

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
Overexpression of NFE2 in MPNs is associated with H3Y41 phosphorylation by JAK2V617F.
JMJD1C is an NFE2 target gene and acts in a positive feedback loop contributing to NFE2 overexpression in MPNs.
Abstract
The transcription factor “nuclear factor erythroid 2” (NFE2) is overexpressed in the majority of patients with myeloproliferative neoplasms (MPNs). In murine models, elevated NFE2 levels cause an MPN phenotype with spontaneous leukemic transformation. However, both the molecular mechanisms leading to NFE2 overexpression and its downstream targets remain incompletely understood. Here, we show that the histone demethylase JMJD1C constitutes a novel NFE2 target gene. JMJD1C levels are significantly elevated in polycythemia vera (PV) and primary myelofibrosis patients; concomitantly, global H3K9me1 and H3K9me2 levels are significantly decreased. JMJD1C binding to the NFE2 promoter is increased in PV patients, decreasing both H3K9me2 levels and binding of the repressive heterochromatin protein-1α (HP1α). Hence, JMJD1C and NFE2 participate in a novel autoregulatory loop. Depleting JMJD1C expression significantly reduced cytokine-independent growth of an MPN cell line. Independently, NFE2 is regulated through the epigenetic JAK2 pathway by phosphorylation of H3Y41. This likewise inhibits HP1α binding. Treatment with decitabine lowered H3Y41ph and augmented H3K9me2 levels at the NFE2 locus in HEL cells, thereby increasing HP1α binding, which normalized NFE2 expression selectively in JAK2V617F-positive cell lines.
Visual Abstract
Introduction
Despite the characterization of a large number of aberrations in myeloproliferative neoplasm (MPN) patients during the past decade, the molecular pathogenesis of these disorders remains incompletely understood. Besides the acquisition of various mutations, MPN patients also display changes in gene expression. For example, we have demonstrated that levels of the transcription factor “nuclear factor erythroid 2” (NFE2) are significantly elevated in patients with MPNs.1,2 Increased NFE2 expression is likewise seen in polycythemic disorders with augmented hypoxia-inducible factor signaling.3 However, both our understanding of the changes leading to altered gene expression in MPNs as well as our awareness of their pathophysiological consequences is limited. This impedes our ability to develop interventions to counteract these changes.
We have investigated the consequences of altered NFE2 expression in vivo. In 2 murine models, increased NFE2 levels, either by introduction of a transgene (tg) or by expression of mutated versions of NFE2, found in a subset of MPN patients, led to the development of an MPN phenotype.4,5 Moreover, augmented NFE2 activity in mice predisposes these animals to develop acute leukemia at a rate similar to that observed in MPN patients.
Because the downstream effectors through which elevated NFE2 levels effect either an MPN phenotype or its leukemic transformation are not known, we have followed various strategies to identify novel NFE2 target genes and asked whether they contribute to the pathophysiology of these disorders. In a first series of experiments, we modulated NFE2 levels in purified human CD34+ hematopoietic stem cells (HSCs).6 NFE2 levels were increased in HSCs by lentiviral overexpression of the complementary DNA or decreased by expression of a short hairpin RNA (shRNA). These data identified the proinflammatory cytokine interleukin 8 (IL8) as a novel NFE2 target.6 As IL8 levels constitute an independent prognostic factor for survival in primary myelofibrosis (PMF) patients,7 these data underline the role of altered NFE2 activity in the pathophysiology of MPNs.
However, this detailed approach limited the list of candidate genes to those expressed in CD34+ HSCs and modulated within 24 hours of increasing NFE2 levels. To identify additional NFE2 targets, we used an alternative strategy, interrogating published chromatin immunoprecipitation (ChIP) sequencing (ChIPseq) data of NFE2 binding.8 Among the potential candidates, we concentrated on epigenetic modifiers, as it has become apparent that a large subset of genes mutated in MPN patients affect histone modifications.9,10 As these possess enzymatic activity, they constitute druggable targets. Here, we test the hypothesis that the histone demethylase JMJD1C constitutes a novel NFE2 target gene.
Materials and methods
Transient transfection and luciferase assays
Transient transfection and luciferase assays were conducted as previously described.6
Plasmid constructs
The JMJD1C promoter element and the −16-kb enhancer were amplified by polymerase chain reaction (PCR) from total cellular DNA of HEL cells using the following primers: −1032/+190-bp promoter (forward primer, 5′GCAGAGGTTGCAGTGAGTTGAGAT3′; reverse primer, 5′TGAAGCACTACTGCATCCTCCGATC3′) and −16-kb enhancer region (forward primer, 5′GTAGAAATGGGATCTCCCCAGGTT3′; reverse primer, 5′CTTCGAAATAAGGACTCAAAGCACA3′).
