm streptavidin-coated magnetic beads (MyOne; Invitrogen) and anchored in a digoxigenin-coated fluidic cells (38,39). R-loop formation/dissociation experiments were carried out as previously described (32) in 20 mM Tris–HCl pH 8, 150 mM NaCl and 0.1 mg/ml BSA at 37°C. Experiments using the target with CCG PAM were also performed in presence of 5 mM MgCl2. Mg2+ facilitates the R-loop formation, thus providing a more stringent control. R-loop formation was detected at about –7 turns of negative supercoiling and a force of 0.4 pN, corresponding to a torque of –6.7 ± 0.5 pN nm (39). Rotational shifts were estimated from the linear part on the left side of the rotation curve (10). R-loop dissociation experiments were performed at about +12 turns of supercoiling and a force of 5 pN, corresponding to a torque of +32 ± 3 pN nm. Each target variant was characterized with at least two repeats, i.e. on at least two different molecules.
Data analysis was carried out using customized software code written in Labview and Matlab as well as Origin 9 (32). Mean R-loop formation and dissociation times were determined from exponential fits to cumulative distributions of the data (10) (see Supplementary Figures S1 and S2 for measured distributions). Each mean-time value was calculated from ∼25 events.
Permanganate probing
The target g8 DNA fragment (213 bp) and its mutant variants were amplified by PCR of M13mp18 phage DNA (wild-type and engineered escape mutants (34)) using g8-dir 5′-agtctttagtcctcaaagcctctg-3′ and g8-rev 5′-agcttgctttcgaggtgaatttc-3′ primers. For radioactive labeling, 3–5 pmol of the target DNA fragments were combined with 8 pmol of [γ-32P]-ATP (3000 Ci/mmol) and 10 units of T4 polynucleotide kinase (NEB) in 20 μl of the reaction buffer containing 70 mM Tris–HCl (pH 7.6), 10 mM MgCl2, 5 mM DTT, and incubated for 30 min at 37°C. 32P-labeled DNA fragments were purified by micro Bio-Spin™ chromatography on columns packed with Bio-Gel P-30 (Bio-Rad) and used for permanganate probing reactions performed as described before (40). Target binding was performed in 10 μl of binding buffer (40 mM Tris–HCl, pH 8.0, 50 mM NaCl, 10 mM MgCl2, 0.5 mM TCEP, 50 μg/ml BSA) using 15 nM labeled DNA fragment and 2 μM Cascade. After 30 min incubation at 37°C, the probing reaction was initiated by adding KMnO4 to a final concentration of 2.5 mM. The reaction was quenched after 15 s by the addition of 10 μl 1% 2-mercaptoethanol. The reaction products were extracted using a phenol–chloroform mixture, followed by an ethanol precipitation. The DNA pellets were dissolved in 100 μl of freshly prepared 1 M piperidine solution and placed in a 90 °C water bath for 10 min. After chloroform extraction DNA was ethanol precipitated. The pellets were dissolved in 8 μl of formamide loading buffer. The reaction products were separated using an 8% denaturing PAGE gel and visualized with a Typhoon 9400 phosphorimager.
Cas3-mediate DNA degradation
Cas3-mediated DNA cleavage experiments were carried out in 20 mM HEPES–KOH pH 7.5 supplemented with 35 mM KCl, 10 mM MgCl2, 10 μM CoCl2, 1.5 mM ATP and 1 mM TCEP. First, Cascade binding to the target plasmids was tested by incubating 5 or 10 nM plasmid with 100 nM Cascade at 37°C for 30 min. The reaction products were separated on 1% agarose gels. Cascade binding was seen as a small but noticeable shift towards lower mobility (Supplementary Figure S3). To measure DNA degradation, 100 nM Cas3 was added to the Cascade-bound plasmid. The reaction was allowed to proceed at 37°C for variable times and was stopped by adding 30 mM EDTA and rapid cooling on ice. Reaction products were separated on 1% agarose gel and visualized by ethidium bromide staining using a Bio-Rad gel imaging system. For each target variant, the intensity of Cascade-bound plasmid in absence of Cas3 was taken as zero-time reference. The processed fraction of plasmid was calculated from the intensity decrease of the supercoiled plasmid species normalized by the zero-time reference.
Detection of primed adaptation in vivo
Primed adaptation in vivo was studied using E. coli KD263 cells (K-12 F+, lacUV5-cas3 araBp8-cse1, CRISPR I: repeat-spacer g8-repeat) as described in (26,41). Cells were transformed with pUC19 carrying the corresponding target variant. Single colonies were picked inoculated in LB medium containing 100 μg/ml of ampicillin and grown overnight. The cultures were then used to inoculate fresh LB without antibiotic and cells were grown for few additional hours until an OD600 of 0.4 was reached. Expression of cas genes was induced by addition of 1 mM IPTG (induction of the cas3 gene) and 1 mM arabinose (induction of operon containing genes encoding Cascade subunits and Cas1–Cas2). At various times, 10-μl culture aliquots were withdrawn and diluted 1:10 in deionized water. 1 μl of diluted cultures was used in a 20-μl PCR reaction with Taq polymerase using the 5′-aaggttggtgtcttttttac and 5′-gtcgctgccgtgacgttatg primers to amplify CRISPR array (including part of the leader and all repeats and spacers). The PCR product was 308 bp long without a newly incorporated spacer and 369 bp long with one newly incorporated spacer. The PCR products were analyzed on 2% agarose gels. Gel images were quantified using Image Lab 5.0 software. An average of at least two repeats for each time point and each target variant was used to calculate the priming efficiency.
