

Abstract
In most mammalian cells, the primary cilium is a microtubule‐enriched protrusion of the plasma membrane and acts as a key coordinator of signaling pathways during development and tissue homeostasis. The primary cilium is generated from the basal body, and cancerous inhibitor of protein phosphatase 2A (CIP2A), the overexpression of which stabilizes c‐MYC to support the malignant growth of tumor cells, is localized in the centrosome. Here, we show that CIP2A overexpression induces primary cilia disassembly through the activation of Aurora A kinase, and CIP2A depletion increases ciliated cells and cilia length in retinal pigment epithelium (RPE1) cells. CIP2A depletion also shifts metabolism toward the glycolytic pathway by altering the expression of metabolic genes related to glycolysis. However, glycolytic activation in CIP2A‐depleted cells does not depend on cilia assembly, even though enhanced cilia assembly alone activates glycolytic metabolism. Collectively, these data suggest that CIP2A promotes primary cilia disassembly and that CIP2A depletion induces metabolic reprogramming independent of primary cilia.
Keywords: Aurora A, CIP2A, glycolysis, metabolic reprogramming, primary cilia
Subject Categories: Cancer; Cell Adhesion, Polarity & Cytoskeleton; Metabolism
Introduction
The primary cilium, a microtubule‐enriched protrusion of the plasma membrane in most eukaryotic cells, has been proposed as a key coordinator of signaling pathways during development and tissue homeostasis. Therefore, primary cilia defects may cause human diseases and a developmental disorder known as ciliopathy 1. Most ciliopathies appear as genetic syndromes, such as Joubert syndrome, Bardet–Biedl syndrome (BBS), Alström syndrome (ALMS), Meckel–Gruber syndrome (MKS), and nephronophthisis (NPHP) 2. Interestingly, certain subclasses of ciliopathies, such as BBS and ALMS, are linked to obesity and diabetes 3, 4, 5. It was recently revealed that primary ciliary signaling at the hypothalamus controls body weight through the regulation of satiety signaling and food intake 6, 7. However, a role for primary cilia in metabolic homeostasis remains unknown.
Several studies have shown that primary cilia deficiency plays a role in cancer 8, 9. Primary cilia are decreased or lost in a variety of cancer types, including breast, prostate, ovarian, skin, and pancreatic cancer and in renal cell carcinoma and basal cell carcinoma 10, 11, 12, 13, 14, 15. In general, it was considered that the appearance and disappearance of primary cilia passively depend on the cell cycle 16, 17. However, recent studies have shown that primary cilia actively act as a checkpoint to prevent cell cycle re‐entry 18, 19. Therefore, it is assumed that the disassembly of the primary cilia promotes tumorigenesis by dysregulating the cell cycle. Indeed, it is known that Aurora A and HEF1 oncoproteins dysregulate cell cycle and also promote ciliary disassembly 20. In addition, most kinases, such as Aurora A, Plk1, and NEK2, which spatiotemporally regulate correct cell division, are highlighted as negative regulators of ciliogenesis 20, 21, 22, 23. These results suggest that ciliogenesis and cell cycle are closely linked to each other and that they have bidirectional aspects.
A decade ago, cancerous inhibitor of protein phosphatase 2A (CIP2A) was revealed as an oncogenic protein overexpressed in most human cancers and is involved in the progression of several human malignancies via the inhibition of protein phosphatase 2A (PP2A) activity, leading to c‐MYC stabilization 24. CIP2A also interacts with NEK2 during G2/M phase and enhances NEK2 kinase activity to facilitate centrosome separation 25. Because NEK2 regulates cilia disassembly through a concerted mechanism involving Kif24‐mediated microtubule depolymerization, we investigated whether CIP2A promotes cilia disassembly. On the other hand, cancer cells efficiently reprogram their metabolism to acquire necessary nutrients and utilize those nutrients for growth and for cellular building blocks, even if oxygen is not limited 26, 27. This metabolic shift is mediated by the aberrant activation of signaling pathways and oncogenic factors, leading to the altered expression of genes involved in glucose import, catabolism, and anabolism 28. In this study, since c‐MYC is considered a master modulator of growth and cellular metabolic pathways 29 and CIP2A mediates cancer progression through the metabolic pathway 30, we investigated whether CIP2A plays a role in primary cilia disassembly, whether CIP2A affects metabolic reprogramming, and whether cilia assembly is able to shift metabolism toward the glycolytic pathway.
Results
CIP2A overexpression induces cilia disassembly, and CIP2A depletion increases both the number of ciliated cells and cilia length in RPE1 cells
Because immortalized human retinal pigment epithelial (hTERT‐RPE1) cells assemble primary cilia at a high frequency, this cell line was used to determine whether CIP2A affects cilia assembly or disassembly (ciliogenesis). First, we observed location of CIP2A at the end of primary cilia. To address where CIP2A localizes exactly, CIP2A location was compared with the location of various markers including γ‐tubulin (basal body), CEP164 (distal end of centriole), CEP290 (proximal end of centriole), and TMEM67 (transition zone). CIP2A co‐localized with TMEM67 at transition zone (TZ) (Fig 1A). Because primary cilia assembly is enhanced under serum‐starved conditions, and cilia disassembly occurs when cells are re‐stimulated by serum‐containing media 20, we evaluated whether the protein level of CIP2A is regulated by those conditions. CIP2A expression was downregulated after 24 h under serum‐starved condition and slowly elevated after serum re‐stimulation (Fig 1B). We also confirmed the decreased level of CIP2A after serum starvation (S.S.) and increased level of CIP2A after serum re‐stimulation by immunostaining (Fig EV1A). As CIP2A localized at TZ and serum re‐stimulation elevated the expression of CIP2A, we tested whether CIP2A accumulates more at TZ after re‐stimulation. CIP2A gradually increased after serum re‐stimulation and accumulated at TZ to a higher degree at 5 h post‐re‐stimulation (Figs 1C and EV1B). These data indicate that CIP2A is involved in ciliogenesis under the physiological conditions.
Figure 1. CIP2A localizes at the transition zone and regulates ciliogenesis.

- Immunofluorescence images of endogenous CIP2A and primary cilia in RPE1 cells. Cells were fixed with methanol and stained with antibodies specific for CIP2A (red) and ARL13b or γ‐tubulin, CEP164, CEP290, and TMEM67 (green). DNA was stained with DAPI (blue). Shown are the maximum projections from z stacks; scale bar = 5 μm.
- Cells were serum‐starved (SS) for the indicated time, and serum was added for 1 or 2 h. The expression of CIP2A was analyzed by immunoblot and β‐actin served as the loading control.
- RPE1 cells were serum‐starved (SS) for 24 h, and serum was added for the indicated times. For endogenous co‐immunoprecipitation analysis, cell lysates were immunoprecipitated (IP) with anti‐CIP2A at each time point and IP proteins were analyzed by immunoblot.
- Immunofluorescence images of RPE1 cells transfected with GFP or GFP‐CIP2A following serum starvation for 48 h. Primary cilia were stained with antibodies specific for ARL13b (red). DNA was stained with DAPI (blue). Shown are the maximum projections from z stacks of representative GFP‐ or GFP‐CIP2A‐transfected cells. Scale bar = 10 μm. The percentage of GFP‐positive cells with primary cilia was determined (n > 50 cells/condition). The average of three independent experiments is shown, with error bars representing s.d. **P < 0.01 compared with GFP‐transfected cells (one‐tailed Student's t‐test).
- Cilium length of each cell was measured using ImageJ software. Mean of each cilium length from 13 cells for GFP‐transfected condition and 7 cells for GFP‐CIP2A transfected condition is presented with error bars representing s.d. as a graph made using GraphPad Prism software. Data points are derived from a single experiment. *P < 0.05 compared to GFP‐transfected cells (one‐way ANOVA).
- RPE1 cells stably expressing Smo‐EGFP (RPE1‐Smo‐EGFP) transiently transfected with control, CIP2A #1, CIP2A #2, or CIP2A #3 siRNA, were serum‐starved for 48 h, fixed, and stained with antibodies against γ‐tubulin (red) and DAPI (blue) for immunofluorescence analysis. Shown are the maximum projections from z stacks of representative cells for each condition. Scale bar = 10 μm. The siRNAs against CIP2A were validated by immunoblot.
- Smo‐EGFP fluorescence was used to measure cilium length. The average of measured cilium length is presented with error bars representing s.d. as a graph made using GraphPad Prism software (n > 86 cells per condition). Data points are derived from a single representative experiment. *P < 0.05, **P < 0.01, ***P < 0.0001 compared with siControl (one‐way ANOVA).
- RPE1 cells transfected with control, CIP2A #2, CIP2A #3, or NEK2 siRNA were cultured in each indicated condition. For growing conditions, cells were cultured in complete media; for serum starvation, cells were cultured in serum‐starved media for 48 h. Serum was added for 24 h after serum starvation. Cells were fixed and stained with antibodies against ARL13b. The percentage of cells with primary cilia was determined for each condition (n > 20 cells/condition). The average of three independent experiments is shown, with error bars representing s.d. *P < 0.05, **P < 0.01, ***P < 0.0001 compared to siControl cells of each condition (one‐way ANOVA). The knockdown of siRNAs against CIP2A and NEK2 was validated by immunoblot.
Source data are available online for this figure.
Figure EV1. CIP2A is involved in ciliogenesis under the physiological conditions.