The PCR fragments were cloned into the pGL4.10 luciferase reporter vector (Promega). Mutations of NFE2-binding sites were inserted by using the GeneArt site-directed mutagenesis system (Invitrogen). NFE2-binding sites at −16-kb “TAAGTCA” and at −120-bp “TGAGTAA” were mutated to “GCCGCAG” and “GTCGCGG,” respectively.
ChIP
ChIP was performed as previously described2 using antibodies listed in supplemental Table 1 (available on the Blood Web site). Input DNA was diluted 1/10.
The primers used for DNA amplification are depicted in supplemental Table 2.
Lentiviral transduction
HEL cells were tranducted with pLEGO-iG-U6 NFE2 shDNA or a scrambled control as previously described.11 Green fluorescent protein (GFP)–expressing cells were sorted to obtain pure populations.
Immunoblotting
Protein expression was determined by western blotting.2 Immune complexes were detected using chemiluminescence (GE Healthcare). Densitometry was performed using National Institutes of Health ImageJ software. The antibodies used for immunoblotting are shown in supplemental Table 3.
Real-time quantitative PCR
Reverse transcription was performed using the TaqMan Reverse Transcription Kit (Applied Biosystems).
Quantitative RT-PCR was performed using the murine Jmjd1c (Mm01150329_m1) assay (Applied Biosystems) and the murine β-2-microglobulin primer-probe combination in supplemental Table 2. Jmjd1c expression was determined using the ΔΔ cycle threshold method.12 The primers that were used for quantitative RT-PCR are depicted in supplemental Table 4.
Study approval
Peripheral blood samples were obtained from polycythemia vera (PV), PMF, and essential thrombocythemia (ET) patients fulfilling the World Health Organization (WHO) criteria for diagnosis13 and from buffy coats of healthy volunteer blood donors. The study protocol was approved by the Ethics Committee at the University Medical Center Freiburg. Informed consent was obtained from all patients included in the study.
Granulocyte isolation
Peripheral blood granulocytes were isolated by dextran sedimentation and Ficoll gradient centrifugation as previously described.14
Decitabine treatment of cell lines
Decitabine was added to the culture medium at the indicated concentrations; medium was renewed every 24 hours.
Statistics
The Student t test and the paired Student t test were applied to determine whether a significant (P < .05) difference existed between 2 groups.
The 1- and 2-way analysis of variance (ANOVA) with the post hoc Tukey multiple comparison test was used to compare multiple groups.
Results
Because the downstream effectors through which elevated NFE2 levels promote either an MPN phenotype or its leukemic transformation are not known, we sought to identify novel NFE2 target genes, especially those encoding epigenetic modifiers. We used the following strategy to interrogate in silico data (Encyclopedia of DNA Elements [ENCODE] Consortium) of ChIPseq analysis of NFE2 binding in an erythroid cell line8 (supplemental Figure 1): NFE2 binding was observed at 5070 sites in the genome, of which 4773 were within 200 kb of a transcriptional start site.15 Of these peaks, 2242 were considered highly significant (peak score above 450), associated with 1986 genes. We compared these 1986 genes containing NFE2 peaks to a list of epigenetic modifiers, published by Plass and colleagues.16,17 From the 60 epigenetic regulators that constitute potential novel NFE2 target genes (supplemental Table 5), we reexamined the top 20 candidates. Using positive and negative controls, we demonstrated that our method indeed specifically predicted gene loci bound by NFE2 in vivo (supplemental Figure 2). We concentrated on JMJD1C as it showed an additional, uncalled NFE2 peak close to the transcriptional start site (Figure 1A), indicating active NFE2-mediated transcriptional regulation.
Figure 1.

Identification of JMJD1C as a novel NFE2 target gene. (A) NFE2 binding to the JMJD1C locus. Top: schematic representation of potential binding sites at the JMJD1C locus determined by in silico analysis.8 Bottom: HEL cell lysates were chromatin immunoprecipitated with antibodies against NFE2 or an IgG control. PCR was performed with primers flanking the 3 predicted binding sites as well as on control sites at +3.2 kb in the NFE2 gene and in the myogenin locus. Data are representative of 3 independent experiments. (B) Jmjd1c mRNA expression in NFE2tg mice. RNA was isolated from bone marrow cells of wild-type (wt) and hNFE2tg mice4 and subjected to quantitative RT-PCR for Jmjd1c expression. Expression levels were normalized to mB2M expression. Bars represent the mean. ***P < .001 by the Student t test. (C) Jmjd1c mRNA levels upon expression in CB3 cells. CB3 cells were transfected with pLego-iG-NFE2wt or an empty control vector and sorted for GFP positivity. Jmjd1c expression was quantified as described in panel B. Mean values and standard error of the mean (SEM) are shown. *P < .05 by the Student t test.
