

Abstract
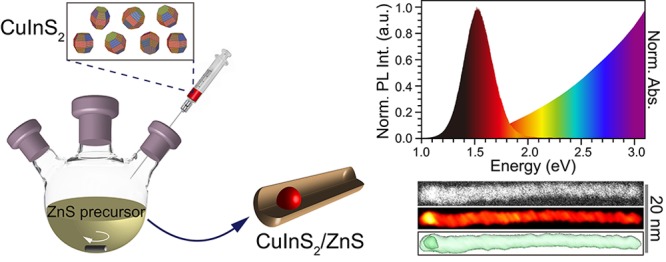
Synthesis protocols for anisotropic CuInX2 (X = S, Se, Te)-based heteronanocrystals (HNCs) are scarce due to the difficulty in balancing the reactivities of multiple precursors and the high solid-state diffusion rates of the cations involved in the CuInX2 lattice. In this work, we report a multistep seeded growth synthesis protocol that yields colloidal wurtzite CuInS2/ZnS dot core/rod shell HNCs with photoluminescence in the NIR (∼800 nm). The wurtzite CuInS2 NCs used as seeds are obtained by topotactic partial Cu+ for In3+ cation exchange in template Cu2–xS NCs. The seed NCs are injected in a hot solution of zinc oleate and hexadecylamine in octadecene, 20 s after the injection of sulfur in octadecene. This results in heteroepitaxial growth of wurtzite ZnS primarily on the Sulfur-terminated polar facet of the CuInS2 seed NCs, the other facets being overcoated only by a thin (∼1 monolayer) shell. The fast (∼21 nm/min) asymmetric axial growth of the nanorod proceeds by addition of [ZnS] monomer units, so that the polarity of the terminal (002) facet is preserved throughout the growth. The delayed injection of the CuInS2 seed NCs is crucial to allow the concentration of [ZnS] monomers to build up, thereby maximizing the anisotropic heteroepitaxial growth rates while minimizing the rates of competing processes (etching, cation exchange, alloying). Nevertheless, a mild etching still occurred, likely prior to the onset of heteroepitaxial overgrowth, shrinking the core size from 5.5 to ∼4 nm. The insights provided by this work open up new possibilities in designing multifunctional Cu-chalcogenide based colloidal heteronanocrystals.
Introduction
Ternary CuInX2 (X = S, Se, Te) nanocrystals (NCs) have attracted increasing attention as promising alternatives for CdX and PbX NCs in applications such as solar cells,1,2 luminescent solar concentrators,3,4 bioimaging,5,6 and light-emitting devices,7,8 due to their lower toxicity, large absorption cross sections across a broad spectral range and wide photoluminescence (PL) tunability.9 To achieve properties that are inaccessible to single component NCs (such as high PL quantum yields and photostability, spatial charge carrier separation, reduced blinking, etc.),10−12 researchers have been developing synthesis methods for colloidal CuInX2-based hetero-NCs (HNCs) (e.g., CuInS2/ZnS concentric core/shell HNCs,13,14 CuInS2/CdS tetrapods,15 CuInSe2/CuInS2 dot-in-rod HNCs16). Anisotropic CuInX2-based HNCs are particularly interesting, since they are expected to exhibit novel properties, such as polarized NIR PL and spatial charge separation, which are attractive for many applications (e.g., polarized LEDs,17 photocatalysts,18,19 etc.).
Nevertheless, reports on the synthesis of anisotropic CuInX2-based HNCs are scarce. This is most likely related to the difficulty in balancing the reactivities of multiple precursors and the high solid-state diffusion rates of all the cations involved in the CuInX2 lattice. These difficulties have been circumvented in recent works by performing cation exchange (CE) in NC templates, which yielded a variety of otherwise inaccessible anisotropic HNCs, such as Cu2–xS/CuInS2 core/crown nanoplatelets,20 CuInSe2/CuInS2 dot core/rod shell nanorods,16 axially segmented Cu2–xS/ZnS,21 and Cu2–xS/CuAS2 (A = In, Ga) heteronanorods.22 CE-based protocols are however limited by the availability of suitable template NCs or HNCs, by the difficulty in completely removing the native cations, and by atom economy (and toxicity) considerations, particularly when using sequential CE protocols starting from Cd-based HNC templates.
In this work, we report a multistep synthesis protocol that yields CuInS2/ZnS dot core/rod shell HNCs. The method is based on the synthesis of wurtzite CuInS2 NCs by topotactic partial Cu+ for In3+ cation exchange in template Cu2–xS NCs, and their subsequent injection together with sulfur in a hot solution of Zn(oleate)2 and suitable coordinating ligands (hexadecylamine). This results in heteroepitaxial growth of wurtzite ZnS primarily on the anion-terminated polar facet of the CuInS2 NC seeds. The resulting colloidal wurtzite CuInS2/ZnS dot-in-rod HNCs have large molar extinction coefficients, PL in the NIR (∼800 nm) with a quantum yield of ∼20%, and are readily dispersible in a variety of solvents.
Results and Discussion
Wurtzite CuInS2 NCs by CE in Template Cu2–xS NCs
The wurtzite CuInS2 (CIS) NCs used as seeds were obtained by topotactic partial Cu+ for In3+ CE in template Cu2–xS NCs, following previously reported procedures (see Experimental Section for details).14,23Figure 1 shows that the product CIS NCs inherit the shape, size (∼5.5 nm) and polydispersity (∼7%) of the template Cu2–xS NCs. Moreover, as previously reported,14,23 the anionic sublattice of the hexagonal high chalcocite Cu2–xS NC templates is preserved after the partial Cu+ for In3+ exchange, and therefore the product CIS NCs adopt the hexagonal wurtzite crystal structure (Figure 1c). The concentration (5.68 × 10–5 M) and composition (Cu1.63In0.79S2) of the product CIS NCs were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) measurements (see Experimental Section for details).
Figure 1.
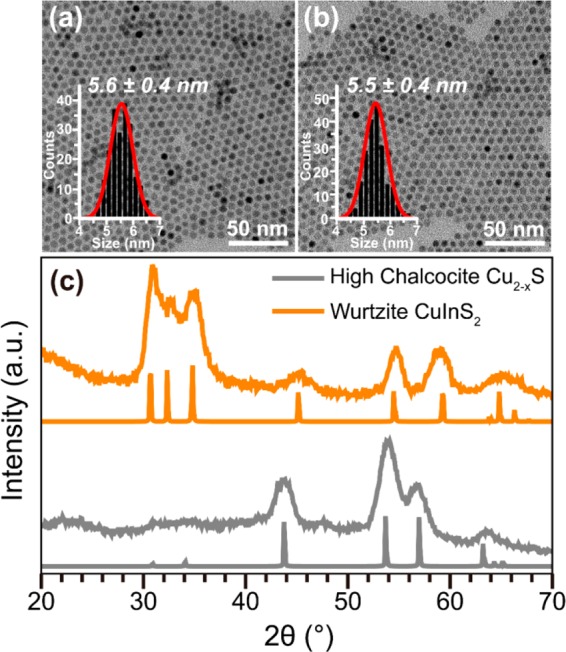
(a,b) TEM images and corresponding size histograms of CIS NCs (b) obtained by partial Cu+ for In3+ CE in template Cu2–xS NCs (a). The size histograms are constructed by measuring over 200 NCs and are fitted to a Gaussian distribution function. (c) X-ray diffraction patterns of template Cu2–xS NCs and product CIS NCs. The gray lines indicate the high chalcocite Cu2S diffraction pattern (JCPDS Card 00-026-1116). The orange lines indicate the wurtzite CIS diffraction pattern (JCPDS Card 01-077-9459).