The efficiency of primed adaptation was measured using qPCR. To this end the amount of CRISPR arrays that acquired a particular plasmid-derived spacer (hotspot 1, HS1) was quantified and normalized by the amount of the GyrA gene on the bacterial genome (Supplementary Figure S4A). The qPCR amplification of the extended CRISPR arrays used the primers 5′-catgagtgataacactgcggcc being complementary to HS1 and 5′-aaggttggtgggttgtttttatgg being complementary to the CRISPR array leader. The qPCR amplification of the GyrA gene used the primers 5′-cggtcaacattgaggaagagc and 5′-tacgtcaccaacgacacgg. DNA amounts were obtained from the qPCR cycle threshold using calibration curves from diluted DNA samples (Supplementary Figures S4A and S4B). The adaptation score was calculated as the percentage of CRISPR arrays that adapted the HS1 spacer over all CRISPR arrays in the sample (see test measurement in Supplementary Figure S4C). In a primed adaptation experiment, the adaptation score is significantly <100% since the spacers from HS1 represent only a subset of all acquired spacers. The adaptation score is nonetheless proportional to the overall level of adaptation, since all targets supported comparable levels of HS1 incorporation (see high throughput sequencing experiments below).
High throughput determination of acquired spacers sequences
KD263 E. coli cells harboring pUC19 plasmids with various target variants were collected 6 h after induction of the cas genes expression. 1 μl of 10-fold diluted culture was used in a 20-μl PCR reaction with 5′-aaggttggtgggttgtttttatgg-3′ and 5′-ggatcgtcaccctcagcagcg-3′ primers to amplify sequences between the leader region to the priming g8 spacer (118 bp in KD263 with unexpanded array). PCR products were separated on 2% agarose gels and a 179 bp band that corresponds to a single acquired spacer was excised and purified using a GeneJet Gel Extraction and DNA Cleanup kit (Thermo Scientific). ∼100 ng of purified DNA amplicons from each target were sequenced using Illumina MiniSeq system according to the recommended protocol of the manufacturer. Each amplicon was read for 150 nucleotides from each side. Results were trimmed and paired using the CLCgenomics software. Further analysis of the results was done using in house developed R scripts. Acquired spacer sequences were identified, counted for frequency and mapped onto the pUC19 plasmid backbone. Graphical visualization of the results was done using the EasyVisio1500 software.
RESULTS
E. coli Cascade forms R-loop complexes on targets that escape CRISPR interference in vivo
In order to relate priming to target binding and Cas3-mediated DNA degradation we used E. coli Cascade with a crRNA harboring the previously studied g8 spacer (34) and characterized its biochemical properties for a number of different DNA target variants (see Figure 1A, Supplementary Table S1). All target variants were derived from a fully matching M13-derived protospacer g8 with interference-proficient ATG PAM. This initial protospacer is further referred to as ‘wild type’ (‘WT’). Cells expressing g8 spacer crRNA efficiently interfere with wild-type M13 phage infection (34). In addition to the WT target we investigated two targets with altered PAM: CCG, which corresponds to the spacer-adjacent trinucleotide of the CRISPR repeat and prevents self-recognition by the CRISPR machinery (24), and ATT (named G-1T as it is derived from wild-type ATG PAM by a single substitution at the –1 position). Additionally, we tested ATG PAM targets carrying single substitutions at four consecutive seed positions: C1T, T2A, G3T and T4G (named according to nucleotide introduced in the non-targeted DNA strand and the relative position from the PAM). In the context of the M13 phage infection, each of these mutations allows bacteriophages to escape CRISPR interference (34). The G-1T and C1T variants were also shown to support primed adaptation in vivo (8).
Figure 1.

Principle of the magnetic tweezers assay used to study R-loop complexes. (A) Schematic representation of the R-loop complex formed by the E. coli Cascade (gray) bound to the wild-type g8 protospacer target (blue). The crRNA is shown in black. (B) A scheme of the magnetic tweezers assay to detect formation and dissociation of single R-loops (see text for detailed explanation). The left side illustrates R-loop formation on negatively supercoiled DNA. DNA unwinding by R-loop formation absorbs about 3 turns of negative supercoiling, leading to a DNA length increase (see green arrow in sketch on top and example trajectory on the bottom). The right side illustrates R-loop dissociation on positively supercoiled DNA (see red arrow in sketch). DNA rewinding upon R-loop dissociation absorbs about 3 turns of positive supercoiling, also leading to DNA length increase. The plot at the bottom, center, shows the characteristic DNA supercoiling curve (DNA length vs. turns) in the absence (gray) and the presence (green) of bound Cascade. The curves are shifted relative to each other due to unwinding of helical turns by the Cascade. Green and red arrows mark R-loop formation and dissociation at negative and positive supercoiling, respectively.
To dissect the impact of the mutations on target recognition, we first studied their influence on R-loop formation. To this end we employed a previously described magnetic tweezer assay (10,32) that allows one to directly monitor R-loop formation in a time-resolved manner on single supercoiled DNA molecules (see Figure 1B for a detailed scheme). In brief, one end of a linear DNA molecule containing a protospacer is tethered to the surface inside a fluidic cell, while the other end is attached to a magnetic bead. This allows one to apply controlled forces using the field of a pair of permanent magnets (37) and to stretch the DNA. The DNA length is measured in real-time using microscopy and automated analysis of the recorded images (36). Rotating the magnets allows one to twist the DNA, i.e. to introduce positive or negative supercoils. When DNA is negatively or positively supercoiled at low force, its length decreases since the molecule absorbs twist by writhing around itself.
