

Abstract
Cenp‐F is a multifaceted protein implicated in cancer and developmental pathologies. The Cenp‐F C‐terminal region contains overlapping binding sites for numerous proteins that contribute to its functions throughout the cell cycle. Here, we focus on the nuclear pore protein Nup133 that interacts with Cenp‐F both at nuclear pores in prophase and at kinetochores in mitosis, and on the kinase Bub1, known to contribute to Cenp‐F targeting to kinetochores. By combining in silico structural modeling and yeast two‐hybrid assays, we generate an interaction model between a conserved helix within the Nup133 β‐propeller and a short leucine zipper‐containing dimeric segment of Cenp‐F. We thereby create mutants affecting the Nup133/Cenp‐F interface and show that they prevent Cenp‐F localization to the nuclear envelope, but not to kinetochores. Conversely, a point mutation within an adjacent leucine zipper affecting the kinetochore targeting of Cenp‐F KT‐core domain impairs its interaction with Bub1, but not with Nup133, identifying Bub1 as the direct KT‐core binding partner of Cenp‐F. Finally, we show that Cenp‐E redundantly contributes together with Bub1 to the recruitment of Cenp‐F to kinetochores.
Keywords: Cenp‐F, in silico modeling, kinetochores, mitosin, nuclear pore
Subject Categories: Cell Cycle, Structural Biology
Introduction
Cenp‐F (centromere protein F, also known as mitosin in human and Lek1 in mouse) is a large protein that contributes to multiple cellular processes including cell division, gene expression, cell morphogenesis, vesicular transport, and ciliogenesis (reviewed in 1, 2, 3; see also 4, 5, 6). Overexpression of this multifaceted protein has been linked to various cancers, whereas its inactivation or mutations lead to diverse developmentally related disorders (reviewed in 1, 2; see also 5, 6, 7). Cenp‐F has a complex and dynamic expression and localization pattern 8, 9, 10: its expression peaks in G2 when it is mainly detected within the nuclear matrix, with a minor fraction detected at the centrosomes and mitochondria 11, 12. At the G2/M transition, Cenp‐F relocalizes to the nuclear envelope (NE) where it contributes via NudE/EL to dynein/dynactin recruitment to the NE. This facilitates the positioning of the centrosomes close to the NE in prophase cells 13 and the migration of the nucleus toward the centrosomes prior to mitotic entry in rat neural stem cells 14, 15. In prophase, Cenp‐F is recruited to the outer kinetochores (KT) where it persists until early anaphase when it localizes to the spindle midzone before being degraded 8, 9, 10. Cenp‐F depletion by RNA interference in human cell lines revealed its implication in kinetochore–microtubule attachment and its contribution to the sustained activation of the spindle assembly checkpoint 16, 17, 18, 19, 20, 21. Some of these phenotypes may reflect kinetochore‐dependent functions of Cenp‐F, involving interactions with microtubules or its other kinetochore partners such as NudE/EL or Cenp‐E 20, 22, 23, 24, 25. However, Cenp‐F implication in mitosis remains controversial as no major viability or proliferation defect was observed upon its knockout in mouse embryonic fibroblasts, or upon CRISPR/Cas9‐induced depletion in HeLa cells 7, 26.
The localization of Cenp‐F at kinetochores involves multiple domains including a C‐terminal KT‐core, an internal repeated sequence, and a C‐terminal CAAX farnesylation site 27, 28 (see Fig 1A). In addition, RNA interference experiments have shown that kinetochore localization of Cenp‐F is impaired upon depletion of various kinetochore proteins including the Bub1 kinase and the kinesin‐like motor protein, Cenp‐E (29, 30; reviewed in 1). However, its direct tether(s) at kinetochores is yet unknown. In contrast, Cenp‐F recruitment to the NE in late G2 cells was shown to rely on its direct interaction with a nuclear pore constituent, Nup133 13, but also implies its farnesylation 28 and its Cdk1‐dependent nuclear exit 15. Nup133 is a subunit of the Nup107‐160 complex (Y‐complex), a key structural element of the nuclear pore complex (NPC). In interphase, repeated copies of the Y‐complex assemble to form the cytoplasmic and nuclear rings of the NPCs 31, 32 although a fraction of this complex can also be found inside the nucleoplasm 33. In mitosis, a pool of the Y‐complex is recruited to the kinetochores where it is required for proper chromosome segregation 34, 35. Noteworthy, kinetochore localization of the Y‐complex relies on both Cenp‐F and the Ndc80 complex 34, showing a dual function of the Nup133/Cenp‐F interaction at both nuclear pores and kinetochores. Our previous studies revealed that the N‐terminal domain (NTD) of Nup133 interacts with a C‐terminal region of Cenp‐F (hCenp‐F; [aa 2,644–3,065]; Fig 1A) 13, 34, but how this interaction is regulated in time and space remains largely unknown. Moreover, this region of Cenp‐F encompasses one of its two microtubule binding domains 20 and binding sites to diverse partners including Rb 36, ATF4 37, and Miro 12, but also its KT‐core domain 27 (Fig 1A).
Figure 1. Minimal domains required for the Y2H interaction between Nup133 and Cenp‐F.
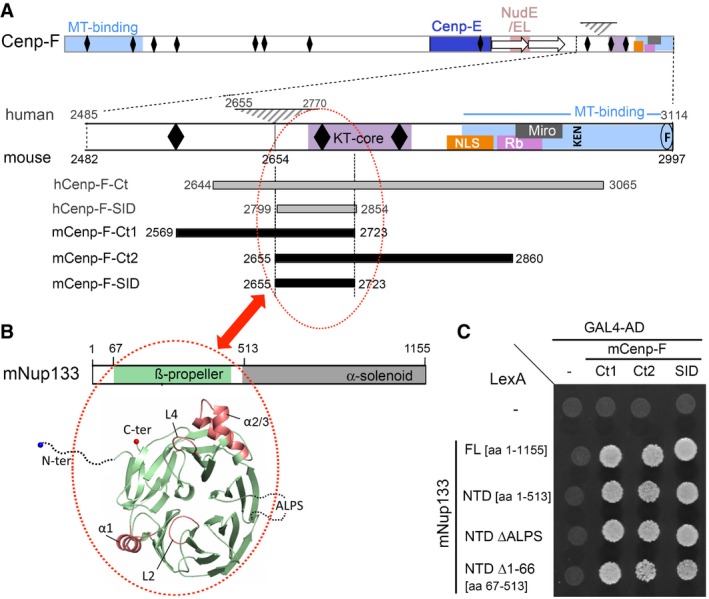
- Schematics of Cenp‐F and its C‐terminal domain based on data gathered for human and mouse Cenp‐F. The 115‐aa domain found in human but not mouse Cenp‐F is indicated by a gray triangle (see the corresponding sequence alignment in Fig EV1 for details). Predicted leucine zippers (black diamonds), internal repeated sequence (arrows), binding sites for microtubules (MT‐binding), Cenp‐E, NudE/EL, Rb and Miro, the core region required for kinetochore binding (KT‐core), nuclear localization signal (NLS), the conserved KEN7 degradation motif (KEN), and farnesylation site (F) are indicated (see text for references). Numbers refer to amino acid positions for human and mouse Cenp‐F. The position of hCenp‐F‐Ct, mCenp‐F‐Ct1, and mCenp‐F‐Ct2 and of human and mouse Cenp‐F‐SID segments is also shown.
- Scheme of mNup133 protein organization, and structure of its N‐terminal β‐propeller. The dashed lines correspond to the unstructured N‐terminal residues (aa 1–66) and the extremities of the ALPS loop (aa 251–268) not included in the model. The four helices or loop regions (α1, L2, α2/3, L4) protruding out of the modeled mNup133 β‐propeller structure are colored in red.
- Interactions between the indicated mNup133 fragments cloned into pB27‐LexA and mCenp‐F C‐terminal fragments cloned into pP6‐Gal4‐AD were assayed by Y2H based on growth on ‐LWH medium as described in Materials and Methods. Empty bait and prey vectors were used as negative controls (−). Western blots demonstrating the proper expression of the LexA and GAL4‐AD fusions are presented in Appendix Fig S2A(i) and B(i).
In view of the multiplicity of Cenp‐F partners within its C‐terminal region, and considering the interplay between Nup133 and Cenp‐F at nuclear pores and kinetochores, functional studies of Cenp‐F in mitosis critically require the generation of separation‐of‐function mutations that discriminate Cenp‐F interaction with Nup133 from its other targeting determinants, notably at kinetochores.
By combining in silico modeling and yeast two‐hybrid (Y2H) assays, we have now identified critical surfaces and specific amino acids required for Cenp‐F/Nup133 interaction. We could thus define mutations that differentially impact Cenp‐F localization at the NE in prophase and kinetochores in mitosis. Moreover, we show that Bub1 is the direct KT‐core binding partner of Cenp‐F that contributes, along with Cenp‐E, to the recruitment of full‐length Cenp‐F to kinetochores.
Results and Discussion
In silico modeling of Cenp‐F/Nup133 interaction
To specifically disrupt the Cenp‐F/Nup133 interaction without affecting the other functions of these proteins, we aimed at refining their assembly mode and identifying critical amino acids specifically required for their interaction.
We used ULTImate Y2H screens (Hybrigenics) to precisely delineate the Nup133 binding domain on both human and mouse Cenp‐F (see Materials and Methods). Pairwise protein sequence alignment revealed that the predicted minimal domain of Cenp‐F interacting with Nup133 (selected interacting domain, SID® as defined by Hybrigenics) independently identified in these two species corresponded to nearly the same conserved domain within the Cenp‐F C‐terminal region (Figs 1A and EV1). We validated this predicted binding domain by demonstrating that a construct encompassing aa 2,655–2,723 of mCenp‐F (subsequently named mCenp‐F‐SID) interacted with mNup133‐N‐terminal domain (NTD) in the Y2H assay as did the original fragments isolated in the screen (mCenp‐F‐Ct1 and mCenp‐F‐Ct2; Fig 1A and C).
Figure EV1. Alignment of human (h) and mouse (m) Cenp‐F C‐terminal domains.
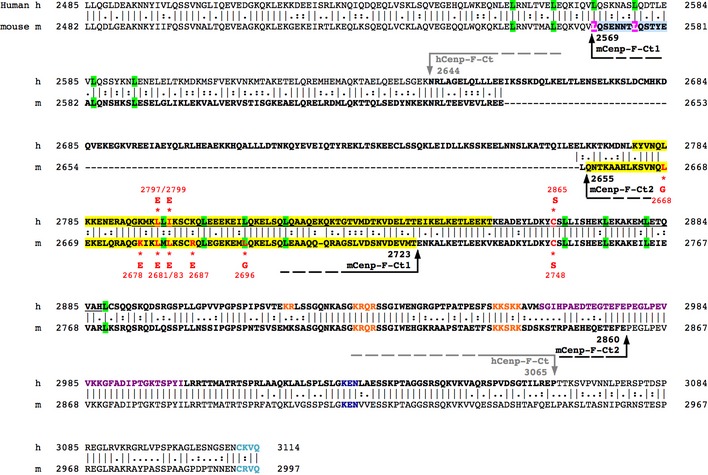
Alignments between human and mouse Cenp‐F (NCBI Reference Sequence: NP_057427.3 and NP_001074832.2, respectively) were performed based on EMBOSS Matcher. Arrows indicate the boundaries of hCenp‐F‐Ct, mCenp‐F‐Ct1, and mCenp‐F‐Ct2 fragments used in this study (bold font). Human and mouse Cenp‐F‐SID are in highlighted in yellow. Leucine residues involved in the leucine zippers 10 are highlighted in green; residues involved in the NLSs are in orange 10, 21, KEN7 protein degradation motif in dark blue 49, Rb‐binding domain 36 in purple, and C‐terminal CAAX farnesylation site 64 in cyan. The minimal kinetochore‐binding domain of human Cenp‐F is underlined in black 27. Residues mutated in this study are in red, and the amino acid they are replaced by is indicated below (for mCenp‐F) or above (for human Cenp‐F) the sequence, along with their position within the sequence.