Colloidal CIS/ZnS HNCs by Seeded Injection
A solution of wurtzite CIS NC seeds in ODE (100 μL, 5.68 × 10–5 M) combined with 100 μL of sulfur/1-octadecene (S/ODE, 0.5 M) was swiftly injected into a solution of Zn(oleate)2 and different coordinating ligands (2 mmol) in ODE at 210 °C (see Experimental Section and Supporting Information Table S1 for details). The reaction was allowed to proceed at this temperature for 10 min. Transmission electron microscopy (TEM) images show that the shape of the product HNCs strongly depends on the coordinating ligands present in the reaction medium (Supporting Information, Figure S1). The use of aliphatic primary amines results in nanorods (Figure S1c,e) with PL at ∼800 nm (Figure S2), while the other ligands investigated lead to either NCs with ill-defined shapes (OA, TOA, DDT), nanorods with no PL (TOPO), or simply no growth at all (TOP). We have therefore selected hexadecylamine (HDA) as coordinating ligand for further optimization of the synthesis protocol.
To investigate whether the size and shape control over the product HNCs could be improved, we studied the influence of the injection sequence in the reaction outcome (Figure 2). TEM images reveal that the injection sequence has a pronounced impact on the size and shape of the product HNCs (Figure 2). Injecting either the CIS NC seeds mixed with the S-precursor or first the CIS NC seeds and later the S-precursor leads to a mixture of shorter misformed nanorods and irregularly shaped NCs (Figure 2 II and III), while injecting the S-precursor before the CIS NC seeds yields longer and generally better shaped nanorods (Figure 2 IV). The HNCs obtained by injection method IV (i.e., S-precursor followed by CIS NC seeds) exhibit not only better size and shape characteristics, but also superior optical properties (viz., higher PL intensity at 810 nm and absorption spectrum without additional strong bands in the NIR, Figure S3). The strong and broad NIR absorption bands observed in the spectra of the product CIS/ZnS HNCs obtained by using injection methods II and III (Figure S3) are assigned to localized surface plasmon resonances (LSPR), which are often observed in Cu-chalcogenide NCs.9,24,25 LSPR bands involve excess charge carriers, which are due to stoichiometry deviations, typically Cu-vacancies, which lead to excess holes in the valence band.9,24 However, LSPRs due to In-vacancies have also been reported.25 These observations imply that methods II and III lead to product HNCs that are rich in cation (Cu or In) vacancies, in contrast to method IV.
Figure 2.
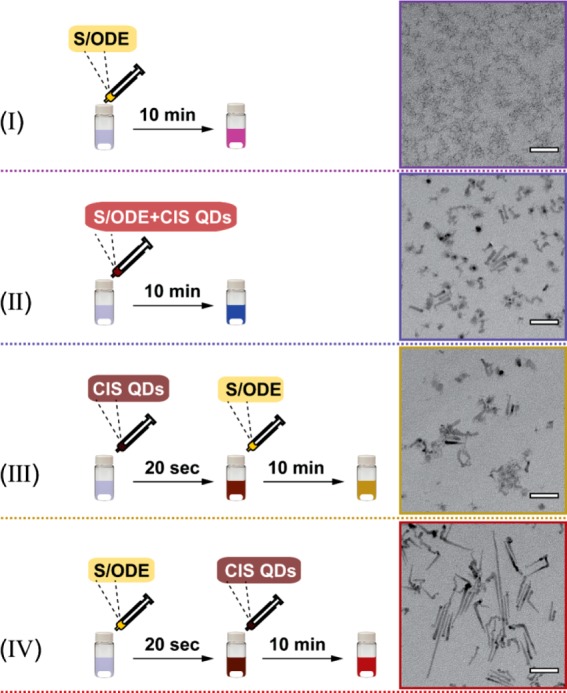
Schematic illustration of four seeded injection methods. All the experimental conditions are the same unless otherwise specified. (I) S/ODE (control experiment, no CIS NC seeds were added), (II) a mixture of S/ODE and CIS NC seeds is injected, (III) CIS NC seeds followed by S/ODE (injection interval time, 20 s), (IV) S/ODE followed by CIS NC seeds (injection interval time, 20 s). The corresponding TEM images of purified products are displayed on the right side of the panel. Scale bars are 50 nm.
It should be noted that the control experiments (injection of S-precursor only, Figure 2I) yielded only small ZnS NCs, clearly demonstrating that the product NCs isolated from reaction protocols II, III and IV are indeed formed by seeded heteroepitaxial growth, and are thus CIS/ZnS HNCs. The control experiments also demonstrate that the used reaction conditions promote the conversion of the S- and Zn-precursors into [ZnS] monomers, indicating that the different outcomes of the three seeded injection methods reflect the balance between the [ZnS] monomer formation rates and a number of other competing processes that interfere with ZnS heteroepitaxial overgrowth on the CIS seed NCs. This is also clearly evidenced by the high concentration of cation vacancies in the products obtained from protocols II and III (see Figure S3 and discussion above). The significance of these observations will be discussed in more detail later (Mechanism Section).
Considering the success of injection method IV, we further investigated the injection interval time (viz., 0, 20, 60, 300 and 600 s), while keeping all other reaction parameters constant (Supporting Information, Figures S4 and S5). The results show that longer intervals (≥60 s) result in irregular and shorter nanorods (Figure S4). The simultaneous injection of S and CIS NC seeds also deteriorates the size and shape control, yielding primarily small and irregularly shaped HNCs (Figure S4a), which display a pronounced LSPR band in the absorption spectrum (Figure S5). We have therefore selected an injection interval of 20 s for further optimization of the HNC growth, by investigating the effect of the Zn/S ratio (1, 2, 4 and 8, 10 min at 210 °C), reaction time (1–60 min at 210 °C and Zn/S = 4), and reaction temperature (190, 210, and 230 °C, at Zn/S = 4 and 10 min reaction time). We observed that increasing the Zn/S ratio up to 4 improves the shape control, but further increase leads to shape distortions (Figure S6). We note that increasing the Zn/S ratio also results in larger blue-shifts in the optical spectra (Figure S7), suggesting that Zn2+ partially interdiffuses in the CIS seed NCs, as previously observed for isotropic CIS/ZnS core/shell HNCs.14 The optimum reaction temperature is found to be 210 °C, since lower temperatures result in a larger fraction of misformed nanorods, while higher temperatures increase the magnitude of the spectral blue-shift (Supporting Information, Figures S8 and S9). Our studies reveal that the growth of the CIS/ZnS HNCs is very fast, being essentially completed in 5 min at 210 °C (Figure S10). Longer reaction times do not lead to significant changes in size and shape, but increase the magnitude of the spectral blue-shift (Figure S11), implying that the interdiffusion of Zn2+ continues after the heteroepitaxial growth is finished. Control experiments using wurtzite CIS seed NCs with the same size and shape of the NCs used in the experiments described above but a different stoichiometry (In/Cu = 0.83 instead of 0.48) show that the stoichiometry of the seed NCs does not have a pronounced impact on the outcome of the seeded growth reaction, since heteronanorods are obtained in both cases (Supporting Information, Figure S12). A possible mechanism for the formation and growth of CIS/ZnS heteronanorods by injection of wurtzite CIS NC seeds will be discussed in more detail below (Mechanism Section). It is worth noting that the seeded injection method developed in our work can be scaled up by a factor 10 by increasing the concentration of precursors while keeping all other reaction parameters fixed (see Experimental Section for details, Figure S13).
Optical Properties of Colloidal CIS/ZnS HNCs
Figure 3 shows the TEM image, the size distribution, and the optical properties of a representative sample of CIS/ZnS HNCs obtained by the optimized seeded injection method described above using the CIS seeds NCs shown in Figure 1b (viz., Method IV: S-precursor injected 20 s before the CIS NC seeds at 210 °C, HDA as ligand, Zn/S= 4, 10 min reaction time). The CIS/ZnS nanorods are 79 ± 11 nm long and have a diameter of 4.2 ± 0.7 nm (Figure 3a,b). They exhibit a featureless absorption spectra extending up to the NIR (∼1.5 eV) and a broad (fwhm = 308 meV) PL band with maximum at 780 nm (1.59 eV) (Figure 3c). The PL quantum yield (QY) of the CIS/ZnS HNCs (20%) is greatly improved with respect to that of the CIS seed NCs (∼0.2%). This is in line with the behavior previously reported for isotropic ZnS shelling of both chalcopyrite and wurtzite CIS NCs.9,13,14 The significant enhancement of the PLQYs upon overgrowth of ZnS on the CIS NC seeds indicates that recombination through surface defects is the major nonradiative decay pathway,26−28 and that the anisotropic ZnS shell is effectively passivating these defects. The molar extinction coefficient (1.4 × 106 M–1 cm–1) of the CIS/ZnS HNCs at 3.54 eV was determined by ICP-OES measurements combined with absorption spectrometry analysis. Reliable PL decay curves could not be obtained for the CIS NC seeds due to their very inefficient PL (PLQY = 0.2%). The CIS/ZnS HNCs exhibit multiexponential PL decay that is initially rather fast (a few ns), and then slows down to several hundreds of ns (Figure 3d, and Supporting Information, Figure S14), similar to the behavior previously reported for both chalcopyrite and wurtzite isotropic CIS/ZnS core/shell HNC.9,14,23,26−32 The slow PL decay dynamics is potentially beneficial for photovoltaic33 and photocatalytic applications,34 since long carrier lifetimes are of great importance for effectively extracting charge carriers.