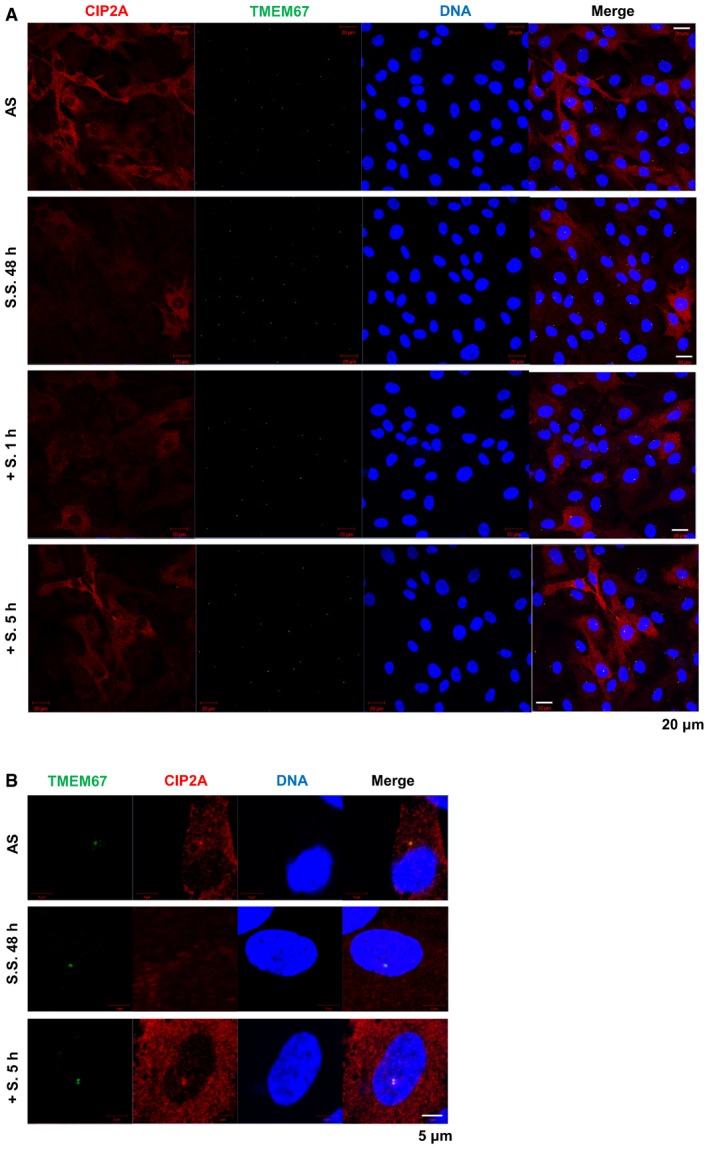
-
A, BImmunofluorescence images of endogenous CIP2A and TMEM67 in RPE1 cells. Cells under following conditions, asynchronized (AS), serum‐starved for 48 h (S.S. 48 h), serum re‐stimulation for 1 h after S.S. 48 h (S. 1 h), serum re‐stimulation for 5 h after S.S. 48 h (S. 5 h) were fixed with methanol and stained with antibodies specific for CIP2A (red), TMEM67 (green) and DNA was stained with DAPI (blue). Shown are the maximum projections from z stacks; scale bar = 20 μm (A) or 5 μm (B).
To determine the role of CIP2A in ciliogenesis, ciliogenesis was investigated in CIP2A‐overexpressing or CIP2A‐depleted cells. Compared with GFP‐transfected cells, cells overexpressing GFP‐CIP2A showed a decreased number of ciliated cells and decreased ciliary length (Fig 1D and E). In contrast, compared with control cells, CIP2A‐depleted cells mostly showed an increased ciliary length (Fig 1F and G, median ciliary length in control cells: 4.033, in CIP2A‐depleted cells: siCIP2A #1; 5.033, siCIP2A #2; 6.692, siCIP2A #3; 4.832 μm). When CIP2A was depleted, the percentage of ciliated cells increased under growing conditions. Under serum‐starved conditions, the basal level of ciliated cells increased, and CIP2A depletion additionally increased the number of ciliated cells. As we previously reported that the interaction between CIP2A and NEK2 enhances NEK2 kinase activity 25, and NEK2 promotes primary cilia disassembly through Kif24‐mediated microtubule depolymerization 31, ciliation in NEK2‐depleted cells was also examined. Consistent with previous reports, NEK2 depletion increased the levels of ciliation in both growing and serum‐starved conditions. Next, we determined whether CIP2A is involved in cilia disassembly by observing the percentage of ciliated cells after serum re‐stimulation in CIP2A‐ or NEK2‐depleted cells. The percentage of ciliated cells decreased to 43.9% after 24 h of serum re‐stimulation in control cells, indicating that cilia disassembly occurred. However, the percentage of ciliated cells was 67.4–77.2% in CIP2A‐depleted cells and 65.6% in NEK2‐depleted cells, and these percentages were significantly higher than those of control cells. There was also no additive increase in the percentage of ciliated cells when both CIP2A and NEK2 were co‐depleted (Fig 1H). These results indicate that CIP2A and NEK2 play a role in cilia disassembly following serum re‐stimulation. Altogether, these results indicate that CIP2A acts as a regulator of cilia disassembly and ciliary length.
CIP2A‐mediated ciliogenesis is not associated with the cell cycle and autophagy
We next determined whether CIP2A‐mediated cilia dynamics regulate the cell cycle. No significant change in cell cycle distribution was observed in either CIP2A‐depleted (Fig 2A) or CIP2A‐overexpressing (Fig 2B) cells. Because ciliary dynamics are regulated by the cell cycle as well as other factors, it is conceivable that CIP2A‐mediated ciliogenesis is not closely related to cell cycle progression. Meanwhile, autophagy promotes primary ciliogenesis 32, and CIP2A inhibits autophagy 33. Thus, it is possible that CIP2A induces cilia disassembly via autophagy inhibition. However, no significant change in LC3‐II was observed in either CIP2A‐depleted or CIP2A‐overexpressing cells (Fig 2C). Moreover, the percentage of ciliated cells in CIP2A‐depleted cells persisted, even when autophagy was inhibited following treatment with bafilomycin and chloroquine (Fig 2D). Collectively, these results indicate that CIP2A‐mediated ciliogenesis is not affected or regulated via cell cycle status or autophagy.
Figure 2. Induced primary cilia assembly in CIP2A‐depleted cells is not associated with the cell cycle and autophagy.
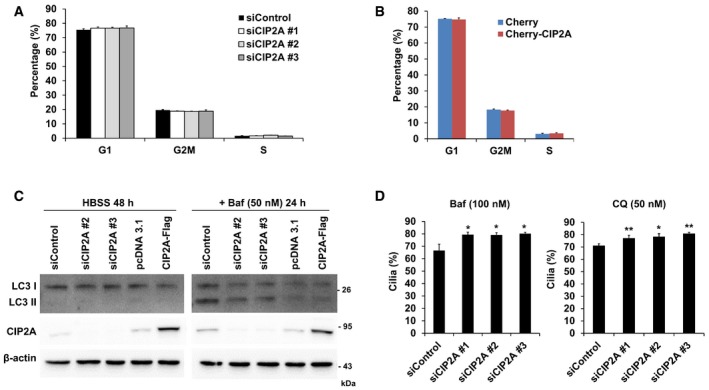
- RPE1 cells transfected with control, CIP2A #1, CIP2A #2, or CIP2A #3 siRNA were fixed and stained with propidium iodide solution for DNA and analyzed by flow cytometry to determine DNA content. The percentage of cells in G1, G2/M, and S phases of the cell cycle was analyzed using FlowJo software. The average of three independent experiments is shown, with error bars representing s.d.
- RPE1 cells transfected with Cherry or Cherry‐CIP2A were fixed and stained to analyze DNA content as described in (A). The average of three independent experiments is shown, with error bars representing s.d.
- RPE1 cells transfected with control siRNA, CIP2A #2 siRNA, CIP2A #3 siRNA, pcDNA3.1, or CIP2A‐Flag were cultured in HBSS for 48 h. Next, 50 nM bafilomycin A1 (Baf) was added for 24 h. Cell extracts were immunoblotted with antibodies against LC3, CIP2A, and β‐actin.
- RPE1 cells transfected with control, CIP2A #1, CIP2A #2, or CIP2A #3 siRNA were serum‐starved for 24 h, followed by treatment with 100 nM Baf or 50 nM chloroquine (CQ) for 24 h. The percentage of cells with primary cilia was determined for each condition (n > 44 cells/condition). The average of three independent experiments is shown, with error bars representing s.d. *P < 0.05, **P < 0.01 compared with siControl cells (one‐tailed Student's t‐test).
Source data are available online for this figure.
CIP2A regulates cilia disassembly through the activation of Aurora A kinase
NEKs play roles in ciliogenesis and/or function 34. Specifically, mutations in NEK1 and NEK8 have been identified in human ciliopathy patients 35, 36, and NEK4 is implicated in cilium stability 37. Hence, to elucidate the underlying molecular mechanism of CIP2A‐induced cilia disassembly, the interaction between CIP2A and NEK family members was examined. Among NEKs, NEK2 interacted most strongly with CIP2A. In addition, CIP2A bound to NEK6, NEK8, NEK9, and NEK11 (Fig 3A). Aurora A and Plk1 are well‐known kinases that reside in the centrosome and enhance ciliary resorption 20, 22, 38. CIP2A is also located in the centrosome. Thus, we investigated whether CIP2A forms a complex with Aurora A and Plk1 and found that CIP2A interacted with both kinases (Fig 3B).
Figure 3. CIP2A interacts with and activates Aurora A, which disassembles primary cilia.