We then sought to model the structure of the mCenp‐F‐SID segment. This region of mCenp‐F has not been crystallized so far, but the C‐terminal domain of Cenp‐F features potential leucine zippers and was reported to homodimerize in a Y2H assay 9, 10. Bioinformatics analyses indeed predicted that most of this mCenp‐F segment adopts coiled‐coil conformations (9; Fig EV2A). We first confirmed the dimerization of the mCenp‐F‐Ct2 domain by in vivo crosslinking of HeLa cells transfected with HA‐tagged‐mCenp‐F‐Ct2 (Fig EV2C). Because the mCenp‐F‐Ct2 segment encompasses two predicted leucine zippers, we next characterized the oligomeric status of the most conserved region in Cenp‐F‐SID domain (Cenp‐F‐miniSID, aa 2,663–2,706 in mCenp‐F; Fig EV2A and B) using SEC‐MALS (size‐exclusion chromatography–multi‐angle light scattering; Fig EV2D). This approach revealed the propensity of this short wild‐type (WT) peptide to dimerize, notably at higher salt concentration (Fig EV2D(ii)). This trend is consistent with the high isoelectric point of the studied peptide (pI = 9), whose numerous positively charged residues induce repulsive electrostatic forces counteracting the stability of the coiled‐coil.
Figure EV2. Coiled‐coil analysis and dimerization properties of mCenp‐F C‐terminal domain.
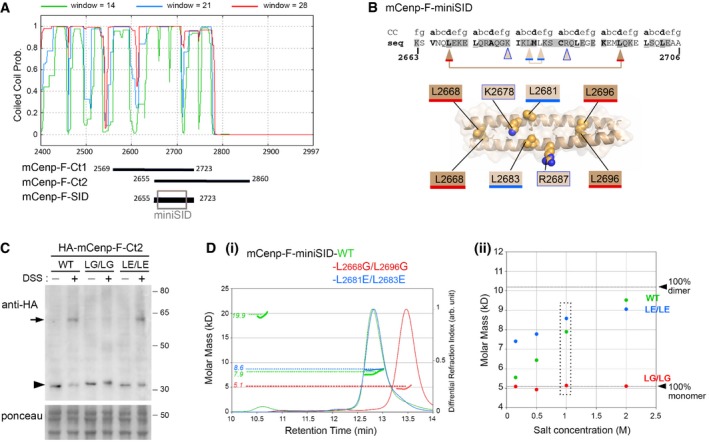
- COILS/PCOILS on Bioinformatics Toolkit 55 was launched for mCenp‐F [AA 2,401–2,997(end)]. Probabilities of being coiled‐coil region based on a window of 14, 21, and 28 residues are shown in green, blue, and red, respectively. The position of mCenp‐F‐Ct1, mCenp‐F‐Ct2, and mCenp‐F‐SID segments is represented. The gray box corresponds to the modeled peptide (mCenp‐F‐miniSID [aa 2,663–2,706]) presented in (B) and used in (D).
- Top: Heptad position information is shown above the amino acid sequence of mCenp‐F‐miniSID. Residues in buried positions a and d of the coiled‐coil heptad are highlighted in bold, and underlined when they correspond to leucine (L). Conserved residues (ConSurf server score greater than 6 65) are colored in gray. Arrowheads point to the residues mutated in this study using the same color code as in the model. Bottom: Model of the 3D‐structure of mCenp‐F [AA 2,663–2,706] coiled‐coil domain, with the a‐ and d‐positions of the coiled‐coil heptad highlighted as sticks. The side chains of all the residues mutated in the study are shown as spheres on the visible face of the coiled‐coil: (i) buried residues L2668 and L2696 which contact themselves in the symmetric molecule of the parallel coiled‐coil, (ii) residues L2681 and L2683 which are exposed and conserved hydrophobic residues, and (iii) residues K2678 and R2687 which form potentially stabilizing interactions in the interface of InterEvDock Model 3.
- In vivo crosslinking analyses. Whole‐cell extracts from HeLa cells transiently expressing HA‐tagged mCenp‐F‐Ct2‐WT, mCenp‐F‐Ct2‐LG/LG (L2668G/L2696G), or mCenp‐F‐Ct2‐LE/LE (L2681E/L2683E) and treated or not with DSS were analyzed by Western blot using anti‐HA antibodies. The Ponceau red staining is used as loading control. Molecular masses are indicated (kilodaltons). The arrowhead and arrow point to the position of the mCenp‐F‐Ct2 monomers and crosslinked dimers, respectively. Source data are available for this Western blot.
- (i) SEC‐MALS analysis at 20°C of mCenp‐F‐miniSID‐WT (in green), mCenp‐F‐miniSID‐LE/LE (in blue), or mCenp‐F‐miniSID‐LG/LG (in red) in 1 M NaCl‐containing buffer. Differential refractive index curves normalized between 0 and 1 (right y‐axis) are plotted for the three peptides as a function of the elution volume. The calculated molar masses (in kD, left y‐axis) are plotted as bold lines. For each elution peak, the dashed horizontal line indicates the mass of the molecular species measured (indicated in the corresponding color on the left y‐axis). Note that the calculated SEC‐MALS molar mass of the WT and LE/LE peptides is intermediate between the theoretical molar mass expected for a monomer (5.1 kD) and a dimer (10.2 kD), most likely reflecting a fast exchange between monomeric and dimeric forms. The WT peptide also exhibits the presence of a minor (˜5%) fraction of a tetrameric form. In contrast, the molar mass of the L2668G/L2696G mutant is consistent with a monomeric form only. (ii) Calculated molar masses in kD as a function of NaCl concentration (0.15, 0.5, 1, and 2 M), reported as colored circles for the three peptides with the same color code as in panel (i). The box indicates the points that were extracted from the 1 M NaCl experiment shown in panel (i).
Source data are available online for this figure.
We could therefore confidently model the Cenp‐F‐miniSID domain as a continuous parallel coiled‐coil (see Materials and Methods; Fig EV2B). Consistent with this model, mutation of two buried leucines (d‐position in the coiled‐coil heptad; Fig EV2B) to glycine (mCenp‐FL2668G/L2696G) disrupted the stability of the coiled‐coil as revealed both by in vivo crosslinking and by SEC‐MALS assays. In contrast, the L2681E/L2683E mutant targeting surface‐exposed residues did not affect the dimerization properties in vivo, and even stabilized the miniSID dimeric coiled‐coil, likely by counteracting the repulsive electrostatic forces (Fig EV2C and D).
With regard to mNup133, we modeled the structure of its NTD based on the experimental structure of the hNup133 N‐terminal β‐propeller (PDB: 1XKS; see Materials and Methods). Two regions were not resolved in this structure and could therefore not be modeled 38: (i) the first 66 residues predicted as disordered by MobiDB server 39, and (ii) a disordered loop (aa 244–266 of mNup133) encompassing an ALPS motif (Amphipathic Lipid Packing Sensors) 40 (Fig 1B). However, deletion of either of these domains preserved the interaction between mNup133‐NTD and mCenp‐F (Fig 1C and Appendix Fig S2).
Because individual reliable structural models could be generated for both the mNup133 β‐propeller and mCenp‐F C‐terminal dimeric coiled‐coil segment, we could then use protein–protein docking simulations to explore their possible binding modes. We therefore performed a free docking simulation based on the 3D structures modeled for both domains. We used the InterEvDock server 41, which is based on a pipeline that previously yielded the best predictions in the 6th community‐wide CAPRI (Critical Assessment of PRediction of Interactions) evaluation meeting 42, 43. By default, the 10 most probable binding modes are proposed by InterEvDock. For each of these 10 models, the position of the center of mass of the Cenp‐F dimer with respect to Nup133 β‐propeller is illustrated by a colored sphere in Fig 2A(i) and (ii).
Figure 2. Modeling of mNup133 recognition by the mCenp‐F and validation by Y2H of mutants designed to impair Cenp‐F interaction with Nup133.
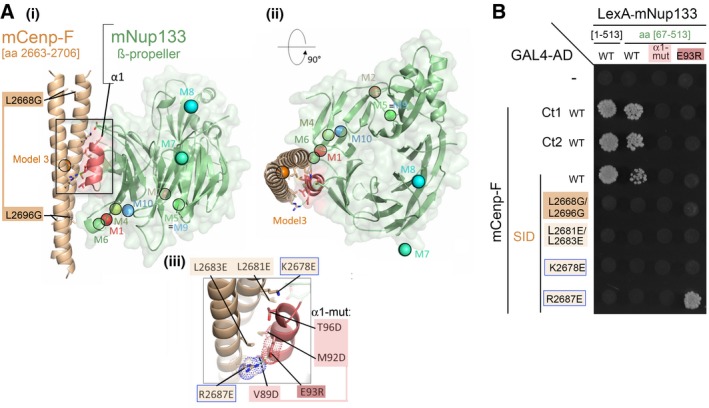
- Predicted Model 3, involving interaction of mCenp‐F‐SID (parallel coiled‐coil, brown) with helix α1 (red) of mNup133 β‐propeller (green). Two orthogonal views are presented in panels (i) and (ii). In (i), Cenp‐F mutated residues impairing dimerization L2669G/L2696G are highlighted. (iii) presents a detailed view of the interface, highlighting the interaction between mCenp‐FR2687 and mNup133E93. Mutations designed to test this model are indicated as sticks and labeled in panels (i) and (iii). The colored spheres in (i) and (ii) represent the center of mass of Cenp‐F dimeric segments for the 10 most likely binding modes of Cenp‐F to Nup133 as predicted by InterEvDock server (M1 to M10) 41.
- Y2H interactions between the indicated fragments of mNup133 and mCenp‐F (‐Ct1, ‐Ct2, or ‐SID), either WT or bearing the indicated mutations, were assayed based on growth on ‐LWH medium. The corresponding Western blots are presented in Appendix Fig S2A(ii) and B(i).