Figure 3.
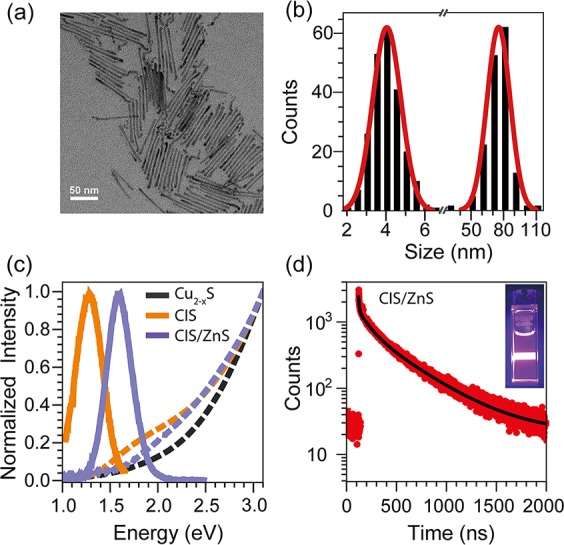
(a,b) TEM image and corresponding size histogram of CIS/ZnS HNCs prepared by injection of wurtzite CIS NC seeds and S/ODE into a solution of Zn(oleate)2 and HDA in ODE at 210 °C following injection protocol IV described above. The size histograms were constructed by measuring the diameter (4.2 nm with a polydispersity of 17%) and length (79 nm with a polydispersity of 14%) of over 200 HNCs and are independently fitted to Gaussian distribution functions. (c) Absorption (dashed lines) and PL (solid lines) spectra of the template Cu2–xS NCs, the product CIS NCs obtained by CE, and the final CIS/ZnS HNCs obtained by seeded injection and shown in (a). As the emission of CIS/ZnS HNCs (780 nm) is at the limit of both the UV–vis and the NIR detector, the full PL spectra were acquired by a combination of the two detectors (excitation wavelength 450 nm). (d) PL decay curve of the CIS/ZnS HNCs shown in (a). The detected wavelength was set at 780 nm. The data is best fit by a triple exponential decay (τ1 = 7.5 ns (1.85%), τ2 = 107 ns (18.93%), τ3 = 410 ns (79.22%)) (see Supporting Information, Figure S14 for details). Inset shows a digital image of a CIS/ZnS HNCs suspension in toluene illuminated by a 405 nm diode laser.
The band gap values of the seed CIS NCs (1.43 eV) and the CIS/ZnS HNCs (1.81 eV) were extracted from the absorption spectra (Supporting Information, Figure S15). A significant spectral blue-shift (viz., 380 and 304 meV in the absorption and PL spectra, respectively) occurs after overgrowth of ZnS on the CIS NC seeds. Spectral blue-shifts have been widely observed after isotropic ZnS shelling of both chalcopyrite and wurtzite CIS NCs,13,14,27,28,31 and have been attributed to a variety of reasons (viz., interdiffusion of Zn2+ into the CIS cores after either CE31 or heteroepitaxial shell overgrowth,14 reduction of the core diameter due to either etching prior to the shell overgrowth28 or shell ingrowth by CE13,35). Possible causes for the spectral blue-shift observed for the CIS/ZnS HNCs prepared in the present work will be presented below, after the discussion of the structural characterization of the HNCs and in conjunction with the proposed growth mechanism.
Structural Characterization of Colloidal CIS/ZnS HNCs
The composition of the CIS/ZnS HNCs was determined by ICP-OES. As shown in Table S2, the In/Cu ratio of the product CIS/ZnS HNCs (In/Cu = 0.43) is almost the same as that of the CIS seed NCs (In/Cu = 0.48). The slight excess of Zn compared to S may be ascribed to capping of the HNCs surface by Zn(oleate)2. The CIS seed NCs and the product CIS/ZnS HNCs obtained by seeded injection method IV (see Figure 3 above for TEM image and optical properties) were analyzed by high-resolution TEM and high angle annular dark field scanning transmission electron microscopy (HAADF-STEM). High-resolution TEM shows that the CIS seed NCs have the wurtzite structure with a thickness of 14 to 15 atomic columns, and have a shape that can be approximated to a hexagonal bifrustum (Figure 4a and Figure S16). High-resolution HAADF-STEM images (Figure 4c,e and Figure S17) and XRD patterns (Figure S18) demonstrate that the CIS/ZnS HNCs inherited the wurtzite structure of the CIS seed NCs, since the Fourier Transform (FT) analysis shows the characteristic (010) and (002) wurtzite ZnS lattice planes with a ⟨001⟩ growth direction, along the c-axis of the hexagonal close-packed ZnS structure. This confirms the heteroepitaxial nature of the ZnS growth.
Figure 4.

(a) High-resolution TEM image of the CIS seed NCs. The Fourier transform (FT) analysis shows that the CIS NCs have the wurtzite crystal structure (see details in the Supporting Information, Figure S16). (b) Overview HAADF-STEM image of the CIS/ZnS HNCs. The line profile in the inset reveals that the intensity is higher at one end of the nanorods. (c–f) High resolution HAADF-STEM images. FT analyses of the regions indicated with red squares are shown in the insets. Cell views of the CIS/ZnS HNCs are given in (d,f). The FT patterns in (c) and (e) are consistent with the [420] and the [100] zone axis of the wurtzite, respectively. For clarity, the cell views simulated from FT patterns in (c) and (e) are presented in (d) and (f), respectively (red represents Cu/In/Zn atoms while yellow denotes S atoms).
The thickness of a single nanorod is approximately 6 to 16 atomic layers, which is similar to the thickness of the CIS seed NCs, implying that the CIS core is likely located at the thicker side of the nanorods. Elemental EDX line scans were not possible due to the e-beam sensitivity of the nanorods, which could not withstanding the large electron doses required to obtain reliable elemental maps. Instead, HAADF-STEM imaging was used to determine the location of the CIS core, since the differences in atomic number between the CIS and ZnS constituents are sufficiently large to ensure significant intensity differences (ZCu = 29, ZIn = 49, ZZn = 30). A line profile confirms that the intensity is much higher at one side of the rod (Figure 4b and Figure S19). The heavier CIS appears brighter in the HAADF-STEM images, from which it can be deduced that the CIS NC seed is likely located at one end of the nanorod (Figure 4b and Figure S19). However, since the intensity in HAADF-STEM images scales with both the projected thickness and the atomic number of the elements present, 2D images cannot unambiguously confirm core–shell architectures. Therefore, electron tomography was performed to study the internal structure of the CIS/ZnS HNCs. The electron tomography reconstruction of a single CIS/ZnS core/shell HNC shows that the CIS NC core is indeed located at one side of the nanorod, since the highest intensity occurs near the thickest tip, but is not equal in regions of equal thickness (Figure 5, and Supporting Information, Figures S21–S24), and therefore must originate from the larger Z-number of In in comparison to Zn (49 and 30, respectively). The CIS core appears to be slightly prolate with a diameter of ∼4 nm, which is smaller than that of the CIS seed NCs (5.5 nm). We also performed electron tomography measurements on other CIS/ZnS nanorods, including misformed ones (Supporting Information, Figures S21–S24). In all cases, the CIS cores are located at one end of the nanorods, and are smaller than the original seed NCs and slightly prolate. We note that the size reduction of the CIS NC seeds after ZnS anisotropic heteroepitaxial overgrowth offers a plausible explanation for the blue-shifts observed in the absorption and PL spectra (see Figure 3 and discussion above). We note that the HAADF-STEM overview images also show the presence of smaller nanoparticles (Figure 4b, Figure S19 and S20), which are likely unintentional byproducts of the baking procedure used to reduce the carbon contamination (this is particularly evident in the image shown in the Supporting Information, Figure S20).