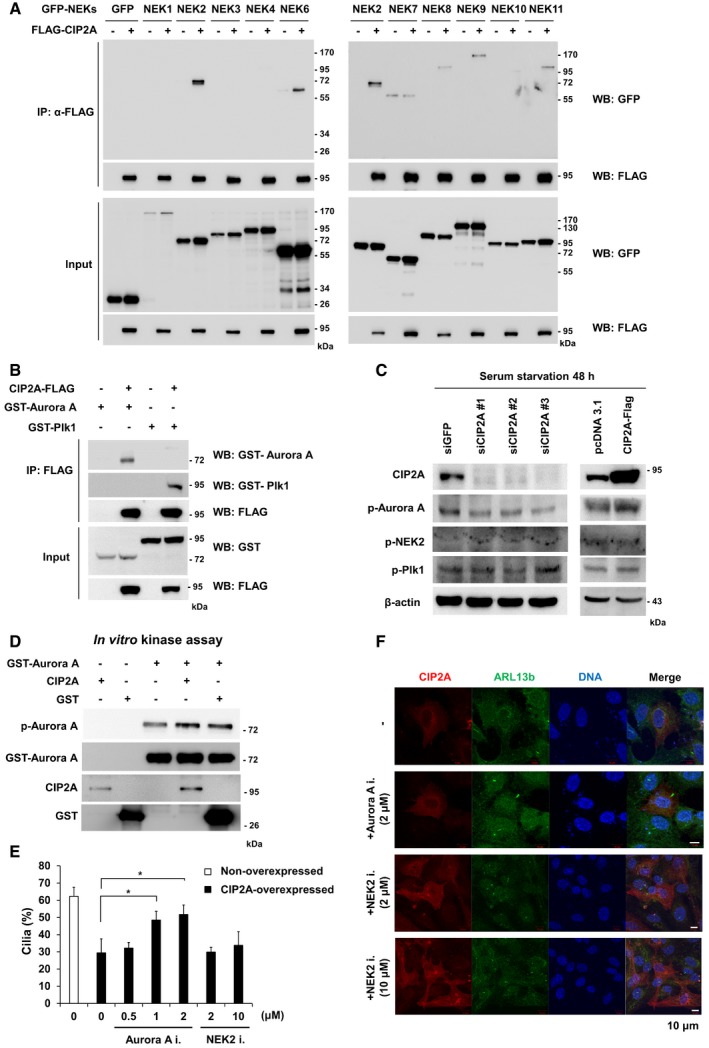
- HEK293 cells were used for a co‐immunoprecipitation analysis between FLAG‐tagged CIP2A and GFP‐tagged NEK family proteins. HEK293 cells lysates were immunoprecipitated with anti‐FLAG antibodies, and IP proteins were analyzed by immunoblot.
- FLAG‐tagged CIP2A constructs were coexpressed with the GST‐tagged Plk1 construct or the GST‐tagged Aurora A construct in HEK293 cells. FLAG‐tagged proteins were immunoprecipitated with anti‐FLAG antibodies, and the co‐immunoprecipitation of GST‐tagged proteins was determined by immunoblot.
- RPE1 cell lysates after siRNA transfection and serum starvation for 48 h were used for immunoblot analyses.
- Aurora A activity was measured using an in vitro kinase assay with recombinant proteins. After the kinase reaction, samples were subjected to SDS–PAGE. Samples were transferred onto a nitrocellulose membrane and processed for immunoblot using phosphorylation‐specific Aurora A antibodies.
- The percentage of CIP2A‐overexpressed and non‐overexpressed cells with primary cilia was evaluated after treatment with the Aurora A inhibitor MLN 8237, or the NEK2 inhibitor Rac‐CCT 250863 (n > 14 cells/condition) under serum‐starved condition. The average of three independent experiments is shown, with error bars representing s.d. *P < 0.05 compared with MLN 8237 non‐treated cells (one‐tailed Student's t‐test).
- Immunofluorescence images of RPE1 cells transfected with CIP2A‐FLAG following serum starvation for 48 h and treatment with MLN 8237 or Rac‐CCT 250863 for 24 h. Primary cilia were stained with antibodies specific for ARL13b (red). DNA was stained with DAPI (blue). Shown are the maximum projections from z stacks of representative non‐treated or treated transfected cells. Scale bar = 10 μm.
Source data are available online for this figure.
To determine whether CIP2A affects the activity of kinases, including Aurora A, Plk1, and NEK2, the phosphorylation level of those kinases was examined in CIP2A‐depleted cells. The level of phosphorylation of Aurora A at T288 was reduced in CIP2A‐depleted cells, whereas no alteration in Plk1 or NEK2 phosphorylation was observed (Fig 3C). Next, to determine whether CIP2A directly regulates Aurora A kinase activity, recombinant CIP2A was incubated with Aurora A. Aurora A phosphorylation was elevated by interaction with CIP2A (Fig 3D). Thus, we determined whether CIP2A‐mediated Aurora A activation is related to cilia disassembly. RPE1 cells overexpressing CIP2A were treated with the Aurora A inhibitor MLN 8237, and the number of ciliated cells was counted. CIP2A overexpression reduced the percentage of ciliated cells to 29.4% compared with non‐overexpressed cells, but treatment with the Aurora A inhibitor restored the percentage of ciliated cells to 51.5%. However, when CIP2A‐overexpressing cells were treated with the NEK2 inhibitor Rac‐CCT 250863, the percentage of ciliated cells did not change (Fig 3E and F). Furthermore, to address whether CIP2A localization at TZ is NEK2 dependent, CIP2A localization in NEK2‐depleted cells or NEK2 inhibitor‐treated cells was examined. The CIP2A localization did not change in both NEK2 knockdown or NEK2 inhibition (Fig EV2). These results indicate that CIP2A regulates primary cilia disassembly through the activation of Aurora A kinase, but not NEK2.
Figure EV2. CIP2A localization is NEK2 independent.
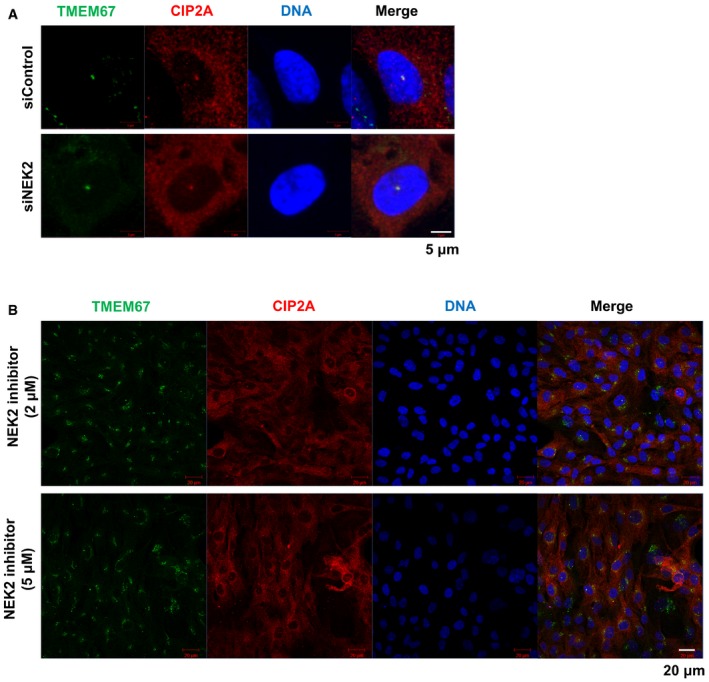
- Cells were transfected with control or NEK2 siRNAs for 48 h, and then fixed for staining and imaging. Shown are the maximum projections from z stacks; scale bar = 5 μm.
- Cells were treated with the NEK2 inhibitor Rac‐CCT 250863 for 24 h and then fixed for staining and imaging. Shown are the maximum projections from z stacks; scale bar = 20 μm.
CIP2A depletion activates glycolytic metabolism by altering the expression of glucose metabolism‐related genes
Because primary cilia play an essential role in Warburg‐like metabolic reprogramming 39, and CIP2A is involved in ciliogenesis, it is possible that CIP2A exerts a metabolic reprogramming function. To test this hypothesis, XF analysis was performed to measure the extracellular acidification rate (ECAR) in CIP2A‐depleted cells. ECAR measurement showed increased glycolytic activity, capacity, and reserve in CIP2A‐depleted cells compared with control cells (Fig 4A and B). Indeed, the extracellular level of lactate, a metabolic marker of enhanced glycolysis, was also elevated in the culture media of CIP2A‐depleted cells (Fig 4C). Gene expressions of HK2 and PKM2, which are associated with glycolysis, were elevated in CIP2A‐depleted cells. In contrast, the expression levels of PDK1 and LDHB, which accelerate the TCA cycle, were decreased in CIP2A‐depleted cells (Fig 4D). Additionally, we screened a panel of ~90 carbon energy sources to metabolically fingerprint CIP2A‐depleted cells. Cells adopted in fuel‐depleted medium were cultured with a spectrum of fuel sources to measure the preference of substrate utilization. Compared with control cells, CIP2A‐depleted cells grew rapidly in the presence of fuel, such as glucose, acetoacetate, and butyrate (Fig 4E). Together, these data suggest that CIP2A is strongly associated with metabolic reprogramming and substrate preference.
Figure 4. Depletion of CIP2A activates glycolysis by altering the expression of glucose metabolism‐related genes.
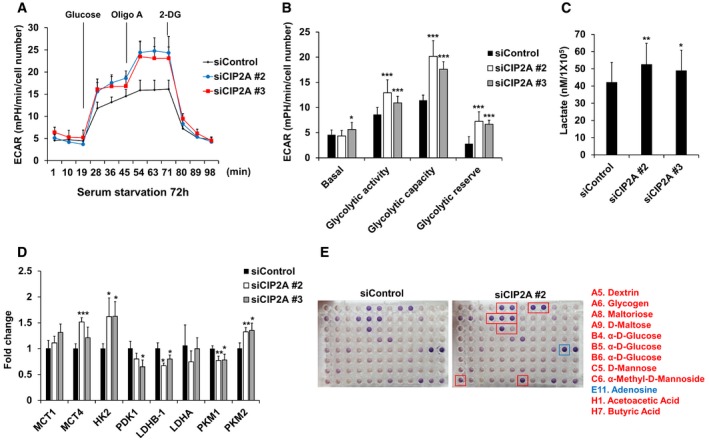
- RPE1 cells were seeded onto Seahorse Bioscience V7 cell culture plates (4 × 104/well) after siRNA transfection followed by serum starvation for 72 h. Time course for the measurement of ECAR indicates the basal conditions, followed by the sequential addition of glucose (10 mM), oligomycin (2 μM), and 2‐DG (20 mM). Data points are the average of a single representative experiment with error bars representing s.d. (n = 3 wells per condition). Experiments were independently repeated at least three times.
