

Abstract
Scar formation after brain injury is still poorly understood. To further elucidate such processes, here, we examine the interplay between astrocyte proliferation taking place predominantly at the vascular interface and monocyte invasion. Using genetic mouse models that decrease or increase reactive astrocyte proliferation, we demonstrate inverse effects on monocyte numbers in the injury site. Conversely, reducing monocyte invasion using CCR2−/− mice causes a strong increase in astrocyte proliferation, demonstrating an intriguing negative cross‐regulation between these cell types at the vascular interface. CCR2−/− mice show reduced scar formation with less extracellular matrix deposition, smaller lesion site and increased neuronal coverage. Surprisingly, the GFAP + scar area in these mice is also significantly decreased despite increased astrocyte proliferation. Proteomic analysis at the peak of increased astrocyte proliferation reveals a decrease in extracellular matrix synthesizing enzymes in the injury sites of CCR2−/− mice, highlighting how early key aspects of scar formation are initiated. Taken together, we provide novel insights into the cross‐regulation of juxtavascular proliferating astrocytes and invading monocytes as a crucial mechanism of scar formation upon brain injury.
Keywords: aryl hydrocarbon receptor, astrogliosis, monocytes, scar formation, sonic hedgehog pathway, traumatic brain injury
Subject Categories: Immunology, Neuroscience, Vascular Biology & Angiogenesis
Introduction
Mammalian brain injury results in permanent damage and scar formation, as opposed to scarless wound healing in other vertebrates 1, 2. Notably, adult neural stem cells (NSCs) react to injury in zebrafish and contribute to regeneration and repair 3, 4. In mammals, however, the closest equivalent cell type are proliferative reactive astrocytes that are either recruited from stem cell compartments 5, 6, 7, 8 or originate locally and resume proliferation and NSC hallmarks upon injury 9, 10, 11, 12. While beneficial functions of these reactive astrocytes have been reported 6, 7, 8, 13, 14, 15, increased proliferation of reactive astrocytes has also been suggested to contribute to the persisting astroglial scar at the injury site 15, 16, besides many more factors affecting scar formation after central nervous system (CNS) injury 17. Intriguingly, the subpopulation of astrocytes that proliferates after stab wound injury in the murine cerebral cortex is largely located with their soma directly at blood vessels 18. This specific location is consistent with increased inflammation and delayed closure of the blood–brain barrier (BBB) upon ablating proliferating astrocytes 13, 14. To which extent these effects are due to excessive cell death and how they may be mediated are still poorly understood.
In addition to the function of reactive astrocytes during scar formation is the role of invading immune cells. When blocking CCR2+ monocyte invasion into the injured brain 19, 20, 21, 22, 23, 24, several dissimilar and partially opposite consequences on behavioral outcome and lesion size have been reported in distinct injury conditions. For example, monocyte‐derived macrophages are needed for long‐term recovery after stroke 23, 25. In contrast, conditions without monocyte invasion using the CCR2−/− mice resulted in improved cognitive functions 20, a reduced cavity size after traumatic brain injury (TBI) 22, and reduced brain edema and expression of inflammatory mediators after stroke 24. These latter results suggest an adverse influence of invading monocytes in response to brain injury, while the former results demonstrate a beneficial role. Region‐ and lesion‐specific heterogeneity of glial cells and their accordingly differential reactions have recently been highlighted as a likely cause for discrepancies in the field of scar formation 26. Notably, none of the above studies performed in CCR2−/− mice examined glial cell reaction or proliferation, even though in vitro experiments suggest that cytokines and growth factors secreted by infiltrating immune cells modulate the proliferative response in resident glial cells 27. Toward a better understanding of the cross‐talk between monocytes and astrocytes after traumatic brain injury in vivo, here, we examined how modulating astrocyte proliferation affects monocyte invasion and how blocking monocyte invasion affects proliferation of astrocytes.
Results
Dynamics of astrocyte proliferation and immune cell infiltration after cerebral cortex ischemia or traumatic injury
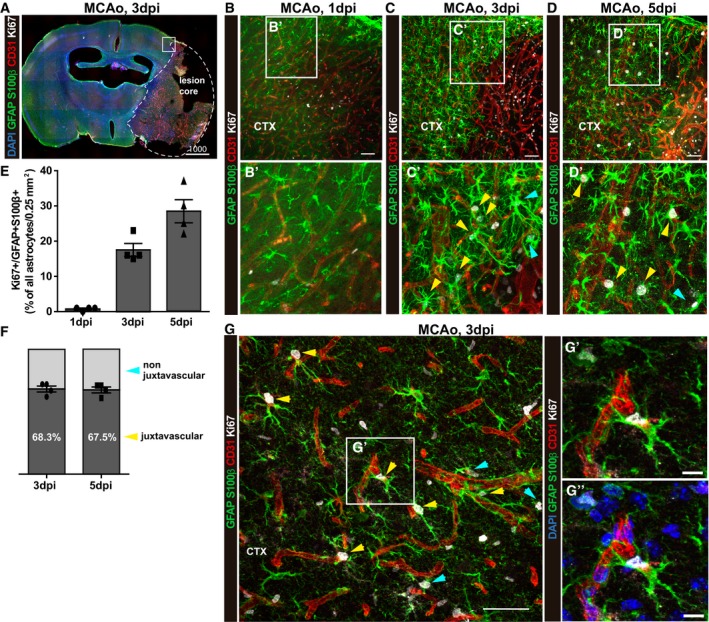
Prior to addressing the possible function of proliferating astrocytes, we first asked whether their location is biased to the vicinity of blood vessels, that is, takes predominantly place at juxtavascular or perivascular position: (i) in different injury conditions and (ii) over time in one injury condition. As most analyses to date had been done after stab wound injury, we examined an ischemic model, middle cerebral artery occlusion (MCAo). Using Ki67 immunostaining, we monitored proliferation at 1, 3, or 5 days post‐injury (dpi) and astrocytes were detected by co‐labeling for S100β and GFAP (Fig EV1A–D). Note that in the vicinity of the injury site virtually, all S100β+ astrocytes were also GFAP+ (see, e.g., Ref. 10). At 1 dpi, virtually no astrocytes were Ki67+ in the injured cerebral cortex grey matter (GM, Fig EV1B), while this percentage increased at 3 dpi to 17 and 29% at 5 dpi (Fig EV1E). To determine the position of the proliferating astrocytes with regard to the blood vessels, we triple stained for CD31 and performed 3D analysis using confocal stacks as described previously 10, 11, 18 to identify astrocytes whose somata are directly adjacent to blood vessels (Fig EV1G, G′ and G′′). Interestingly, at both 3 and 5 dpi, the majority of proliferating astrocytes were juxtavascular in direct vicinity to a CD31+ blood vessel (Fig EV1C, D, F and G). Thus, also after stroke, a relatively small proportion of all astrocytes (up to one‐third) proliferate and predominantly located at the blood vessel.
Figure EV1. Proliferation of astrocytes after MCAo is enriched at juxtavascular positions.
-
AStitched tiles epifluorescence micrographs of a representative brain slice at 3 dpi post‐MCAo. The lesion core is demarcated with a dashed line. A square shows the region of interest for quantifications.
-
B–DConfocal images from immunostaining for GFAP and S100β, Ki67, and blood vessels (CD31) from the penumbra of the ischemic lesion at 1, 3, and 5 dpi. Arrows point to proliferating astrocytes at juxtavascular positions (yellow) and not juxtavascular positions (cyan). The cell nuclei were counterstained with DAPI.
-
E, FHistograms depict the proportion of proliferating astrocytes among all (GFAP+S100β+) astrocytes in the lesioned GM (E) and the proportion of astrocytes proliferating at juxtavascular positions at 3 and 5 dpi (F). All data (individual data points, i.e., animals, are indicated as dots, triangles or squares) are represented as mean ± SEM per independent experiments (n = 4). Significance of differences between means was analyzed using one‐way ANOVA followed by Tukey's multiple comparison test.
-
GHigh‐power confocal micrographs of proliferating juxtavascular astrocytes (yellow arrows) with higher magnification in (G′ and G′′) showing the maximum projection of 10 single optical planes of the Z‐stack from the image shown in (G). Arrows point to proliferating astrocytes at juxtavascular positions (yellow) and not juxtavascular positions (cyan).
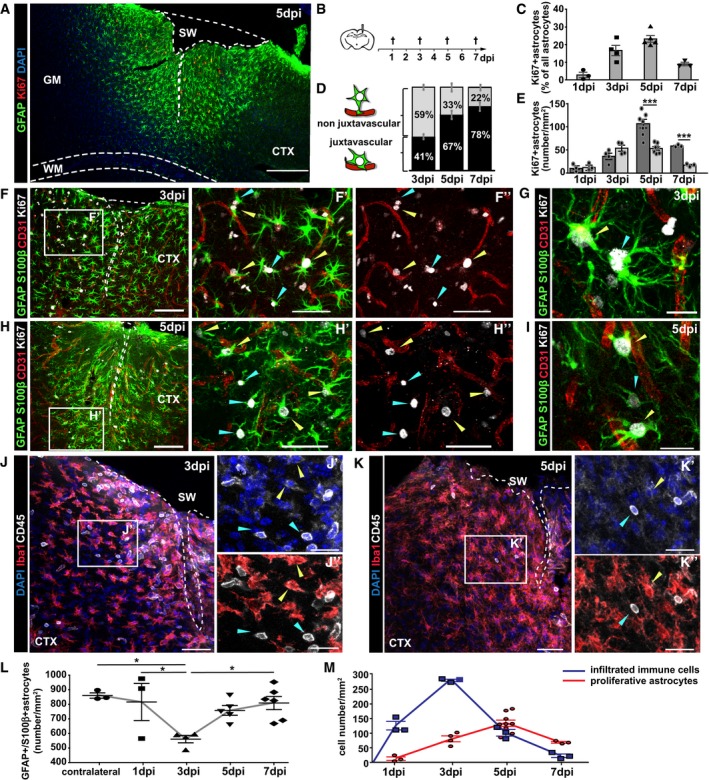
As the juxtavascular bias of astrocyte proliferation after MCAo was reached at 3 dpi, we examined the temporal profile after stab wound injury (Fig 1A and B). At 1 dpi, few astrocytes were Ki67+ in the injury site of the cerebral cortex GM, and their proportion significantly increased until 5 dpi when about a quarter of astrocytes were Ki67+ (Fig 1C and F–I). After this time point, the proliferation of reactive astrocytes decreased again, with virtually no Ki67+ astrocytes detectable at 2 weeks post‐injury (Fig 1C, at 10 dpi: 9 ± 4 Ki67+ astrocytes/mm2; at 28 dpi: 5 ± 3 Ki67+ astrocytes/mm2; see also Ref. 28). At 5 and 7 dpi, juxtavascular astrocytes were significantly increased among proliferating astrocytes, while this bias was not present at earlier stages (Fig 1D and E).
Figure 1. Inverse correlation between astrocyte proliferation and immune cell infiltration in the injured GM .
-
ARepresentative photomicrograph of frontal brain section immunostained with GFAP (astrocytes), Ki67 (proliferating cells) displaying the reactive border in the injured cerebral cortex (CTX) grey matter (GM) at 5 dpi. Note that the site of stab wound injury (SW) demarcated with white dashed line is restricted to the GM and does not involve the white matter (WM, demarcated by cyan dashed line).
-
BSchematic outline of the experiment.
-
CThe histogram depicts changes in the percentage of proliferating astrocytes among all astrocytes (GFAP+S100β+) per 0.25 mm2 of lesioned cortex.
-
D, EProportion of juxtavascular proliferating astrocytes (D) and their numbers (black bars) relative to the non‐juxtavascular proliferating astrocytes (gray bars) (E) at different time points after injury.
-
F–IExamples of immunostaining with GFAP, S100β, Ki67, and CD31 at 3 dpi (F, G) and 5 dpi (H, I). Reactive proliferating astrocytes at juxtavascular (yellow arrowheads) and non‐juxtavascular positions (cyan arrowheads).
-
J–LImmunolabeling of microglia (Iba1+CD45+, yellow arrowheads) and leukocytes (Iba1−CD45+, cyan arrowheads) in the penumbra at 3 dpi (J) and 5 dpi (K). The dashed lines indicate the site of injury. The cell nuclei were counterstained with DAPI. Changes in the total numbers of GFAP+S100β+ astrocytes at the injury site during the first week after lesion are summarized in (L).
-
MThe numbers of immune cells (CD45+Iba1−) peak at 3 dpi and decrease when the astrocyte proliferation reaches its peak at 5 dpi.
These data prompt the question whether the total number of astrocytes at this position indeed increases or whether their preferential proliferation compensates a predominant loss of astrocytes at the vascular interphase. Consistent with previous reports about astrocyte death after injury 29, astrocytes were significantly reduced in number at 3 dpi but recovered again at 5 dpi (Fig 1L) 30. The proportion of juxtavascular astrocytes was comparable to the contralateral hemisphere at 1–3 dpi (38%, Fig EV2), suggesting that cell death affects both astrocyte fractions equally. At 7 dpi, however, the proportion of juxtavascular astrocytes increased to 45% (Fig EV2B). Thus, the preferential transition of juxtavascular astrocytes into proliferative states starts around 4 dpi in the injured GM and helps to replenish astrocyte numbers with a preferential location at the juxtavascular interface.