The DNA length dependence on the applied supercoiling is revealed by a characteristic rotation curve (gray in Figure 1B). Upon R-loop formation by Cascade, ∼30 bp of DNA are unwound. This absorbs about three turns of supercoiling, such that the rotation curve in presence of the R-loop is shifted by same amount of turns towards lower supercoiling (green curve in Figure 1B). R-loop formation on a substrate with a fixed amount of negative turns is thus seen as a sudden increase in DNA length (green arrows in Figure 1B). One can quantify the kinetics of R-loop formation by monitoring the time between applying negative supercoiling and the DNA length increase due to R-loop formation. Moreover, the extent of DNA unwinding in the R-loop complex can be quantified, since it linearly corresponds to the extent of the rotational shifts (10). Furthermore, as shown in previous studies, R-loops formed by S. thermophilus Cascade would typically dissociate when sufficient positive supercoiling (torque) is applied to the DNA molecule by magnetic tweezers (10). R-loop dissociation (DNA rewinding) absorbs positive supercoiling resulting in a back-shift of the rotation curve. Thus, R-loop dissociation for a fixed amount of positive supercoils introduced is seen also as a sudden increase in the DNA length (red arrows in Figure 1B).
When investigating R-loop formation on the WT target carrying a fixed number of negative turns, a sudden DNA length increase in the time-trajectory as well as a shift of the whole rotation curve (when subsequently rotating the magnets) are detected and indicate DNA unwinding by R-loop formation (Figure 2). As expected, DNA unwinding was not detected when the substrate contained a fully matching protospacer but a CCG PAM (Figure 2). These experiments were carried out at 37°C in the presence of Mg2+—conditions that support rapid R-loop formation by E. coli Cascade on the WT sequence (32). Surprisingly and in strong contrast to Cascade from S. thermophilus (10), R-loops of E. coli Cascade formed on the WT target could not be dissociated (rotation curve did not back-shift) even at maximal applicable positive supercoiling/torque at which the B-form DNA structure collapses (see Figure 1B right side and the trajectory in Figure 2, top row, right panel). Thus, conformational locking (11–13) by E. coli Cascade appears to be strong and practically irreversible.
Figure 2.

Example trajectories of R-loop formation and dissociation by E. coli Cascade measured with magnetic tweezers. Data is shown for all investigated target variants (nomenclature, mutation positions and base-pairing schemes, with mutations indicated in red, are shown on the left side). R-loop formation is seen as a sudden DNA length jump at negative supercoiling (left trajectories recorded at 0.4 pN force and about –7 turns) and as a shift of the supercoiling curve (middle trajectories, grey and green curves refer to unbound and bound DNA). R-loop dissociation is seen as a length jump at positive supercoiling (right trajectories including mean dissociation times taken at 5 pN force and about +12 turns). Shown trajectories were smoothed with a sliding average filter of 1 s.
When investigating target variants bearing single point mutations, R-loop formation was observed on each of these targets as a single-step DNA length increase (Figure 2). Possible R-loop intermediates, if they existed, were too short-lived to be detected. Measured rotational shifts were between 2 to 3 turns, suggesting that the R-loops on all targets covered very similar regions of unwound DNA. Thus, the ability of E. coli Cascade to form R-loops on the set of escape point-mutant targets was directly demonstrated. However, in contrast to the WT target, most of the mutant variants exhibited R-loop dissociation, i.e. less stable locking (Figure 2, see also below).
The ability of Cascade to form R-loops on relaxed linear DNA substrates as well as the extent of R-loops formed was also tested in bulk assays. To this end, potassium permanganate probing that reveals thymine residues in single-stranded form was used (Figure 3). The results were in full agreement with the magnetic tweezers assay: the wild-type target, as well as the seed mutants in positions 1–4 and the PAM variant with single substitution at position -1 all showed Cascade-dependent localized melting of DNA and the extent of this melting was identical for all targets (the differences in patterns of permanganate sensitivity between the wild-type and some of the mutant complexes are due to introduction/removal of thymine residues by substitutions). In reactions containing a DNA substrate with a fully matching g8 protospacer and a CCG PAM, the permanganate sensitivity was weak and the pattern that was detected was markedly different compared to other complexes indicating the absence of an R-loop.
Figure 3.

KMnO4 probing of mutant g8 protospacers bound by Cascade–g8–crRNA. (A) Probing of complexes formed on WT, PAM (CCG, G-1T), and indicated protospacer mutant templates. Positions of permanganate-sensitive thymines are indicated by dark red numbers on the right side of the gel. (B) A schematic model of R-loop formed upon the recognition of g8 protospacer target. Thymines sensitive to permanganate oxidation are shown in red (see also panel A).
R-loop formation on escape targets is slower than on wild-type target
We further investigated how strongly the R-loop formation kinetics differed between WT and mutant targets. Since measurements of R-loop formation rates by magnetic tweezers require analysis of multiple individual binding/dissociation events, it was practically impossible to obtain such data for substrates on which highly stable R-loop formed. We, therefore, sought conditions that prevent locking but not R-loop formation. To this end, we introduced six consecutive mismatches with the crRNA at the PAM-distal end of the WT protospacer (Figure 4A) (note that all other tweezers and ensemble measurements use targets without PAM-distal mismatches). The same number of mismatches abolished locking by the S. thermophilus Cascade by preventing zipping of the R-loop until the end of the protospacer (10). As anticipated, all R-loops formed by E. coli Cascade on the target with six PAM-distal mismatches dissociated instantaneously upon applying mild positive supercoiling. This indicates the absence of locking (Figure 4B), such that multiple R-loop formation-dissociation cycles could be carried out. To measure R-loop formation times for the target variants, a set of substrates was produced where single PAM or seed substitutions were combined with the PAM-distal mutation introducing 6 mismatches (see Figure 1B and Supplementary Table S1). Mean R-loop formation times were obtained from the analysis of consecutive single R-loop formation events (see Figure 4C and Supplementary Figure S1). R-loop formation on the target with 26 fully matching base pairs from the PAM (i.e. a shortened version of the wild type target) was the fastest (6.7 ± 1.7 s at defined supercoiling conditions with a torque of – 6.7 ± 0.5 pN nm). The G-1T PAM mutation and the C1T seed mismatch retarded the R-loop formation strongly, by 35- to 50-fold. The T2A, G3T and T4G exhibited a moderate 5- to 15-fold retardation of R-loop formation.