- Graph of the results described in (A). Basal indicates basal levels; glycolytic activity indicates glucose‐stimulated ECAR; glycolytic capacity indicates oligomycin‐stimulated ECAR; and glycolytic reserve indicates the difference between the maximum glycolytic capacity and the basal glycolytic rate. Data were normalized to cell number. Data points are the average of a single representative experiment with error bars representing s.d. (n = 12 per condition). Experiments were independently repeated at least three times. *P < 0.05, **P < 0.01, ***P < 0.001 compared with siControl cells (one‐tailed Student's t‐test).
- RPE1 cells were transfected with control, CIP2A #2, or CIP2A #3 siRNA. Cells were serum‐starved for 48 h, followed by glucose and amino acid starvation for 1 h. Glucose (10 mM) was added, and after 1 h, cell culture supernatants were collected and lactate production was measured using a Biovision kit. The average of three independent experiments is shown, with error bars representing s.d. *P < 0.05, **P < 0.01 compared with siControl cells (one‐tailed Student's t‐test).
- Quantitative RT–PCR (qRT–PCR) analysis of the expression of MCT1, MCT4, HK2, PDK1, LDHB‐1, LDHA, PKM1, and PKM2 in cells transfected with control or CIP2A #2 or CIP2A #3 siRNA for 24 h and then serum‐starved for 48 h. 18S rRNA was used as a normalization control. The average of three independent experiments is shown, with error bars representing the s.d. *P < 0.05, **P < 0.01, ***P < 0.001 compared with siControl cells (one‐tailed Student's t‐test).
- RPE1 cells were seeded onto BIOLOG plates PM‐M1 for 48 h after siRNA transfection followed by serum starvation for 24 h. The provided redox reagent was added to quantify substrate use at 590 and 750 nm. Red boxes indicate higher measurement, and blue box indicates lower measurement in siCIP2A #2 cells compared with siControl cells.
Primary cilia are necessary for glycolytic activity
Next, we determined whether increased cilia assembly correlates with elevated glycolysis, because CIP2A depletion increases cilia assembly. To address this question, XF analysis was performed with two cilia‐deficient cell lines, Kif3a‐KO MEF cells and CEP290‐KO RPE1 cells, which were confirmed by immunofluorescence staining of primary cilia using antibodies specific for ARL13b (Fig 5A and B). Both cilia‐deficient cell lines showed decreased glycolysis, and the difference in the ECAR level between ciliated and nonciliated cells was greatly enhanced under serum‐starved conditions. However, no significant difference in the oxygen consumption rate (OCR) was observed between ciliated and nonciliated cells (Fig 5C). In addition to the ECAR decrease in cilia‐deficient cells, the expression levels of glycolytic genes, including HK2 and LDHA, and the extracellular level of lactate were also diminished in nonciliated cells (Fig 5D and E). Collectively, these results indicate that primary cilia are necessary for glycolytic activity, especially under serum‐starved conditions.
Figure 5. Both glycolysis and gene expression associated with glucose metabolism are decreased in cilia‐deficient cell lines.
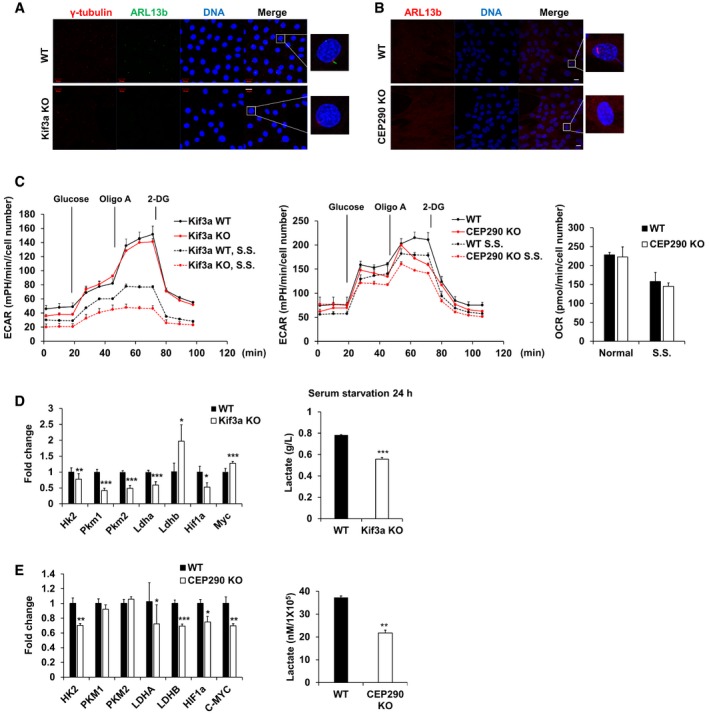
-
AImmunofluorescence images of WT and Kif3a KO MEF cells were serum‐starved for 24 h. Centrosome was stained with antibodies specific for γ‐tubulin (red), and primary cilia were stained with antibodies specific for ARL13b (green) and DNA was stained with DAPI (blue). Shown are the maximum projections from z stacks of representative cells. Scale bar = 20 μm.
-
BImmunofluorescence images of WT and CEP290 KO RPE1 cells which were serum‐starved for 24 h. Primary cilia were stained with antibodies specific for ARL13b (red), and DNA was stained with DAPI (blue). Shown are the maximum projections from z stacks of representative cells. Scale bar = 20 μm.
-
CKif3a WT and KO MEF cells or RPE1 WT and CEP290 KO cells were seeded onto Seahorse Bioscience V7 cell culture plates (4 × 104/well). For serum‐starved conditions, the media were changed for serum deprived media for 24 h. Time course for the measurement of ECAR indicates the basal conditions, followed by the sequential addition of glucose (10 mM), oligomycin (2 μM), and 2‐DG (20 mM). OCR indicates the average of oxygen consumption rate during the measurement of ECAR. Data were normalized to cell number. Data points are the average of a single representative experiment with error bars representing s.d. (n = 4 wells per condition). Experiments were independently repeated at least three times. S.S., serum starvation.
-
D, EqRT–PCR analysis of the expression of Hk2, Pkm1, Pkm2, Ldha, Ldhb, Hif1a, and Myc in Kif3a WT or Kif3a KO MEF cells cultured in the absence of serum for 24 h. Gapdh was used as a normalization control. Kif3a WT or Kif3a KO MEF cells were serum‐starved for 24 h, and then, cell culture media were collected and the lactate level was measured using YSI 2300 biochemical analyzer (YSI Life Science). The average of three independent experiments is shown, with error bars representing the s.d. *P < 0.05, **P < 0.01, ***P < 0.001 compared with WT MEF cells (one‐tailed Student's t‐test).
Glycolytic metabolism activation is primary cilia independent in CIP2A‐depleted cells
Next, we determined whether the metabolic shift toward the glycolytic pathway in CIP2A‐depleted cells depends on cilia assembly. To address this question, CIP2A was depleted in CEP290‐KO RPE1 cells, which cannot generate cilia. Surprisingly, CIP2A‐depleted CEP290‐KO RPE1 cells showed improved ECAR, regardless of cilia assembly (Fig 6A). Moreover, the mRNA expression levels of HK2 and MCT4 increased, while the expression level of LDHB decreased in CIP2A‐depleted CEP290‐KO cells, consistent with WT RPE1 cells (Fig 6B), indicating that the cilia‐mediated and CIP2A‐mediated glycolytic metabolic shifts are not interconnected.
Figure 6. Activation of glycolysis in CIP2A‐depleted cells is primary cilia independent.
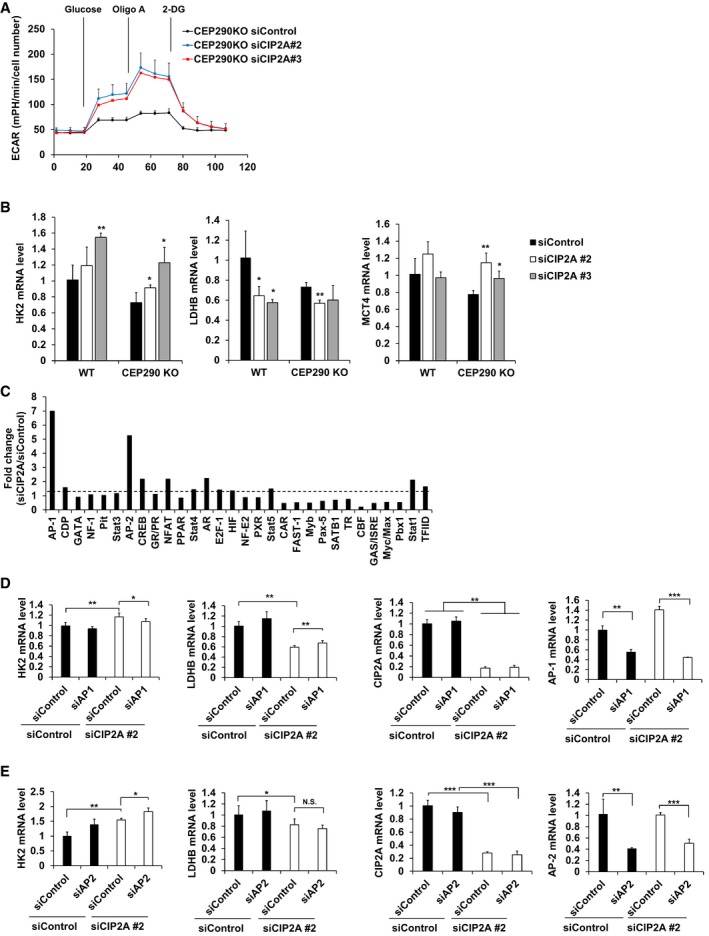
-
ARPE1 CEP290 KO cells were seeded onto Seahorse Bioscience V7 cell culture plates (4 × 104/well) after siRNA transfection followed by serum starvation for 48 h. Time course for the measurement of ECAR indicates the basal conditions, followed by the sequential addition of glucose (10 mM), oligomycin (2 μM), and 2‐DG (20 mM). Data points are the average of a single representative experiment with error bars representing s.d. (n = 4 wells per condition). Experiments were independently repeated at least three times.