Figure EV2. Proportion of juxtavascular astrocytes in the GM .
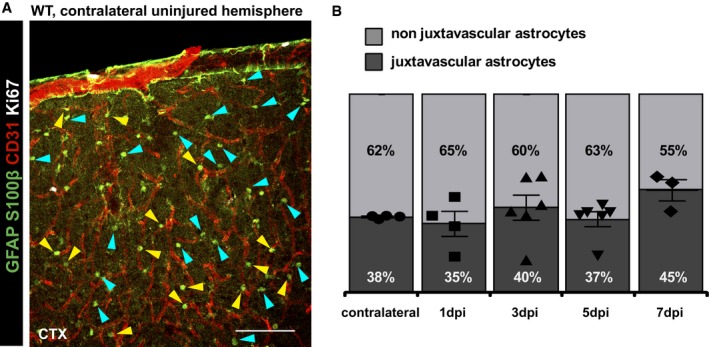
- Confocal images of S100β and GFAP immunostaining labeling all astrocytes in the GM of the uninjured contralateral hemisphere co‐stained for CD31 (vasculature) and Ki67 (proliferating cells). Arrowheads point to juxtavascular (yellow) and non juxtavascular (cyan) astrocytes in the uninjured GM parenchyma. Note that virtually no astrocytes proliferate in the uninjured site. Scale bar: 100 μm.
- Percentages of juxtavascular astrocytes among all GFAP/S100β immunolabeled astrocytes at different time points. All data (individual data points, i.e., animals, are indicated as separate symbols) are represented as mean ± SEM per independent experiments (n = 4 for the contralateral side and 1 dpi, n = 5 for 3 and 5 dpi, and n = 3 for 7 dpi). Significance of differences between means was analyzed using Kruskal–Wallis test, P = 0.0935.
To determine the temporal relation between juxtavascular astrocyte proliferation and monocyte invasion, we stained for CD45 (which is expressed at high levels by monocytes and lymphocytes 31, 32) and Iba1, enabling the distinction between recently infiltrated leukocytes (CD45+Iba1−) and reactive resident or previously infiltrated microglia (CD45+Iba1+; Fig 1J and K). CD45+Iba1− leukocytes were detectable within an area of 250 μm surrounding the injury site from as early as 1 dpi (Fig 1M), with their numbers peaking by 3 dpi (Fig 1J and M) and decreasing thereafter (Fig 1K and M) with virtually no CD45+Iba1− leukocytes detectable in the brain parenchyma at 7 dpi (Fig 1M). To identify the nature of the invading CD45+ cells after injury, we collected tissue at 3 dpi with a biopsy punch, dissociated the cells, and isolated CD45+ cells by magnetic‐activated cell sorting (MACS). Fluorescence‐activated cell sorting (FACS) was used to classify the identity of CD45+ cells by double‐staining for CD11b or T‐ and B‐cell markers (CD19 and CD3, respectively) (Appendix Fig S1). While lymphocytes were readily detectable in samples taken from spleen (Appendix Fig S1A), they were absent in brain tissue isolated from the lesion site where CD45+ cells were exclusively CD11b and CD45 high, that is, monocytes (Appendix Fig S1B). Notably, CD45+Iba1− cells did not proliferate as we detected virtually no Ki67+ cells. Conversely, the number of Iba1+ microglia (resident and recently infiltrated) that were also Ki67+ peaked 3 dpi (when monocyte invasion is highest) and had virtually vanished by 5 dpi (28 and Appendix Fig S2). Taken together, the temporal profile of monocyte invasion is close to 24 h slower than after controlled cortical impact (CCI) (a kinetic model of traumatic brain injury in the hippocampus 21) and parallels microglia proliferation but shows a negative correlation to proliferating juxtavascular astrocytes (Fig 1M, compare red and blue lines).
Loss of function: increased number of CD45+Iba1− cells in cerebral cortex after stab wound injury in mice with reduced proliferation of astrocytes
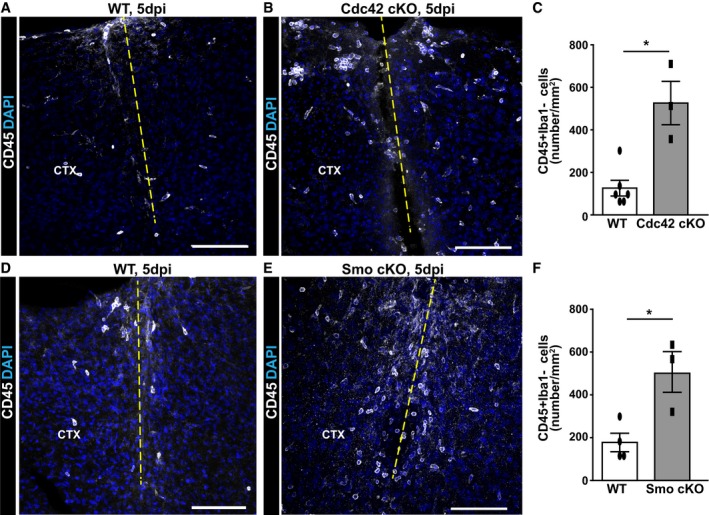
In order to examine to which extent reduced astrocyte proliferation would influence leukocyte invasion, we took advantage of two previously generated mouse models that have reduced proliferation of astrocytes. For example, astrocyte‐specific conditional deletion of cdc42 18, 33 by GLASTCreERT2 induced by tamoxifen reduces astrocyte proliferation to 40% of the control value at 5 dpi with the injury applied 3 weeks after tamoxifen treatment 18, 33. Using our injury model, the number of CD45+Iba1− cells at the injury site was increased 4× in the GM parenchyma of GLASTCreERT2/cdc42fl/fl mice compared to the tamoxifen‐treated controls (GLASTCreERT2/cdc42WT/WT; Fig 2A–C). This increase was not yet apparent at 3 dpi, but still persisted at 7 dpi (CD45+ cells/mm2 in WT 22.67 ± 6.12 vs. 77.00 ± 14.50 in Cdc42 cKO, unpaired t‐test, P = 0.026, n = 3). Importantly, control animals also received the same tamoxifen treatment, demonstrating increased recruitment of CD45+Iba1− cells depending on the genotype (cdc42 deletion in astrocytes) rather than the tamoxifen application.
Figure 2. Reduced astrocyte proliferation results in increased immune cell infiltration into the injured GM .
-
A–FMicrographs depict representative examples of immunostaining with CD45 in the injured GM of WT mice (A, D), conditional astrocyte‐specific Cdc42 knockout mice (B) and conditional astrocyte‐specific Smo knockout mice (E) at 5 dpi. The dashed lines indicate the site of injury. The cell nuclei were counterstained with DAPI. Quantitative analysis of these experiments is shown in (C) and (F). All data (dots and squares depict individual data points, i.e. animals) are represented as mean ± SEM from independent experiments, that is, tamoxifen‐treated animals. Significance of differences between means was analyzed using Mann–Whitney test and is indicated based on the P‐value (*P = 0.0275, n = 6 for WT and n = 3 for Cdc42 cKO in C; *P = 0.0498, n = 4 for WT and n = 3 for Smo cKO in F). Scale bars: 100 μm (A, B, D, E).
To rule out any non‐specific consequences resulting from the deletion of cdc42, we assessed the consequences of tamoxifen‐induced deletion of the sonic hedgehog (Shh) pathway receptor smoothened (smo) in astrocytes on monocyte invasion in the GLASTCreERT2/smofl/fl mice after injury. Specifically, when GLASTCreERT2/smofl/fl mice were injured 3 weeks after tamoxifen treatment, reactive astrocyte proliferation was reduced to 40%, as shown previously 11. When invading CD45+Iba1− monocytes were examined at 5 dpi, we noted a significant increase (4×) at the stab wound injury site compared to control (Fig 2D–F). Notably, recombination driven by tamoxifen in GLASTCreERT2 mice affects no more than half of all astrocytes in the above mice 18, 33. Thus, in conditions with only subtle changes in astrocyte proliferation (if 50% astrocytes reduce their proliferation by 60%, this is a total of 30% only), the number of invading monocytes was 4× increased. These results suggest that these cells are sensitive to changes in astrocyte proliferation, independent of the gene (cdc42 or smo) that was deleted.
Gain of function: increased proliferation of astrocytes results in diminished number of CD45+Iba1− cells in cerebral cortex after injury
The above observations in genetically modified mice with reduced proliferation of reactive astrocytes would predict that an increase in reactive astrocyte proliferation should result in reduced leukocyte infiltration. To test this, we induced expression of SMOM2‐YFP, a constitutively active form of smoothened 34, 35, selectively in adult astrocytes by GLASTCreERT2‐mediated recombination deleting a stop cassette 34 and performed stab wound injury 3 weeks after tamoxifen application. At 5 dpi, many astrocytes in the GM of GLASTCreERT2‐SMOM2 mice were YFP+. The number of Ki67+ cells and proliferating astrocytes was increased by more than 2× (Fig 3A–F). By 5 dpi, this lead to an overall increase of astrocyte numbers (Fig 3F), most of which were juxtavascular (57%). Thus, despite the random expression of SMOM2 in astrocytes, the proliferative reaction is still biased to the juxtavascular population. Importantly, the number of CD45+Iba1− cells at the injury site of mice with conditional SMOM2‐YFP activation was significantly reduced to about half the number in controls (Fig 3G–K), indicating that increased juxtavascular astrocyte proliferation strongly reduces CD45+Iba1− cell numbers within the injured GM. Notably, CD45+Iba1− cells were often detected within the blood vessels or meninges rather than in the parenchyma (Fig EV3A and B). Thus, the increase in astrocyte proliferation reduces monocyte numbers in the injury site probably by interfering with their invasion (see Discussion).
Figure 3. Increased astrocyte proliferation is paralleled with reduced infiltration of immune cells in mice with inducible SMOM2 expression in astrocytes at 5 dpi.
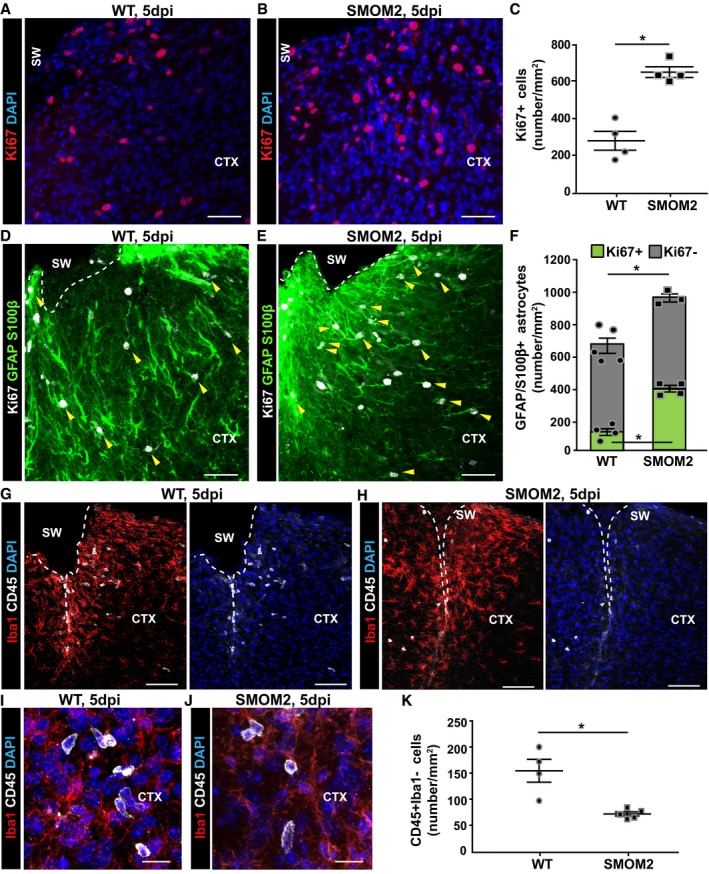
-
A, BDistribution of Ki67+ cells in the injured GM from WT (A) and conditional SMOM2 mice (B) at 5 dpi.
-
CQuantification of Ki67+ cells in the GM parenchyma at this time point after injury (*P = 0.029, n = 4).
-
D–FRepresentative micrographs of cortical sections obtained from WT (D) or SMOM2 (E) mice and immunostained for GFAP, S100β, and Ki67 at 5 dpi. Arrowheads point to proliferating reactive astrocytes (GFAP/ S100β+Ki67+). The numbers of proliferating (green bars, *P = 0.029, n = 4) and non‐proliferating astrocytes (gray bars) per area of the injured cortical GM are shown in (F). Note that the total numbers of astrocytes within the penumbra are significantly increased in the injury site in SMOM2 mice (*P = 0.036, n = 5 for WT and n = 3 for SMOM2).
-
G–JMicrographs depict examples of double‐immunostaining for Iba1 and CD45 in the cortical GM of WT (G, I) and SMOM2 (H, J) mice at 5 dpi.
-
KQuantitative analysis showing the numbers of CD45+ Iba1− cells within the injured GM from WT and SMOM2 mice at 5 dpi (*P = 0.016, n = 4 for WT and n = 5 for SMOM2).