Figure 4.

Cascade binding kinetics measured on the shortened targets. (A) Sketch of Cascade bound to the DNA targets containing 6 PAM distal mismatches that prevent locking of the shortened 26-bp long R-loop. (B) Representative trajectory from magnetic tweezers measurement of repetitive R-loop formation-dissociation events obtained with E. coli Cascade on the shortened target. Jumps corresponding to R-loop formation are indicated. Dissociation of R-loops is obtained as soon as positive supercoiling (positive turns) is applied. The R-loop formation time is measured from the moment the desired negative supercoiling was reached. (C) Average R-loop formation times for the different protospacer/PAM variants obtained from exponential fits to of the recorded kinetics (Supplementary Figure S1). R-loop formation for seed and PAM mutants is at least five times slower than for the WT target.
Target mutations at PAM and PAM-proximal positions attenuate R-loop locking
While R-loop locking on the WT target was irreversible even at high positive supercoiling, R-loop dissociation could be observed for most of the target variants, in particular the G-1T PAM mutation as well as seed mutations at positions +1, +2 and +3 (Figure 2, seen as sudden DNA length increase at conditions of positive supercoiling). To quantify R-loop dissociation we measured the times required for R-loop dissociation after introduction of positive supercoiling (torque of +32 ± 3 pN nm). Fitting of the cumulative time distributions provided mean dissociation times of 9 ± 1.9 s for the G-1T PAM variant and 20 ± 4.2 s, 252 ± 56 s and 1950 ± 540 s for targets with single seed mutations at positions +1, +2 and +3, respectively (Figure 5). R-loops on the WT target as well on the target with a seed mutation at position +4 did not dissociate under the conditions of the experiment over the course of at least one hour (Figure 2). Overall, these results clearly indicate that the presence of mutations in the PAM or in the seed region very close to the PAM (positions +1 and +2) strongly attenuates R-loop locking by E. coli Cascade without altering the extent of DNA unwinding. The presence of more distant seed mutations (positions +3 and +4) supports, however, strong WT-like R-loop locking.
Figure 5.
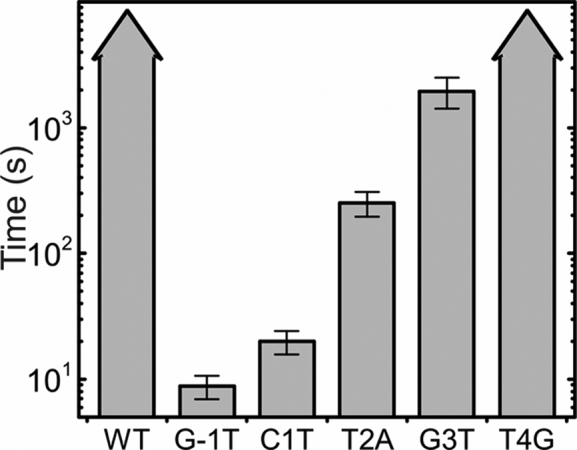
Mean times required to dissociate single R-loops at high positive torque. Arrowheads on bars indicate that no R-loop dissociation events were observed in the course of 1-h observation.
For the weakly locked target variants (positions –1, +1 and +2) we were able to perform repetitive formation-dissociation cycles and to quantify the R-loop formation kinetics of complexes with full-sized R-loops. The R-loop formation times on these substrates agreed with the times measured for corresponding substrates with six PAM-distal mismatches (Supplementary Figure S5) thus validating the previous approach.
Cas3 recruitment to Cascade complexes on escape targets correlates with the R-loop locking strength
The results presented above indicate that with the exception of the target with CCG-PAM, all tested mutated targets that escape CRISPR interference in vivo are bound by E. coli Cascade in vitro and form full-length R-loops. We therefore wondered whether the escape phenotype could be due to attenuated recruitment of the Cas3 nuclease to R-loop complexes with different Cascade conformations. To test this idea, we investigated Cas3-mediated degradation of plasmids containing g8 protospacer variants that were bound by Cascade. Cascade binding to supercoiled plasmids could be seen as a shift in in the plasmid mobility during agarose gel electrophoresis (Figure 6 and Supplementary Figure S3). In agreement with the experiments described above, binding was detected for all plasmids but the CCG-PAM variant. Cascade-bound plasmids were next combined with an excess of Cas3 at conditions optimized for target degradation (24,29,41). DNA cleavage was seen as the conversion of supercoiled Cascade-bound plasmid DNA into relaxed and/or linearized forms as well as the appearance of a smear of shorter degradation products at longer reaction times (Figure 6) (13,29). For the WT target, most of the supercoiled plasmid species disappeared (corresponding to a processed fraction of 0.84, Figure 6C) with a mean reaction time of ∼8 min. Cas3-catalyzed DNA degradation using targets with seed mutations at positions +3 and +4 was similarly fast. In contrast, DNA degradation of targets bearing the G-1T PAM mutation or seed mutations at position +1 and +2 was greatly decreased. The strong attenuation of DNA cleavage in these cases was not caused by dissociation of less stable R-loop complexes, since all supercoiled (and, therefore, uncut) DNA remained bound to Cascade during the entire 120-min time course of the reaction (Figure 6A). It is interesting to note that the rate and the efficiency of Cas3 cleavage strongly correlates with the R-loop dissociation time, i.e. locking strength, on a particular target (compare Figure 5 with Figure 6C and D, Pearson correlation coefficients r of 0.88 and 0.94 with probability values P of 0.021 and 0.006 for rate and efficiency, respectively).