-
BqRT–PCR analysis of the expression of HK2, LDHB, and MCT4 in CEP290 KO RPE1 cells transfected with control or CIP2A #2, CIP2A #3 siRNAs for 24 h, and then serum‐starved for 48 h cells. 18S rRNA was used as a normalization control.
-
CRPE1 cells were transfected with CIP2A #2 or control siRNAs, and nuclear extracts were prepared and subjected to TF profiling assay I. Fold change of activated transcription factors using TF activation profiling plate array assay.
-
D, EqRT–PCR analysis of the expression of HK2, LDHB, CIP2A, and AP‐1 (D) or AP‐2 (E) in RPE1 cells transfected with control or CIP2A #2 and AP‐1 or AP‐2 siRNAs for 24 h, and then serum‐starved for 48 h. 18S rRNA was used as a normalization control.
Because the genes involved in the glycolytic pathway are regulated in CIP2A‐depleted cells, we performed a transcription factor profiling plate array to identify transcription factors that play a role in the regulation of gene expression. The AP‐1 and AP‐2 transcription factors were activated in CIP2A‐depleted cells compared with control cells (Fig 6C). Next, we examined whether activation of the glycolytic pathway in CIP2A‐depleted cells could be restored by transcription factor knockdown. AP‐1 and AP‐2 were depleted, and the expression levels of glycolytic genes were observed; HK2 and LDHB expression was rescued by AP‐1 depletion but not by AP‐2 depletion (Fig 6D and E). This result indicates that AP‐1 activation is required for the elevated expression of glycolytic genes in CIP2A‐depleted cells.
Discussion
Because the primary cilium plays a role in the regulation of intracellular signaling pathways, including oncogenic signaling, it is conceivable that disturbances in cilia‐mediated signaling pathways can result in tumor formation 40, 41. Moreover, the loss of primary cilia has been detected in a variety of cancers, including breast, ovarian, melanoma, renal cell carcinoma, basal cell carcinoma, and pancreatic cancers. Trio kinases, including Aurora A kinase, Plk1, and NEK2, are localized in the centrosome and play an important role in cell cycle regulation. These kinases are also established as regulators of ciliary disassembly 20, 22, 23. From this perspective, identifying a novel regulator of the primary cilium could provide a better understanding of and a new molecular mechanism underlying primary cilia disassembly in many types of cancer cells. In this study, we showed that CIP2A overexpression stimulated the disassembly of primary cilia through the activation of Aurora A kinase. Because CIP2A is overexpressed in most human cancers, including breast, cervical, pancreatic ductal adenocarcinoma, lung, and gastric cancer 42, 43, 44, 45, 46, it is possible that enhanced CIP2A expression accelerates the loss of primary cilia in those cancer cells. Therefore, the inhibition of CIP2A may suppress tumor growth via the induction of cilia assembly.
In contrast to the observation that primary cilia are lost in many types of cancer, recent studies have shown the presence of primary cilia in several cancer types. Yasar et al 47 showed that primary cilia frequency is significantly elevated in lung adenocarcinoma, colon adenocarcinoma, follicular lymphoma, and pancreatic adenocarcinoma (PDAC). In addition, the presence of primary cilia is required for tumor initiation in addition to the activation of Hedgehog or Wnt signaling in medulloblastoma 48 and is significantly associated with poor prognosis in PDAC 49. However, it remains controversial whether primary cilia exert a pro‐ or anti‐tumor effect during tumor initiation and progression. Accumulating studies on the role of primary cilia in tumor cells will provide new insights into the relationship between ciliogenesis and tumor progression.
There are several reports that primary cilia are associated with metabolic regulation. Systemic ablation of Ift88 or Kif3a in adult mice results in hyperphagia‐induced obesity with increased leptin, glucose, and insulin levels 6. Primary cilia on hypothalamic neurons, particularly POMC neurons, contain receptors for the orexigenic neuropeptide Y, leptin, and melanin‐concentrating hormone receptor 1 (Mchr1), which are involved in food intake regulation 50, 51, 52. In addition to obesity, ciliopathies, including BBS and ALMS, are characterized by metabolic complications, such as type 2 diabetes mellitus (T2DM). The primary cilium of pancreatic β‐cells is necessary for insulin signaling; thus, when the cilium is disrupted, insulin receptor downstream signaling is impaired, leading to the development of T2DM 53. Moreover, Hedgehog (Hh) signaling rewires cellular metabolism through the cilium‐dependent Smo–Ca2+–Ampk axis in muscle and brown fat 39. Nonetheless, in studies in which the primary cilium was determined to be associated with metabolic diseases, it was not reported whether the primary cilium plays a direct role in glucose metabolism or is the underlying molecular mechanism of primary cilia‐mediated cellular metabolic regulation described. Here, we discovered that the primary cilium itself is necessary for enhancing glycolytic activity under serum‐starved conditions by showing that both the lactate level and the expression of glucose metabolism‐related genes were decreased in two cilia‐deficient cell lines, Kif3a KO MEF and CEP290 KO RPE1 (Fig 5C–E). Thus, we provide additional evidence that the primary cilia itself play a pivotal role in maintaining cellular metabolism homeostasis because the absence of primary cilia could lead to an aberrant state of cellular metabolism. Further studies will focus on elucidating the molecular mechanism underlying how the primary cilia are involved in the regulation of glycolytic activity. There are two possibilities. First, Hh signaling, which is heavily coupled to primary cilia in vertebrates 54, activates glioma (Gli) transcription factors (Gli2 and Gli3). The activated Gli shifts cell metabolism toward aerobic glycolysis by upregulating HK2 and PKM2 mRNA levels 55, 56. Thus, the Hh‐dependent transcriptional program could contribute to decreased glycolysis in cilia‐deficient cells. Second, conversely, the primary cilium plays an inhibitory role in canonical Wnt signaling through Dvl 57, 58, and the loss of primary cilia or an impaired basal body leads to hyperresponsiveness to external canonical Wnt stimuli 59, 60. As several studies have revealed that Wnt signaling directs a metabolic program of glycolysis 61, 62, 63, there could be a connection between altered glycolytic metabolism and Wnt signaling in cilia‐deficient cells.
CIP2A has attracted attention because of its oncogenic potential and tumor‐promoting role in several xenograft studies 24, 42, 64, 65, 66. It was recently revealed that CIP2A exerts an inhibitory effect on macroautophagy by acting as an allosteric inhibitor of mammalian target of rapamycin complex 1 (mTORC1)‐associated PP2A 33 and was identified as a degradation target of chaperone‐mediated autophagy (CMA), which is responsible for MYC stabilization 67. In addition, Peng et al 30 revealed that CIP2A depletion decreased the expression of LDHA and lactate production. Although these studies revealed a correlation between CIP2A and cancer metabolism, how CIP2A contributes to metabolic regulation remained unknown. In this study, we showed that CIP2A upregulates the expression of glucose metabolism‐related enzymes through the activation of the AP‐1 transcription factor (Fig 6D and E).
Because CIP2A‐depleted cells show a preference for butyrate and acetoacetate, it is likely that CIP2A is involved in substrate selectivity to carbon energy sources. Butyrate and acetoacetate are converted into acetyl‐CoA and incorporated into the TCA cycle to generate citrate. ACLY cleaves citrate into acetyl‐CoA and oxalate in an anaplerotic reaction, and these products are then used for fatty acid synthesis and replenishing malate for the TCA cycle, respectively. Although CIP2A depletion enhances glycolysis, CIP2A‐depleted cells shift the metabolic program from glycolysis to partial activation of the TCA cycle to fatty acid synthesis when CIP2A‐depleted cells are cultured with butyrate or acetoacetate. Meanwhile, butyrate exhibits histone deacetylase inhibitor (HDACi) activity; thus, treatment with butyrate leads to histone hyperacetylation that results in the inhibition of cell proliferation and the induction of apoptosis 68, 69. This chemoprevention effect of butyrate could be inhibited by low CIP2A expression because of the accelerated use of butyrate. Therefore, this chemopreventive metabolic shift could be another consequence of CIP2A overexpression in cancer cells. Acetoacetate is a ketone body that can be enzymatically reduced to β‐hydroxybutyrate, merging to the metabolic pathway of butyrate. Thus, acetoacetate also exerts a chemopreventive effect by decreasing tumor growth and ATP concentration 70.
It is known that reduction in cilia length in the hypothalamic neurons of mice increases food intake and decreases energy expenditure, leading to obesity 71. They found that leptin reduces cilia length in hypothalamic neuronal cells and cilia length is reduced in diet‐induced obese (DIO) mice in which leptin level is increased. In this study, serum‐starved cells show a decrease in CIP2A protein level and serum addition increases CIP2A level. Because CIP2A and cilia length are under the control of nutrient availability in cell level, if we determine whether CIP2A level is upregulated with cilia length decrease in hypothalamic neurons of DIO mice, it is going to be a good evidence to uncover physiological meaning of CIP2A‐mediated cilia length control.