Figure EV3. Microglia proliferation is increased after expression of SmoM2 in astrocytes by tamoxifen‐mediated recombination in GLASTC re ERT 2/SMOM2‐YFP mice.
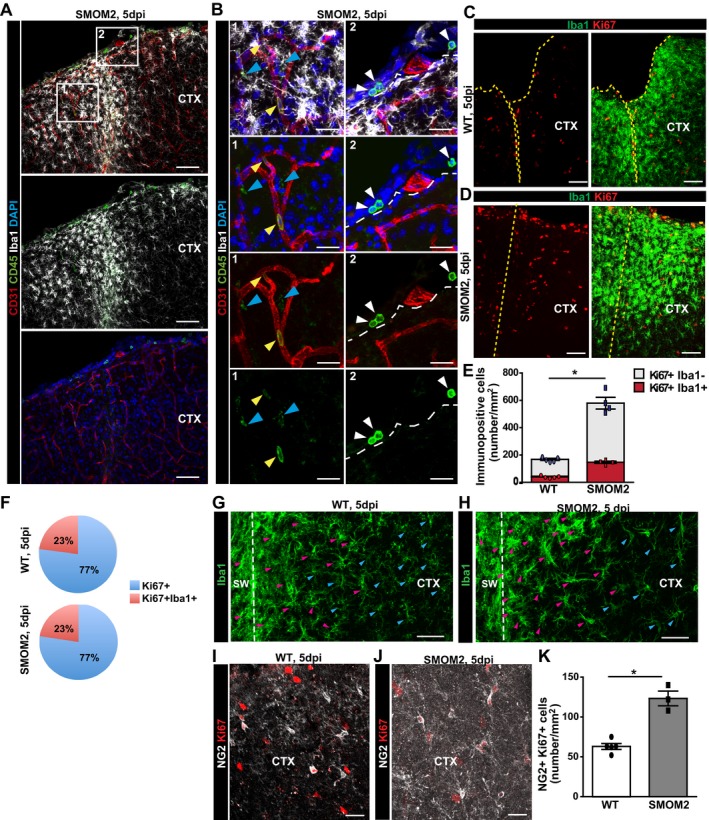
-
AMicrographs depict representative examples of immunostaining with Iba1, CD45, and CD31 in the injured GM of GLASTCreERT2/SmoM2‐YFP mice at 5 dpi.
-
BHigher magnification of the boxed areas (1 and 2 in A) showing CD45+Iba1+ cells (cyan arrowheads) at the injury site and CD45+Iba1− cells accumulating in the vessels within the penumbra (yellow arrowheads in B1) or in the epimeningeal spaces (white arrowheads in B2). The dashed lines indicate the border between the epimeningeal space and cortical parenchyma. The cell nuclei were counterstained with DAPI.
-
C, DRepresentative confocal micrographs of Iba1 and Ki67 immunostaining in the lesioned cortex of WT controls (C) and GLASTCreERT2/SmoM2‐YFP mice (D). The dashed lines indicate the site of injury.
-
EThe histogram displays total numbers of proliferating Ki67+ and Ki67+Iba1+ microglia in both mouse lines at 5 dpi. Significance was tested with Mann–Whitney test (*P = 0.016, n = 5 for WT and n = 4 for SMOM2).
-
FPie chart depicts the proportion of proliferating microglia among all proliferating cells at 5 dpi.
-
G, HConfocal pictures show more pronounced microgliosis toward the center of the lesion (dashed line) in a similar manner in both mice lines. Hypertrophic microglial cell bodies with thick processes are indicated by magenta arrowheads, and cyan arrowheads point to less reactive and more ramified microglial cells.
-
I, JRepresentative confocal images of NG2 and Ki67 immunostaining in the lesioned cortex GM of both lines.
-
KThe histogram depicts the number of proliferating NG2+ cells with a significant increase in GLASTCreERT2/SmoM2‐YFP mice (Mann–Whitney test, *P = 0.035, n = 5 for WT and n = 3 for SMOM2).
SMOM2 expression in astrocytes also affected other glial cell types, supposedly in a paracrine manner. Here, a higher number of Iba1+ cells proliferated and showed stronger activated morphology (Fig EV3C–H). The proliferation rate of NG2+ glia was also increased in the induced GLASTCreERT2‐SMOM2 mice (Fig EV3I–K). These data add further support to the cross‐talk between glial cells after injury, as SMOM2 expression in astrocytes affects NG2 glia and microglia proliferation as well as monocyte invasion.
Reduced infiltration of CCR2+ monocytes increases astrocyte proliferation
Toward a better understanding of the interplay between invading monocytes and juxtavascular astrocyte proliferation, we examined the injury site in the absence of invading monocytes due to the lack of the chemokine (C–C motif) receptor CCR2 replaced by RFP 36 (Fig EV4 for cells targeted by RFP). Consistent with previous work that showed this receptor as essential for trans‐endothelial migration of monocytes 36, 37, virtually no CD45+Iba1− cells had entered the cerebral cortex at 3 dpi (Fig EV4A and B) or 5 dpi in CCR2−/− mice (Figs 4A–D, and EV4C and D). Similar to the observations in GLASTCreERT2‐SMOM2, the very few CD45+Iba1− cells still present (Fig 4C) were typically located within the vessels rather than in the parenchyma (Fig EV4F and G). Notably, recruitment was virtually not affected in the heterozygous CCR2+/− mice (Fig 4B and D). More than 80% of the CD45+Iba1− cells were RFP+ in CCR2+/− mice at 3 and 5 dpi (Fig EV4E), confirming that the infiltrating cells were mostly monocyte‐derived (Fig EV4C–E)—a feature consistent with the FACS data shown above (Appendix Fig S1). There is, however, a small proportion of CD45+Iba1− cells at 3 and 5 dpi that are not RFP+ (Fig EV4E). Given the absence of lymphocytes in the injury site, these may be monocytes with low CCR2 expression levels and thus explain the small number of CD45+Iba1− cells still present in the injury site of CCR2−/− mice (Fig 4D). Taken together, these data show that CCR2+ monocytes, on large, invade the stab wound injury site and fail to do so in CCR2−/− mice. Importantly, RFP was only observed in CD45+ cells, confirming that no other cells in the brain parenchyma express CCR2 (see also Ref. 36).
Figure EV4. CCR2 deficiency impairs immune cell infiltration into the injured parenchyma.
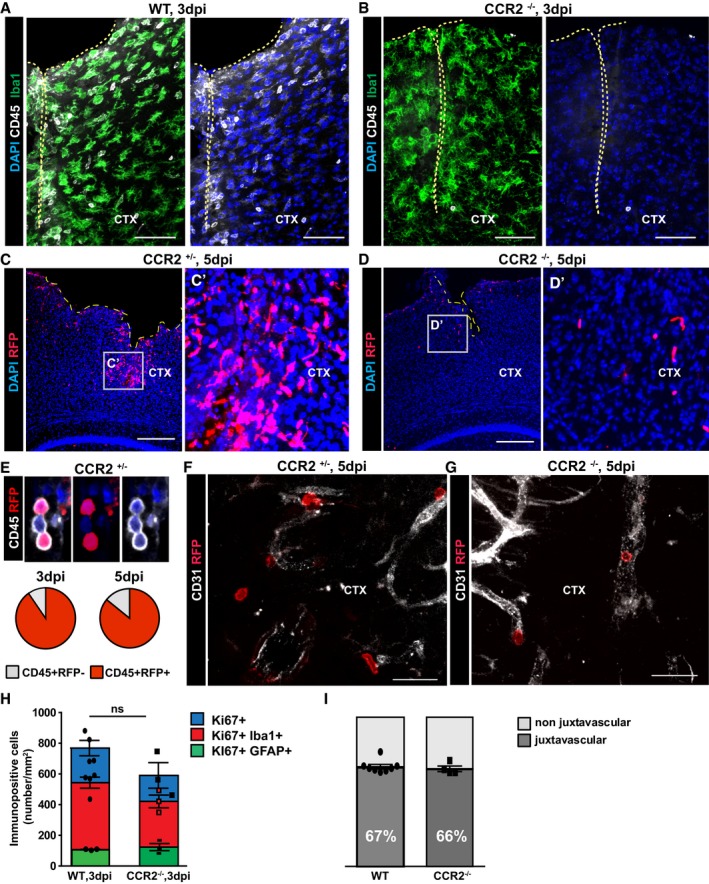
-
A, BConfocal images of Iba1 and CD45 double‐immunostaining in WT (A) and CCR2−/− (B) mice at 3 dpi. The cell nuclei were counterstained with DAPI.
-
C, DOverview of the lesioned cortex in heterozygous CCR2RFP/+ (C) and homozygous CCR2RFP/RFP (D) mice.
-
EConfocal images of immune cell infiltrates at 5 dpi show co‐localization of RFP expression and CD45. Pie graphs display the proportion of CD45 cells that were also RFP+ at 3 and 5 dpi.
-
F, GRFP expressing cells were detected in the parenchyma in heterozygous (F), but not in homozygous CCR2RFP/RFP mice where they remained inside blood vessels (G).
-
H, IHistograms depicting the number of proliferating glial cells at 3 dpi (H) (ns, P = 0.2286 for all Ki67+ cells, n = 4 for WT and n = 3 for CCR2−/−; P = 0.1143 for Iba1+Ki67+, n = 4 WT, n = 3 CCR2−/−; P = 0.600 for GFAP+Ki67+, n = 3) and the proportion of juxtavascular proliferating astrocytes among all proliferating astrocytes at the injury site of WT and CCR2−/− mice at 5 dpi (I). All data (individual data points, i.e., animals, are indicated as dots or squares) are represented as mean ± SEM per independent experiments (n ≥ 3). Significance of differences between means was analyzed using Mann–Whitney test.
Figure 4. Reduction in immune cell infiltration after stab wound injury in CCR2−/− mice increases proliferation at the injury site.
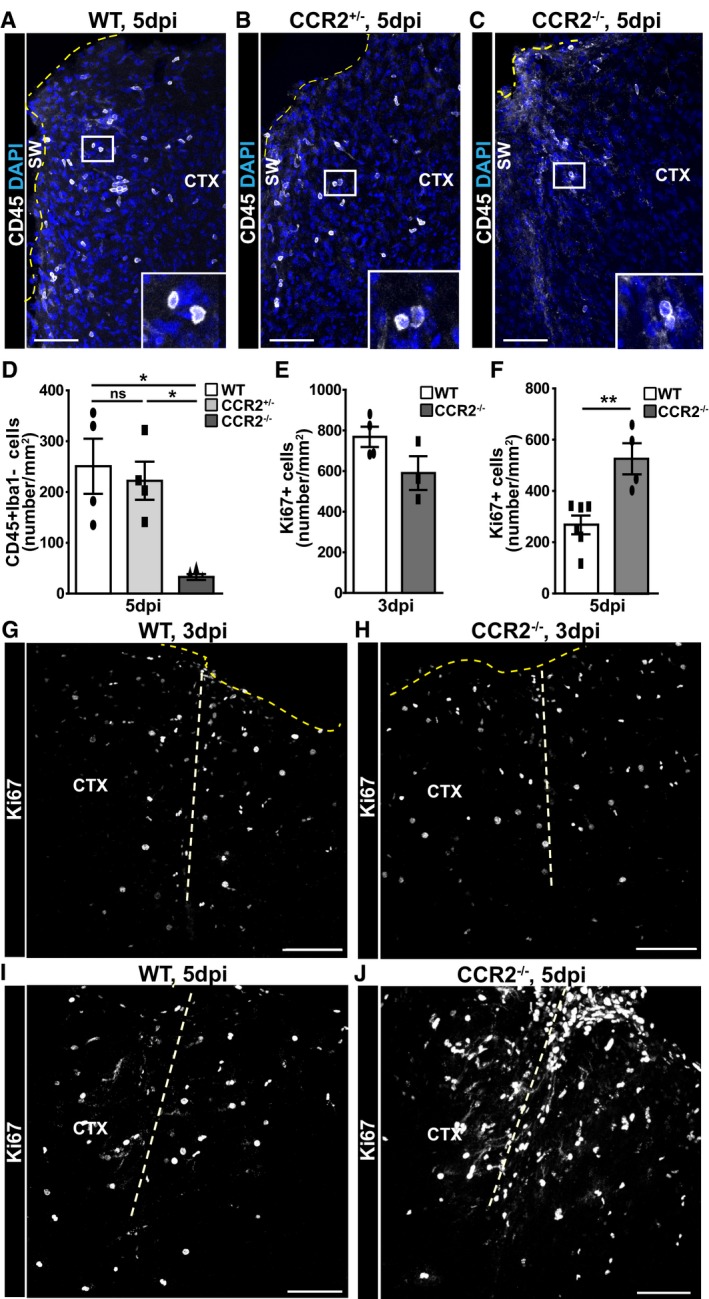
-
A–JConfocal images of CD45+ cells around the lesion site of WT (A), heterozygous CCR2+/− (B), and homozygous CCR2−/− (C) mice at 5 dpi and Ki67+ cells at 3 dpi (G, H) and 5 dpi (I, J) in WT (G, I) and homozygous CCR2−/− (H, J) mice. The dashed thick lines indicate the site of injury, the thin dark yellow dashed lines represent the pial surface. The cell nuclei were counterstained with DAPI. (D–F) Histograms depicting the numbers of CD45+ Iba1− cells (D) and Ki67+ cells in the injured cortical GM from mice of the indicated genotypes at 3 dpi (E) and 5 dpi (F) (one‐way ANOVA, Tukey's multiple comparison test, *P < 0.05, **P < 0.01, D: *P = 0.059, n = 4; E: P = 0.229, n = 4 for WT and n = 3 for CCR2−/−; and F: **P = 0.0087, n = 6 for WT and n = 4 for CCR2−/−), respectively. All data (dots and squares depict individual data points, i.e., animals) are represented as mean ± SEM. Significance of differences between means is indicated based on the P‐value (*P < 0.05, **P < 0.01). Scale bars: 100 μm.