Challenging mNup133/mCenp‐F docking models using Y2H assay
To evaluate whether any of the 10 models represented a relevant binding mode, we first focused on the importance of α‐helices (α1 and α2/3; 38) and loops (L2 and L4) decorating the Nup133 β‐propeller (Figs 1B and EV3A). This choice was motivated by other examples of β‐propellers within the NPC that interact with their partners via loop regions between consecutive blades 44, 45, 46. We first individually replaced each of the four helices or loops within mNup133 β‐propeller with glycine–serine‐rich sequences (Appendix Table S1). Because replacement of helices α1 and α2/3 impaired the expression of the corresponding mNup133 mutant in yeast (Fig EV3B(ii)), we also generated mutants for superficial residues within these two loops. Replacing residues V89, M92, and T96, which form a conserved apolar patch within helix α1 of mNup133, by aspartic acids (α1‐mut: mNup133V89D/M92D/T96D; Fig 2A(iii)) abrogated interaction with mCenp‐F (Figs 2B and EV3B). In contrast, replacing loops L2 and L4 by glycine–serine‐rich sequences or mutating/deleting residues E317, D321, and Y330Δ of helix α2/3 (α2/3‐mut: mNup133E317R/D321R/Y330Δ) did not affect interaction with mCenp‐F (Fig EV3). This indicated that the Nup133 α1 helix is the major binding site for Cenp‐F.
Figure EV3. Model of mNup133 β‐propeller structure and contribution of various loops on Nup133 to its Y2H interaction with Cenp‐F.
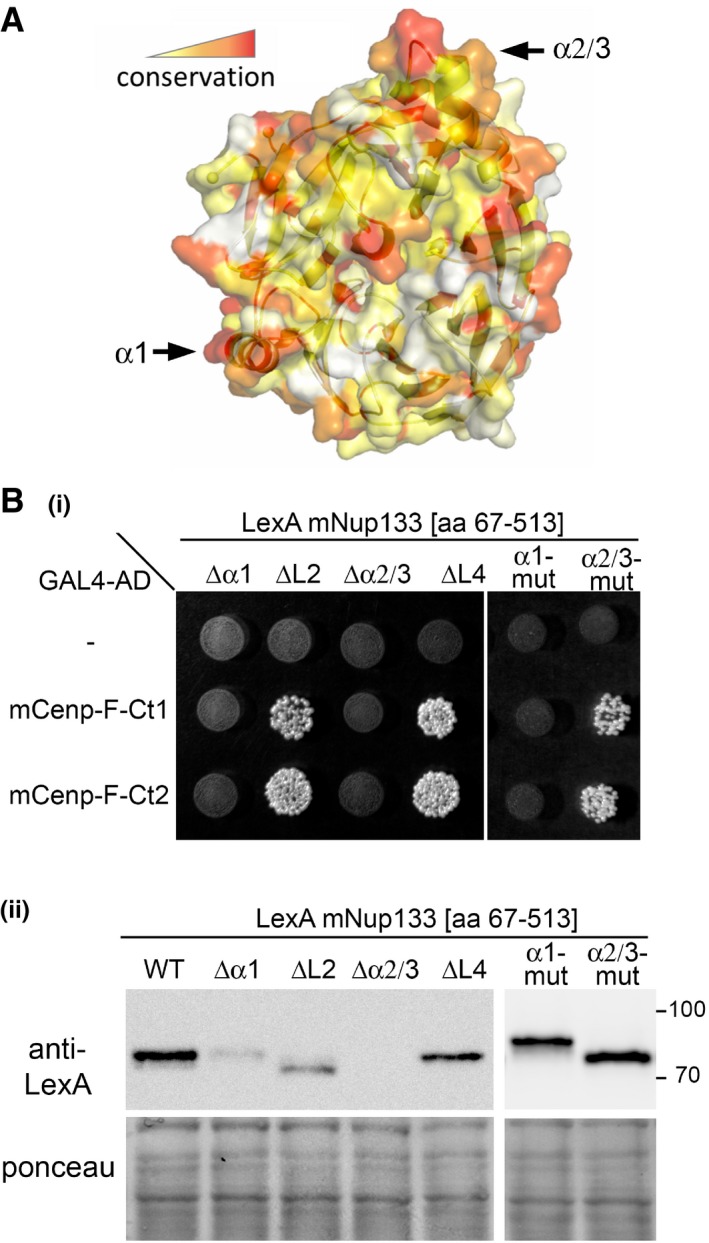
- The model was colored using ConSurf server 65. With default options, the server found 77 non‐redundant proteins homologous to mNup133 with a sequence identity > 35%. Residue conservation is gradient‐colored from white (variable) to red (conserved). The original alignment is provided as Appendix Fig S1.
- (i) Yeast co‐expressing the indicated preys (pP6‐GAL4‐AD vectors; empty or containing Cenp‐F[Ct1] or [Ct2]) and baits (pB27‐LexA vectors, empty (−) or containing various mutant forms of Nup133 β‐propeller [aa 67–513]) were grown on ‐LWH medium. Yeast growth on this medium indicates interaction. The following mutations were introduced within Nup133 [aa 67–513]: ∆α1: aa [87–99] are replaced by GlyGlySerGly; ∆L2: aa [162–171] are replaced by GlyGly; ∆α2/3: aa [313–335] are replaced by GlySerGlySerGly; ∆L4: aa [360–365] are replaced by GlySerGly; α1‐mut: mNup133V89D/M92D/T96D; α2/3‐mut: mNup133E317R/D321R/Y330Δ. (ii) The expression of the various LexA‐Nup133 [aa 67–514] mutants was assessed by Western blot using anti‐LexA antibody. Molecular masses are indicated (kilodaltons). Note that the replacement of helices α1 and α2/3 by short Gly‐Ser linkers (GGSG and GSGSG, respectively, leading to ∆α1 and ∆α2/3) likely impaired the folding of the mutant proteins, ultimately resulting in their instability explaining their very low expression levels in yeast. In contrast, the mutation of these helices (α1‐mut and ∆α2/3‐mut) does not alter the expression of the corresponding LexA‐Nup133 [aa 67–514] fusions. Source data are available for this Western blot.
Source data are available online for this figure.
Among the best 10 models proposed by the InterEvDock server, only Model 3 (evaluated as the second best model by the InterEvScore 47 and SOAP‐PP 48 scoring methods) was consistent with a binding mode involving the α1 helix on mNup133 as the binding site for mCenp‐F (Fig 2A). To further assess the relevance of this model, we next designed a set of mutants within either mCenp‐F‐SID or mNup133 β‐propeller and analyzed them by Y2H (Fig 2B and Appendix Fig S2). Consistent with our model that predicted mCenp‐F dimer formation to be the structural basis for interaction with mNup133, we observed a loss of mNup133 β‐propeller interaction with the mCenp‐F‐SID L2668G/L2696G mutant that disrupts coiled‐coil assembly (Figs 2B and EV2B–D). We then designed mutants targeting surface‐exposed residues of mCenp‐F and predicted to prevent interaction with Nup133 without impairing Cenp‐F dimerization. We generated a double‐point mutant (mCenp‐FL2681E/L2683E) affecting amino acids predicted to interact with Nup133 residues mNup133V89/M92/T96, as well as two single‐point mutants, mCenp‐FK2678E and mCenp‐FR2687E (Fig 2A). As predicted by the InterEvScore model, the three mutants impaired interaction with mNup133‐NTD [aa 1–513] and [aa 67–513] (Fig 2B). Note however that a likely non‐specific interaction was observed when the mCenp‐FR2687E mutant was assayed against full‐length mNup133 (Fig EV4). To further confirm that our selected InterEvScore model represents an accurate picture of mNup133/mCenp‐F interaction, we finally designed the mNup133E93R mutation that affects one of the most conserved residues in the mNup133 N‐terminal domain (Fig EV3A; Appendix Fig S1). Importantly, this mutation abolished the interaction with wild‐type mCenp‐F‐SID but specifically compensated the R2687E mutation on Cenp‐F‐SID (Fig 2B). This compensation points to a direct and fundamental contact between mCenp‐FR2687 and mNup133E93, fully supporting our predicted docking model (Fig 2A(iii) and B).
Figure EV4. The mC enp‐ FR2687E mutant interacts, likely in a non‐specific manner, with full‐length Nup133 but not with its C‐terminal domain and localizes in GLFG bodies.
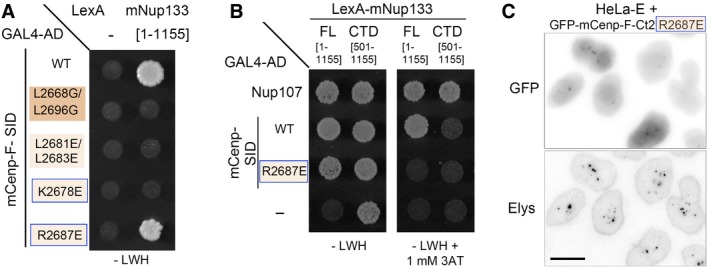
-
A, BY2H interactions between LexA alone (−) or fused to mNup133 full‐length (FL, [aa 1–1,155]) or C‐terminal domain (CTD, [aa 501–1,155]) and GAL4‐AD either alone (−) or fused to mCenp‐F‐SID (WT or bearing the indicated mutations) or Nup107 [aa 784–925] were assayed based on growth on ‐LWH medium supplemented, when indicated, by 1 mM 3‐Aminotriazole (3AT). Note in (A) that the mCenp‐FR2687E mutant, but none of the other mCenp‐F mutants assayed in Fig 2B, interacts with full‐length mNup133. However, unlike WT mCenp‐F‐SID, the interaction of the R2687E mutant with full‐length mNup133 is abrogated by the addition of 1 mM 3‐Aminotriazole, indicating that the mCenp‐FR2687E mutant may interact with the full‐length mNup133 in a rather weak manner. In (B), note that the lexA‐mNup133‐CTD construct is transactivating when used in ‐LWH medium. Under conditions required to prevent this transactivation (i.e., addition of 1 mM 3‐Aminotriazole), the interaction of Nup133‐Cterm with its established C‐terminal partner, Nup107, is preserved, while no interaction was detected between Nup133‐Cterm and mCenp‐F‐SID, either WT or R2687E. This observation is consistent with our previous studies using human Nup133 constructs that indicated that the C‐terminal domain of Nup133 does not interact with Cenp‐F 13.
-
CHeLa‐E cells transiently transfected with GFP‐mCenp‐F‐Ct2 R2687E were fixed 2 days after transfection and stained with an antibody directed against Elys that labels the NPCs and the GLFG bodies in interphase cells. The corresponding Western blot is presented in Appendix Fig S3. Scale bar, 10 μm. Note that consistent with the Y2H interaction observed with full‐length Nup133 [aa 1–1,155], GFP‐mCenp‐F‐Ct2 R2687E localizes in the GLFG bodies.
Together, these data validated our in silico structural model, in which the conserved α1 helix protruding out of the mNup133 β‐propeller (Fig EV3A) binds to a parallel coiled‐coil segment of mCenp‐F, centered around a set of conserved apolar residues exposed to the surface of both partners. This analysis further provided us with specific Cenp‐F C‐terminal mutants affecting its interaction with Nup133.