In conclusion, this study revealed that CIP2A is involved in the regulation of ciliogenesis and metabolic reprogramming toward the glycolytic pathway by altering the expression of metabolic genes related to glycolysis. The primary cilia are also associated with metabolic reprogramming. The aim of the present study was to determine whether CIP2A‐mediated metabolic reprogramming is related to ciliogenesis‐mediated metabolic reprogramming. We found that the two pathways are not directly linked. As understanding cancer metabolism will provide insights into basic cancer pathophysiology and the clinical oncology of cancer 72, further research on the role of CIP2A and cilia in cancer metabolism will be invaluable.
Materials and Methods
Cell culture
The human retinal pigment epithelial cell line (hTERT‐RPE1), the RPE1‐Smo‐EGFP stable cell line, the RPE1‐CEP290 WT cell line, and the RPE1‐CEP290 KO cell line were generated using standard CRISPR/Cas9 system by Professor Joon Kim (Korea Advanced Institute of Science and Technology, KAIST). Detailed characterization of the cell line will be published elsewhere. Cell cultures were maintained in Dulbecco's modified Eagle's medium: Nutrient Mixture F‐12 (DMEM/F12, HyClone) supplemented with 10% FBS (Equitech) at 37°C in a humidified atmosphere of 5% CO2. HEK293T cells were cultured in DMEM (HyClone) supplemented with 10% FBS. The MEF Kif3a‐WT and the MEF Kif3a‐KO cell lines were obtained from Professor Hyuk Wan Ko (Dongguk University), and cell cultures were maintained in DMEM supplemented with 10% FBS. To induce cilia assembly, cells were incubated in serum‐free media for 48 h.
Immunofluorescence microscopy
RPE1 cells were seeded onto 0.1% gelatin‐coated glass coverslips in 12‐well plates. After 24 h, cells were fixed with 4% paraformaldehyde for 15 min for primary cilia staining and fixed with −20°C cold Methanol for 10 min for CIP2A staining, washed with PBS, and then permeabilized with 0.1% Triton X‐100 at room temperature for 1 min. After washing with PBS, the cells were permeabilized and blocked with 3% BSA for 30 min at room temperature. Coverslips were subsequently incubated in the following primary antibodies for 1 h: 1:400 anti‐ARL13b, 1:100 anti‐γ‐tubulin, 1:100 anti‐CIP2A, 1:100 anti‐CEP164, 1:100 anti‐CEP290, and 1:200 anti‐TMEM67. FITC‐ or TRITC‐coupled secondary antibodies (Santa Cruz Biotechnology) diluted in 3% BSA were used, and DNA was stained with DAPI. Images were acquired on an LSM‐700 Confocal Laser Scanning Microscope (Carl Zeiss) using a 63× oil immersion objective lens and ZEN software (Nikon). Fields were selected randomly for each well.
Quantification and statistical analysis
Primary cilia were stained with an antibody specific for ARL13B. To acquire the percentage of ciliated cells, cilia‐stained cells and total cells were quantified manually. At least 100–150 cells were counted in each experiment. Cilia length was measured using ImageJ software (NIH). The statistical data were obtained from at least two independent experiments performed in duplicate. qRT–PCR data were normalized by dividing all control and treatment values by the mean of the control. Differences between experimental values were considered significant when the P‐value was < 0.05 according to Student's t‐test.
Propidium iodide staining and flow cytometry
Cells were harvested at 48 h post‐siRNA transfection by trypsinization and fixed overnight with ice‐cold 70% ethanol. After washing with PBS containing 0.25% Triton X‐100, cells were suspended in PBS containing 1% BSA, 20 g/ml propidium iodide, and 10 g/ml RNase A and incubated for 30 min at 37°C in the dark. Samples were analyzed using a BD Biosciences FACScan, and the data of 10,000 events for each sample were analyzed with FlowJo software (LLC).
siRNA and plasmid transfection
siRNA transfection into RPE1 cells was performed using Lipofectamine RNAiMax Transfection Reagent (Invitrogen) according to the manufacturer's transfection protocol. The following siRNA oligonucleotides were used: to suppress CIP2A expression, CIP2A #1, 5′‐GGA GUG GUU UGU CGG AGC ATT‐3′; CIP2A #2, 5′‐GAC AGA AAC UCA CAC GAC UAU TT‐3′; and CIP2A #3, 5′‐AUU UGU GAC UUC GUA ACA AUA TT‐3′; to suppress AP‐1 expression, 5′‐UCC UGA AAC AGA GCA UGA CCC UGA AUG ATT‐3′; and to suppress AP‐2 expression, 5′‐CCG UCU CCG CCA UCC CUA UUA ACA ATT‐3′. For NEK2 silencing, ON‐TARGETplus SMARTpool (Dharmacon) siRNA oligonucleotides were used. Plasmid transfection into RPE1 cells was performed with X‐tremeGENE HP DNA Transfection Reagent (Roche) according to the manufacturer's transfection protocol. Plasmid transfection into HEK293T cells was performed using PEI solution.
Immunoblot analysis
For immunoblot analysis, cells were lysed in lysis buffer [50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, and 1% NP‐40, supplemented with a protease and phosphatase inhibitor mixture (Roche)]. Cell lysates were obtained by centrifugation for 15 min at 4°C at 20,000 g, and concentrations of the supernatants were quantified using the Pierce BCA Protein Assay Kit (Thermo Scientific). Total protein lysates were prepared using 5× SDS sample buffer and heating at 95°C for 10 min. Proteins were separated electrophoretically on a 12% SDS–polyacrylamide gel and transferred onto a 0.45‐μm pore size nitrocellulose membrane for 2 h. The membrane was incubated overnight with antibodies containing 3% BSA in TBS‐T (150 mM NaCl, 20 mM Tris–HCl (pH 8.0), and 0.05% Tween‐20) at 4°C, followed by incubation with HRP‐conjugated goat anti‐mouse or anti‐rabbit IgG (Fab) (Enzo Life Sciences) in 5% skim milk in TBS‐T at room temperature for 2 h. Proteins were visualized with ECL Western blotting reagent and analyzed on a Fusion Solo‐S image analyzer (Vilber).
Co‐immunoprecipitation assay
Cells were lysed with lysis buffer [50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, and 1% NP‐40, supplemented with a protease and phosphatase inhibitor mixture (Roche)]. Cell lysates were obtained by centrifugation for 15 min at 4°C at 20,000 g. For endogenous protein immunoprecipitation, cell lysates were incubated with 1 μg of antibody for overnight followed by incubation with protein G agarose beads (Amicogen, 2010005) for 2 h at 4°C. For overexpressed protein immunoprecipitation, cell lysates were incubated with FLAG M2 affinity gel (Sigma, A2220) for overnight at 4°C. The immunocomplexes were then washed with lysis buffer for four times, and the immunocomplexes were separated by SDS–polyacrylamide gel and immunoblotting analysis was performed as described above.
Antibodies and chemicals
The following antibodies and chemicals were used: ARL13b (Proteintech, 17711‐1‐AP), CIP2A (Santa Cruz Biotechnology, sc‐80659); NEK2 (BD Biosciences, 610594); γ‐tubulin (Sigma, T1592); CEP164 (Proteintech, 22227‐1‐AP), CEP290 (Bethyl Laboratories, A301‐659A), TMEM67 (Proteintech, 13975‐1‐AP), Aurora A (Cell Signaling, #4718S); p‐Aurora A (Cell Signaling, #2914S); LC3 (Novus Biologicals, NB100‐2331); β‐actin (Santa Cruz Biotechnology, sc‐47778); GFP (Santa Cruz Biotechnology, sc‐9996); FLAG M2 (Sigma, F1804); p‐Plk1 (Cell Signaling, #5472S); GST (Abcam, ab18183); bafilomycin A1 (Sigma, B1793); chloroquine (Sigma, C6628); Aurora kinase inhibitor MLN 8237 (Selleckem, S1133); and NEK2 inhibitor Rac‐CCT 250863 (Tocris, #4546).
XF 24 bioenergetics assay
Transfected or non‐transfected RPE1 cells were seeded at a density of 4 × 104 cells onto XF24 cell culture microplates (Seahorse Bioscience) in 350 μl of growth media and incubated at 37°C in a humidified 5% CO2 incubator overnight. To induce cilia assembly, cells were incubated in serum‐free media for 24 or 48 h, and OCR or ECAR was measured according to the manufacturer's protocol. OCR and ECAR values were normalized to each cell number and analyzed according to the manual.
Production and purification of recombinant CIP2A and in vitro kinase assay
pET28a‐CIP2A constructs were transformed into Escherichia coli strain BL21 cells, and recombinant protein was induced with 0.2 mM isopropyl‐beta‐d‐thiogalactopyranoside (IPTG) treatment overnight at 15°C. Cells were harvested by centrifugation at 3,000 g for 15 min at 4°C, resuspended in 0.1% Triton X‐100 in PBS, and sonicated with 10 cycles for 10 s. After centrifugation at 20,200 g for 15 min at 4°C, the lysates were incubated with Ni‐NTA beads for 24 h at 4°C. His‐CIP2A‐coupled Ni‐NTA beads were collected by centrifugation at 14,500 g for 1 min and washed three times with washing buffer [50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 0.5 mM EDTA, and 1% NP‐40]. Recombinant CIP2A was eluted with 500 μl of 20 mM imidazole and subsequently concentrated using a Centrifugal Filter column (Amicon, Millipore). For the in vitro measurement of Aurora A kinase, 0.3 μg of active recombinant GST‐tagged Aurora A (SignalChem) and purified His‐tagged CIP2A were used. For the in vitro measurement of NEK2 kinase activity, 0.5 μg of active recombinant GST‐tagged NEK2 (Upstate), purified His‐tagged CIP2A, and 1 μg of β‐casein (substrate) were used. The kinase reaction was performed in the presence of 5 μM ATP with 5 μCi γ32P‐ATP at 30°C for 30 min. The reaction was stopped by the addition of 5× SDS sample buffer and subjected to SDS–PAGE analysis. Samples were transferred onto a nitrocellulose membrane and processed for immunoblotting using a phosphorylation‐specific Aurora A antibody or autoradiography to detect phosphorylated NEK2.