Next, we examined whether the absence of invading monocytes in the CCR2−/− mice affects glial cell proliferation after stab wound injury. At 3 dpi, the number of Ki67+ cells was comparable between KO and WT genotypes (Fig 4E, G, and H). As this was also the case for proliferating astrocytes (Ki67+GFAP/S100β+) and Ki67+ NG2 glia (Fig EV4H), the initiation of macroglia proliferation does not depend on invading monocytes. At 5 dpi, however, we found a pronounced difference between the KO and WT genotypes with many more Ki67+ cells in the stab wound injury site of CCR2−/− compared to WT littermates (Fig 4F, I, and J). Moreover, at this time point, the number of proliferating astrocytes in the stab wound injury site of CCR2−/− was also significantly increased compared to WT (Fig 5A–C), suggesting an intriguing negative regulation between astrocytes and invading monocytes. Interestingly, astrocyte proliferation still occurs mostly at juxtavascular positions in CCR2−/− mice with 66 ± 3% of all proliferating astrocytes located with their soma at the blood vessels (Fig EV4I).
Figure 5. Reduction of immune cell infiltration after stab wound injury in CCR2−/− mice increases macroglia proliferation and microglia reactivity at the injury site.
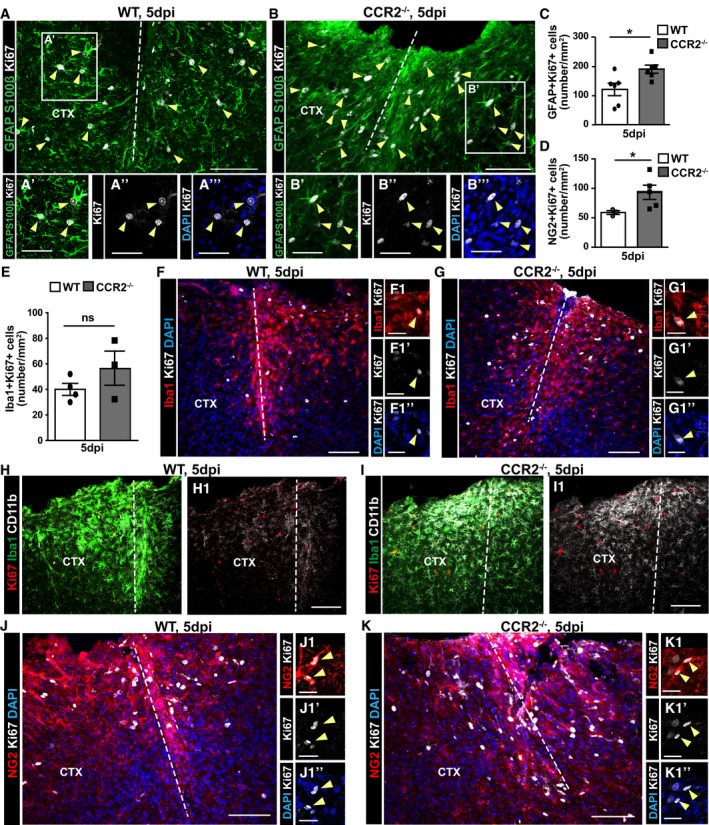
-
A–KMicrographs depicting immunostaining as indicated on the left side of the panels to identify proliferating Ki67+ glial cells in the injured GM of WT and CCR2−/− mice at 5 dpi (A, B, F–K). Examples of proliferating astrocytes (A, B: GFAP+S100β+Ki67+), microglia (F, G: Iba1+Ki67+), and NG2 glia (J, K: NG2+Ki67+) are indicated by yellow arrowheads and higher magnifications in the insets shown in (A′–A‴, B′–B‴, F1–F1″, G1–G1″, J1–J1″, K1–K1″). Panels (F1, G1, J1, and K1) do not show the same section as (F, G, J, and K), respectively, but were derived from the same experiment (same animal and same staining). The dashed lines indicate the site of injury. The cell nuclei were counterstained with DAPI. (C–E) Histograms depicting the number of proliferating astrocytes (C), NG2 glia (D), and microglia (E) in the penumbra at 5 dpi [Mann–Whitney test, C: *P = 0.016, n = 6; D: *P = 0.036, WT (n = 3) and CCR2−/− (n = 6); E: P = 0.400, WT (n = 4) and CCR2−/− (n = 3)]. All data (data points, i.e., individual animals, are depicted by dots and squares) are represented as mean ± SEM. Significance of differences between means is indicated based on the P‐value (*P < 0.05, **P < 0.01). Note the increase in CD11b+ microglia around the injury site in CCR2−/− mice (H, I). Scale bars: 100 μm (A, B, F–K), 25 μm (A′, B′, F1, G1, J1, K1).
We also examined microglial cells as key contributors to the reactive gliosis in WT and CCR2−/− mice. Interestingly, the proliferation of Iba1+ cells was not increased in the CCR2−/− mice compared to WT at 5 dpi (Fig 5E–G), but their activation state was affected with higher CD11b immunoreactivity and slightly elevated Tmem119 immunostaining observed in CCR2−/− mice at 5 dpi (Fig 5H and I; Appendix Fig S3), that is, at the time when the proliferation of astrocytes and NG2 glia was increased (Fig 5D and J–K). Thus, the failure of monocyte invasion elicits an increase in macroglia proliferation, apparently accompanied by enhanced microglia activation.
As astrocytes at juxtavascular positions may also affect the BBB closure after injury, we injected fluorescently labeled cadaverine at 3 dpi, a time when the BBB is still open after stab wound injury. Interestingly, the area covered by cadaverine was significantly reduced in the lesion site of CCR2−/− mice compared to WT (Appendix Fig S4), suggesting faster closure of BBB leakage in the absence of invading monocytes.
Reduced scar formation and improved neuroprotection in the injured GM of CCR2−/− mice
Next, we examined the long‐term consequences of the above described changes in the CCR2−/− mice. As the gliotic scar has been suggested to be derived from proliferating reactive astrocytes after spinal cord injury 14, 16, 38, we stained for GFAP at 28 dpi in both genotypes (Fig 6A and B). Surprisingly, GFAP immunoreactivity around the injury site was reduced in CCR2−/− mice (Fig 6B). Indeed, measuring the GFAP+ area at the injury site 28 dpi showed a significant reduction in CCR2−/− compared to WT mice (Fig 6E). Notably, not only the area but also the levels of GFAP immunostaining were reduced in the CCR2−/− mice compared to WT controls, clearly demonstrating a reduced astroglial scar in the cerebral cortex of CCR2−/− mice, despite the increased astrocyte proliferation at 5 dpi.
Figure 6. Reduced scar formation in the injured GM of CCR2−/− mice.
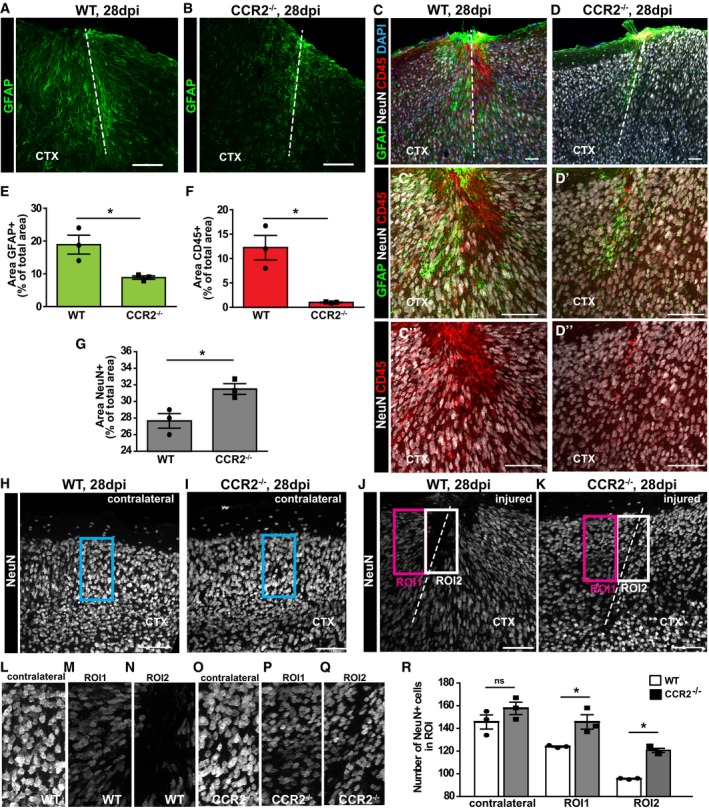
-
A, BRepresentative images depict the GFAP immunoreactive area at 28 dpi in WT (A) and CCR2−/− mice (B).
-
C, DTriple immunolabeling with NeuN, CD45, and GFAP in WT (C) and CCR2−/− mice (D) with higher magnifications to depict the NeuN‐free areas in the injured GM at 28 dpi (C′, D′) that are colonized by CD45+ macrophages (C′–D″).
-
E–GHistograms in (E–G) depict percentages of the area covered by immunoreactivity to GFAP (Unpaired t‐test, *P = 0.027, n = 3) (E), NeuN (Unpaired t‐test, *P = 0.038, n = 3) CD45 (Unpaired t‐test, *P = 0.011, n = 3) (F) and CD45 (Unpaired t‐test, *P = 0.011, n = 3) NeuN (Unpaired t‐test, *P = 0.038, n = 3) (G) normalized to the acquired area for each slice.
-
H–KRepresentative images of NeuN staining of the uninjured contralateral hemisphere (H, I) and ipsilateral lesion site (J, K), displaying the regions of interest (ROIs) selected for quantification of the number of NeuN+ cells.
-
L–QHigher magnifications of the ROIs.
-
RHistogram depicting the number of NeuN+ cells in ROI of the uninjured contralateral hemisphere, as well as ROI1 and ROI2 at the injury site. Statistical significance was examined with one‐way ANOVA (P < 0.0001) with Tukey's post hoc test, *P < 0.05, ns P > 0.05, n = 3.
We also stained for NeuN+ neurons and CD45 to scrutinize the remnant lesion site. In WT GM, the lesion is still visible at 28 dpi as a neuron free, NeuN‐negative zone filled with CD45+ cells (Fig 6C). Virtually no CD45+ area was detectable in the CCR2−/− mice at 4 weeks after injury (Fig 6D and F). Accordingly, the area covered by NeuN immunostaining was significantly larger in the CCR2−/− mice (Fig 6C, D, and G). After injury, neurons die or downregulate NeuN as evident in comparison with the contralateral site (compare, e.g., Fig 6L–N). To determine whether the number of NeuN+ neurons is changed in the CCR2−/− lesion site, we quantified these in defined regions of interest (ROI) of the border (ROI1, no CD45+ cells in WT) and the core of the lesion site, where CD45+ cells accumulate in WT (ROI2, Fig 6H–K). In the border, the number of NeuN+ neurons is 20% higher in CCR2−/− mice compared to WT controls (Fig 6M, P, and R). The same effect was observed in the lesion core (ROI2, Fig 6N, Q, and R). No difference in NeuN+ neurons was detectable in a comparable ROI on the contralateral uninjured side (Fig 6L, O, and R), demonstrating that the differences observed are due to the differential reaction to the injury. In both WT and CCR2−/− mice, the number of NeuN+ cells at the lesion site (ROI1, 2) was reduced compared to the contralateral site, but this reduction was less pronounced in the CCR2−/− (Fig 6R). These data, therefore, demonstrate a neuroprotective effect after stab wound injury upon lack of monocyte invasion and increased macroglial proliferation. Indeed, the faster recovery of astrocyte numbers may help protect neurons from damage given their functional relevance after injury 39.
Given the improvements at the stab wound lesion site in the cerebral cortex GM of CCR2−/− mice, we next examined components of the extracellular matrix (ECM) as further indicators of scar formation. Immunostaining for glycosaminoglycans such as in chondroitin sulfate proteoglycans (CSPGs) with the CS‐56 antibody was very low on the contralateral side (Fig 7A), but high in the scarred GM region close to the pial surface at 28 dpi in WT mice (Fig 7B). Interestingly, however, CS‐56 immunostaining was reduced in the stab wound region of CCR2−/− compared to WT mice (Fig 7C and D). Collagen I and IV immunoreactivity was also upregulated upon stab wound injury in WT mice 28 dpi, particularly strong surrounding the blood vessels and within the stab wound injury site (Fig 7E, Appendix Fig S5). Interestingly, collagen IV was barely detectable in the injured CCR2−/− cerebral cortex (Fig 7F), while less differences were observed in collagen I immunostaining (see e.g. Ref. 40; Appendix Fig S5). However, there were fewer reactive astrocytes at the collagen‐I‐positive scar and the collagen‐I‐positive vasculature around the injury site in the CCR2−/− compared to WT mice (Appendix Fig S5). Taken together, all the above described parameters show reduced scar formation and increased neuroprotection after stab wound injury in CCR2−/− cerebral cortex, suggesting a rather detrimental role for invading monocyte in these processes.