Figure 6.

Kinetics of Cas3-mediated cleavage of plasmid DNA bound by Cascade. (A) Target plasmids containing indicated g8 protospacer variants were preincubated with Cascade–g8–crRNA. DNA cleavage reaction was initiated by the addition of Cas3. At indicated times after Cas3 addition reactions were terminated. Products were separated by agarose gel electrophoresis. Sketches next to gel images illustrate positions of the DNA topoisomers (supercoiled, nicked and linear plasmid). (B) Kinetics of DNA cleavage quantified from the disappearance of the supercoiled DNA species in the agarose gels. Error bars represent standard deviations from two to three repeat measurements. (C and D) Initial cleavage rate and cleavage efficiency obtained from the fits of the data shown in B.
Escape targets induce primed adaptation independently of R-loop locking
In order to determine whether there is a link between the locking state of target-bound Cascade and priming, we tested whether target variants studied here support primed adaptation in vivo (only the weakly locked PAM mutant G-1T and the +1 seed mutant were previously tested and shown to support priming) (8). E. coli KD263 cells harboring cas genes under inducible promoters and a CRISPR array with g8 spacer were transformed with plasmids harboring the WT g8 target or its variants (Figure 7A). The cas gene expression was induced in plasmid-bearing cell cultures, and adaptation was followed by PCR amplification of CRISPR array at various times post-induction (Figure 7B). In agreement with previous work (24,42), no adaptation was detected in induced cultures of cells harboring plasmids without protospacer, with the WT target or with the CCG PAM variant, while robust adaptation was observed in cultures transformed with plasmids carrying targets with a G-1T PAM and the +1 seed mutation. Notably, targets with seed mutations at positions +2, +3 and +4 also supported adaptation at comparable rates (Supplementary Figure S6) and apparently comparable levels (Figure 7B).
Figure 7.

Priming by g8 target variants. (A) Scheme of the E. coli KD263 CRISPR locus. The cas gene expression is controlled by inducible promoters. The CRISPR array consists of a single g8 spacer (blue boxes) surrounded by two repeats (black boxes). Priming is induced by transforming the cells with pUC19 plasmids carrying the protospacer variants. Incorporation of new spacers (green box) is revealed using PCR amplification of the CRISPR array and agarose gel electrophoresis. (B) Incorporation of new spacers probed at different times after induction for the indicated g8 protospacer variants. (C) Mapping of spacers acquired from the G-1T variant target protospacer plasmid to the pUC19 backbone (see Supplementary Figure S7 for other target variant plasmids). The height of the histogram bars corresponds to the number of HTS reads found for a particular position. The location of the priming protospacer and the PAM is shown as a blue-red box. The histogram entry in orange marks the hotspot HS1, which was used for semi-quantitative measurements of the primed adaptation efficiency (see E). (D) Position-dependent acquisition frequency for targets with seed mutation plotted over the acquisition frequency for the G-1T PAM mutation target. A high correlation between spacer acquisition patterns of all tested target variants (see Supplementary Figure S7B for correlation coefficients) is apparent. (E) Relative frequency of priming (i.e. CRISPR array extension) probed by qPCR with a primer specific for the frequently incorporated protospacer HS1 (see C) for the different target variants. Error bars represent the standard deviation of three repeat measurements.
We further tested whether the locking state of Cascade impacts the specificity to select particular spacers during priming. For all target variants that supported priming, PCR fragments corresponding to expanded CRISPR arrays were subjected to Illumina sequencing. After filtering, the acquired spacer sequences were extracted and mapped onto the donor plasmid backbone (Figure 7C, Supplementary Figure S7A). No significant differences among the target variants could be detected: all of them displayed the previously reported strand-biased hot-spot pattern of protospacers from which newly acquired spacers originated (8,18,22,23,25,26). These patterns were for all targets highly correlated to each other (Figure 7D) with Pearson correlation coefficients >0.97 (Supplementary Figure S7B).
The identical use of protospacers during primed adaptation for the different targets allowed us to design a semi-quantitative assay to measure the adaptation efficiency. The assay involved qPCR reactions where one of the primers was specific to a frequently acquired spacer (from hotspot HS1, see Figure 7C). By normalizing the qPCR signal from HS1 by the qPCR signal from a genomic gyrA gene, an adaptation score could be calculated (Figure 7E, see Materials and Methods for details). All targets that supported priming had a similar adaptation score, i.e. priming occurred for all of them at a comparable level. We therefore conclude that the locking state of Cascade, while well correlated with the in vitro ability to recruit Cas3 for target degradation, does neither influence the extent of primed adaptation (insignificant correlation with r = −0.21 and P = 0.7) nor the sequence preferences during spacer selection in vivo. In other words, in vivo priming occurs independently of the particular locking state of target-bound Cascade.