Lactate production assay
After serum starvation for 48 h, cells were washed with PBS and incubated with Seahorse XF Base medium for 1 h. Glucose (10 mM) was added. After 1 h, cell culture supernatants were collected, and lactate production was measured using the Lactate Fluorometric Assay Kit according to the manufacturer's instructions (Biovision).
RNA extraction and qRT–PCR
Total RNA was extracted using Trizol (Takara) according to the manufacturer's instructions. Isolated total RNA was quantified on an Epoch 2 microplate Spectrophotometer (BioTek), and 1 μg of total RNA was reverse transcribed into cDNA using M‐MLV Reverse Transcriptase (Thermo Scientific). qRT–PCR was performed using Maxima SYBR Green (Thermo Scientific) with primers optimized for real‐time PCR. The sequences of all primers used in this study are provided in Table EV1. qRT–PCR was performed on an ABI7500 or a Quantstudio 3 real‐time PCR detection system (Applied Biosystems). Reaction specificity was confirmed by melting curve analysis. Relative gene expression was analyzed using the comparative C t method.
Transcription factor activity array
The profile of transcription factors activated in CIP2A‐depleted cells was screened using the TF Activation Profiling Array I (Signosis). After a 24‐h transfection, nuclear extracts (5 μg) from control and CIP2A‐depleted cells were extracted using the Nuclear Extraction Kit (Signosis). Transcription factor activity was profiled according to the manufacturer's instructions.
Phenotype microarrays
Phenotype microarray experiments were performed following the manufacturer's instructions provided by Biolog Inc. (http://www.biolog.com/). Briefly, control and CIP2A siRNAs were transfected into RPE1 cells, followed by serum starvation for 24 h and grown as described above and inoculated in BIOLOG plates PM‐M1 for 48 h at 37°C containing 5% CO2. The provided redox reagent was added to quantify substrate use at 590 and 750 nm on an Epoch Microplate reader (BioTek).
Data availability
All data generated during this study are included in this published article and provided as Source Data for each figure.
Author contributions
ALJ planned the research, performed the experiments, and wrote the manuscript. HIK, SH, and SL performed experiments and analyzed data. SJS, HJJ, BS, and JYP prepared constructs and key materials. E‐WL performed Fig 3A experiments and discussed the results. J‐SL, JHP, and MSL contributed to write the manuscript. YY supervised the entire project, co‐wrote, and proofread the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
We thank professors Joon Kim (Korea Advanced Institute of Science and Technology, KAIST) for providing RPE1‐Smo‐EGFP, RPE1‐CEP290 WT, RPE1‐CEP290 KO cell lines and Hyuk Wan Ko (Dongguk University) for providing MEF Kif3a‐WT and the MEF Kif3a‐KO cell lines. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (NRF‐2015M3A9B6027818, 2016R1A5A1011974, 2016R1A2B2011683, 2015R1D1A4A01016662).
EMBO Reports (2018) 19: e45144
References
- 1. Satir P, Pedersen LB, Christensen ST (2010) The primary cilium at a glance. J Cell Sci 123: 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gerdes JM, Davis EE, Katsanis N (2009) The vertebrate primary cilium in development, homeostasis, and disease. Cell 137: 32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. M'Hamdi O, Ouertani I, Chaabouni‐Bouhamed H (2014) Update on the genetics of bardet‐biedl syndrome. Mol Syndromol 5: 51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rahmouni K, Fath MA, Seo S, Thedens DR, Berry CJ, Weiss R, Nishimura DY, Sheffield VC (2008) Leptin resistance contributes to obesity and hypertension in mouse models of Bardet‐Biedl syndrome. J Clin Invest 118: 1458–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Collin GB, Marshall JD, King BL, Milan G, Maffei P, Jagger DJ, Naggert JK (2012) The Alstrom syndrome protein, ALMS1, interacts with alpha‐actinin and components of the endosome recycling pathway. PLoS One 7: e37925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, Yoder BK (2007) Disruption of intraflagellar transport in adult mice leads to obesity and slow‐onset cystic kidney disease. Curr Biol 17: 1586–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kwon O, Kim KW, Kim MS (2016) Leptin signalling pathways in hypothalamic neurons. Cell Mol Life Sci 73: 1457–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hassounah NB, Bunch TA, McDermott KM (2012) Molecular pathways: the role of primary cilia in cancer progression and therapeutics with a focus on Hedgehog signaling. Clin Cancer Res 18: 2429–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seeger‐Nukpezah T, Little JL, Serzhanova V, Golemis EA (2013) Cilia and cilia‐associated proteins in cancer. Drug Discov Today Dis Mech 10: e135–e142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuan K, Frolova N, Xie Y, Wang D, Cook L, Kwon YJ, Steg AD, Serra R, Frost AR (2010) Primary cilia are decreased in breast cancer: analysis of a collection of human breast cancer cell lines and tissues. J Histochem Cytochem 58: 857–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hassounah NB, Nagle R, Saboda K, Roe DJ, Dalkin BL, McDermott KM (2013) Primary cilia are lost in preinvasive and invasive prostate cancer. PLoS One 8: e68521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim J, Dabiri S, Seeley ES (2011) Primary cilium depletion typifies cutaneous melanoma in situ and malignant melanoma. PLoS One 6: e27410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schraml P, Frew IJ, Thoma CR, Boysen G, Struckmann K, Krek W, Moch H (2009) Sporadic clear cell renal cell carcinoma but not the papillary type is characterized by severely reduced frequency of primary cilia. Mod Pathol 22: 31–36 [DOI] [PubMed] [Google Scholar]
- 14. Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M (2009) Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res 69: 422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH Jr, Dlugosz AA, Reiter JF (2009) Primary cilia can both mediate and suppress Hedgehog pathway‐dependent tumorigenesis. Nat Med 15: 1055–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nigg EA, Stearns T (2011) The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol 13: 1154–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uetake Y, Loncarek J, Nordberg JJ, English CN, La Terra S, Khodjakov A, Sluder G (2007) Cell cycle progression and de novo centriole assembly after centrosomal removal in untransformed human cells. J Cell Biol 176: 173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim S, Zaghloul NA, Bubenshchikova E, Oh EC, Rankin S, Katsanis N, Obara T, Tsiokas L (2011) Nde1‐mediated inhibition of ciliogenesis affects cell cycle re‐entry. Nat Cell Biol 13: 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li A, Saito M, Chuang JZ, Tseng YY, Dedesma C, Tomizawa K, Kaitsuka T, Sung CH (2011) Ciliary transition zone activation of phosphorylated Tctex‐1 controls ciliary resorption, S‐phase entry and fate of neural progenitors. Nat Cell Biol 13: 402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA (2007) HEF1‐dependent Aurora A activation induces disassembly of the primary cilium. Cell 129: 1351–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seeger‐Nukpezah T, Liebau MC, Hopker K, Lamkemeyer T, Benzing T, Golemis EA, Schermer B (2012) The centrosomal kinase Plk1 localizes to the transition zone of primary cilia and induces phosphorylation of nephrocystin‐1. PLoS One 7: e38838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee KH, Johmura Y, Yu LR, Park JE, Gao Y, Bang JK, Zhou M, Veenstra TD, Yeon Kim B, Lee KS (2012) Identification of a novel Wnt5a‐CK1varepsilon‐Dvl2‐Plk1‐mediated primary cilia disassembly pathway. EMBO J 31: 3104–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spalluto C, Wilson DI, Hearn T (2012) Nek2 localises to the distal portion of the mother centriole/basal body and is required for timely cilium disassembly at the G2/M transition. Eur J Cell Biol 91: 675–686 [DOI] [PubMed] [Google Scholar]
- 24. Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, Ala‐aho R, Nielsen C, Ivaska J, Taya Y et al (2007) CIP2A inhibits PP2A in human malignancies. Cell 130: 51–62 [DOI] [PubMed] [Google Scholar]
- 25. Jeong AL, Lee S, Park JS, Han S, Jang CY, Lim JS, Lee MS, Yang Y (2014) Cancerous inhibitor of protein phosphatase 2A (CIP2A) protein is involved in centrosome separation through the regulation of NIMA (never in mitosis gene A)‐related kinase 2 (NEK2) protein activity. J Biol Chem 289: 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Warburg O (1956) On the origin of cancer cells. Science 123: 309–314 [DOI] [PubMed] [Google Scholar]
- 27. Cairns RA, Harris IS, Mak TW (2011) Regulation of cancer cell metabolism. Nat Rev Cancer 11: 85–95 [DOI] [PubMed] [Google Scholar]