Figure 7. Reduced extracellular matrix deposition at the injured GM of CCR2−/− mice.

-
A–FThe injured GM parenchyma (ipsilateral) at 28 dpi is characterized by upregulation of CSPG, as detected by immunoreactivity for CS‐56 (A′–D′) and GFAP (A–D), when compared with the uninjured GM parenchyma (contralateral). Of note, WT mice (A–B′) display increased levels of CS‐56 immunostaining compared to CCR2−/− mice (C–D′). Collagen IV staining of cortical sections reveals an increased deposition predominantly in the basement membrane surrounding the vasculature in regions of GFAP upregulation (E–E″) that was much reduced in CCR2−/− mice (F–F″) at 28 dpi. The site of injury is indicated by a dashed line. The cell nuclei were counterstained with DAPI. Scale bars: 100 μm.
Most importantly, these improvements seen at the injury site in CCR2−/− cortices persisted into later stages with it being difficult to locate remnants of the lesion in these mice 3 months post‐injury. Indeed, the GFAP+ lesion site was only detectable in few sections (Appendix Fig S6B). This is in contrast to WT mice where we could readily detect the GFAP+ scar in the cerebral cortex GM 3 months post‐injury (Appendix Fig S6A). Thus, the astrogliotic scar is permanently reduced in the CCR2−/− mice despite the increase in astrocyte proliferation. The latter also prompted us to examine the total numbers of astrocytes in the lesion site to determine whether the increase in astrocyte proliferation leads to a permanent increase in the former lesion site of CCR2−/− mice. Interestingly, this is not the case and astrocyte numbers were well comparable at the lesion to corresponding regions in the contralateral cortex of CCR2−/− mice (Appendix Fig S6C), indicating that astrocyte homeostasis is restored.
Proteome analysis of the lesion site at 5 dpi in WT and CCR2−/− mice
Given the strong reduction in scar formation, we aimed to determine whether changes at earlier stages, when glial cell proliferation and reactivity are affected, may relate to reduced scarring at later stages. Toward this aim, we took an unbiased proteomic approach to understand the sum of all the differences between the injury region in CCR2−/− mice compared to WT. In tissue collected 5 dpi with a biopsy punch of 2.5 mm diameter, we detected a total of 4,656 proteins (Database: Swissprot mouse). This quantitative analysis showed that GFAP was upregulated in both WT and CCR2−/− by 4× and no difference in GFAP levels were detectable 5 dpi. This suggests that the reduction in the GFAP+ area at later stages (28 dpi) is not due to an initially reduced upregulation of this protein. We further noted much higher levels (more than 100×) of tenascin‐C (Tn‐C) in WT compared to CCR2−/−, an intriguing observation given the adverse effects Tn‐C has on the lesion size and scar formation 41. Reduced Tn‐C levels surrounding the injury site in CCR2−/− mice at 5 dpi was confirmed by immunostaining (Fig EV5). Likewise, we observed reduced levels of Stat1 in proteomics from WT compared to CCR2−/− mice, a key transcription factor regulating the reaction to injury and neurodegeneration 42.
Figure EV5. Tenascin‐C is reduced in the penumbra of CCR2−/− mice after stab wound injury.
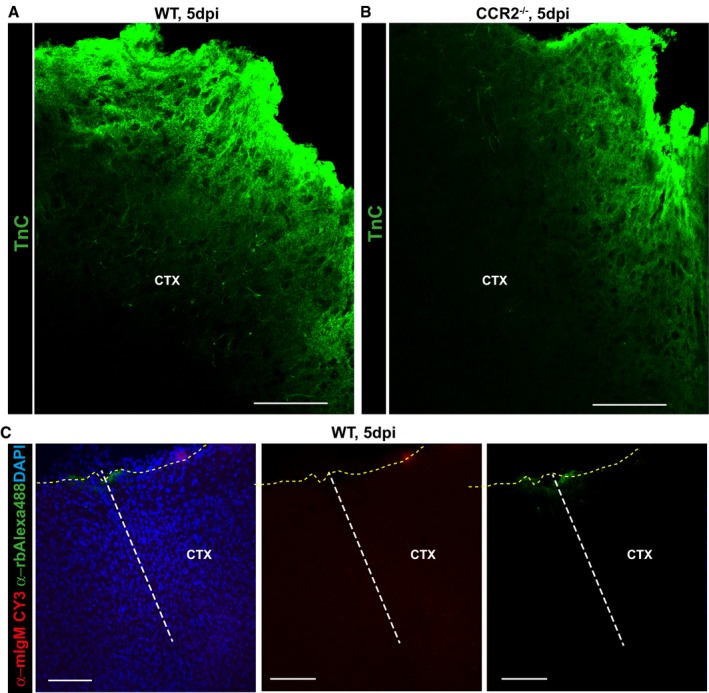
-
A–CConfocal images displaying tenascin‐C (Tn‐C) immunostaining in the injured GM from WT (A) and CCR2−/− (B) mice at 5 dpi. Confocal images of control staining with only the secondary antibodies as indicated in the panel used for immunolabeling of ECM components showing absence of non‐specific binding of these antibodies at injury site (C). The dashed lines indicate the site of injury (white) and the pial surface (yellow). The cell nuclei were counterstained with DAPI. Scale bars: 75 μm (A, B), 150 μm (C).
These differences, however, were not significant due to the variation between samples not prepared and analyzed at the same time. For a more stringent assessment, we only compared samples run simultaneously (n = 4 per genotype) and only significant differences (P ≤ 0.05, n = 4) were included in the analysis. This revealed 311 proteins significantly different in their normalized abundance (Appendix Table S1) among the biopsies at 5 dpi from WT and CCR2−/− mice (Fig 8A). Among these are interesting individual proteins, such as Prominin1, a stem cell marker, or GSK3a, a core signaling component, significantly increased more than 2× in the injury site of CCR2−/− mice (Fig 8A, blue, B). Conversely, the glycosyltransferase B3gat2 involved in synthesis of gylcosaminoglycans is 8× higher in WT compared to CCR2−/− mice (Fig 8A, red, B), suggesting an early start of the differences in ECM observed at 4 weeks after injury, as described above. Likewise, inter‐alpha‐trypsin inhibitor heavy chain H3 (Itih3) is detected at higher levels in the WT lesion site (Fig 8A, red, B) and has been shown to bind hyaluronic acid and stabilize ECM 43, further supporting differences regulating ECM composition already at 5 dpi between WT and CCR2−/− mice. Thus, the lack of monocyte invasion strongly affects ECM formation at early stages after lesion.
Figure 8. Proteome analysis of the lesion site at 5 dpi in WT and CCR2−/− mice.
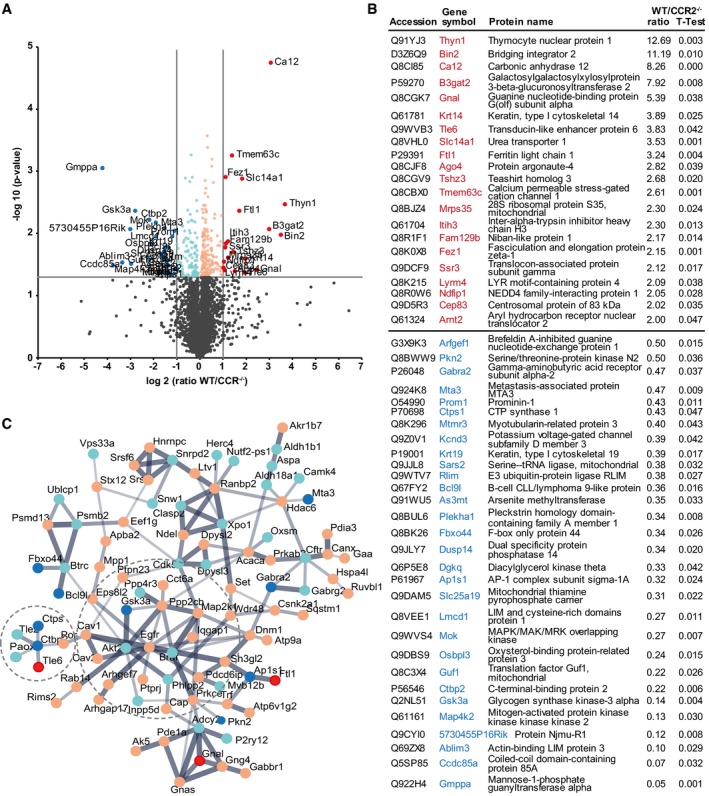
- Volcano plot showing data from proteomics of the injured GM parenchyma from WT and CCR2−/− mice at 5 dpi. Proteins that were found significantly more abundant in WT mice (−log10 of P‐value, y‐axis) and showed a fold change of more than twofold are displayed as red dots, with light red dots indicating significant proteins with fold changes below twofold. Accordingly, proteins that were found significantly more abundant in CCR2−/− mice (−log10 of P‐value, y‐axis) and showed a fold change of more than twofold are displayed as blue dots, with light blue dots indicating significant proteins with fold changes below twofold. Non‐significantly altered proteins below the horizontal gray line at P = 0.05 are indicated in gray.
- List of proteins with significant (P ≤ 0.05) and twofold different levels of abundance at the injury site of the different genotypes.
- String protein network of the significantly different proteins of the lesion site at 5 dpi in WT and CCR2−/− mice. Proteins are displayed as dots with coloring as described in (A) and are connected based on confidence, with line thickness indicating strength of data support. Two prominent subnetworks are indicated by circles.
To better understand the interaction of the proteins differentially regulated, we performed STRING analysis. Many of the significantly regulated proteins are connected by interaction or complex formation, centering around epidermal growth factor receptor (EGFR) and Gsk3a (Fig 8C). Interestingly, Gsk3a may be responsible for the increased proliferation, given it influences many signaling pathways regulating glial cell proliferation after injury, such as EGF, Wnt, retinoic acid, PDGF, insulin/IGF, and interleukin 3 signaling (Appendix Tables S2 and S3).
To gain a more comprehensive view on pathways significantly enriched in the proteome, we focused on the proteins that were at least 2× more abundant in WT or CCR2−/− mice, respectively (Fig 8A and B, and Appendix Table S1). These were then used for Gene Ontology (GO) enrichment analysis 44 to identify overrepresented GO terms among regulated proteins (Appendix Tables S2 and S3). Interestingly, GO terms associated with cell proliferation and the biosynthetic process of pyrimidine nucleotides and their metabolism were significantly enriched in the CCR2−/− lesion biopsy (Appendix Table S3), further supporting the change in key signaling pathways promoting astrocyte and NG2 glia proliferation in the CCR2−/− lesion site.
Among the GO terms significantly enriched in WT compared to CCR2−/− were several related to the immune response (Appendix Table S2). One central protein here is the NEDD4 family‐interacting protein 1 (Ndfip1) that negatively regulates leukocyte‐mediated immunity and the type 2 immune response (Fig 8A and B). Ndfip1 has also been described to be expressed in surviving neurons after TBI 45 and is involved in the regulation of cell proliferation 46. Most relevant for the later effects on scar formation are, however, several GO terms significantly higher in WT related to glycosaminoglycan synthesis and metabolic processes (Appendix Table S2), indicating that scar formation processes involving ECM deposition are already differentially regulated between WT and CCR2−/− at 5 dpi. Thus, the proteome analysis reveals not only pathways regulating astrocyte proliferation in the CCR2−/− lesion site, but also shows that already after 5 dpi, CSPG synthesis is reduced; highlighting that key aspects of scar formation including ECM deposition are determined at these early stages when astroglial cell proliferation peaks.