DISCUSSION
In this work we comprehensively characterized Cascade binding and Cas3 mediated DNA degradation on targets that contain single mutations in the PAM or the seed region and correlated the observed behavior with the ability to prime spacer acquisition in vivo. A central finding of our study is that all tested target variants with single substitutions readily support R-loop formation, as well as Cas3-mediated DNA degradation. However, the kinetics of both processes was partially reduced for the mutant targets compared to the WT target. R-loop formation was slowed down for all mutant targets, but mostly affected by the G-1T PAM mutation and the +1 seed mutation (up to 50-fold). During Cas3-mediated DNA degradation of preformed R-loops, the degradation kinetics was slowed down most significantly for the G-1T PAM and the +1 seed mutation and, to a lesser extent, for the +2 seed mutation. However, seed mutations at positions +3 and +4 showed a WT-like Cas3 cleavage rate. The observed cleavage kinetics correlated well with the strength of R-loop locking measured in the magnetic tweezers assay, i.e. targets with longer dissociation times of R-loops under high positive torque were cleaved faster by Cas3 in bulk experiments.
It has been previously shown that a +1 seed mutation causes escape from CRISPR interference and stimulates primed adaptation in E. coli cultures (8). The Cse1 subunit of Cascade for an R-loop on such a target (as well as for single PAM mutations) adopted an open conformation, while it was in a closed conformation on a WT substrate (29). A +1 seed mutation was also reported to support Cas3 DNA degradation at significantly reduced rate. The magnetic tweezers assay measures a global effect of conformational changes that contribute to locking. Given that we observe extremely strong locking on the WT target and weak locking for a +1 seed mismatch target, we can conclude that full locking requires a closed Cse1 conformation. Thus, the attenuated locking and DNA degradation that we observe for targets with the G-1T PAM and with the +1 as well as the +2 seed mutations indicates a predominantly open Cse1 conformation on these substrates. Locking involves a large movement of Cse1 and the Cse2 dimer – the latter establishing PAM-distal DNA contacts (11). The crystal structure of Cascade with bound single-stranded DNA (43), as well as a combination of Cryo-EM and molecular dynamics simulations (44), suggest that on the weakly locked targets the Cse2 dimer adopts a locked position that stabilizes the R-loop on the PAM-distal side, while Cse1 remains in the open conformation. The open conformation of Cse1 fails to support full R-loop locking, leaving the R-loop in a ‘semi-locked’ state (see model Figure 8). The differences in R-loop stabilities between the weakly locked targets may be due to Cse1 being in dynamic equilibrium between the predominantly adopted open and the closed conformations. Targets with seed mismatches more distal to the PAM (from position +3 onwards) support WT-like locking and thus Cse1 should adopt here a closed conformation, which is additionally supported by the WT-like DNA degradation rates. The fact that locking is practically irreversible on these substrates suggests that the closed state is almost exclusively occupied. We note that there may be slight differences in the occupancy of the closed Cse1 conformation for the WT and the +4 mismatch target, since we cannot quantitatively evaluate the differences in locking strength between these two targets.
Figure 8.

A model of conformation states of Cascade at different target variants. The Cse1 subunit is predominantly in the open state for the –1 and +1 substitutions resulting in a weak (semi-locked) R-loop that is not able to support efficient Cas3 recruitment. For other seed substitutions the locking strength and the ability to recruit Cas3 increase with increasing distance from the PAM, reaching the WT level for +4 substitution (suggesting a closed WT-like Cse1 conformation that readily recruits Cas3).
Dual control of DNA degradation by (i) triggering locking upon R-loop expansion until the PAM-distal end of the target combined with (ii) additional verification of the PAM by Cse1 seems to be a shared mechanism at least for Cascade complexes of Type I-E CRISPR–Cas systems. It has been shown for S. thermophilus Cascade (7) that R-loop degradation is impeded for PAM mutants, while R-loops with a +2 seed mismatch (corresponding to the +1 position in E. coli Cascade) were cleaved at WT rate (7). For T. fusca Cascade, Cas3-recruitment is impeded by PAM and +1 seed mutations but not for more PAM-distal mutations (45). The relative involvement of the first base pairs of the seed in this additional verification step seems, however, to vary between these species.
The strong differences in R-loop degradation for target variants tested in our work did not lead to changes in primed adaptation, which occurred at comparable levels for all mutant targets. This result is difficult to reconcile with a model in which priming is triggered by the open conformation of Cse1 (29). The open-form Cascade is thought to represent a specific priming signal for a Cas3–Cas1–Cas2 complex, inducing a distinct mode of Cas3 movement along the DNA molecule with less DNA degradation and concomitant spacer acquisition (28). While this model can explain the behavior of protospacers with mutations at positions -1, +1 and +2, the priming behavior of +3 and +4 mismatches is not explained.
An alternative kinetic model (31) previously showed that the persistent presence of target plasmid DNA at conditions of reduced interference allows bulk levels of spacer acquisition in cultures that by far exceed the acquisition levels that can be attained during a restricted time window as in the case of rapid interference. According to this model high yields of primed adaptation are a consequence of a steady slow Cas3-based production of target DNA fragments at low interference rates such that the loss of invader DNA can be compensated by its ongoing replication. Since R-loop complexes with mismatches at positions +3 and +4 readily recruit Cas3 and support rapid DNA degradation, their ability to promote priming should arise only from the slower R-loop formation kinetics detected in vitro. Likewise the kinetic model can explain priming for the targets with mutations at positions –1, +1 and +2 since they also exhibit a low overall rate of target degradation. Thus, for all tested target variants, invader DNA should, at conditions of ongoing replication, persist over longer durations, as is indeed evidenced by the escape phenotype of phages and plasmids carrying these mutations. Together with the ongoing degradation of the foreign DNA a constant production of substrates for spacer acquisition by Cas1–Cas2 should be ensured. Thus, our data—while clearly supporting the existence of multiple conformations of the Cascade complex on target protospacers - are more consistent with a minimalistic kinetic model for primed adaptation.