- 28. Dang CV (2012) Links between metabolism and cancer. Genes Dev 26: 877–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dang CV (2013) MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med 3: a014217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peng B, Lei N, Chai Y, Chan EK, Zhang JY (2015) CIP2A regulates cancer metabolism and CREB phosphorylation in non‐small cell lung cancer. Mol BioSyst 11: 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim S, Lee K, Choi JH, Ringstad N, Dynlacht BD (2015) Nek2 activation of Kif24 ensures cilium disassembly during the cell cycle. Nat Commun 6: 8087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q (2013) Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502: 254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Puustinen P, Rytter A, Mortensen M, Kohonen P, Moreira JM, Jaattela M (2014) CIP2A oncoprotein controls cell growth and autophagy through mTORC1 activation. J Cell Biol 204: 713–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fry AM, O'Regan L, Sabir SR, Bayliss R (2012) Cell cycle regulation by the NEK family of protein kinases. J Cell Sci 125: 4423–4433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Otto EA, Trapp ML, Schultheiss UT, Helou J, Quarmby LM, Hildebrandt F (2008) NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol 19: 587–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thiel C, Kessler K, Giessl A, Dimmler A, Shalev SA, von der Haar S, Zenker M, Zahnleiter D, Stoss H, Beinder E et al (2011) NEK1 mutations cause short‐rib polydactyly syndrome type majewski. Am J Hum Genet 88: 106–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Coene KL, Mans DA, Boldt K, Gloeckner CJ, van Reeuwijk J, Bolat E, Roosing S, Letteboer SJ, Peters TA, Cremers FP et al (2011) The ciliopathy‐associated protein homologs RPGRIP1 and RPGRIP1L are linked to cilium integrity through interaction with Nek4 serine/threonine kinase. Hum Mol Genet 20: 3592–3605 [DOI] [PubMed] [Google Scholar]
- 38. Wang G, Chen Q, Zhang X, Zhang B, Zhuo X, Liu J, Jiang Q, Zhang C (2013) PCM1 recruits Plk1 to the pericentriolar matrix to promote primary cilia disassembly before mitotic entry. J Cell Sci 126: 1355–1365 [DOI] [PubMed] [Google Scholar]
- 39. Teperino R, Amann S, Bayer M, McGee SL, Loipetzberger A, Connor T, Jaeger C, Kammerer B, Winter L, Wiche G et al (2012) Hedgehog partial agonism drives Warburg‐like metabolism in muscle and brown fat. Cell 151: 414–426 [DOI] [PubMed] [Google Scholar]
- 40. Oh EC, Katsanis N (2013) Context‐dependent regulation of Wnt signaling through the primary cilium. J Am Soc Nephrol 24: 10–18 [DOI] [PubMed] [Google Scholar]
- 41. Goetz SC, Anderson KV (2010) The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11: 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Come C, Laine A, Chanrion M, Edgren H, Mattila E, Liu X, Jonkers J, Ivaska J, Isola J, Darbon JM et al (2009) CIP2A is associated with human breast cancer aggressivity. Clin Cancer Res 15: 5092–5100 [DOI] [PubMed] [Google Scholar]
- 43. Huang LP, Adelson ME, Mordechai E, Trama JP (2010) CIP2A expression is elevated in cervical cancer. Cancer Biomark 8: 309–317 [DOI] [PubMed] [Google Scholar]
- 44. Wang L, Gu F, Ma N, Zhang L, Bian JM, Cao HY (2013) CIP2A expression is associated with altered expression of epithelial‐mesenchymal transition markers and predictive of poor prognosis in pancreatic ductal adenocarcinoma. Tumour Biol 34: 2309–2313 [DOI] [PubMed] [Google Scholar]
- 45. Dong QZ, Wang Y, Dong XJ, Li ZX, Tang ZP, Cui QZ, Wang EH (2011) CIP2A is overexpressed in non‐small cell lung cancer and correlates with poor prognosis. Ann Surg Oncol 18: 857–865 [DOI] [PubMed] [Google Scholar]
- 46. Li W, Ge Z, Liu C, Liu Z, Bjorkholm M, Jia J, Xu D (2008) CIP2A is overexpressed in gastric cancer and its depletion leads to impaired clonogenicity, senescence, or differentiation of tumor cells. Clin Cancer Res 14: 3722–3728 [DOI] [PubMed] [Google Scholar]
- 47. Yasar B, Linton K, Slater C, Byers R (2017) Primary cilia are increased in number and demonstrate structural abnormalities in human cancer. J Clin Pathol 70: 571–574 [DOI] [PubMed] [Google Scholar]
- 48. Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez‐Buylla A (2009) Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med 15: 1062–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emoto K, Masugi Y, Yamazaki K, Effendi K, Tsujikawa H, Tanabe M, Kitagawa Y, Sakamoto M (2014) Presence of primary cilia in cancer cells correlates with prognosis of pancreatic ductal adenocarcinoma. Hum Pathol 45: 817–825 [DOI] [PubMed] [Google Scholar]
- 50. Loktev AV, Jackson PK (2013) Neuropeptide Y family receptors traffic via the Bardet‐Biedl syndrome pathway to signal in neuronal primary cilia. Cell Rep 5: 1316–1329 [DOI] [PubMed] [Google Scholar]
- 51. Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K (2008) Bardet‐Biedl syndrome proteins are required for the localization of G protein‐coupled receptors to primary cilia. Proc Natl Acad Sci USA 105: 4242–4246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stepanow S, Reichwald K, Huse K, Gausmann U, Nebel A, Rosenstiel P, Wabitsch M, Fischer‐Posovszky P, Platzer M (2011) Allele‐specific, age‐dependent and BMI‐associated DNA methylation of human MCHR1. PLoS One 6: e17711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gerdes JM, Christou‐Savina S, Xiong Y, Moede T, Moruzzi N, Karlsson‐Edlund P, Leibiger B, Leibiger IB, Ostenson CG, Beales PL et al (2014) Ciliary dysfunction impairs beta‐cell insulin secretion and promotes development of type 2 diabetes in rodents. Nat Commun 5: 5308 [DOI] [PubMed] [Google Scholar]
- 54. Huangfu D, Anderson KV (2006) Signaling from Smo to Ci/Gli: conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development 133: 3–14 [DOI] [PubMed] [Google Scholar]
- 55. Di Magno L, Manzi D, D'Amico D, Coni S, Macone A, Infante P, Di Marcotullio L, De Smaele E, Ferretti E, Screpanti I et al (2014) Druggable glycolytic requirement for Hedgehog‐dependent neuronal and medulloblastoma growth. Cell Cycle 13: 3404–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, Wickramasinghe R, Scott MP, Wechsler‐Reya RJ (2003) Transcriptional profiling of the Sonic hedgehog response: a critical role for N‐myc in proliferation of neuronal precursors. Proc Natl Acad Sci USA 100: 7331–7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A et al (2005) Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet 37: 537–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bergmann C, Fliegauf M, Bruchle NO, Frank V, Olbrich H, Kirschner J, Schermer B, Schmedding I, Kispert A, Kranzlin B et al (2008) Loss of nephrocystin‐3 function can cause embryonic lethality, Meckel‐Gruber‐like syndrome, situs inversus, and renal‐hepatic‐pancreatic dysplasia. Am J Hum Genet 82: 959–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, DeMartino GN, Fisher S et al (2007) Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet 39: 1350–1360 [DOI] [PubMed] [Google Scholar]
- 60. Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen MH, Chuang PT, Reiter JF (2008) Kif3a constrains beta‐catenin‐dependent Wnt signalling through dual ciliary and non‐ciliary mechanisms. Nat Cell Biol 10: 70–76 [DOI] [PubMed] [Google Scholar]
- 61. Lee SY, Jeon HM, Ju MK, Kim CH, Yoon G, Han SI, Park HG, Kang HS (2012) Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res 72: 3607–3617 [DOI] [PubMed] [Google Scholar]
- 62. Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, Long F (2013) WNT‐LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab 17: 745–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pate KT, Stringari C, Sprowl‐Tanio S, Wang K, TeSlaa T, Hoverter NP, McQuade MM, Garner C, Digman MA, Teitell MA et al (2014) Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J 33: 1454–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Laine A, Sihto H, Come C, Rosenfeldt MT, Zwolinska A, Niemela M, Khanna A, Chan EK, Kahari VM, Kellokumpu‐Lehtinen PL et al (2013) Senescence sensitivity of breast cancer cells is defined by positive feedback loop between CIP2A and E2F1. Cancer Discov 3: 182–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ma L, Wen ZS, Liu Z, Hu Z, Ma J, Chen XQ, Liu YQ, Pu JX, Xiao WL, Sun HD et al (2011) Overexpression and small molecule‐triggered downregulation of CIP2A in lung cancer. PLoS One 6: e20159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mathiasen DP, Egebjerg C, Andersen SH, Rafn B, Puustinen P, Khanna A, Daugaard M, Valo E, Tuomela S, Bottzauw T et al (2012) Identification of a c‐Jun N‐terminal kinase‐2‐dependent signal amplification cascade that regulates c‐Myc levels in ras transformation. Oncogene 31: 390–401 [DOI] [PubMed] [Google Scholar]
- 67. Gomes LR, Menck CFM, Cuervo AM (2017) Chaperone‐mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy 13: 928–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Davie JR (2003) Inhibition of histone deacetylase activity by butyrate. J Nutr 133: 2485S–2493S [DOI] [PubMed] [Google Scholar]
- 69. Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ (2012) The Warburg effect dictates the mechanism of butyrate‐mediated histone acetylation and cell proliferation. Mol Cell 48: 612–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fine EJ, Miller A, Quadros EV, Sequeira JM, Feinman RD (2009) Acetoacetate reduces growth and ATP concentration in cancer cell lines which over‐express uncoupling protein 2. Cancer Cell Int 9: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Han YM, Kang GM, Byun K, Ko HW, Kim J, Shin MS, Kim HK, Gil SY, Yu JH, Lee B et al (2014) Leptin‐promoted cilia assembly is critical for normal energy balance. J Clin Invest 124: 2193–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vander Heiden MG, DeBerardinis RJ (2017) Understanding the intersections between metabolism and cancer biology. Cell 168: 657–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Data Availability Statement
All data generated during this study are included in this published article and provided as Source Data for each figure.