Aryl hydrocarbon receptor expression is highest in juxtavascular astrocytes after stab wound injury
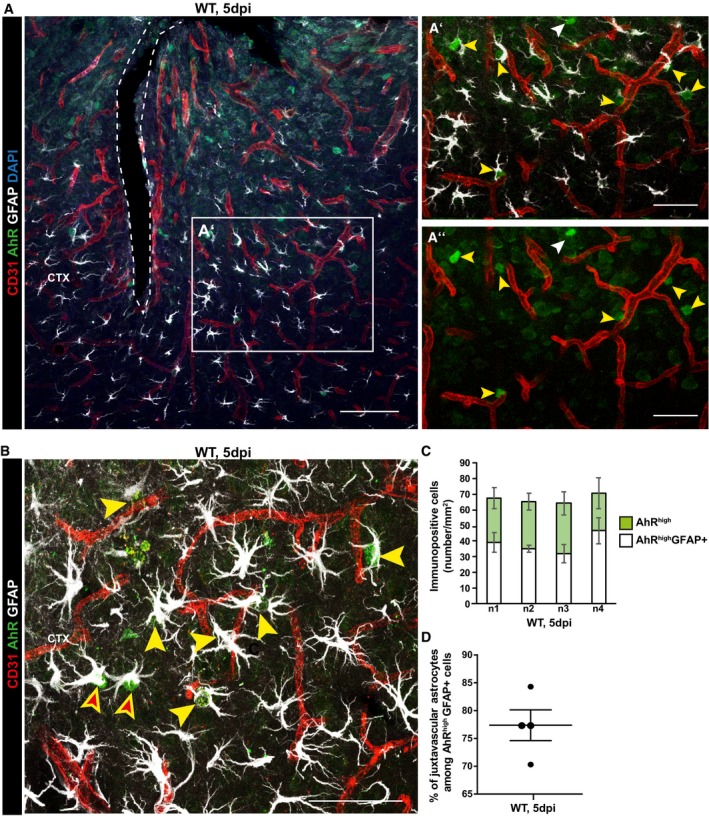
While the above proteomic analysis highlighted some mechanisms regulating the improved scar formation in the absence of invading monocytes, we also aimed to identify candidate pathways that may explain the opposite side of regulation, namely how juxtavascular astrocyte proliferation could contribute to restrict monocyte invasion. We reasoned that this pathway may be less active in the condition when monocytes do not invade and, hence, be higher in WT mice in our proteome. One candidate that caught our attention is ARNT2 (Fig 8B), an essential co‐factor for the canonical signaling pathway of the Aryl hydrocarbon receptor (AhR) signaling 47, 48, 49. As AhR has previously been shown to repress Ccl2 and other chemokines in astrocytes 50, and given Ccl2 attracts CCR2+ monocytes 51, 52, 53, we stained for AhR and reactive astrocytes at 5 dpi (Fig 9). Notably, AhR staining was nuclear (Fig 9A and B) indicating active signaling that leads to nuclear translocation 54. The majority of AhR+ nuclei were found in GFAP+ reactive astrocytes (Fig 9C) and the vast majority of AhR+ astrocytes was at juxtavascular positions (almost 78%; Fig 9D). Thus, increased proportions of juxtavascular AhR+ astrocytes may form a sheet of astrocytes with low cytokine levels 50 surrounding the blood vessels and thereby contribute to reduce monocyte invasion.
Figure 9. Juxtavascular astrocytes are enriched in nuclear Aryl hydrocarbon receptor immunostaining at the stab wound injury site in WT mice.
-
A–DConfocal micrographs depicting AhR immunostaining together with GFAP for reactive astrocytes and CD31 for blood vessels (A, B) showing many examples of juxtavascular proliferating astrocytes containing nuclear AhR indicated by yellow arrowheads (A′, A″, B) surrounding the stab wound injury site at 5 dpi. White arrowheads point GFAP negative cell containing nuclear AhR in A′‐A′′, and non juxtavascular reactive AhR+ astrocytes are indicated by red filled arrowheads in (B). Histogram depicts the number of AhR+ astrocytes separately for each animal (n1–4) (C) and their proportion (individual data points, i.e. animals, are indicated as dots) located at juxtavascular positions (D). All data are represented as mean ± SEM per independent experiments (n = 4). The injury site is indicated by a dashed line, and nuclei were stained with DAPI. Scale bars: (A): 100 μm; (A′, A″): 50 μm, (B): 25 μm.
Discussion
Here, we showed a crucial cross‐regulation between juxtavascular astrocyte proliferation and invading monocytes—each of them inhibiting the other. By using different mouse models targeting adult astrocytes, we found that reduced astrocyte proliferation significantly increased the number of CD45+Iba1− cells at the injury site, while increased juxtavascular astrocyte proliferation was accompanied by a reduction of CD45+Iba1− cells. Given the virtual complete absence of CD45+Iba1− cells in the CCR2−/− mice, we conclude that almost all CD45+Iba1− cells are invading monocytes dependent on the CCR2 receptor, that is, Ly6ChighCCR2+CXCR1low 55. Indeed, no T or B cells were detectable in the cerebral cortex GM after stab wound injury (as shown above and Ref. 56). The finding that increased juxtavascular astrocyte proliferation reduces monocyte numbers in the lesion site is particularly important, because so far the effects of reduced numbers of proliferating astrocytes by cell ablation 13 or Stat3 deletion were examined only after spinal cord injury 15, 38 or neurotoxic brain injury 57. Our new data now show a positive effect of increased astrocyte proliferation in limiting monocyte invasion. This concept is further supported by the reduced scar formation in the absence of monocyte invasion that was accompanied by increased astrocyte proliferation. Most importantly, these data revise the concept about astrocytes in scar formation as the GFAP+ scar was reduced in the CCR2−/− mice despite increased astrocyte proliferation.
Astrocyte proliferation limits monocyte invasion
Generally, proliferating astrocytes could regulate CCR2+ monocytes either by reducing their invasion, proliferation, differentiation, or survival in the brain parenchyma. As we could not detect proliferating CD45+Iba1− in the injury site and their differentiation into microglia is disputed 58, proliferating astrocytes may rather affect their survival or recruitment. Given the juxtavascular position of most (70%) proliferating astrocytes, they may well affect monocyte recruitment at this interface. Indeed, we observed CD45+ cells accumulating in the vessels in conditions with increased astrocyte proliferation, consistent with their failure to infiltrate. Interestingly, juxtavascular astrocytes share a common ancestor during development, as shown by clonal analysis 59, suggesting that they may differ from other astrocytes not only in their position, but also their intrinsic makeup.
But which could be the factors of juxtavascular astrocytes regulating monocyte invasion? One possibility would be their special coupling by tight junctional proteins which has been recently shown to regulate immune cell invasion 60. Another possibility would be expression of repellent factors or reduced levels of factors attracting monocytes. The latter is suggested by the heterogeneity in nuclear AhR observed here in reactive astrocytes after stab wound injury, but not in the intact cortex where AhR is hardly detectable (see also Ref. 61). Most AhR+ astrocytes are indeed at juxtavascular positions at the peak of astrocyte proliferation. This is interesting, as previous work showed that AhR represses a variety of chemokines (Ccl2, Ccl20, Cxcl10), as well as cytokines (IL6, 12 and 23) and pro‐inflammatory markers 50, supporting the idea that these astrocytes may contribute to limit further inflammation and monocyte invasion (for astrocyte‐derived Ccl2 effects see e.g. Ref. 62). It will now be important to determine genome‐wide expression differences between juxta‐ and non‐juxtavascular astrocytes, and the AhR signaling provides a marker for this subset of astrocytes after injury. When examining the transcriptome of all GFAP+ reactive astrocytes after stab wound 10, or in other brain injury conditions 10, 63, 64, many genes involved in immune cell regulation are altered in expression. This not only highlights the key role of astrocytes in this interplay, but also prompts further analysis as to which of these pathways are enriched at the juxtavascular interface.
Invading monocytes regulate macroglia proliferation and scar formation
Importantly, we observed a crucial feedback regulation among invading monocytes on astrocyte proliferation with much increased astrocyte and NG2 glia proliferation in the CCR2−/− lesioned area. In addition, microglia activation appeared to be stronger in the absence of monocyte invasion, while their proliferation was not different. These data highlight not only the multiple effects that the absence of invading monocytes elicits, but also that the higher proliferation and microglia activation set the stage for a later strikingly diminished scar formation. At 4 weeks post‐injury, astrogliosis as well as ECM components of the scar, such as collagen IV, Tn‐C, and CSPG, are reduced at the injury site in the absence of CCR2+ invading monocytes. These findings are particularly intriguing in regard to the correlation between reduced astrocyte proliferation and diminished scar formation after spinal cord injury 14, 15, 16, 38. As we observed the opposite in the brain, namely less persistent GFAP reactivity in the CCR2−/− mice where more reactive astrocytes proliferated, this suggests a profound difference in the role of proliferating and other reactive astrocyte populations. Our data are well in agreement with the concept that reactive astrocyte proliferation serves a beneficial role 14, 15, 16, 30, 38 as increased astrocyte proliferation correlates with reduced lesion size and scar formation in the CCR2−/− mice. Thus, reactive astrocyte proliferation in the acute phase of cerebral cortex injury does not lead to increased astrocyte numbers nor to increased GFAP+ scar. In the CCR2−/− mice, the number of astrocytes was not different between WT and CCR2−/− mice in the lesion site 28 dpi despite the increased proliferation, and the astrocytes with persisting GFAP levels were even reduced in number over several months. Thus, the scar‐forming astrocytes may rather correspond to astrocyte subtypes different from the proliferating subset, such as the recently discovered neurotoxic subset of astrocytes 65. However, markers for neurotoxic astrocytes, such as C3, were not detectably altered in the proteome at 5 dpi. Also, GFAP was not notably altered at these earlier stages, but only reduced later, at 4 and 12 weeks post‐injury. Interestingly, at the earlier time point of 5 dpi, many signaling pathways were differentially regulated in the lesion site of WT and CCR2−/− mice, including EGF, Wnt, PDGF, RA, and IL3.
These findings also raise the interesting possibility that not only astrocyte proliferation is increased in the absence of monocyte invasion, but they may be more generally similar to an immature state that has been shown to be beneficial in various injury paradigms (see, e.g., Refs 66, 67, 68). As we observed no obvious difference in abundance of fibrinogen in the proteome, we conclude that its potent effect on TGFβ signaling and astrocyte activation 69 may not be central to the differences observed between WT and CCR2−/− mice. However, the proteome analysis may miss smaller differences in proteins including those entering from blood or cerebrospinal fluid. As significant differences in BBB leakage were observed between the genotypes 3 dpi, these changes may likely contribute to some of the phenotypes observed, such as reduced activation of pathways involved in promoting astrocyte scar formation, for example, via TGFβ signaling 69. To dissect the origin of factors influencing juxtavascular astrocyte proliferation, it will now be important to identify the secretome and surface proteome of invading monocytes toward a better understanding of the interaction between these cell types.
However, many of these pathways affected by the absence of monocyte invasion in CCR2−/− mice regulate not only astrocyte or NG2 glia proliferation but also microglia activation and mediate neuroprotection, showing the multitude of signaling changes elicited in the absence of monocyte invasion. Most importantly, the proteome analysis unraveled early effects on ECM deposition, which is already profoundly downregulated in the absence of invading monocytes at this early stage. These findings thus demonstrate that key aspects of scar formation are determined at this early stage.
Thus, invading CCR2+ cells play direct and indirect roles in regulating proliferation of other glial cells and promoting scar formation after stab wound injury. This highlights that, irrespective of the different phenotypes of macrophages ranging from more pro‐inflammatory to more anti‐inflammatory, their entire absence reduces scar formation and improves neuroprotection in this injury condition. CCR2+ monocytes change their phenotype depending on the brain environment they encounter (see, e.g., Refs 23, 55, 58). While their anti‐inflammatory role has been suggested to be important in improving neuronal survival and function 23, our results show that the total absence of invading monocytes results in less scar formation. Interestingly, there may be a difference between traumatic and ischemic injury, as mild TBI performed in CCR2−/− mice also revealed a smaller lesion size 22, even though the amount of tau phosphorylation is considerably increased in the absence of invading monocytes. Moreover, CCR2+ monocytes were identified as the main source of Nox2‐mediated neurotoxic ROS after TBI 21, a role similar to some results obtained in spinal cord injury 70. It will now be important not only to tease out the signaling events underlying the adverse effects of monocyte invasion, but also determine whether the different effects obtained by blocking CCR2+ cell recruitment are due to differential effects on glial cell proliferation, which was not monitored in any of the previous studies. Indeed, proliferation of astrocytes is important to restore astrocyte numbers after injury and differential effects on astrocyte proliferation may well contribute to different outcomes. As reactive astrocytes influence ion and transmitter homeostasis, they directly affect neuronal function and survival in the injury site 39. Thus, it is crucial to consider astrocyte proliferation when assessing brain injury, given their importance for restoring astrocyte numbers and limiting monocyte invasion which has profound effects on scar formation.
Materials and Methods
Animals
Experiments were performed with 2‐ to 3‐month‐old mice from mixed genders. For inducible deletion of cdc42 in astrocytes, mice carrying floxed alleles (cdc42 fl/fl) 71 were crossed with GLASTCreERT2 mice 72 and the CAG‐eGFP reporter line 73. For inducible deletion of Smo in astrocytes, homozygous mice carrying floxed Smo alleles (Smo fl/fl, 73), GLAST CreERT2 mice 72, and the CAG‐eGFP reporter line 74 were used. CCR2 RFP mice 36 (Jackson laboratories, Cat 017586) were crossed with C57BL/6J mice (Charles River Laboratories; Sulzfeld, Germany) to obtain heterozygous and WT controls. For constitutive expression of the Smo/EYFP fusion gene and unrestrained hedgehog signaling in cortical astrocytes, homozygous Gt(ROSA)26Sor tm1(Smo/EYFP)Amc/J mice (Jackson laboratory, Cat 005130) were crossed with GLAST CreERT2 mice. Animals were allocated to experimental groups regarding their genotype and kept under standard conditions with access to water and food ad libitum. Animal handling and experimental procedures were performed in accordance with German and European Union guidelines and were approved by the State of Upper Bavaria.
Tamoxifen‐induced recombination
For the induction of Cre‐mediated recombination in GLAST CreERT2 mice, tamoxifen (Sigma) was administered orally (40 mg/ml in corn oil/10% ethanol) divided in three applications of 250 μl each, every second day within 5 days to achieve a total dose of 30 mg. Mice were analyzed 3 weeks after the last induction of recombination to ensure clearance of endogenous targeted protein.