Figure 5.
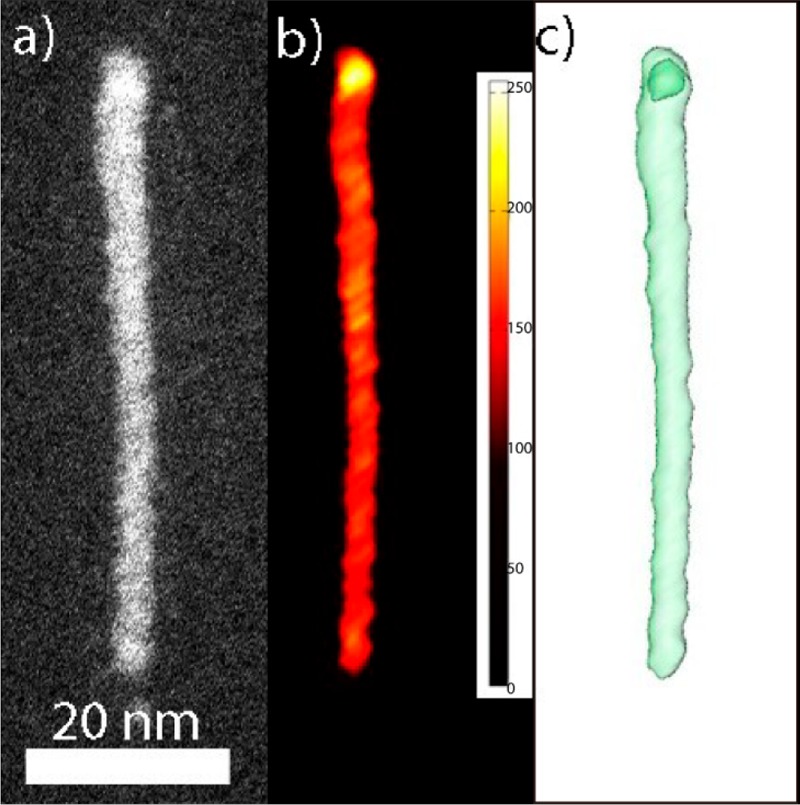
Electron tomography reconstruction of a single CIS/ZnS HNCs. The core appears brighter in the 2D image (a) and the orthoslice (b). (c) A threshold is used to distinguish the core from the shell in 3D. Additional measurements are provided in the Supporting Information (Figures S21–S24).
Growth Mechanism of Colloidal CIS/ZnS Dot Core/Rod Shell HNCs
Seeded growth protocols have been widely used to synthesize HNCs with a diversity of compositions, morphologies, and heteroarchitectures (e.g., CdSe/CdS dot core/rod shell nanorods and tetrapods, and CdSe/CdS quasi-spherical concentric core/shell quantum dots).10,11,36−39 This synthesis technique is based on coinjecting one of the shell precursors (typically the chalcogen precursor) and preformed NC cores into a hot solution containing adjuvant coordinating ligands and the second shell precursor (typically the metal precursor).10 The preformed NCs act as seeds for heterogeneous nucleation and heteroepitaxial growth, thereby suppressing homogeneous nucleation and directing the shape evolution of the product heteronanocrystal.10 The outcome of the reaction is dictated by the shape and crystal structure of the seed NCs and the growth conditions.10 For example, under conditions that favor anisotropic growth (viz., high monomer concentrations, suitable surfactants and sufficiently high temperatures),10 coinjection of wurtzite CdSe seed NCs and S-precursors into a hot solution of Cd-precursors leads to CdSe/CdS dot core/rod shell heteronanorods,10,37,38 while zinc-blende CdSe seed NCs yield CdSe/CdS dot core/multipod shell heterotetrapods.10,37 The different morphologies of the resulting HNCs can be understood by considering the faceting of the seeds.10,37,38 Zinc-blende CdSe NCs expose four equivalent (111) facets, leading to the anisotropic growth of four equivalent wurtzite CdS rods (i.e., tetrapods). In contrast, wurtzite CdSe NCs contain two (002) polar facets, which have a higher free-energy than the other facets, thereby showing lower activation energies for heterogeneous nucleation and faster growth rates.10,37,38 Consequently, the heteroepitaxial growth rates along the c-axis direction are much faster than in the other directions, leading to CdSe/CdS dot-in-rod core/shell heteronanorods. Interestingly, the growth on the Se-terminated polar facet is faster than on the Cd-terminated polar facet, resulting in core/shell heteronanorods in which the seed NC ends up closer to one side of the rod, rather than in its center.10,37,38 This implies that the NC growth proceeds by addition of monomer units (e.g., [CdS]) to the facets, since the layer-by-layer adsorption of atomic species would periodically reverse the termination of the polar facets, leading to identical growth rates in both directions of the nanorod axis.10 This inference is further supported by recent work on wurtzite CdSe/CdS core/shell HNCs (both quasi-spherical39 and bullet-shaped40), which suggests that heteroepitaxial shells grow by incorporation of monomer units, even when the SILAR strategy is employed.40
The formation of wurtzite CIS/ZnS dot core/rod shell heteronanorods by seeded injection can thus be understood by considering that the CIS NCs used as seeds in our work have the wurtzite structure (Figure 1, Figure 4a, Figure S16). The information obtained from high resolution TEM measurements can be used to model the shape of the CIS seed NCs (Supporting Information, Figure S25), showing that the top and bottom facets of the hexagonal bifrustrum shaped seed NCs correspond to the (00–2) and (002) polar facets of the wurtzite structure, which have different chemical compositions and polarities (Cu+/In3+ terminated and S2– terminated, respectively). As discussed above, heterogeneous nucleation and heteroepitaxial growth on the polar facets is favored, especially considering the small lattice mismatch (2.8%) between the {002} planes of wurtzite CIS and wurtzite ZnS, thereby leading to heteroepitaxial growth primarily in the c-direction (Figure 6). Under the optimized conditions used to produce the CIS/ZnS heteronanorods displayed in Figures 3–5 above, the ZnS growth rate in the polar direction is very fast (∼21 nm/min, Figure S10), being comparable to that observed for CdS on wurtzite CdSe seed NCs (∼19 nm/min).36 Remarkably, in contrast to the case of CdSe/CdS dot core/rod shell heteronanorods, we observe that fast heteroepitaxial growth of ZnS occurs primarily on one of the polar facets of the wurtzite CIS seed NCs, even in misformed nanorods (Figures 4 and 5, Supporting Information Figures S19–S24), since growth on both polar facets would result in the cores being far from both nanorod tips, albeit off-centered in case of different growth rates. We propose that this preferred facet is the anion terminated polar facet, which is unpassivated under the conditions prevalent in our experiments, since the ligands present in the reaction medium, viz., OA and HDA, are both hard Lewis bases and therefore cannot bind to sulfide anions (which are also hard Lewis bases). This facet is thus fully accessible for deposition of [ZnS] monomer units. The lack of ligands on the S-terminated polar facet of hexagonal bifrustum shaped wurtzite NCs has also been demonstrated for ZnS NCs obtained by Cu+ for Zn2+ CE in template Cu2–xS NCs, which were observed to adsorb at a toluene-air interface primarily through the anion-terminated (002) facet.41 The fact that the ZnS nanorod growth remains axially asymmetric implies that it proceeds by addition of [ZnS] monomer units to the anion terminated (002) facet, so that its polarity is preserved throughout the growth, consistent with the growth mechanism recently proposed for other heteronanocrystals and discussed above.10,39,40 Heteroepitaxial growth on the other facets, including the cation terminated (002) facet, is prevented by the presence of ligands (OA and/or HDA). The dramatic impact of ligand passivation on the accessibility of surface sites, and consequently on the heteroepitaxial growth rates, is demonstrated by a control experiment in which all the reaction conditions were kept unchanged, and the seed NCs were replaced by wurtzite CuInS2 quantum dots with the same size and shape of the previously used seeds, but capped with 1-dodecanethiol (DDT). Remarkably, the product NCs obtained from this reaction were still nearly spherical and had roughly the same size of the seed NCs (Supporting Information, Figure S26), showing that DDT prevented fast ZnS heteroepitaxial growth on all facets. This is most likely due to the strong surface passivation of CuInS2 by DDT, which can bind to both Cu(I) and In(III) sites and also form the sulfur terminated polar facet.42,43 The blue-shift observed in the optical spectra of the product NCs can be attributed to slow etching and alloying reactions following slow ZnS overgrowth. These competing processes will be discussed in more detail below.