Recruitment of Cenp‐F C‐terminal fragments to intranuclear GLFG bodies underlines spatiotemporal regulations of Nup133/Cenp‐F interaction
To validate the impact of the characterized Cenp‐F mutants in a cellular context, we next transfected HeLa cells with GFP‐mCenp‐F‐Ct1 and GFP‐mCenp‐F‐Ct2 constructs. Both fusions lack the KEN box that leads to Cenp‐F degradation after anaphase onset (49; Fig 1A) and are thus stably expressed throughout the cell cycle. In interphase cells, both fusions localize in the nucleus (Figs 3A and EV5A). In mitotic cells, a faint albeit clear enrichment at kinetochores is observed in cells expressing the GFP‐mCenp‐F‐Ct2 fusion that encompasses the previously described KT‐core domain of Cenp‐F (Figs 3B and EV5B(ii); 27). In contrast, we never detected the GFP‐mCenp‐F‐Ct1 fusion at kinetochores, a result consistent with the fact that it lacks both the NLS and the second leucine zipper present in the KT‐core region (27; our unpublished data). Finally, unlike endogenous Cenp‐F, neither GFP‐mCenp‐F‐Ct1 nor GFP‐mCenp‐F‐Ct2 was detected at the NE in late G2/prophase cells, possibly because they lack the CAAX farnesylation site 28 and/or determinants required for their nuclear export at mitotic onset 15.
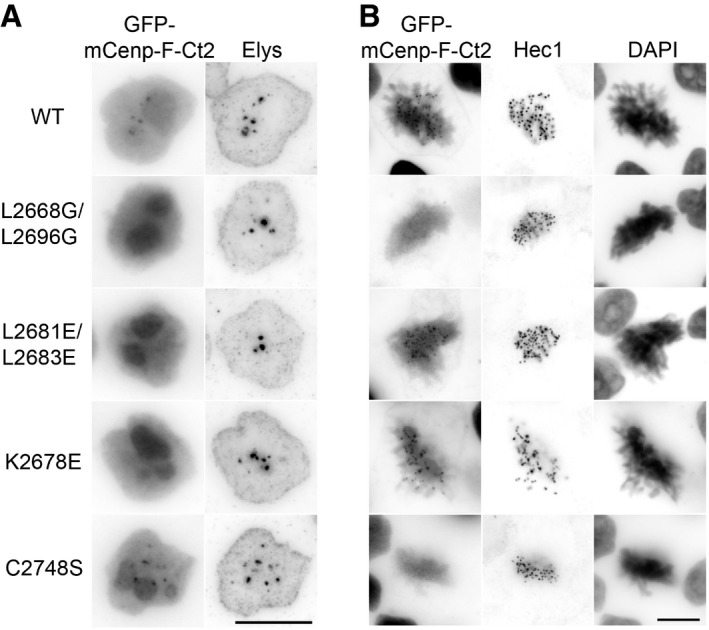
Figure 3. Mutations within GFP‐mCenp‐F‐Ct2 can differentially affect its targeting to intranuclear GLFG bodies and to kinetochores.
-
A, BHeLa‐E cells transiently transfected with GFP‐mCenp‐F‐Ct2, either WT or bearing the indicated mutations, were fixed 2 days after transfection and stained with (A) an antibody directed against Elys, a Y‐complex partner, that labels the NPCs and the GLFG bodies in interphase cells or (B) an anti‐Hec1 antibody that stains kinetochores in mitotic cells. Cells were also stained with DAPI. For each transfected construct, a representative interphase cell (3D reconstruction covering 1.6–2 μm, A) and a mitotic cell (single focal plane, B) are presented. The corresponding Western blots and larger fields of transfected cells are presented in Appendix Fig S3. Scale bars, 10 μm.
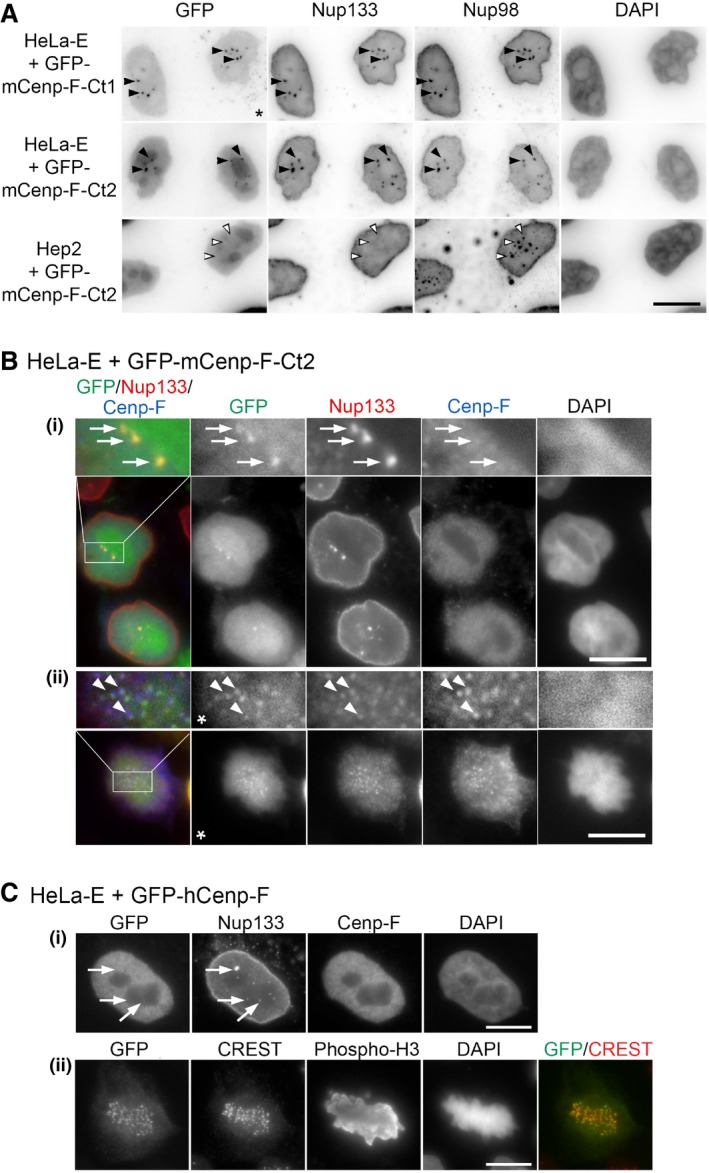
Figure EV5. Unlike GFP‐mCenp‐F‐Ct1 and GFP‐mCenp‐F‐Ct2, full‐length Cenp‐F is not targeted to Nup133‐labeled intranuclear GLFG bodies present in HeLa‐E cells.
-
AHeLa‐E or HEp‐2 cells transiently transfected with GFP‐mCenp‐F‐Ct1 or GFP‐mCenp‐F‐Ct2 were fixed 2 days after transfection and immunolabeled with rabbit anti‐Nup133 and anti‐Nup98 antibodies. DNA was stained with DAPI. Projections of three consecutive z‐sections covering 0.6 μm are shown. The star indicates that exposure time was increased in the GFP channel for cells transfected with GFP‐mCenp‐F‐Ct1 as compared to GFP‐mCenp‐F‐Ct2. Note that GFP‐mCenp‐F‐Ct1 may reach the nucleus by passive diffusion through the NPC due to its rather small size (47 kDa), whereas the GFP‐mCenp‐F‐Ct2 fusion that features a bipartite NLS 10, 21 (Fig 1A) further accumulates in the nucleolus. Black and white arrowheads indicate GLFG bodies containing or not Nup133. Scale bar, 10 μm.
-
B, CHeLa‐E cells transfected with GFP‐mCenp‐F‐Ct2 (B) or GFP‐hCenp‐F (C) were fixed 2 and 1 day after transfection, respectively, and immunolabeled with the indicated antibodies. A single plane is shown except for panels (C, ii) where a projection of 11 z‐sections is presented to better visualize the kinetochores (marked by the CREST serum). In (B), threefold magnifications of the marked areas are also presented. Arrows in the G2 cells shown in (B, (i)) and (C, (i)) (identified based on their strong Cenp‐F levels) point to GLFG bodies stained by anti‐Nup133 that contain GFP‐mCenp‐F‐Ct2 but neither endogenous Cenp‐F (the anti‐Cenp‐F antibody used here does not recognize the transfected GFP‐mCenp‐F‐Ct2 construct) nor GFP‐hCenp‐F. Arrowheads in the metaphase cell (B, ii) highlight co‐localization of GFP‐mCenp‐F‐Ct2, Nup133, and endogenous Cenp‐F at kinetochores. Note that a clear localization of the KT‐core domain of hCenp‐F was previously reported to require either the internal repeats or a longer C‐terminal extension including the C‐terminal CAAX farnesylation site that are absent from our construct 27, 28. This minor discrepancy likely reflects the improved detection due to our pre‐extraction conditions and the use of a GFP tag that does not rely on antibody detection. The star indicates that the gamma was altered on that image to improve the visualization of GFP‐mCenp‐F‐Ct2 kinetochore localization. Scale bars, 10 μm.
Noteworthy, a fraction of both GFP‐mCenp‐F‐Ct1 and GFP‐mCenp‐F‐Ct2 was detected along with Nup133 within discrete intranuclear structures, stained by the GLFG repeat‐containing Nup98 nucleoporin and thus corresponding to the GLFG bodies present in HeLa‐E cells 33, 50 (Figs 3A and EV5A). In contrast, GFP‐mCenp‐F‐Ct2 was not recruited to the Nup98‐positive but Nup133‐negative GLFG bodies present in HEp‐2 cells 51 (Fig EV5A). This indicates that the recruitment of GFP‐mCenp‐F‐Ct2 to the GLFG bodies present in HeLa‐E cells relies on the Y‐complex, via Nup133. The observation that these C‐terminal fragments of Cenp‐F can interact with Nup133 in GLFG bodies throughout the cell cycle demonstrates that cell‐cycle‐dependent posttranslational modifications are not strictly required for Nup133/Cenp‐F interaction. Conversely, unlike GFP‐mCenp‐F‐Ct1 and GFP‐mCenp‐F‐Ct2, full‐length Cenp‐F is not recruited to Nup133‐containing GLFG bodies (Fig EV5B and C) most likely because retention of full‐length Cenp‐F at other intranuclear sites may restrict its diffusion within the nucleoplasm (as also revealed by its exclusion from the nucleoli). These data, along with previous studies revealing the implication of Cdk1‐dependent nuclear exit of Cenp‐F for its targeting to the NE 15, 52, point to a crucial role of compartmentalization in the regulation of Cenp‐F/Nup133 interaction.
Mutations within Cenp‐F C‐terminal domain discriminate NPC/nuclear bodies from kinetochore targeting
We next used the dual localization of the GFP‐mCenp‐F‐Ct2 domain at both intranuclear bodies in interphase and kinetochores in mitosis as readout to analyze in human cells the effect of the mutations impairing mCenp‐F‐SID interaction with Nup133. Indeed, since these mutations were clustered around the previously described KT‐core domain of Cenp‐F 27, 37 (Figs 1A and EV1), it was critical to determine whether they impaired KT‐targeting of Cenp‐F. Upon transient transfection in HeLa‐E cells, all the GFP‐mCenp‐F‐Ct2‐derived mutants assayed were similarly expressed (Appendix Fig S3A) and accumulated in the nucleus and nucleolus as the wild‐type construct (Fig 3A). The GFP‐mCenp‐F‐Ct2 L2668G/L2696G fusion, bearing mutations predicted to prevent dimerization, localized neither to the GLFG bodies nor to kinetochores, pointing that in vivo the coiled‐coil folding and dimerization are prerequisite for the correct localization of this Cenp‐F domain at both locations (Fig 3A and B). In agreement with our Y2H data, the GFP‐mCenp‐F‐Ct2 L2681E/L2683E and K2678E mutants that displayed impaired interaction with Nup133 did not localize to the GLFG bodies in HeLa‐E cells (Fig 3A). Importantly, these mutants were targeted to kinetochores, indicating that they are properly folded and able to dimerize (Fig 3B).