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge the generous gift of Cas3 by Dr Scott Bailey. We are indebted to Dr Ekaterina Savitskaya for the library preparation and HTS sequencing. We acknowledge the advice from and discussion with Dr Ekaterina Semenova. We are grateful to Ekaterina Rubtsova for developing the EasyVisio1500 software.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Skoltech Ph.D. program in the Life Sciences (to A.K.); European Research Council consolidator grant [GA 724863 to R.S].; NIH [R01 GM10407] and Russian Science Foundation [14-14-00988] grants to K.S.; UMNIK grant 8115GU/2015 to O.M., and institutional support from Skoltech to K.S. Funding for open access charge: Skolkovo Institute of Science and Technology internal funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P.. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007; 315:1709–1712. [DOI] [PubMed] [Google Scholar]
- 2. Mojica F.J.M., Díez-Villaseñor C., García-Martínez J., Almendros C.. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009; 155:733–740. [DOI] [PubMed] [Google Scholar]
- 3. Yosef I., Goren M.G., Qimron U.. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012; 40:5569–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brouns S.J.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J.H., Snijders A.P.L., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J.. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008; 321:960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jore M.M., Lundgren M., van Duijn E., Bultema J.B., Westra E.R., Waghmare S.P., Wiedenheft B., Pul Ü., Wurm R., Wagner R. et al. . Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat. Struct. Mol. Biol. 2011; 18:529–536. [DOI] [PubMed] [Google Scholar]
- 6. Wiedenheft B., Lander G.C., Zhou K., Jore M.M., Brouns S.J.J., van der Oost J., Doudna J.A., Nogales E.. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature. 2011; 477:486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rutkauskas M., Sinkunas T., Songailiene I., Tikhomirova M., Siksnys V., Seidel R.. Directional R-loop formation by the CRISPR-cas surveillance complex cascade provides efficient off-target site rejection. Cell Rep. 2015; 10:1534–1543. [DOI] [PubMed] [Google Scholar]
- 8. Datsenko K.A., Pougach K., Tikhonov A., Wanner B.L., Severinov K., Semenova E.. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 2012; 3:945. [DOI] [PubMed] [Google Scholar]
- 9. Fineran P.C., Gerritzen M.J.H., Suarez-Diez M., Kunne T., Boekhorst J., van Hijum S.A.F.T., Staals R.H.J., Brouns S.J.J.. Degenerate target sites mediate rapid primed CRISPR adaptation. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E1629–E1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szczelkun M.D., Tikhomirova M.S., Sinkunas T., Gasiunas G., Karvelis T., Pschera P., Siksnys V., Seidel R.. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:9798–9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hayes R.P., Xiao Y., Ding F., van Erp P.B.G., Rajashankar K., Bailey S., Wiedenheft B., Ke A.. Structural basis for promiscuous PAM recognition in type I–E Cascade from E. coli. Nature. 2016; 530:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xiao Y., Ng S., Nam K.H., Ke A.. How type II CRISPR–Cas establish immunity through Cas1–Cas2-mediated spacer integration. Nature. 2017; 550:137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sinkunas T., Gasiunas G., Fremaux C., Barrangou R., Horvath P., Siksnys V.. Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J. 2011; 30:1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sinkunas T., Gasiunas G., Waghmare S.P., Dickman M.J., Barrangou R., Horvath P., Siksnys V.. In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J. 2013; 32:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Westra E.R., van Erp P.B.G., Künne T., Wong S.P., Staals R.H.J., Seegers C.L.C., Bollen S., Jore M.M., Semenova E., Severinov K. et al. . CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by cascade and Cas3. Mol. Cell. 2012; 46:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gong B., Shin M., Sun J., Jung C.-H., Bolt E.L., van der Oost J., Kim J.-S.. Molecular insights into DNA interference by CRISPR-associated nuclease-helicase Cas3. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:16359–16364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li M., Wang R., Zhao D., Xiang H.. Adaptation of the Haloarcula hispanica CRISPR–Cas system to a purified virus strictly requires a priming process. Nucleic Acids Res. 2014; 42:2483–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Richter C., Dy R.L., McKenzie R.E., Watson B.N.J., Taylor C., Chang J.T., McNeil M.B., Staals R.H.J., Fineran P.C.. Priming in the Type I-F CRISPR–Cas system triggers strand-independent spacer acquisition, bi-directionally from the primed protospacer. Nucleic Acids Res. 2014; 42:8516–8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li M., Wang R., Xiang H.. Haloarcula hispanica CRISPR authenticates PAM of a target sequence to prime discriminative adaptation. Nucleic Acids Res. 2014; 42:7226–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li M., Gong L., Zhao D., Zhou J., Xiang H.. The spacer size of I-B CRISPR is modulated by the terminal sequence of the protospacer. Nucleic Acids Res. 2017; 45:4642–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rao C., Chin D., Ensminger A.W.. Priming in a permissive type I-C CRISPR–Cas system reveals distinct dynamics of spacer acquisition and loss. RNA. 2017; 23:1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Savitskaya E., Semenova E., Dedkov V., Metlitskaya A., Severinov K.. High-throughput analysis of type I-E CRISPR/Cas spacer acquisition in E. coli. RNA Biol. 2013; 10:716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Swarts D.C., Mosterd C., van Passel M.W.J., Brouns S.J.J.. CRISPR interference directs strand specific spacer acquisition. PLoS One. 2012; 7:e35888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xue C., Seetharam A.S., Musharova O., Severinov K., Brouns S.J.J., Severin A.J., Sashital D.G.. CRISPR interference and priming varies with individual spacer sequences. Nucleic Acids Res. 2015; 43:10831–10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Staals R.H.J., Jackson S.A., Biswas A., Brouns S.J.J., Brown C.M., Fineran P.C.. Interference-driven spacer acquisition is dominant over naive and primed adaptation in a native CRISPR–Cas system. Nat. Commun. 