Surgical procedures
Stab wound injury was performed as previously described 9, 11. Briefly, animals were anesthetized and subjected to a parasagittal stab wound injury (0.6 mm deep and 1 mm long) on the right cortical hemisphere using an ophthalmological knife and stereotactic apparatus. Animals were sacrificed at 1, 3, 5, 7, 28, and 90 days after the surgery.
Transient focal ischemia was performed as previously described 11. Briefly, male C57BL/6 mice (body weight 18–22 g, Charles River, Sulzfeld, Germany) were anesthetized with 4% halothane, 30% O2, and 66% N2O. Anesthesia was then maintained with 1% halothane, 30% O2, and 69% N2O for the whole duration of surgery (< 20 min). Body temperature was kept at 37.0 ± 0.1°C with a feedback‐controlled heating pad (Heater Control Module, FHC, Bowdoinham, ME, USA). A flexible laser Doppler probe was glued onto the exposed left parietal skull over the territory of the middle cerebral artery (MCA) for continuous monitoring of regional cerebral blood flow (rCBF; Perimed 4001 Master, Perimed, Järfälla, Sweden). Thereafter, the left common and external carotid arteries were exposed and ligated. The common carotid artery was incised and a silicone‐coated 8‐0 nylon monofilament was pushed into the internal carotid artery until the laser Doppler signal indicated occlusion of the middle cerebral artery (MCAo). Wounds were sutured, and the animals transferred to an incubator (32°C) and allowed to wake up. Forty minutes after MCAo, mice were shortly re‐anesthetized and ischemia was terminated by removal of the intraluminal suture. For maintenance of body temperature after surgery, animals were re‐transferred to the incubator for another 2 h. Mice were killed 24 h or 3 days after MCAo in deep halothane anesthesia. All experimental procedures were performed in accordance with the policies of and approved by the state of Bavaria.
Immunohistochemistry
After transcardial perfusion with phosphate‐buffered saline (PBS) followed by 4% paraformaldehyde in PBS, the brains were removed, post‐fixed overnight at 4°C, and cryoprotected in 30% sucrose. Brains were cut at 40 μm, and sections were stained with primary and secondary antibodies according to previously described protocols 28 or stored at −20°C for future analysis. For staining with anti‐collagen I antibody, sections were pre‐incubated in 0.5% Pepsin in 5 mM HCl at 37°C for 15 min with subsequent 10 min wash in PBS. Rabbit polyclonal antibody to Tn‐C (batch KAF14, 1:200) was generously provided by Prof. Dr. Andreas Faissner (Ruhr‐University Bochum, Germany). List of primary and secondary antibodies and their dilution used for immunohistochemical analysis are listed in Appendix Table S4. Nuclei were visualized with DAPI (0.1 μg/ml; Sigma Aldrich).
Analysis of the blood–brain barrier permeability
Blood–brain barrier leakage following stab wound injury was determined by using Alexa Fluor 555–cadaverine (A30677, Thermo Fischer Scientific), as described in 75, 76. Briefly, mice were injected in the tail vein with Alexa Fluor 555–cadaverine (250 μg dissolved in 200 μl NaCl 0.9%). At 3 h after injection, mice were deeply anesthetized and perfused for ca. 7 min with ice‐cold PBS (pH 7.4). After dissection, brains were post‐fixed for 48 h in PFA 4% and then cryoprotected for 24 h in 30% sucrose in PBS. Coronal sections of 40 μm were cut, and immunohistochemistry was performed as described above. Single‐channel immunofluorescence confocal images were collected with equal settings for all experimental groups, and the surface of the Alexa Fluor 555–cadaverine area was measured using NIH ImageJ software.
Isolation and fluorescence‐activated cell sorting (FACS) of immune cells
The GM from a biopsy punch (Ø 0.25 cm) around the lesion area was dissociated using the adult brain dissociation kit mouse and rat from (MiltenyiBiotec) following the manufacturer's instructions except the red blood cell removal step. This was not necessary as the meninges were removed during tissue preparation. Immune cells were isolated by CD45 magnetic associated cell separation (MACS) according to the manufacturer's protocol (anti‐CD45 MicroBead Kit, mouse; Miltenyi) and then stained with the following antibodies for flow cytometry (Biolegend): anti‐Gr‐1 (BV421; 1:500); anti‐ Ly6c (PE) anti‐CD45 APCCy7 (clone 30‐F11; 1:100); anti‐CD11b BV421 (clone M1/70; 1:500); anti‐CD3 Alexa‐488 (clone 145‐2C11; 1:400); anti‐CD19 Alexa‐647 (clone 6D5; 1:500); and anti F4/80 PeCy7 (clone BM8; 1:500). FACS measurements were performed on a FACS Canto II (BD), and data were analyzed using FlowJo software.
LC‐MSMS and label‐free quantitative analysis
For proteomic analysis, tissue samples (∅ 0.25 cm) were punched from the lesioned and contralateral uninjured somatosensory GM at 5 dpi and immediately frozen at −80°C for posterior analysis. Tissue samples were lysed in 100 μl urea/thiourea Buffer (9 M urea, 2 M thiourea, 4% CHAPS, 65 mM DTT) in a Precellys homogenizator (VWR). 20 μg per sample was digested using a modified FASP procedure 77, 78, resulting in two fractions after sequential proteolysis with first Lys‐C (Wako Chemicals) followed by trypsin (Promega). LC‐MSMS analysis was performed on a Q Exactive HF mass spectrometer (ThermoFisher Scientific) online coupled to a Ultimate 3000 RSLC nano‐HPLC (Dionex). Acquired raw data were loaded to the Progenesis QI software for MS1 intensity based label‐free quantification (Nonlinear Dynamics, Waters), separately for the two different proteolyzed fractions. After alignment in order to reach a maximum overlay of peptide features, filtering of singly charged features and features with charges > 7 and normalization to correct for systematic experimental variation, all MSMS spectra were exported and searched against the Swissprot mouse database (16772 sequences, Release 2016_02) using the Mascot search engine. Search settings were: enzyme trypsin or Lys‐C, respectively, 10 ppm peptide mass tolerance and 0.02 Da fragment mass tolerance, one missed cleavage allowed, carbamidomethylation was set as fixed modification, methionine oxidation and asparagine or glutamine deamidation were set as variable modifications. A Mascot‐integrated decoy database search calculated an average false discovery of < 1% when searches were performed with a mascot percolator score cutoff of 13 and significance threshold of 0.05. Peptide assignments were re‐imported into the Progenesis QI software. The abundances of all unique peptides allocated to each protein were summed up. The two datasets were combined, and the resulting normalized abundances of the single proteins were then used for calculation of fold changes of proteins and significance values by a Student's t‐test.
Generation of protein network
A protein network analysis of all 311 significantly different proteins from the proteomics analysis was performed using STRING database of known and predicted protein–protein interactions (version 10.0) 79, with the following settings: Interaction sources were experiments and databases, and minimum required interaction score was set to medium confidence. Proteins were connected based on confidence, with line thickness indicating strength of data support. Disconnected nodes and small networks with < 5 proteins were discarded, and the resulting protein network is given in Fig 7B.
Pathway enrichment analysis
Pathway enrichment analysis of GO terms of biological processes was performed separately for the 20 proteins which were significantly at least twofold more abundant in WT and the 29 proteins which were significantly twofold more abundant in CCR2−/− mice, using the Generanker tool (Genomatix GmbH, Munich). Significantly overrepresented (P < 0.05) biological processes with their respective GO terms, significance values, enrichment factors, and list of proteins are given in Appendix Tables S1–S3.
Data availability
The mass spectrometry data from this publication have been deposited to the ProteomeXchange via the PRIDE 80 partner repository with the dataset identifier PXD008906.
Quantification and statistical analysis
Quantification of immunostaining was performed on ≥ 3 sections per animal from each experimental group (3–6 animals) on multi‐channel, confocal 3D stacks, using Zeiss ZEN 2010 software and Cell Counter plugin of NIH ImageJ software. The levels of individual immunostaining were adjusted independently for optimal visualization and are represented as means ± standard error of the mean (SEM) calculated between different animals. Sections stained for GFAP, NeuN, CD45, or CSPG as well as the area of the Alexa Fluor 555–cadaverine were documented using constant exposure settings. Single‐channel immunofluorescence confocal images were thresholded, and the surface of the immunopositive area was measured using NIH ImageJ software. Areas are shown in graphs as mean values ± SEM. GraphPad Prism 7 was used to perform statistical analysis. Unpaired two‐tailed Student's t‐test or Mann–Whitney U‐test were used for single comparisons depending of data distribution, assessed with the D'Agostino and Pearson's omnibus normality test. ANOVA followed by Tukey's post‐test or Kruskal–Wallis test were used for multiple comparisons. In all statistical analyses, significance was accepted at P < 0.05. The group size was determined to allow statistical evaluation of differences between two groups with a power higher than 0.8.
Author contributions
MG, SS, and JF conceptualized the study. JF, SS, NM, SMH, JKj, and JKr, NP involved in methodology. JF, SS, and JM‐P formally analyzed the date. JF, NM, and SS involved in experimental investigation. MG, SMH, and NP collected the resources. JF and MG wrote the original draft of the manuscript. SS, JM‐P, SMH, NM, NP, and RMG involved in review writing and editing. JF, SS, and JM‐P visualized the study. MG involved in funding.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We are particularly grateful to Gabi Jäger and Manja Thorwirth for excellent technical assistance. We thank Giacomo Masserdotti for help with statistics and Adam O'Neill for excellent suggestions on the manuscript. We are also thankful to Prof. Dr. Andreas Faissner (Ruhr‐University Bochum, Germany) for generously providing the KAF14 antibody and to Elisabetta Petroziello for help with identification of invading immune cells by MACS and FACS. This work was supported by the German Research Foundation to MG via the SFB870, Synergy Excellence Cluster, and the Priority Program 1757, a Scholarship to JF from the ALEARG Program co‐funded by the Ministry of Education, Science and Technology of Argentina and the German Academic Exchange Service (DAAD) and to JKj by Swedish Society for Medical Research (SSMF).
EMBO Reports (2018) 19: e45294
Contributor Information
Swetlana Sirko, Email: swetlana.sirko@med.uni-muenchen.de.
Magdalena Götz, Email: magdalena.goetz@helmholtz-muenchen.de.