Because the C‐terminal fragments of Cenp‐F are not targeted to the NE in prophase, we next introduced mutations impairing Nup133 interaction in the context of full‐length GFP‐hCenp‐F (L2797E/I2799E in human Cenp‐F, corresponding to L2681E/L2683E in mCenp‐F; Fig EV1). Consistent with a previous study 49, the WT form of GFP‐hCenp‐F mimicked the localization of endogenous Cenp‐F (Figs EV5C and 4A). In particular, a clear NE staining was observed in nearly all (n = 23 out of 27 cells) prophase cells in which GFP‐hCenp‐F is already localized at kinetochores (Fig 4A). In contrast, the L2797E/I2799E mutant form of GFP‐hCenp‐F, while properly targeted to kinetochores, was in most cases either not detectable or barely enriched at the NE in late G2/prophase cells (n = 18 and 12, respectively, out of 32 cells), even in cells displaying a clear kinetochore staining (Fig 4B and C). Together, these functional data thus validated in vivo the mutants generated based on our in silico modeling of Nup133/Cenp‐F interaction.
Figure 4. L2797E/I2799E mutations prevent GFP‐hCenp‐F targeting to the NE in late G2/prophase cells.
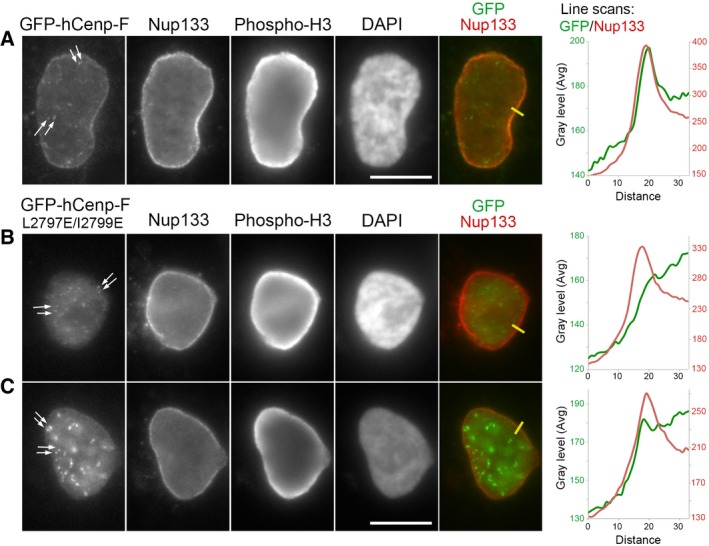
-
A–CHeLa‐E cells transiently transfected with GFP‐hCenp‐F‐WT (A) or GFP‐hCenp‐F‐L2797E/I2799E (B, C) were fixed 1 day after transfection and immunolabeled with the indicated antibodies. A single plane from widefield acquisitions of prophase cells (identified based on phospho‐histone H3 staining and persistence of a NE) is shown. Scale bars, 10 μm. Arrows point to kinetochores labeled by GFP‐hCenp‐F. Line scans (yellow lines on images, plotted from the cytoplasm toward the nucleoplasm, distances in pixels) measuring the intensity of GFP‐hCenp‐F (green lines) and Nup133 (red lines) signals in late G2/prophase cells reveal the peak of GFP‐hCenp‐F‐WT that co‐localizes with Nup133 at the NE (A). In contrast, either no enrichment (B) or a very minor accumulation (C) is detectable in GFP‐hCenp‐F‐L2797E/I2799E‐transfected cells.
Bub1 is the direct KT‐core binding site for Cenp‐F that contributes, along with Cenp‐E, to its recruitment to kinetochores
A previous study had identified a conserved cysteine essential for the kinetochore targeting function of Cenp‐F KT‐core region (C2865 in hCenp‐F, corresponding to C2748 in mCenp‐F; 27; Fig EV1). To evaluate a possible contribution of Nup133 (possibly via a distinct domain) to the kinetochore targeting of Cenp‐F, we mutated this cysteine in the mCenp‐F‐Ct2 segment and tested its interaction with Nup133. In the Y2H assay, the C2748S mutation did not prevent the interaction of mCenp‐F‐Ct2 with full‐length mNup133 (Fig 5A). Consistently, the GFP‐mCenp‐F‐Ct2 C2748S mutant still co‐localized with Nup133 within the GLFG bodies in HeLa‐E cells (Fig 3A), while as anticipated 27, it was not detectable at kinetochores (Fig 3B).
Figure 5. Cenp‐F direct interaction with Bub1 is inhibited by mutation of a conserved cysteine and is involved, together with Cenp‐E, in its recruitment to kinetochores.
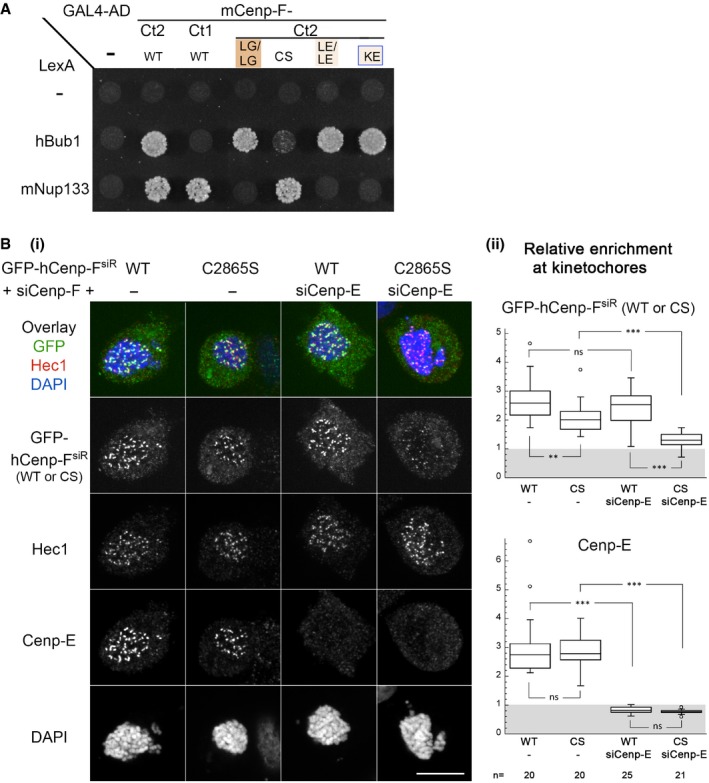
- Y2H interactions between full‐length mNup133 or hBub1 and mCenp‐F (‐Ct1 or ‐Ct2), either WT or bearing the indicated mutations, were assayed based on growth on ‐LWH medium supplemented with 5 mM 3AT. The corresponding Western blots are presented in Appendix Fig S2A(iii) and B(ii).
- (i) HeLa‐E cells were transfected with siRNA duplexes targeting Cenp‐F (−) or both Cenp‐F and Cenp‐E (+siCenp‐E) along with a plasmid directing the expression of a Cenp‐F‐siRNA‐resistant form of GFP‐hCenp‐F (GFP‐hCenp‐FsiR), either WT or bearing the C2865S mutation (CS). Two days after transfection, cells were treated with Nocodazole (20 μM for 4 h), fixed, and immunolabeled with the indicated antibodies and DAPI. A projection of 11 z confocal sections of the individual stainings and the overlay between the GFP signal and Hec1 staining (used to identify and quantify kinetochores) are presented. Scale bar, 10 μm. (ii) The relative enrichment of GFP‐Cenp‐F and Cenp‐E at kinetochores was quantified as described in Materials and Methods for n cells from two independent experiments. Box plots were generated using KaleidaGraph (Synergy Software): Each box encloses 50% of the normalized values obtained, centered on the median value. The bars extending from the top and bottom of each box mark the minimum and maximum values within the dataset falling within an acceptable range. Values falling outside of this range are displayed as an individual point. Statistical analyses were performed using Wilcoxon–Mann–Whitney rank‐sum test. Standard conventions for symbols indicating statistical significance are used: ns, not significant: P > 0.05; **P ≤ 0.01; ***P ≤ 0.001.
We thus aimed to determine which other kinetochore partner of Cenp‐F would rely on this conserved cysteine. Among the constituents whose depletion affects the targeting of Cenp‐F to kinetochores, Bub1 kinase was reported to interact with Cenp‐F in a Y2H assay (unpublished results from T.J. Yen cited in 22). This prompted us to assess whether Bub1 could specifically interact with the KT‐core fragment of Cenp‐F. Y2H analysis revealed that unlike Nup133, hBub1 interacts with mCenp‐F‐Ct2 but not with mCenp‐F‐Ct1 that lacks the second leucine zipper motif present in Cenp‐F KT‐core domain (Figs 1A and 5A). Moreover, the C2748S mutation strongly inhibits Cenp‐F interaction with Bub1 (Fig 5A). These data indicate that Bub1 represents the main KT‐core‐dependent tether for Cenp‐F to kinetochores. Note that a direct interaction between the Bub1 kinase domain and a dimeric coiled‐coil in Cenp‐F C‐terminal domain has been meanwhile demonstrated through biochemical reconstitution [preprint: 53]. Importantly, when introduced within mCenp‐F‐Ct2, none of the mutations impairing Cenp‐F association with Nup133 affected its Y2H interaction with Bub1 (Fig 5A). This demonstrates that the lack of interaction with Nup133 or Bub1 is not merely a consequence of global misfolding of the corresponding mutants and that Cenp‐F features two distinct binding sites for Nup133 and Bub1 located on two consecutive leucine zipper‐containing segments.
To evaluate the functional implication of the Bub1/Cenp‐F interaction, we next generated siRNA‐resistant forms of full‐length GFP‐hCenp‐F and cotransfected HeLa cells with the resulting GFP‐hCenp‐FsiR‐WT or ‐C2865S plasmids along with Cenp‐F siRNA duplexes. To increase the percentage of mitotic cells and prevent a potential contribution of the MT‐binding sites of Cenp‐F to its kinetochore localization, cells were treated with Nocodazole prior to immunolabeling. Both qualitative and quantitative fluorescence image analyses revealed that even in the absence of endogenous Cenp‐F (that may have contributed to the GFP‐transgene localization via heterodimerization), GFP‐hCenp‐FsiR‐C2865S was largely retained at kinetochores (Fig 5B). This differed from the behavior of the corresponding GFP‐mCenp‐F‐Ct2 C2748S mutant that is not detectable at kinetochores (Fig 3). This result was also unexpected since depletion of Bub1 or the lack of its C‐terminal tail was reported to cause an efficient mislocalization of Cenp‐F from kinetochores [30, 54, preprint: 53]. Together, these data pointed to the existence of redundant targeting determinants of Cenp‐F, involving Bub1‐mediated interactions outside of the Cenp‐F‐Ct2 domain. A potential candidate was the kinesin‐like motor protein, Cenp‐E. Indeed, Cenp‐E and Cenp‐F directly interact with each other 22 (see Fig 1A) and were reported to stabilize each other at kinetochores 30. Moreover, Cenp‐E targeting to kinetochores also indirectly relies on Bub1 30. We thus analyzed the localization of GFP‐hCenp‐FsiR‐WT or ‐C2865S in HeLa cells co‐depleted for endogenous Cenp‐F and Cenp‐E. Under our experimental conditions, the kinetochore targeting of GFP‐hCenp‐FsiR‐WT was not significantly affected by Cenp‐F/Cenp‐E co‐depletion. In contrast, this RNAi treatment nearly entirely abolished the kinetochore localization of GFP‐Cenp‐FsiR‐C2865S (Fig 5B). Our data thus demonstrate that Bub1 is the direct KT‐core binding partner of Cenp‐F that contributes, along with Cenp‐E, to the localization of full‐length Cenp‐F to kinetochores.