Figure 6.

Schematic illustration of the mechanism proposed for the multistep seeded growth protocol used in this work to synthesize colloidal CIS/ZnS dot core/rod shell HNCs.
As discussed above, controlled anisotropic heteroepitaxial ZnS growth on wurtzite CIS seed NCs requires a very specific set of conditions. The different products obtained from the three different seeded injection methods (II, III, and IV in Figure 2 above) clearly demonstrate that the outcome of the seeded growth reaction is determined by a delicate balance between the [ZnS] monomer formation from the precursors and a number of competing processes (etching, alloying, cation exchange), that either consume the precursors prior to their conversion to monomers or directly interfere with the ZnS heteroepitaxial overgrowth on the CIS seed NCs by making the surface too dynamic or inaccessible. This is in agreement with a recent study in which the intricate interplay between these competing processes and their impact on the outcome of ZnS shelling reactions was investigated in detail using chalcopyrite CIS NCs in a seeded growth approach without delayed injection (equivalent to injection protocol II in Figure 2 above).44 This study showed that under most conditions etching, selective cation extraction, and alloying prevailed, and that ZnS heteroepitaxial shell overgrowth is only dominant if reactive S- and Zn-precursors and high reaction temperatures (210 °C) are used.44 The prevalence of etching and selective cation extraction is also evident in injection protocols II and III in the present work, since the product HNCs are characterized by large spectral blue-shifts and high concentration of cation vacancies (see Figure S2 and discussion above). To examine potential etching effects, the CIS seed NCs were exposed to an ODE solution containing different ligands (OA, HDA, Zn(oleate)2, and S/ODE) at 210 °C for 10 min (Supporting Information, Figure S27). We observe that HDA and OA induce ripening, while S/ODE and Zn(oleate)2 lead to etching of CIS NCs. The etching induced by S/ODE can be ascribed to side-reactions with the Sulfur precursor, since elemental sulfur is known to react with ODE/alkylamines forming a number of reactive S-species (e.g., H2S),25,45 which have been shown to extract In3+ (and to a lesser extent also Cu+) from CIS NCs.25 The etching induced by Zn(oleate)2 can be attributed to a reaction through which Zn2+ in solution and In3+/Cu+ on the surface of CIS NCs exchange coordinating molecules, resulting in In/Cu oleate complex and S/S2– in solution.46 Cation exchange is also likely in Protocol III, since the CIS NC seeds are directly injected in a hot solution containing only Zn-precursors.31 The injection of S/ODE in the hot solution containing Zn(oleate)2 and HDA prior to the injection of the CIS seed NCs is thus beneficial for a number of reasons. First, it minimizes etching, cation exchange and alloying processes because the sulfur species formed upon reaction of elemental S with HDA will quickly react with Zn(oleate)2 (activated by HDA)47 to form [ZnS] monomers. Consequently, the injected CIS seed NCs will be exposed to lower concentrations of both active S-species and Zn-precursors. Second, the concentration of [ZnS] monomers will be higher, thereby promoting fast anisotropic growth. The precursor to monomer conversion has been shown to be the rate-limiting step in the formation of a variety of binary metal chalcogenide NCs (viz., CdX, PbX, Cu2S, X= S, Se),10,47−51 and therefore it is advantageous in the present case to allow the [ZnS] monomer concentration to build up prior to injection of the CIS seeds. This explains why a delay of 20 s between the injections of S/ODE and CIS seed NCs was found necessary to improve the size and shape control over the product CIS/ZnS heteronanorods. Longer delays are less effective because too high monomer concentrations will favor homogeneous nucleation of ZnS NCs.
Nevertheless, the delayed injection of the CIS seed NCs was not sufficient to completely suppress etching, since size reduction of the CIS cores from 5.5 to ∼4 nm was still observed (Figure 5, Figure S21–S24). It is probable that most of the etching occurs at early stages of the reaction, before the onset of heteroepitaxial ZnS overgrowth. The thin (∼1 monolayer) ZnS shell that overcoats all other facets is likely formed also at the onset of the ZnS heteroepitaxial growth, preventing further etching of the CIS cores. However, ZnS overgrowth on these facets does not proceed beyond the early stages, possibly because the fast growing S-terminated (002) polar facet outcompetes them for the limited [ZnS] monomer supply and because access to these facets is hindered by a dense layer of ligands (both OA and HDA) strongly bound to the Zn-rich surface. We note that it is unlikely that CE is a relevant competing process, since the Cu/In ratio of the CIS seed NCs remains essentially unchanged in the product CIS/ZnS heteronanorods (Table S2). However, slow ZnS diffusion into the CIS cores, causing spectral blue-shifts, seems to occur throughout the reaction, continuing even after the ZnS heteroepitaxial growth has stopped, particularly at high reaction temperatures and Zn/S ratios. This process can be minimized by using short reaction times (10 min) and the optimal reaction temperature of 210 °C. Longer reaction times are also undesirable because they favor internal ripening of the nanorods, which become shorter and thicker (length decreases from ∼100 to ∼70 nm, diameter increases from ∼4 to 4.2 nm, see Supporting Information, Figure S10). Internal ripening of nanorods is a commonly observed free-energy minimization process, which sets in when the monomer concentrations in solution are no longer able to sustain anisotropic growth.10,52 Under these conditions mass transport from high-energy to low-energy facets becomes favorable.
It is interesting to note that, even under the optimized conditions described above, the fraction of CIS/ZnS core/shell heteronanorods displaying deviations from the ideal linear nanorod shape is significant (Figures 3 and 4, and Supporting Information Figures S17, S19–S24). These deviations from linearity can be ascribed to stacking faults in the nanorods, which arise when zinc blende layers form during the growth of the predominantly wurtzite lattice. This type of planar defect is very common in nanorods and nanowires of II–VI and III–V semiconductors and result from the very small activation energy required to interconvert wurtzite and zinc blende, and from the fact that the (001) facets of wurtzite are indistinguishable from the (111) facets of zinc blende.53−57 Therefore, small fluctuations in the local growth conditions (e.g., thermal fluctuations, differences in concentration gradients, transient proximity with other growing NCs, etc.) can lead to switching from wurtzite to zincblende (and vice versa), resulting in phase alternations and stacking faults.53−57 Future work will thus be directed toward further optimization of the growth conditions, in order to minimize local fluctuations in the growth conditions.