2016; 7:12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shmakov S., Savitskaya E., Semenova E., Logacheva M.D., Datsenko K.A., Severinov K.. Pervasive generation of oppositely oriented spacers during CRISPR adaptation. Nucleic Acids Res. 2014; 42:5907–5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Semenova E., Savitskaya E., Musharova O., Strotskaya A., Vorontsova D., Datsenko K.A., Logacheva M.D., Severinov K.. Highly efficient primed spacer acquisition from targets destroyed by the Escherichia coli type I-E CRISPR–Cas interfering complex. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:7626–7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Redding S., Sternberg S.H., Marshall M., Gibb B., Bhat P., Guegler C.K., Wiedenheft B., Doudna J.A., Greene E.C.. Surveillance and processing of foreign DNA by the Escherichia coli CRISPR–Cas system. Cell. 2015; 163:854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xue C., Whitis N.R., Sashital D.G.. Conformational control of cascade interference and priming activities in CRISPR immunity. Mol. Cell. 2016; 64:826–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Künne T., Kieper S.N., Bannenberg J.W., Vogel A.I.M., Miellet W.R., Klein M., Depken M., Suarez-Diez M., Brouns S.J.J.. Cas3-derived target DNA degradation fragments fuel primed CRISPR adaptation. Mol. Cell. 2016; 63:852–864. [DOI] [PubMed] [Google Scholar]
- 31. Severinov K., Ispolatov I., Semenova E.. The influence of Copy-Number of targeted extrachromosomal genetic elements on the outcome of CRISPR–Cas defense. Front. Mol. Biosci. 2016; 3:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rutkauskas M., Krivoy A., Szczelkun M.D., Rouillon C., Seidel R.. Single-molecule insight into target recognition by CRISPR–Cas complexes. Methods Enzymol. 2017; 582:239–273. [DOI] [PubMed] [Google Scholar]
- 33. Beloglazova N., Kuznedelov K., Flick R., Datsenko K.A., Brown G., Popovic A., Lemak S., Semenova E., Severinov K., Yakunin A.F.. CRISPR RNA binding and DNA target recognition by purified Cascade complexes from Escherichia coli. Nucleic Acids Res. 2015; 43:530–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Semenova E., Jore M.M., Datsenko K.A., Semenova A., Westra E.R., Wanner B., van der Oost J., Brouns S.J.J., Severinov K.. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:10098–10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klaue D., Seidel R.. Torsional stiffness of single superparamagnetic microspheres in an external magnetic field. Phys. Rev. Lett. 2009; 102:028302. [DOI] [PubMed] [Google Scholar]
- 36. Huhle A., Klaue D., Brutzer H., Daldrop P., Joo S., Otto O., Keyser U.F., Seidel R.. Camera-based three-dimensional real-time particle tracking at kHz rates and Ångström accuracy. Nat. Commun. 2015; 6:5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Daldrop P., Brutzer H., Huhle A., Kauert D.J., Seidel R.. Extending the range for force calibration in magnetic tweezers. Biophys. J. 2015; 108:2550–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schwarz F.W., Toth J., van Aelst K., Cui G., Clausing S., Szczelkun M.D., Seidel R.. The Helicase-Like domains of type III restriction enzymes trigger Long-Range diffusion along DNA. Science. 2013; 340:353–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maffeo C., Schöpflin R., Brutzer H., Stehr R., Aksimentiev A., Wedemann G., Seidel R.. DNA-DNA interactions in tight supercoils are described by a small effective charge density. Phys. Rev. Lett. 2010; 105:158101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuznedelov K., Mekler V., Lemak S., Tokmina-Lukaszewska M., Datsenko K.A., Jain I., Savitskaya E., Mallon J., Shmakov S., Bothner B. et al. . Altered stoichiometry Escherichia coli Cascade complexes with shortened CRISPR RNA spacers are capable of interference and primed adaptation. Nucleic Acids Res. 2016; 44:10849–10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Musharova O., Klimuk E., Datsenko K.A., Metlitskaya A., Logacheva M., Semenova E., Severinov K., Savitskaya E.. Spacer-length DNA intermediates are associated with Cas1 in cells undergoing primed CRISPR adaptation. Nucleic Acids Res. 2017; 45:3297–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Westra E.R., Semenova E., Datsenko K.A., Jackson R.N., Wiedenheft B., Severinov K., Brouns S.J.J.. Type I-E CRISPR–Cas systems discriminate target from Non-Target DNA through base Pairing-Independent PAM recognition. PLoS Genet. 2013; 9:e1003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mulepati S., Heroux A., Bailey S.. Crystal structure of a CRISPR RNA-guided surveillance complex bound to a ssDNA target. Science. 2014; 345:1479–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Van Erp P.B.G., Jackson R.N., Carter J., Golden S.M., Bailey S., Wiedenheft B.. Mechanism of CRISPR-RNA guided recognition of DNA targets in Escherichia coli. Nucleic Acids Res. 2015; 43:8381–8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jung C., Hawkins J.A., Jones S.K., Xiao Y., Rybarski J.R., Dillard K.E., Hussmann J., Saifuddin F.A., Savran C.A., Ellington A.D. et al. . Massively parallel biophysical analysis of CRISPR–Cas complexes on next generation sequencing chips. Cell. 2017; 170:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.