In conclusion, our study provides a successful example of a hybrid analysis using Y2H interaction studies and in silico structural modeling. With this approach, we could elaborate an interaction model between the N‐terminal domain of Nup133 and a short dimeric coiled‐coil segment of Cenp‐F, which was validated in an in vivo context upon expression of either short fragments or the full‐length Cenp‐F protein. This robust experimental pipeline demonstrates that despite being located within a very short domain, the determinants required for the interaction of Cenp‐F with Nup133 and Bub1 can be functionally separated. At kinetochores, Cenp‐F may thus simultaneously interact with both Bub1 and Nup133. However, our localization studies demonstrate that affecting Cenp‐F/Nup133 interaction specifically impairs its prophase NE localization. In contrast, Bub1 appears as the major KT‐core tether for Cenp‐F that operates in a redundant manner with Cenp‐E to localize full‐length Cenp‐F to kinetochores. While previous functional studies were solely based on Cenp‐F depletion, our separation‐of‐function mutations will be instrumental in the future to determine the extent to which the kinetochore and/or NE localization of Cenp‐F contribute to its multiple functions in physiological and pathological situations.
Materials and Methods
Structural models of Nup133 and Cenp‐F domains
Pairwise sequence alignment of human/mouse Cenp‐F proteins was performed using EMBOSS Matcher (http://www.ebi.ac.uk/Tools/psa/emboss_matcher/).
Coiled‐coil conformation predictions on mCenp‐F C‐terminal domain were performed using COILS/PCOILS on the Bioinformatics Toolkit server 55. Because the structural deformation induced by kinks in phase‐shifting regions is difficult to predict, a model for the entire segment spanning mCenp‐F‐Ct1 and mCenp‐F‐Ct2 could not be generated with high confidence. In contrast, most of mCenp‐F‐SID segment was predicted as a canonical, continuous, and conserved coiled‐coil region (miniSID, gray box in Fig EV2A). This stretch consists of 44 residues [aa 2,663–2,706] containing six heptads in which half of the (a/d) coiled‐coil positions are leucine residues. This is consistent with previous predictions 10, which suggested that this coiled‐coil region is a leucine zipper. Since the vast majority of leucine zippers dimerize in a parallel mode 56, we used a canonical parallel coiled‐coil template to model residues [2,663–2,706] using CCBuilder server 57 with default options.
mNup133 was modeled by a conservative template‐based modeling method using the Swissmodel server 58 based on the structure of the human ortholog (PDB: 1xks, exhibiting 81% sequence identity to mNup133).
Docking simulation for the Nup133 and Cenp‐F domains
The rigid‐body docking step was performed using the InterEvDock server that takes into account both the physicochemical nature of protein surfaces 48, 59 and the co‐evolutionary information between two binding partners 41, 47 (http://bioserv.rpbs.univ-paris-diderot.fr/services/InterEvDock/). We took as input the structural models of Nup133 and Cenp‐F and used the advanced options allowing to specify the multiple sequence co‐alignments. Indeed, Cenp‐F is a homodimer and its multiple sequence alignment had to be adapted to contain a copy of every sequence accounting for the dimeric structure. Besides, both monomers in the Cenp‐F PDB file were changed to the same chain label. The structures of each of the 10 consensus rigid‐body models recovered from the server were relaxed to remove steric clashes and optimize side‐chain contacts using Rosetta following the protocol described in 41. The 3D coordinates of the structural model shown in Fig 2A are provided in Appendix Fig S6.
Size‐exclusion chromatography with multi‐angle laser light scattering (SEC‐MALS)
N‐ter acetylated and C‐ter amidated peptides were purchased from custom synthesis service (ProteoGenix; > 95% purity) and verified by MALDI. Peptide solutions at a concentration of 5 mg/ml were injected on Superdex 75 10/300 GL (GE Healthcare) equilibrated in 50 mM Tris pH 7.4 with four different salt conditions, 0.15, 0.5, 1, or 2 M NaCl, at a flow rate of 0.5 ml/min (20°C). Separation and ultraviolet detection were performed using Shimadzu HPLC system, light scattering was monitored using mini DAWN TREOS system (Wyatt Technology), and concentration was measured by Optilab T‐rEX differential refractometer (Wyatt Technology). Molar masses of proteins were calculated using ASTRA 6.1 software (Wyatt Technology).
Plasmids
Plasmids used in this study are listed in Appendix Table S1. They were either previously published or generated using standard molecular cloning techniques including PCR amplification using proofreading DNA polymerases (Phusion HF, NEB) and Clontech In‐Fusion HD Cloning Kit or NEBuilder HiFi DNA Assembly Cloning Kits. For all constructs, PCR‐amplified fragments and junctions were checked by sequencing. Plasmid maps are available upon request.
Yeast two‐hybrid
The minimal Nup133‐binding site of human Cenp‐F (defined as SID®, Selected Interaction Domain; Hybrigenics) was identified by reanalyzing the outcome of a previous Y2H screen performed using hNup133‐NTD [aa 1–500] as bait to screen a human breast tumor epithelial cells RP1 cDNA library 13.
The pB27‐LexA‐mNup133 [aa 1–1,156] plasmid was used to screen a random primed mouse Embryonic Stem Cell_RP1 cDNA library cloned into the pP6‐Gal4‐AD plasmid using a high‐throughput proprietary yeast two‐hybrid‐based technology (ULTImate Y2H Screen; Hybrigenics). To investigate pairwise interactions between mNup133 and mCenp‐F by yeast two‐hybrid, the wild‐type or mutant mNup133 and mCenp‐F fragments were cloned in the pB27‐LexA and pP6‐Gal4‐AD plasmid, respectively. pB27‐LexA‐derived plasmids were transformed in L40ΔGal4 strain, while pP6‐Gal4‐AD‐based plasmids were transformed into the Y187 strain as described previously 31. Bait and prey strains were mated in rich medium, and diploids were grown for 2–3 days on minimum medium lacking leucine and tryptophan (‐LW, as control to ensure mating efficiency) or lacking leucine, tryptophan, and histidine (‐LWH) supplemented, when indicated, with 1 or 5 mM 3‐Aminotriazole (3AT) a competitive inhibitor of the HIS3 gene product that increases the stringency of the Y2H assay.
To analyze the expression of the yeast two‐hybrid constructs, total proteins were extracted from yeast cells using the NaOH–TCA lysis method essentially as described 60. Briefly, three OD600 values of exponentially growing yeast cells were collected by centrifugation, washed once in water, resuspended in 550 μl of 0.17 M NaOH, and incubated for 10 min on ice. 50 μl of 50% TCA was then added, and the samples were incubated again for 10 min on ice. After a 2‐min centrifugation at 12,000 g, pellets were resuspended in 60 μl of Laemmli 2×, 15 μl 1 M Tris, and 2% β‐mercaptoethanol, denatured 5 min at 95°C, and analyzed by Western blot (Appendix Fig S2).
Cell culture, plasmid and siRNA transfections, crosslinking, and whole‐cell extracts
HeLa‐E and HEp‐2 are subclones of HeLa cells initially received from F. Perez (Institut Curie, Paris, France) and Luc Mouthon (Institut Cochin, Paris, France), respectively. These cell lines were grown at 37°C in DMEM (Life Technologies) supplemented with 10% fetal calf serum, 1% l‐glutamine, 100 μg/ml streptomycin, and 100 U/ml penicillin. For plasmid and siRNA transfections, ~0.4–0.5 million cells plated on 35‐mm dishes were treated with 10 μl Lipofectamine 2000 (Invitrogen) and (i) 4 μg of HA‐mCenp‐F‐Ct2, GFP‐mCenp‐F‐Ct1, or GFP‐mCenp‐F‐Ct2 WT or mutant plasmids, (ii) 2 μg of the pUG6SP‐tTA activation plasmid (gift from M.N. Boddy; Addgene plasmid #41029; 61) and 2 μg of GFP‐hCenp‐F full‐length WT or L2797E/L2799E plasmids (for Fig 4), or (iii) 4 μg of GFP‐mCenp‐FsiR‐WT or GFP‐mCenp‐FsiR‐C2865S plasmid DNA and 240 μg of Cenp‐F siRNA duplex (5′‐CAGAATCTTAGTAGTCAAGTA‐3′; 13) either alone or combined with 240 μg of Cenp‐E siRNA duplex (5′‐CCACUAGAGUUGAAAGAUAUU‐3′; 62; for Fig 5B). The transfection medium was removed after 4 h, and the cells were analyzed by Western blot or immunofluorescence 24 or 48 h after transfection.
For in vivo crosslinking, HeLa‐E cells transfected with HA‐mCenp‐F‐Ct2 WT or mutant plasmids were treated 48 h after transfection with 0.5 mM disuccinimidyl suberate (DSS, a non‐cleavable and membrane permeable homobifunctional NHS‐ester crosslinker; Thermo Scientific) in PBS for 5 min and then quenched with 10 mM Tris pH 8.6 for an additional 15 min prior to cell lysis.
To prepare whole‐cell lysates, transfected human cells were lysed in 2× Laemmli lysis buffer [150‐mM Tris–HCl (pH 6.8), 5% (wt/vol) SDS, 25% (vol/vol) glycerol, and 0.01% (wt/vol) bromophenol blue]. Lysates were incubated for 3 min at 95°C, clarified by sonication [Bioruptor (Diagenode): 3 cycles of 30 s on/off, high power], and denatured again for 3 min at 95°C. Except for crosslinking experiments, protein concentration was then determined using a BCA assay kit (Thermo Scientific). Total protein extracts supplemented with β‐mercaptoethanol (750 mM final) were analyzed by Western blot (~15 μg per lane).
Western blot analyses
For Western blot analysis, yeast or human cell lysates were separated on 8% Tris–glycine SDS–PAGE gels or on NuPAGE™ 4–12% Bis‐Tris gels (using MOPS as running buffer) and transferred to nitrocellulose membranes. The resulting blots were stained using Ponceau, saturated with TBS, 0.1% Tween, and 5% dried milk, and probed with affinity‐purified monoclonal mouse anti‐LexA (Santa Cruz SC‐7544; 1:5,000), polyclonal rabbit anti‐Gal4AD (SIGMA G9293; 1:2,000), mouse monoclonal antibody HA.11 (clone 16B12; Eurogentec #MMS‐101R; 1:2,000), or mouse monoclonal anti‐GFP antibody (Roche Diagnostic #11814460001; 1:1,000). Incubations of the membrane with primary and HRP‐conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) were done in TBS buffer (0.1% Tween, 5% dried milk), and signals were detected by enhanced chemiluminescence (SuperSignal® Pico or Femto; Thermo Scientific).