Conclusions
To summarize, we developed a multistep seeded growth protocol that yields CIS/ZnS dot core/rod shell HNCs with photoluminescence in the NIR. The method is based on the synthesis of wurtzite CIS NCs by topotactic partial Cu+ for In3+ CE in template Cu2–xS NCs, and their subsequent use as seed NCs. The CIS seed NCs are injected in a hot solution of zinc oleate and hexadecylamine in octadecene, 20 s after the injection of sulfur in octadecene. The delayed injection of the seed NCs is crucial to allow the concentration of [ZnS] monomers to build up, thereby maximizing the anisotropic heteroepitaxial growth rates while minimizing the rates of competing processes (etching, cation exchange, alloying). This results in fast (∼21 nm/min) heteroepitaxial growth of wurtzite ZnS primarily on the Sulfur-terminated polar facet of the CuInS2 seed NCs, the other facets being overcoated only by a thin (∼1 monolayer) shell. The asymmetric axial growth of the nanorod proceeds by addition of [ZnS] monomer units, so that the polarity of the terminal (002) facet is preserved throughout the growth. The colloidal wurtzite CIS/ZnS dot core/rod shell HNCs obtained in our work have a large molar extinction coefficient (1.4 × 106 M–1 cm–1 at 3.54 eV) and show PL in the NIR (∼800 nm) with PLQYs of ∼20%. The synthesis approach presented here can be upscaled by a factor 10. Our findings provide new insights on the growth mechanism of anisotropic CIS/ZnS heteronanocrystals, which open up new possibilities in designing multifunctional Cu-chalcogenide based colloidal heteronanocrystals.
Experimental Section
Materials
Copper(I) acetate (CuOAc, 97%), indium nitrate hydrate (In(NO3)3·H2O, 99.99%), 1-dodecanethiol (DDT, 98%), trioctylphosphine oxide (TOPO, 99%), 1-octadecene (ODE, 90%), trioctylphosphine (TOP, 90%), zinc acetate (Zn(OAc)2, 99.99%), oleic acid (OA, 90%), hexadecylamine (HDA, 90%), oleylamine (OLAM, 70%), trioctylamine (TOA, 98%), sulfur (S, 99.998%), nitric acid (HNO3, 69.5%), anhydrous toluene, methanol and butanol were purchased from Sigma-Aldrich. Lumogen red 305 (Article No.: 94720) was from Kremer Pigmente GmbH & Co. KG. TOPO, ODE, OA, HDA, OLAM and TOA were degassed at 120 °C for overnight prior to synthesis. Other reagents were used as received. The chemicals were weighted and handled inside a glovebox.
Synthesis of Cu2–xS NCs
Colloidal high-chalcocite Cu2–xS NCs were synthesized following a previously reported method.14 CuOAc (0.253 g, 2 mmol), 3.667 g of TOPO (3.667g, 9.3 mmol), and 20 mL ODE were degassed at 100 °C for 1 h. Subsequently, the reaction flask was purged by N2 and the temperature was set to 210 °C with heating speed ∼20 °C/min. At 160 °C, 5 mL of DDT were swiftly injected into the flask. A gradual change in solution color indicated nucleation and growth of Cu2–xS NCs. These NCs were allowed to grow at 210 °C for 40 min, and quenched by naturally cooling down to room temperature. The crude products (∼30 mL) were mixed with isometric butanol and methanol, followed by centrifugation at 5000 rpm for 15 min. This washing step was repeated twice to remove residual precursors. After that, the purified Cu2–xS pellet was dispersed into 10 mL of anhydrous toluene.
Synthesis of CIS NCs
Wurtzite CIS NCs were obtained from template Cu2–xS NCs by partial Cu+ for In3+ CE following previously reported procedures.14 10 mL of previously purified Cu2–xS NCs were diluted by adding 70 mL of toluene. The In-precursor solution was prepared by dissolving 0.640 g In(NO3)3·H2O (2 mmol) in a mixture of 40 mL methanol and 990 μL TOP (∼2 mmol), which was subsequently added to the diluted Cu2–xS NCs solution and vigorously stirred at room temperature (∼20 °C) for 3 days. The product CIS NCs were purified by using aforementioned washing procedure. Finally, the product CIS NCs were redispersed into degassed ODE (20 mL) and stored in a glovebox for further use. The concentration of CIS NCs (5.68 × 10–5 M) was determined by ICP-OES measurements (see Supporting Method 1 for details).
Preparation of S and Zn Stock Solution
0.5 M S stock solution was prepared by dissolving sulfur flakes (0.321 g, 10 mmol) in ODE (20 mL) at 180 °C for 2 min under N2 protection. 0.25 mmol/g Zn stock solution was obtained by heating the mixture of Zn(OAc)2 (2.19 g, 10 mmol), OA (6.21 g, 22 mmol) and ODE (33 g) in a three-neck flask at 140 °C for 1 h followed by degassing at 100 °C for another 1 h to remove generated acetic acid and water.
Synthesis of CIS/ZnS HNCs
In a typical optimized synthesis, a mixture of Zn stock solution (0.8 g), HDA (690 μL, 2 mmol) and ODE (3.39 mL) was heated to 210 °C under N2. When the temperature stabilized at 210 °C, 100 μL of S stock solution were swiftly injected. After 20 s, 100 μL of a solution of CIS NCs in ODE (5.68 × 10–6 mmol) were swiftly injected into the hot reaction mixture under vigorous stirring. The reaction was allowed to proceed at 210 °C for 10 min, followed by naturally cooling down to room temperature. The crude reaction mixture was purified by using the same washing procedure described above. Finally, the washed CIS/ZnS HNCs were dispersed into 4 mL of toluene, and stored in a glovebox.
Scale-up Synthesis of CIS/ZnS HNCs
The seeded injection approach can be scaled up ten times by increasing the amount of each precursor while keeping all the ratios fixed. Briefly, Zn(oleate)2 (8 g), HDA (6.9 mL, 20 mmol) and ODE (33.9 mL) were loaded into a 100 mL three-neck flask, and then heated to 210 °C in a glovebox. At 210 °C, 1 mL of S stock solution was rapidly injected. After 20 s, 1 mL of a solution of CIS NCs in ODE (5.68 × 10–5 mmol) was swiftly injected into the hot reaction mixture under vigorous stirring. The reaction was allowed to proceed at 210 °C for 10 min, and then quenched by injecting 10 mL of butanol. The crude reaction mixture was purified by using the same washing procedure described above. Finally, the washed CIS/ZnS HNCs were dispersed into 10 mL of toluene, and stored in a glovebox.
Optical Spectroscopy
Samples for optical measurements were prepared by dispersing them into 3 mL anhydrous toluene in sealed quartz cuvettes under N2 protection. Absorption spectra were recorded on a PerkinElmer Lambda 950 UV–vis–NIR spectrometer. PL spectra were measured by an Edinburgh Instruments FLS920 spectrofluorometer equipped with a 450 W Xe lamp as excitation source and double grating monochromators for both the excitation and the emission. As the emission of samples (∼800 nm) stands at the edges of UV–vis and NIR detector, the full PL spectra were obtained by separately using a Hamamatsu R928 photomultiplier tube (250–800 nm) and a liquid N2 cooled Hamamatsu R5509–72 photomultiplier tube (750–1600 nm) as detectors. The spectra were corrected for the instrumental response. PL decay curves were acquired by time-correlated single-photon counting via time-to-amplitude conversion using a Hamamatsu photosensor module H7422 as a detector, while a pulsed diode laser (EPL-445 Edinburgh Instruments, 441.4 nm, 80.2 ps pulse width, 0.02–20 MHz repetition rate) as the excitation source.
Photoluminescence Quantum Yields (PLQYs)
The PLQYs were measured using Lumogen red 305 (PLQY = 95%) in anhydrous toluene as a standard. To avoid inner filter effects, the absorbances of QD and lumogen red 305 solutions at and above the excitation wavelength (442 nm) below 0.1 (see Supporting Method 2, and Figure S28 for details).
X-ray Diffraction (XRD)
XRD results were recorded on Bruker D2 Phaser, equipped with a Co Kα X-ray source (1.79026 Å). Samples were washed at least 3 times, dried under vacuum overnight, and uniformly dispersed on a silicon wafer prior the XRD measurements.
Transmission Electron Microscopy (TEM)
TEM images were acquired using a FEI Tecnai-12 microscope operating at 120 kV. Samples for TEM imaging were prepared by drop-casting a toluene solution of purified NCs onto a carbon-coated 200 mesh copper TEM grid.
High Angle Annular Dark Field Scanning Transmission Electron Microscopy (HAADF-STEM)
HAADF-STEM images were acquired with a Tecnai Osiris electron microscope operated at 200 kV and high resolution images were acquired on an aberration corrected “cubed” FEI Titan 60–300 electron microscope operated at 300 kV. The sample was drop-casted on an ultrathin grid to reduce the background signal from the carbon support and thereby improve the image quality.