References
- 1. Tanaka EM, Ferretti P (2009) Considering the evolution of regeneration in the central nervous system. Nat Rev Neurosci 10: 713–723 [DOI] [PubMed] [Google Scholar]
- 2. Alunni A, Bally‐Cuif L (2016) A comparative view of regenerative neurogenesis in vertebrates. Development 143: 741–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barbosa JS, Sanchez‐Gonzalez R, Di Giaimo R, Baumgart EV, Theis FJ, Götz M, Ninkovic J (2015) Neurodevelopment. Live imaging of adult neural stem cell behavior in the intact and injured zebrafish brain. Science 348: 789–793 [DOI] [PubMed] [Google Scholar]
- 4. Kroehne V, Freudenreich D, Hans S, Kaslin J, Brand M (2011) Regeneration of the adult zebrafish brain from neurogenic radial glia‐type progenitors. Development 138: 4831–4841 [DOI] [PubMed] [Google Scholar]
- 5. Carlen M, Meletis K, Goritz C, Darsalia V, Evergren E, Tanigaki K, Amendola M, Barnabe‐Heider F, Yeung MS, Naldini L et al (2009) Forebrain ependymal cells are Notch‐dependent and generate neuroblasts and astrocytes after stroke. Nat Neurosci 12: 259–267 [DOI] [PubMed] [Google Scholar]
- 6. Sabelström H, Stenudd M, Reu P, Dias DO, Elfineh M, Zdunek S, Damberg P, Goritz C, Frisen J (2013) Resident neural stem cells restrict tissue damage and neuronal loss after spinal cord injury in mice. Science 342: 637–640 [DOI] [PubMed] [Google Scholar]
- 7. Frisen J (2016) Neurogenesis and gliogenesis in nervous system plasticity and repair. Annu Rev Cell Dev Biol 32: 127–141 [DOI] [PubMed] [Google Scholar]
- 8. Benner EJ, Luciano D, Jo R, Abdi K, Paez‐Gonzalez P, Sheng H, Warner DS, Liu C, Eroglu C, Kuo CT (2013) Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4. Nature 497: 369–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Götz M (2008) Origin and progeny of reactive gliosis: a source of multipotent cells in the injured brain. Proc Natl Acad Sci USA 105: 3581–3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sirko S, Irmler M, Gascon S, Bek S, Schneider S, Dimou L, Obermann J, De Souza Paiva D, Poirier F, Beckers J et al (2015) Astrocyte reactivity after brain injury: the role of galectins 1 and 3. Glia 63: 2340–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sirko S, Behrendt G, Johansson PA, Tripathi P, Costa M, Bek S, Heinrich C, Tiedt S, Colak D, Dichgans M et al (2013) Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. [corrected]. Cell Stem Cell 12: 426–439 [DOI] [PubMed] [Google Scholar]
- 12. Ren Y, Ao Y, O'Shea TM, Burda JE, Bernstein AM, Brumm AJ, Muthusamy N, Ghashghaei HT, Carmichael ST, Cheng L et al (2017) Ependymal cell contribution to scar formation after spinal cord injury is minimal, local and dependent on direct ependymal injury. Sci Rep 7: 41122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV (1999) Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar‐forming, reactive astrocytes in adult transgenic mice. Neuron 23: 297–308 [DOI] [PubMed] [Google Scholar]
- 14. Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV (2004) Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci 24: 2143–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anderson MA, Burda JE, Ren Y, Ao Y, O'Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, Sofroniew MV (2016) Astrocyte scar formation aids central nervous system axon regeneration. Nature 532: 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray‐Thompson Z, Ao Y, Sofroniew MV (2013) Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3‐dependent mechanisms after spinal cord injury. J Neurosci 33: 12870–12886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Filous AR, Silver J (2016) Targeting astrocytes in CNS injury and disease: a translational research approach. Prog Neurobiol 144: 173–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bardehle S, Kruger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H, Snippert HJ, Theis FJ, Meyer‐Luehmann M, Bechmann I et al (2013) Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci 16: 580–586 [DOI] [PubMed] [Google Scholar]
- 19. Semple BD, Kossmann T, Morganti‐Kossmann MC (2010) Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab 30: 459–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hsieh CL, Niemi EC, Wang SH, Lee CC, Bingham D, Zhang J, Cozen ML, Charo I, Huang EJ, Liu J et al (2014) CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J Neurotrauma 31: 1677–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morganti JM, Jopson TD, Liu S, Riparip LK, Guandique CK, Gupta N, Ferguson AR, Rosi S (2015) CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci 35: 748–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gyoneva S, Kim D, Katsumoto A, Kokiko‐Cochran ON, Lamb BT, Ransohoff RM (2015) Ccr2 deletion dissociates cavity size and tau pathology after mild traumatic brain injury. J Neuroinflammation 12: 228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J, Miskinyte G, Ge R, Ahlenius H, Lindvall O et al (2016) Monocyte‐derived macrophages contribute to spontaneous long‐term functional recovery after stroke in mice. J Neurosci 36: 4182–4195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV (2007) Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 38: 1345–1353 [DOI] [PubMed] [Google Scholar]
- 25. Plesnila N (2016) The immune system in traumatic brain injury. Curr Opin Pharmacol 26: 110–117 [DOI] [PubMed] [Google Scholar]
- 26. Adams KL, Gallo V (2018) The diversity and disparity of the glial scar. Nat Neurosci 21: 9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haan N, Zhu B, Wang J, Wei X, Song B (2015) Crosstalk between macrophages and astrocytes affects proliferation, reactive phenotype and inflammatory response, suggesting a role during reactive gliosis following spinal cord injury. J Neuroinflammation 12: 109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simon C, Götz M, Dimou L (2011) Progenitors in the adult cerebral cortex: cell cycle properties and regulation by physiological stimuli and injury. Glia 59: 869–881 [DOI] [PubMed] [Google Scholar]
- 29. Zhao X, Ahram A, Berman RF, Muizelaar JP, Lyeth BG (2003) Early loss of astrocytes after experimental traumatic brain injury. Glia 44: 140–152 [DOI] [PubMed] [Google Scholar]
- 30. Heimann G, Canhos LL, Frik J, Jager G, Lepko T, Ninkovic J, Götz M, Sirko S (2017) Changes in the proliferative program limit astrocyte homeostasis in the aged post‐traumatic murine cerebral cortex. Cereb Cortex 27: 4213–4228 [DOI] [PubMed] [Google Scholar]
- 31. D'Mello C, Le T, Swain MG (2009) Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci 29: 2089–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thomas ML (1989) The leukocyte common antigen family. Annu Rev Immunol 7: 339–369 [DOI] [PubMed] [Google Scholar]
- 33. Robel S, Bardehle S, Lepier A, Brakebusch C, Götz M (2011) Genetic deletion of cdc42 reveals a crucial role for astrocyte recruitment to the injury site in vitro and in vivo . J Neurosci 31: 12471–12482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP (2004) Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev 18: 937–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xie J, Murone M, Luoh S‐M, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam C‐W, Hynes M, Goddard A et al (1998) Activating Smoothened mutations in sporadic basal‐cell carcinoma. Nature 391: 90–92 [DOI] [PubMed] [Google Scholar]
- 36. Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, Charo IF (2010) Selective chemokine receptor usage by central nervous system myeloid cells in CCR2‐red fluorescent protein knock‐in mice. PLoS One 5: e13693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chu HX, Arumugam TV, Gelderblom M, Magnus T, Drummond GR, Sobey CG (2014) Role of CCR2 in inflammatory conditions of the central nervous system. J Cereb Blood Flow Metab 34: 1425–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV (2008) STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci 28: 7231–7243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robel S, Sontheimer H (2016) Glia as drivers of abnormal neuronal activity. Nat Neurosci 19: 28–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hara M, Kobayakawa K, Ohkawa Y, Kumamaru H, Yokota K, Saito T, Kijima K, Yoshizaki S, Harimaya K, Nakashima Y et al (2017) Interaction of reactive astrocytes with type I collagen induces astrocytic scar formation through the integrin‐N‐cadherin pathway after spinal cord injury. Nat Med 23: 818–828 [DOI] [PubMed] [Google Scholar]
- 41. Fujimoto M, Shiba M, Kawakita F, Liu L, Shimojo N, Imanaka‐Yoshida K, Yoshida T, Suzuki H (2016) Deficiency of tenascin‐C and attenuation of blood‐brain barrier disruption following experimental subarachnoid hemorrhage in mice. J Neurosurg 124: 1693–1702 [DOI] [PubMed] [Google Scholar]
- 42. Ly A, Merl‐Pham J, Priller M, Gruhn F, Senninger N, Ueffing M, Hauck SM (2016) Proteomic profiling suggests central role of STAT signaling during retinal degeneration in the rd10 mouse model. J Proteome Res 15: 1350–1359 [DOI] [PubMed] [Google Scholar]
- 43. Bost F, Diarra‐Mehrpour M, Martin JP (1998) Inter‐alpha‐trypsin inhibitor proteoglycan family–a group of proteins binding and stabilizing the extracellular matrix. Eur J Biochem 252: 339–346 [DOI] [PubMed] [Google Scholar]
- 44. GOC (2014) Gene Ontology Consortium: going forward. Nucleic Acids Res 43: D1049–D1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sang Q, Kim MH, Kumar S, Bye N, Morganti‐Kossman MC, Gunnersen J, Fuller S, Howitt J, Hyde L, Beissbarth T et al (2006) Nedd4‐WW domain‐binding protein 5 (Ndfip1) is associated with neuronal survival after acute cortical brain injury. J Neurosci 26: 7234–7244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Howitt J, Low LH, Putz U, Doan A, Lackovic J, Goh CP, Gunnersen J, Silke J, Tan SS (2015) Ndfip1 represses cell proliferation by controlling Pten localization and signaling specificity. J Mol Cell Biol 7: 119–131 [DOI] [PubMed] [Google Scholar]
- 47. Barrow LL, Wines ME, Romitti PA, Holdener BC, Murray JC (2002) Aryl hydrocarbon receptor nuclear translocator 2 (ARNT2): structure, gene mapping, polymorphisms, and candidate evaluation for human orofacial clefts. Teratology 66: 85–90 [DOI] [PubMed] [Google Scholar]
- 48. Mimura J, Ema M, Sogawa K, Fujii‐Kuriyama Y (1999) Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev 13: 20–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nebert DW (2017) Aryl hydrocarbon receptor (AHR): “pioneer member” of the basic‐helix/loop/helix per‐Arnt‐sim (bHLH/PAS) family of “sensors” of foreign and endogenous signals. Prog Lipid Res 67: 38–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, Chao CC, Patel B, Yan R, Blain M et al (2016) Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med 22: 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Conductier G, Blondeau N, Guyon A, Nahon JL, Rovere C (2010) The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol 224: 93–100 [DOI] [PubMed] [Google Scholar]
- 52. Reaux‐Le Goazigo A, Van Steenwinckel J, Rostene W, Melik Parsadaniantz S (2013) Current status of chemokines in the adult CNS. Prog Neurobiol 104: 67–92 [DOI] [PubMed] [Google Scholar]
- 53. Gyoneva S, Ransohoff RM (2015) Inflammatory reaction after traumatic brain injury: therapeutic potential of targeting cell‐cell communication by chemokines. Trends Pharmacol Sci 36: 471–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tomita S, Jiang HB, Ueno T, Takagi S, Tohi K, Maekawa S, Miyatake A, Furukawa A, Gonzalez FJ, Takeda J et al (2003) T cell‐specific disruption of arylhydrocarbon receptor nuclear translocator (Arnt) gene causes resistance to 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin‐induced thymic involution. J Immunol 171: 4113–4120 [DOI] [PubMed] [Google Scholar]
- 55. Katsumoto A, Lu H, Miranda AS, Ransohoff RM (2014) Ontogeny and functions of central nervous system macrophages. J Immunol 193: 2615–2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McKee CA, Lukens JR (2016) Emerging roles for the immune system in traumatic brain injury. Front Immunol 7: 556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. O'Callaghan JP, Kelly KA, VanGilder RL, Sofroniew MV, Miller DB (2014) Early activation of STAT3 regulates reactive astrogliosis induced by diverse forms of neurotoxicity. PLoS One 9: e102003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ransohoff RM (2016) A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19: 987–991 [DOI] [PubMed] [Google Scholar]
- 59. Martin‐Lopez E, Garcia‐Marques J, Nunez‐Llaves R, Lopez‐Mascaraque L (2013) Clonal astrocytic response to cortical injury. PLoS One 8: e74039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Horng S, Therattil A, Moyon S, Gordon A, Kim K, Argaw AT, Hara Y, Mariani JN, Sawai S, Flodby P et al (2017) Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J Clin Invest 127: 3136–3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu K, Yang Z, Shi R, Luo C, Zhang Z (2016) Expression of aryl hydrocarbon receptor in rat brain lesions following traumatic brain injury. Diagn Pathol 11: 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xu J, Dong H, Qian Q, Zhang X, Wang Y, Jin W, Qian Y (2017) Astrocyte‐derived CCL2 participates in surgery‐induced cognitive dysfunction and neuroinflammation via evoking microglia activation. Behav Brain Res 332: 145–153 [DOI] [PubMed] [Google Scholar]
- 63. Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA (2012) Genomic analysis of reactive astrogliosis. J Neurosci 32: 6391–6410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kamphuis W, Kooijman L, Orre M, Stassen O, Pekny M, Hol EM (2015) GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild‐type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer's disease. Glia 63: 1036–1056 [DOI] [PubMed] [Google Scholar]
- 65. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541: 481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim SK, Nabekura J, Koizumi S (2017) Astrocyte‐mediated synapse remodeling in the pathological brain. Glia 65: 1719–1727 [DOI] [PubMed] [Google Scholar]
- 67. Jiang P, Deng W (2016) Regenerating white matter using human iPSC‐derived immature astroglia. Neurogenesis (Austin) 3: e1224453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hayakawa K, Haas C, Fischer I (2016) Examining the properties and therapeutic potential of glial restricted precursors in spinal cord injury. Neural Regen Res 11: 529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, Margolis RU, Akassoglou K (2010) Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF‐beta after vascular damage. J Neurosci 30: 5843–5854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, Ransohoff RM, Popovich PG (2011) Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci 31: 9910–9922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wu X, Quondamatteo F, Lefever T, Czuchra A, Meyer H, Chrostek A, Paus R, Langbein L, Brakebusch C (2006) Cdc42 controls progenitor cell differentiation and beta‐catenin turnover in skin. Genes Dev 20: 571–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mori T, Tanaka K, Buffo A, Wurst W, Kuhn R, Götz M (2006) Inducible gene deletion in astroglia and radial glia–a valuable tool for functional and lineage analysis. Glia 54: 21–34 [DOI] [PubMed] [Google Scholar]
- 73. Nakamura T, Colbert MC, Robbins J (2006) Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ Res 98: 1547–1554 [DOI] [PubMed] [Google Scholar]
- 74. Long F, Zhang XM, Karp S, Yang Y, McMahon AP (2001) Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 128: 5099–5108 [DOI] [PubMed] [Google Scholar]
- 75. Yanagida K, Liu CH, Faraco G, Galvani S, Smith HK, Burg N, Anrather J, Sanchez T, Iadecola C, Hla T (2017) Size‐selective opening of the blood‐brain barrier by targeting endothelial sphingosine 1‐phosphate receptor 1. Proc Natl Acad Sci USA 114: 4531–4536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Watkins S, Robel S, Kimbrough IF, Robert SM, Ellis‐Davies G, Sontheimer H (2014) Disruption of astrocyte‐vascular coupling and the blood‐brain barrier by invading glioma cells. Nat Commun 5: 4196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wisniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362 [DOI] [PubMed] [Google Scholar]
- 78. Grosche A, Hauser A, Lepper MF, Mayo R, von Toerne C, Merl‐Pham J, Hauck SM (2016) The proteome of native adult muller glial cells from murine retina. Mol Cell Proteomics 15: 462–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta‐Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP et al (2015) STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43: D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vizcaino JA, Csordas A, del‐Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez‐Riverol Y, Reisinger F, Ternent T et al (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44: D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The mass spectrometry data from this publication have been deposited to the ProteomeXchange via the PRIDE 80 partner repository with the dataset identifier PXD008906.