Immunofluorescence microscopy
Antibodies used in this study were as follows: affinity‐purified rabbit polyclonal anti‐human Nup133 (#756‐77; 1:100; 31; used in Fig EV5A and B); rat monoclonal anti‐mouse Nup133 (#74; clone 9C2; 1:100; used in Figs 4 and EV5C) generated against recombinant histidine‐tagged mouse Nup133 [aa 66–512] and purified by ProteoGenix (validation of this antibody for IF experiments is provided in Appendix Fig S4); rat monoclonal anti‐Nup98‐Nter (clone 2H10, Abcam ab50610, 1:500); mouse monoclonal anti‐Cenp‐F (clone 11/Mitosin; BD Biosciences #610768; 1:400); rabbit polyclonal anti‐Cenp‐F (Abcam ab5, 1:400); rabbit polyclonal anti‐Elys/Mel28 antibody (FID1 serum, directed against histidine‐tagged human Mel28 protein, aa 1,208–1,800; generously provided by R. Walczak and I. Mattaj, EMBL, Heidelberg, Germany; 1:1,000; 63); mouse monoclonal anti‐HEC1 (clone 9G3; Abcam ab3613; 1:500); rabbit anti‐Cenp‐E antibody (generously provided by T.J. Yen, Philadelphia, USA; 1:1,000; 22); autoimmune CREST serum (Antibodies Incorporated 15‐235; 1:500); and mouse monoclonal anti‐phospho‐H3 Ser10 (Abcam ab14955; 1:4,000). Secondary antibodies were from Jackson ImmunoResearch Laboratories, Inc. (cy3, cy5) and were generally used at 1:500 dilution (except at 1:2,000 dilution when anti‐mouse cy3 was used in combination with anti‐phospho‐H3).
Fluorescence signals within the intranuclear bodies and at kinetochores were best detected upon pre‐extraction of cells, followed by a fixation–extraction procedure as previously described 33. Briefly, cells grown on coverslips were quickly pre‐extracted for 15 s in PHEM buffer (20 mM K–1,4‐piperazinediethanesulfonic acid, 10 mM K–ethylene glycol tetraacetic acid, 1 mM MgCl2, pH 6.8) containing 0.2% Triton and then fixed for 10 min in the same buffer further containing 4% paraformaldehyde supplemented with 0.85% of 1 N NaOH. For Fig 5B, the transfected cells were not pre‐extracted but directly fixed for 20 min with 3% paraformaldehyde in PBS supplemented with 0.85% of 1 N NaOH and then permeabilized with 0.5% Triton X‐100 in PBS for 8 min. In both cases, following a 15‐ to 30‐min saturation in PBS containing 1% BSA, coverslips were then successively incubated for 1 h with primary and secondary antibodies diluted in PBS + 1% BSA. Cells were then stained with 0.1 μg/ml of DAPI and were mounted in Moviol medium.
Image acquisition and analysis
Widefield fluorescence images were acquired using a microscope (DM6000B; Leica) with a 100×, numerical aperture 1.4 (HCX Plan‐Apo) oil immersion objective and a charge‐coupled device (CCD) camera (CoolSNAP HQ; Photometrics). A piezoelectric motor mounted underneath the objective lens enabled rapid and precise Z‐positioning. Spinning disk images were acquired on an inverted microscope (DMI8; Leica) with a CSU‐W1 spinning disk head (Yokogawa) and a sCMOS ORCA‐Flash 4 V2+ camera (Hamamatsu) using 100×/1.4 oil objective. Image stacks were acquired without camera binning, with a plane spacing of 0.2 μm. Either a unique plane or 3D maximum‐intensity projections performed using MetaMorph (Universal Imaging Corp.) or ImageJ/Fiji software are presented as indicated on the figures.
Line scan analyses were performed using ImageJ. A line of 10 pixels width was drawn on the focal plane of the NE (defined based on Nup133 staining), and the average intensity along this line was plotted for each channel using Microsoft Excel. Quantifications of fluorescence intensities at kinetochores were carried out using a Fiji macro (provided in Appendix Fig S5) on 3D projections covering the entire mitotic cells (raw data are included in Appendix Table S2). Briefly, kinetochores were identified based on Hec1 staining. Fluorescence intensities for each kinetochore were then calculated and normalized to the average intensity measured on the entire cell. For each cell, all kinetochores were independently quantified and their intensity was averaged. Box plots were generated using KaleidaGraph (Synergy Software). Statistical analyses were performed using Wilcoxon–Mann–Whitney rank‐sum rest provided by the KaleidaGraph software.
Author contributions
AB, RG, FO, JD, and VD conceived and designed the experiments; AB, SM‐B, FO, and CB performed the wet experiments; JY and RG performed the modeling simulations and designed the mutants; AB, SM‐B, RG, JY, FO, RV, and VD analyzed the data; and VD, AB, and RG wrote the manuscript with contribution from all co‐authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Acknowledgements
We are grateful to I. Bouhlel for the characterization of rat monoclonal Nup133 antibody, to T. Schwartz for help with the design of initial mutations within the Nup133 β‐propeller, to the protein–protein interactions platform of I2BC for their help in running the SEC‐MALS experiment, and to V. Contremoulin from the ImagoSeine core facility for help with kinetochore quantification. We kindly thank I. Mattaj and R. Walczak for sharing Elys antibodies, A. Desai for the hBub1 cDNA construct, and X. Zhu for full‐length human Cenp‐F plasmid. We also acknowledge L. Pintard and B. Palancade for critical reading of the manuscript and other members of the laboratory for helpful advices. Work in the laboratories of VD, RG, and JD is supported by the Centre national de la recherche scientifique (CNRS), the French National Research Agency under Grant No. ANR‐2012‐BSV2‐0008‐01 to VD and ANR‐15‐CE11‐0008‐01 to RG, the “Fondation pour la Recherche Médicale” (Foundation for Medical Research) under Grant No. DEQ20150734355, “Equipe FRM 2015” and the Labex Who Am I? (ANR‐11‐LABX‐0071; Idex ANR‐11‐IDEX‐0005‐02) to VD, the French Infrastructure for Integrated Structural Biology (FRISBI) [ANR‐10‐INSB‐05‐01] to RG and FO, and Fondation ARC pour la Recherche sur le Cancer (PJA 20151203418) and Ligue Contre le Cancer‐Comité d'Ile de France 2015 to JD. AB received PhD fellowships from the “Ministère de l'Enseignement Supérieur et de la Recherche” and the “Ligue Nationale contre le Cancer” and a “Transition Post‐Doc” grant from the Labex Who Am I?. JY received a PhD fellowship from IDEX Paris‐Saclay, ANR‐11‐IDEX‐0003‐02—“IDI 2013”. The ImagoSeine core facility of the Institut Jacques Monod is a member of IBiSA and France‐BioImaging (ANR‐10‐INBS‐04) infrastructures.
EMBO Reports (2018) 19: e44742
References
- 1. Ma L, Zhao X, Zhu X (2006) Mitosin/CENP‐F in mitosis, transcriptional control, and differentiation. J Biomed Sci 13: 205–213 [DOI] [PubMed] [Google Scholar]
- 2. Varis A, Salmela AL, Kallio MJ (2006) Cenp‐F (mitosin) is more than a mitotic marker. Chromosoma 115: 288–295 [DOI] [PubMed] [Google Scholar]
- 3. Goodwin RL, Pabon‐Pena LM, Foster GC, Bader D (1999) The cloning and analysis of LEK1 identifies variations in the LEK/centromere protein F/mitosin gene family. J Biol Chem 274: 18597–18604 [DOI] [PubMed] [Google Scholar]
- 4. Pooley RD, Moynihan KL, Soukoulis V, Reddy S, Francis R, Lo C, Ma LJ, Bader DM (2008) Murine CENPF interacts with syntaxin 4 in the regulation of vesicular transport. J Cell Sci 121: 3413–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dees E, Miller PM, Moynihan KL, Pooley RD, Hunt RP, Galindo CL, Rottman JN, Bader DM (2012) Cardiac‐specific deletion of the microtubule‐binding protein CENP‐F causes dilated cardiomyopathy. Dis Model Mech 5: 468–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Waters AM, Asfahani R, Carroll P, Bicknell L, Lescai F, Bright A, Chanudet E, Brooks A, Christou‐Savina S, Osman G et al (2015) The kinetochore protein, CENPF, is mutated in human ciliopathy and microcephaly phenotypes. J Med Genet 52: 147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pfaltzgraff ER, Roth GM, Miller PM, Gintzig AG, Ohi R, Bader DM (2016) Loss of CENP‐F results in distinct microtubule‐related defects without chromosomal abnormalities. Mol Biol Cell 27: 1990–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rattner JB, Rao A, Fritzler MJ, Valencia DW, Yen TJ (1993) CENP‐F is a.ca 400 kDa kinetochore protein that exhibits a cell‐cycle dependent localization. Cell Motil Cytoskeleton 26: 214–226 [DOI] [PubMed] [Google Scholar]
- 9. Liao H, Winkfein RJ, Mack G, Rattner JB, Yen TJ (1995) CENP‐F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J Cell Biol 130: 507–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhu X, Chang KH, He D, Mancini MA, Brinkley WR, Lee WH (1995) The C terminus of mitosin is essential for its nuclear localization, centromere/kinetochore targeting, and dimerization. J Biol Chem 270: 19545–19550 [DOI] [PubMed] [Google Scholar]
- 11. Moynihan KL, Pooley R, Miller PM, Kaverina I, Bader DM (2009) Murine CENP‐F regulates centrosomal microtubule nucleation and interacts with Hook2 at the centrosome. Mol Biol Cell 20: 4790–4803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanfer G, Courtheoux T, Peterka M, Meier S, Soste M, Melnik A, Reis K, Aspenstrom P, Peter M, Picotti P et al (2015) Mitotic redistribution of the mitochondrial network by Miro and Cenp‐F. Nat Commun 6: 8015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bolhy S, Bouhlel I, Dultz E, Nayak T, Zuccolo M, Gatti X, Vallee R, Ellenberg J, Doye V (2011) A Nup133‐dependent NPC‐anchored network tethers centrosomes to the nuclear envelope in prophase. J Cell Biol 192: 855–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu DJ, Baffet AD, Nayak T, Akhmanova A, Doye V, Vallee RB (2013) Dynein recruitment to nuclear pores activates apical nuclear migration and mitotic entry in brain progenitor cells. Cell 154: 1300–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baffet AD, Hu DJ, Vallee RB (2015) Cdk1 activates pre‐mitotic nuclear envelope dynein recruitment and apical nuclear migration in neural stem cells. Dev Cell 33: 703–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bomont P, Maddox P, Shah JV, Desai AB, Cleveland DW (2005) Unstable microtubule capture at kinetochores depleted of the centromere‐associated protein CENP‐F. EMBO J 24: 3927–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holt SV, Vergnolle MA, Hussein D, Wozniak MJ, Allan VJ, Taylor SS (2005) Silencing Cenp‐F weakens centromeric cohesion, prevents chromosome alignment and activates the spindle checkpoint. J Cell Sci 118: 4889–4900 [DOI] [PubMed] [Google Scholar]
- 18. Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH (2005) FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 7: 126–136 [DOI] [PubMed] [Google Scholar]