Electron Tomography (ET)
Prior to the measurements, the sample was baked at 120 °C for several hours and an additional further plasma treatment of 2 × 10′′ was performed to reduce contamination. ET experiments were performed on a Tecnai G2 electron microscope operated at 200 kV. Series of 15 projection images were acquired with an angular range from −70° to +70° and a tilt increment of 10°.
Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES)
ICP-OES measurements were performed on a PerkinElmer Optima 8300 ICP-OES spectrometer equipped with high-performance Segmented-array Charge-coupled Device (SCD) detector. Samples were carefully dried under vacuum overnight and thoroughly dissolved in HNO3 (69.5%). The digested samples were further diluted 1000 times to reach <1 ppm range for the measurement. The relative standard deviation of Cu (at 327.393 nm), In (at 230.606 nm) and Zn (at 206.200 nm) is less than 1%.
Acknowledgments
Chenghui Xia acknowledges China Scholarship Council (CSC) for financial support (No. 201406330055). S.B and N.W. acknowledge funding from the European Research Council (Starting Grant No. COLOURATOMS 335078). C.d.M.D. acknowledge financial support from the division of Chemical Sciences (CW) of The Netherlands Organization for Scientific Research (NWO) under Grant Number ECHO.712.014.001. The authors thank Xiaobin Xie and Da Wang for some TEM measurements, Donglong Fu for XRD measurements, Christina H. M. van Oversteeg for ICP-OES measurements, and Chun-Che Lin for suggestions regarding the synthesis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b01412.
TEM images, absorption and PL spectra of CIS/ZnS HNCs via a seeded growth approach in different experimental conditions; Experimental band gap estimation of CIS NC seeds and CIS/ZnS HNCs; ICP results of CIS/ZnS HNCs; HRTEM images of CIS NC seeds; HAADF-STEM image of CIS/ZnS HNCs; Electron tomography of CIS/ZnS HNCs; High resolution HAADF-STEM images of CIS/ZnS HNCs; Crystal structure simulation of wurtzite CIS seed NCs; Determination of CIS NCs concentration; Measurement of the PL quantum yields (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Du J.; Du Z.; Hu J.-S.; Pan Z.; Shen Q.; Sun J.; Long D.; Dong H.; Sun L.; Zhong X.; Wan L.-J. J. Am. Chem. Soc. 2016, 138, 4201–4209. 10.1021/jacs.6b00615. [DOI] [PubMed] [Google Scholar]
- Pan Z.; Mora-Seró I.; Shen Q.; Zhang H.; Li Y.; Zhao K.; Wang J.; Zhong X.; Bisquert J. J. Am. Chem. Soc. 2014, 136, 9203–9210. 10.1021/ja504310w. [DOI] [PubMed] [Google Scholar]
- Meinardi F.; McDaniel H.; Carulli F.; Colombo A.; Velizhanin K. A.; Makarov N. S.; Simonutti R.; Klimov V. I.; Brovelli S. Nat. Nanotechnol. 2015, 10, 878–885. 10.1038/nnano.2015.178. [DOI] [PubMed] [Google Scholar]
- Knowles K. E.; Kilburn T. B.; Alzate D. G.; McDowall S.; Gamelin D. R. Chem. Commun. 2015, 51, 9129–9132. 10.1039/C5CC02007G. [DOI] [PubMed] [Google Scholar]
- Pons T.; Pic E.; Lequeux N.; Cassette E.; Bezdetnaya L.; Guillemin F.; Marchal F.; Dubertret B. ACS Nano 2010, 4, 2531–2538. 10.1021/nn901421v. [DOI] [PubMed] [Google Scholar]
- Deng D.; Chen Y.; Cao J.; Tian J.; Qian Z.; Achilefu S.; Gu Y. Chem. Mater. 2012, 24, 3029–3037. 10.1021/cm3015594. [DOI] [Google Scholar]
- Chuang P.-H.; Lin C. C.; Liu R.-S. ACS Appl. Mater. Interfaces 2014, 6, 15379–15387. 10.1021/am503889z. [DOI] [PubMed] [Google Scholar]
- Frohleiks J.; Wefers F.; Wepfer S.; Hong A. R.; Jang H. S.; Nannen E. Adv. Mater. Technol. 2017, 2, 1700154. 10.1002/admt.201700154. [DOI] [Google Scholar]
- van der Stam W.; Berends A. C.; de Mello Donega C. ChemPhysChem 2016, 17, 559–581. 10.1002/cphc.201500976. [DOI] [PubMed] [Google Scholar]
- Donega C. d. M. Chem. Soc. Rev. 2011, 40, 1512–1546. 10.1039/C0CS00055H. [DOI] [PubMed] [Google Scholar]
- Mishra N.; Orfield N. J.; Wang F.; Hu Z.; Krishnamurthy S.; Malko A. V.; Casson J. L.; Htoon H.; Sykora M.; Hollingsworth J. A. Nat. Commun. 2017, 8, 15083. 10.1038/ncomms15083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietryga J. M.; Park Y. S.; Lim J.; Fidler A. F.; Bae W. K.; Brovelli S.; Klimov V. I. Chem. Rev. 2016, 116, 10513–10622. 10.1021/acs.chemrev.6b00169. [DOI] [PubMed] [Google Scholar]
- Zang H.; Li H.; Makarov N. S.; Velizhanin K. A.; Wu K.; Park Y.-S.; Klimov V. I. Nano Lett. 2017, 17, 1787–1795. 10.1021/acs.nanolett.6b05118. [DOI] [PubMed] [Google Scholar]
- Xia C.; Meeldijk J. D.; Gerritsen H. C.; de Mello Donega C. Chem. Mater. 2017, 29, 4940–4951. 10.1021/acs.chemmater.7b01258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto M.; Inoue K.; Okano M.; Saruyama M.; Kim S.; So Y.-G.; Kimoto K.; Kanemitsu Y.; Teranishi T. Nanoscale 2016, 8, 9517–9520. 10.1039/C6NR01097K. [DOI] [PubMed] [Google Scholar]
- van der Stam W.; Bladt E.; Rabouw F. T.; Bals S.; de Mello Donega C. ACS Nano 2015, 9, 11430–11438. 10.1021/acsnano.5b05496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli A.; Meinardi F.; Pasini M.; Galeotti F.; Pinchetti V.; Lorenzon M.; Manna L.; Moreels I.; Giovanella U.; Brovelli S. Nano Lett. 2015, 15, 5455–5464. 10.1021/acs.nanolett.5b01849. [DOI] [PubMed] [Google Scholar]
- Ben-Shahar Y.; Banin U. Top. Curr. Chem. 2016, 374, 54. 10.1007/s41061-016-0052-0. [DOI] [PubMed] [Google Scholar]
- Kalisman P.; Nakibli Y.; Amirav L. Nano Lett. 2016, 16, 1776–1781. 10.1021/acs.nanolett.5b04813. [DOI] [PubMed] [Google Scholar]
- Lee S.; Baek S.; Park J. P.; Park J. H.; Hwang D. Y.; Kwak S. K.; Kim S.-W. Chem. Mater. 2016, 28, 3337–3344. 10.1021/acs.chemmater.6b00323. [DOI] [Google Scholar]
- Zhai Y.; Shim M. ChemPhysChem 2016, 17, 741–751. 10.1002/cphc.201500859. [DOI] [PubMed] [Google Scholar]