- 19. Yang Z, Guo J, Chen Q, Ding C, Du J, Zhu X (2005) Silencing mitosin induces misaligned chromosomes, premature chromosome decondensation before anaphase onset, and mitotic cell death. Mol Cell Biol 25: 4062–4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feng J, Huang H, Yen TJ (2006) CENP‐F is a novel microtubule‐binding protein that is essential for kinetochore attachments and affects the duration of the mitotic checkpoint delay. Chromosoma 115: 320–329 [DOI] [PubMed] [Google Scholar]
- 21. Evans HJ, Edwards L, Goodwin RL (2007) Conserved C‐terminal domains of mCenp‐F (LEK1) regulate subcellular localization and mitotic checkpoint delay. Exp Cell Res 313: 2427–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan GK, Schaar BT, Yen TJ (1998) Characterization of the kinetochore binding domain of CENP‐E reveals interactions with the kinetochore proteins CENP‐F and hBUBR1. J Cell Biol 143: 49–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Soukoulis V, Reddy S, Pooley RD, Feng Y, Walsh CA, Bader DM (2005) Cytoplasmic LEK1 is a regulator of microtubule function through its interaction with the LIS1 pathway. Proc Natl Acad Sci USA 102: 8549–8554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vergnolle MA, Taylor SS (2007) Cenp‐F links kinetochores to Ndel1/Nde1/Lis1/dynein microtubule motor complexes. Curr Biol 17: 1173–1179 [DOI] [PubMed] [Google Scholar]
- 25. Musinipally V, Howes S, Alushin GM, Nogales E (2013) The microtubule binding properties of CENP‐E's C‐terminus and CENP‐F. J Mol Biol 425: 4427–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McKinley KL, Cheeseman IM (2017) Large‐scale analysis of CRISPR/Cas9 cell‐cycle knockouts reveals the diversity of p53‐dependent responses to cell‐cycle defects. Dev Cell 40: 405–420 e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu X (1999) Structural requirements and dynamics of mitosin‐kinetochore interaction in M phase. Mol Cell Biol 19: 1016–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hussein D, Taylor SS (2002) Farnesylation of Cenp‐F is required for G2/M progression and degradation after mitosis. J Cell Sci 115: 3403–3414 [DOI] [PubMed] [Google Scholar]
- 29. Liu ST, Hittle JC, Jablonski SA, Campbell MS, Yoda K, Yen TJ (2003) Human CENP‐I specifies localization of CENP‐F, MAD1 and MAD2 to kinetochores and is essential for mitosis. Nat Cell Biol 5: 341–345 [DOI] [PubMed] [Google Scholar]
- 30. Johnson VL, Scott MI, Holt SV, Hussein D, Taylor SS (2004) Bub1 is required for kinetochore localization of BubR1, Cenp‐E, Cenp‐F and Mad2, and chromosome congression. J Cell Sci 117: 1577–1589 [DOI] [PubMed] [Google Scholar]
- 31. Belgareh N, Rabut G, Bai SW, van Overbeek M, Beaudouin J, Daigle N, Zatsepina OV, Pasteau F, Labas V, Fromont‐Racine M et al (2001) An evolutionarily conserved NPC subcomplex, which redistributes in part to kinetochores in mammalian cells. J Cell Biol 154: 1147–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Beck M, Hurt E (2017) The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol 18: 73–89 [DOI] [PubMed] [Google Scholar]
- 33. Morchoisne‐Bolhy S, Geoffroy MC, Bouhlel IB, Alves A, Auduge N, Baudin X, Van Bortle K, Powers MA, Doye V (2015) Intranuclear dynamics of the Nup107‐160 complex. Mol Biol Cell 26: 2343–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zuccolo M, Alves A, Galy V, Bolhy S, Formstecher E, Racine V, Sibarita JB, Fukagawa T, Shiekhattar R, Yen T et al (2007) The human Nup107‐160 nuclear pore subcomplex contributes to proper kinetochore functions. EMBO J 26: 1853–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Platani M, Santarella‐Mellwig R, Posch M, Walczak R, Swedlow JR, Mattaj IW (2009) The Nup107‐160 nucleoporin complex promotes mitotic events via control of the localization state of the chromosome passenger complex. Mol Biol Cell 20: 5260–5275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ashe M, Pabon‐Pena L, Dees E, Price KL, Bader D (2004) LEK1 is a potential inhibitor of pocket protein‐mediated cellular processes. J Biol Chem 279: 664–676 [DOI] [PubMed] [Google Scholar]
- 37. Zhou X, Wang R, Fan L, Li Y, Ma L, Yang Z, Yu W, Jing N, Zhu X (2005) Mitosin/CENP‐F as a negative regulator of activating transcription factor‐4. J Biol Chem 280: 13973–13977 [DOI] [PubMed] [Google Scholar]
- 38. Berke IC, Boehmer T, Blobel G, Schwartz TU (2004) Structural and functional analysis of Nup133 domains reveals modular building blocks of the nuclear pore complex. J Cell Biol 167: 591–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Potenza E, Di Domenico T, Walsh I, Tosatto SC (2015) MobiDB 2.0: an improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res 43: D315–D320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Drin G, Casella JF, Gautier R, Boehmer T, Schwartz TU, Antonny B (2007) A general amphipathic alpha‐helical motif for sensing membrane curvature. Nat Struct Mol Biol 14: 138–146 [DOI] [PubMed] [Google Scholar]
- 41. Yu J, Vavrusa M, Andreani J, Rey J, Tuffery P, Guerois R (2016) InterEvDock: a docking server to predict the structure of protein‐protein interactions using evolutionary information. Nucleic Acids Res 44: W542–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yu J, Andreani J, Ochsenbein F, Guerois R (2017) Lessons from (co‐)evolution in the docking of proteins and peptides for CAPRI Rounds 28–35. Proteins 85: 378–390 [DOI] [PubMed] [Google Scholar]
- 43. Lensink MF, Velankar S, Wodak SJ (2017) Modeling protein‐protein and protein‐peptide complexes: CAPRI 6th edition. Proteins 85: 359–377 [DOI] [PubMed] [Google Scholar]
- 44. Ren Y, Seo HS, Blobel G, Hoelz A (2010) Structural and functional analysis of the interaction between the nucleoporin Nup98 and the mRNA export factor Rae1. Proc Natl Acad Sci USA 107: 10406–10411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bilokapic S, Schwartz TU (2013) Structural and functional studies of the 252 kDa nucleoporin ELYS reveal distinct roles for its three tethered domains. Structure 21: 572–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu C, Li Z, He H, Wernimont A, Li Y, Loppnau P, Min J (2015) Crystal structure of human nuclear pore complex component NUP43. FEBS Lett 589: 3247–3253 [DOI] [PubMed] [Google Scholar]
- 47. Andreani J, Faure G, Guerois R (2013) InterEvScore: a novel coarse‐grained interface scoring function using a multi‐body statistical potential coupled to evolution. Bioinformatics 29: 1742–1749 [DOI] [PubMed] [Google Scholar]
- 48. Dong GQ, Fan H, Schneidman‐Duhovny D, Webb B, Sali A (2013) Optimized atomic statistical potentials: assessment of protein interfaces and loops. Bioinformatics 29: 3158–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gurden MD, Holland AJ, van Zon W, Tighe A, Vergnolle MA, Andres DA, Spielmann HP, Malumbres M, Wolthuis RM, Cleveland DW et al (2010) Cdc20 is required for the post‐anaphase, KEN‐dependent degradation of centromere protein F. J Cell Sci 123: 321–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Griffis ER, Altan N, Lippincott‐Schwartz J, Powers MA (2002) Nup98 is a mobile nucleoporin with transcription‐dependent dynamics. Mol Biol Cell 13: 1282–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Romana S, Radford‐Weiss I, Lapierre JM, Doye V, Geoffroy MC (2016) Formation of Nup98‐containing nuclear bodies in HeLa sublines is linked to genomic rearrangements affecting chromosome 11. Chromosoma 125: 789–805 [DOI] [PubMed] [Google Scholar]
- 52. Loftus KM, Cui H, Coutavas E, King DS, Ceravolo A, Pereiras D, Solmaz SR (2017) Mechanism for G2 phase‐specific nuclear export of the kinetochore protein CENP‐F. Cell Cycle 16: 1414–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ciossani G, Overlack K, Petrovic A, Huis in ‘t Veld P, Körner C, Wohlgemuth S, Maffini S, Musacchio A (2018) Kinetochore recruitment of CENP‐F illustrates how paralog divergence shapes kinetochore composition and function. BioRxiv https://doi.org/10.1101/276204 [PREPRINT] [Google Scholar]
- 54. Raaijmakers JA, van Heesbeen R, Blomen VA, Janssen LME, van Diemen F, Brummelkamp TR, Medema RH (2018) BUB1 is essential for the viability of human cells in which the spindle assembly checkpoint is compromised. Cell Rep 22: 1424–1438 [DOI] [PubMed] [Google Scholar]
- 55. Alva V, Nam SZ, Soding J, Lupas AN (2016) The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res 44: W410–W415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Johnson PF, McKnight SL (1989) Eukaryotic transcriptional regulatory proteins. Annu Rev Biochem 58: 799–839 [DOI] [PubMed] [Google Scholar]
- 57. Wood CW, Bruning M, Ibarra AA, Bartlett GJ, Thomson AR, Sessions RB, Brady RL, Woolfson DN (2014) CCBuilder: an interactive web‐based tool for building, designing and assessing coiled‐coil protein assemblies. Bioinformatics 30: 3029–3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L et al (2014) SWISS‐MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42: W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Garzon JI, Lopez‐Blanco JR, Pons C, Kovacs J, Abagyan R, Fernandez‐Recio J, Chacon P (2009) FRODOCK: a new approach for fast rotational protein‐protein docking. Bioinformatics 25: 2544–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ulrich HD, Davies AA (2009) In vivo detection and characterization of sumoylation targets in Saccharomyces cerevisiae . Methods Mol Biol 497: 81–103 [DOI] [PubMed] [Google Scholar]
- 61. Zilio N, Wehrkamp‐Richter S, Boddy MN (2012) A new versatile system for rapid control of gene expression in the fission yeast Schizosaccharomyces pombe . Yeast 29: 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim Y, Holland AJ, Lan W, Cleveland DW (2010) Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of CENP‐E. Cell 142: 444–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yokoyama H, Koch B, Walczak R, Ciray‐Duygu F, Gonzalez‐Sanchez JC, Devos DP, Mattaj IW, Gruss OJ (2014) The nucleoporin MEL‐28 promotes RanGTP‐dependent gamma‐tubulin recruitment and microtubule nucleation in mitotic spindle formation. Nat Commun 5: 3270 [DOI] [PubMed] [Google Scholar]
- 64. Ashar HR, James L, Gray K, Carr D, Black S, Armstrong L, Bishop WR, Kirschmeier P (2000) Farnesyl transferase inhibitors block the farnesylation of CENP‐E and CENP‐F and alter the association of CENP‐E with the microtubules. J Biol Chem 275: 30451–30457 [DOI] [PubMed] [Google Scholar]
- 65. Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben‐Tal N (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44: W344–W350 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File