- Zhai Y.; Flanagan J. C.; Shim M. Chem. Mater. 2017, 29, 6161–6167. 10.1021/acs.chemmater.7b02392. [DOI] [Google Scholar]
- van der Stam W.; Berends A. C.; Rabouw F. T.; Willhammar T.; Ke X.; Meeldijk J. D.; Bals S.; de Mello Donega C. Chem. Mater. 2015, 27, 621–628. 10.1021/cm504340h. [DOI] [Google Scholar]
- Bryks W.; Lupi E.; Ngo C.; Tao A. R. J. Am. Chem. Soc. 2016, 138, 13717–13725. 10.1021/jacs.6b08264. [DOI] [PubMed] [Google Scholar]
- Berends A. C.; Meeldijk J. D.; van Huis M. A.; de Mello Donegá C. Chem. Mater. 2017, 29, 10551–10560. 10.1021/acs.chemmater.7b04925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach A. D. P.; Shen X.; Faust A.; Cleveland M. C.; La Croix A. D.; Banin U.; Pantelides S. T.; Macdonald J. E. J. Phys. Chem. C 2016, 120, 5207–5212. 10.1021/acs.jpcc.6b00156. [DOI] [Google Scholar]
- Berends A. C.; Rabouw F. T.; Spoor F. C. M.; Bladt E.; Grozema F. C.; Houtepen A. J.; Siebbeles L. D. A.; de Mello Donegá C. J. Phys. Chem. Lett. 2016, 7, 3503–3509. 10.1021/acs.jpclett.6b01668. [DOI] [PubMed] [Google Scholar]
- Li L.; Pandey A.; Werder D. J.; Khanal B. P.; Pietryga J. M.; Klimov V. I. J. Am. Chem. Soc. 2011, 133, 1176–1179. 10.1021/ja108261h. [DOI] [PubMed] [Google Scholar]
- Coughlan C.; Ibáñez M.; Dobrozhan O.; Singh A.; Cabot A.; Ryan K. M. Chem. Rev. 2017, 117, 5865–6109. 10.1021/acs.chemrev.6b00376. [DOI] [PubMed] [Google Scholar]
- Knowles K. E.; Hartstein K. H.; Kilburn T. B.; Marchioro A.; Nelson H. D.; Whitham P. J.; Gamelin D. R. Chem. Rev. 2016, 116, 10820–10851. 10.1021/acs.chemrev.6b00048. [DOI] [PubMed] [Google Scholar]
- De Trizio L.; Prato M.; Genovese A.; Casu A.; Povia M.; Simonutti R.; Alcocer M. J. P.; D’Andrea C.; Tassone F.; Manna L. Chem. Mater. 2012, 24, 2400–2406. 10.1021/cm301211e. [DOI] [Google Scholar]
- Zhong H.; Lo S. S.; Mirkovic T.; Li Y.; Ding Y.; Li Y.; Scholes G. D. ACS Nano 2010, 4, 5253–5262. 10.1021/nn1015538. [DOI] [PubMed] [Google Scholar]
- McDonald S. A.; Konstantatos G.; Zhang S.; Cyr P. W.; Klem E. J. D.; Levina L.; Sargent E. H. Nat. Mater. 2005, 4, 138–142. 10.1038/nmat1299. [DOI] [PubMed] [Google Scholar]
- Cowan A. J.; Durrant J. R. Chem. Soc. Rev. 2013, 42, 2281–2293. 10.1039/C2CS35305A. [DOI] [PubMed] [Google Scholar]
- Park J.; Kim S.-W. J. Mater. Chem. 2011, 21, 3745–3750. 10.1039/c0jm03194a. [DOI] [Google Scholar]
- Kim D.; Lee Y. K.; Lee D.; Kim W. D.; Bae W. K.; Lee D. C. ACS Nano 2017, 11, 12461–12472. 10.1021/acsnano.7b06542. [DOI] [PubMed] [Google Scholar]
- Talapin D. V.; Nelson J. H.; Shevchenko E. V.; Aloni S.; Sadtler B.; Alivisatos A. P. Nano Lett. 2007, 7, 2951–2959. 10.1021/nl072003g. [DOI] [PubMed] [Google Scholar]
- Carbone L.; Nobile C.; De Giorgi M.; Sala F. D.; Morello G.; Pompa P.; Hytch M.; Snoeck E.; Fiore A.; Franchini I. R. Nano Lett. 2007, 7, 2942–2950. 10.1021/nl0717661. [DOI] [PubMed] [Google Scholar]
- Nakonechnyi I.; Sluydts M.; Justo Y.; Jasieniak J.; Hens Z. Chem. Mater. 2017, 29, 4719–4727. 10.1021/acs.chemmater.7b00354. [DOI] [Google Scholar]
- Bladt E.; van Dijk- Moes R. J. A.; Peters J.; Montanarella F.; de Mello Donega C.; Vanmaekelbergh D.; Bals S. J. Am. Chem. Soc. 2016, 138, 14288–14293. 10.1021/jacs.6b06443. [DOI] [PubMed] [Google Scholar]
- van der Stam W.; Rabouw F. T.; Vonk S. J. W.; Geuchies J. J.; Ligthart H.; Petukhov A. V.; Donega C. d. M. Nano Lett. 2016, 16, 2608–2614. 10.1021/acs.nanolett.6b00221. [DOI] [PubMed] [Google Scholar]
- Gromova M.; Lefrancois A.; Vaure L.; Agnese F.; Aldakov D.; Maurice A.; Djurado D.; Lebrun C.; de Geyer A.; Schulli T. U.; et al. J. Am. Chem. Soc. 2017, 139, 15748–15759. 10.1021/jacs.7b07401. [DOI] [PubMed] [Google Scholar]
- Turo M. J.; Macdonald J. E. ACS Nano 2014, 8, 10205–10213. 10.1021/nn5032164. [DOI] [PubMed] [Google Scholar]
- Berends A. C.; van der Stam W.; Hofmann J. P.; Bladt E.; Meeldijk J. D.; Bals S.; de Mello Donega C. Chem. Mater. 2018, 10.1021/acs.chemmater.8b00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson J. W.; Nagashima K.; Macdonald P. M.; Ozin G. A. J. Am. Chem. Soc. 2011, 133, 5036–5041. 10.1021/ja1109997. [DOI] [PubMed] [Google Scholar]
- Oh N.; Shim M. J. Am. Chem. Soc. 2016, 138, 10444–10451. 10.1021/jacs.6b03834. [DOI] [PubMed] [Google Scholar]
- Li L. S.; Pradhan N.; Wang Y.; Peng X. Nano Lett. 2004, 4, 2261–2264. 10.1021/nl048650e. [DOI] [Google Scholar]
- Owen J. S.; Chan E. M.; Liu H.; Alivisatos A. P. J. Am. Chem. Soc. 2010, 132, 18206–18213. 10.1021/ja106777j. [DOI] [PubMed] [Google Scholar]
- Rempel J. Y.; Bawendi M. G.; Jensen K. F. J. Am. Chem. Soc. 2009, 131, 4479–4489. 10.1021/ja809156t. [DOI] [PubMed] [Google Scholar]
- Abe S.; Čapek R. K.; de Geyter B.; Hens Z. ACS Nano 2012, 6, 42–53. 10.1021/nn204008q. [DOI] [PubMed] [Google Scholar]
- Hendricks M. P.; Campos M. P.; Cleveland G. T.; Plante I. J.; Owen J. S. Science 2015, 348, 1226–1230. 10.1126/science.aaa2951. [DOI] [PubMed] [Google Scholar]
- Jia G.; Banin U. J. Am. Chem. Soc. 2014, 136, 11121–11127. 10.1021/ja505541q. [DOI] [PubMed] [Google Scholar]
- Hughes S. M.; Alivisatos A. P. Nano Lett. 2013, 13, 106–110. 10.1021/nl3036417. [DOI] [PubMed] [Google Scholar]
- Wang F.; Buhro W. E. ACS Nano 2017, 11, 12526–12535. 10.1021/acsnano.7b06639. [DOI] [PubMed] [Google Scholar]
- Lehmann S.; Wallentin J.; Jacobsson D.; Deppert K.; Dick K. A. Nano Lett. 2013, 13, 4099–4105. 10.1021/nl401554w. [DOI] [PubMed] [Google Scholar]
- Wallentin J.; Ek M.; Wallenberg L. R.; Samuelson L.; Deppert K.; Borgström M. T. Nano Lett. 2010, 10, 4807–4812. 10.1021/nl101747z. [DOI] [PubMed] [Google Scholar]
- Glas F.; Harmand J.-C.; Patriarche G. Phys. Rev. Lett. 2007, 99, 146101. 10.1103/PhysRevLett.99.146101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.