The JMJD1C demethylase is a novel NFE2 target gene
We tested the hypothesis that JMJD1C represents a novel NFE2 target gene by conducting ChIP assays to interrogate NFE2 binding to the in silico–identified sites in HEL cells. NFE2 indeed specifically bound 3 sites within the JMJD1C locus in HEL cells, located at −120 bp, −16 kb, and −154 kb relative to the transcriptional start site (Figure 1A). Correspondingly, Jmjd1c messenger RNA (mRNA) expression was significantly upregulated in bone marrow cells of tg mice overexpressing NFE2 (Figure 1B). Moreover, NFE2 and JMJD1C RNA expression were highly correlated (P = .0035) in primary cells from MPN patients (n = 41, PV; n = 19, ET; and n = 9, PMF) as well as healthy controls (supplemental Figure 3). Finally, introduction of NFE2 into CB3 cells, an erythroid cell line devoid of NFE2, was sufficient to induce JMJD1C mRNA expression (Figure 1C). These data demonstrate that JMJD1C expression is directly regulated by the NFE2 transcription factor.
To determine both the functionality and the precise location of NFE2 binding in the JMJD1C locus, we engineered 2 reporter gene constructs, 1 containing the proximal −120-bp sequence and a second containing both the −120 bp as well as the putative −16-kb enhancer sequence (Figure 2A). Reporter gene assays demonstrated that NFE2 in combination with its obligate heterodimer MafG, significantly increased expression of the 120-bp JMJD1C promoter construct in HEK-293T cells (Figure 2B). Inclusion of the −16-kb enhancer led to a further statistically significant increase in reporter gene activity (Figure 2B). Site-directed mutagenesis revealed that the proximal NFE2-binding site is essential for transactivation by NFE2, as its mutation completely abrogated reporter gene activity in HEK-293T cells (Figure 2C). The distal −16-kb site is likewise functional, as its mutation also reduced activity, albeit to a lesser extent (Figure 2C). Finally, we show that NFE2 activity is required for optimal JMJD1C expression as reduction of NFE2 levels by shRNA drastically reduced JMJD1C protein levels in HEL cells (Figure 2D). Taken together, these data show that JMJD1C levels depend on NFE2 activity.
Figure 2.

NFE2 positively regulates JMJD1C by binding regulatory sites. (A-C) Transactivation of the JMJD1C promoter by NFE2. (A) Schematic illustration of the JMJD1C reporter gene constructs. Open circles: predicted NFE2-binding sites as depicted in Figure 1A. The degree of phylogenetic conservation is illustrated. (B) Luciferase reporter vectors were cotransfected into HEK-293T cells with expression plasmids encoding either NFE2 and/or MafG. Data were normalized to the cotransfection with MafG alone, set as 1. Graphs represent mean ± SEM of 4 independent experiments. *P < .05 by Student t test. (C) Potential NFE2-binding sites were disrupted by site-directed mutagenesis in HEK-293T, indicated by crossed circles. Sequences of wt and mutated NFE2 sites are shown (altered bases in bold). Luciferase assays were performed as in panel B. Data were normalized to the −120-bp wt construct cotransfected with MafG only, set as 1. Graphs show mean and SEM of at least 3 independent experiments. ***P < .001, **P < .01 by 1-way ANOVA with the post hoc Tukey comparison test. (D) The effect of NFE2 silencing on JMJD1C expression. HEL cells were transduced with a lentivirus encoding an shRNA against NFE2 (sh) or a scrambled control (scr), sorted for GFP positivity, and analyzed by western blotting. The blot is representative of 3 independent experiments.
JMJD1C expression and global H3K9methylation levels are increased in PV patients
To test whether the increased NFE2 activity present in MPN patients leads to an increase in JMJD1C protein expression, we interrogated primary peripheral blood granulocytes from PV, ET, and PMF patients and healthy controls for JMJD1C protein expression. Although both JAK2V617F-positive and CALR-mutated ET patients expressed JMJD1C levels similar to healthy controls, both PV and PMF patients showed a significant increase in JMJD1C protein expression compared with healthy controls (Figure 3A). The increase in PMF patients was observed in both JAK2V617F-positive and CALR-mutated individuals. We and others have previously reported NFE2 overexpression in ET patients.2,18 However, NFE2 levels in ET are lower than in PV and PMF patients, and a proportion of ET patients show normal NFE2 expression.18 Because JMJD1C was not overexpressed in the ET patients tested here (Figure 3A), we investigated the relationship between NFE2 and JMJD1C protein levels. NFE2 and JMJD1C protein expression were significantly correlated (supplemental Figure 3D, Spearman correlation coefficient, P = .0005).
Figure 3.

Regulation of NFE2 expression by JMJD1C via a positive feedback loop entailing H3K9 demethylation. (A) JMJD1C expression in PV, ET, and PMF patients and healthy controls (HC). Peripheral blood granulocytes from n = 10 PV patients, n = 4 ET patients (3 JAK2V617F, 1 CALR-mutated), n = 6 PMF (2 JAK2V617F, 4 CALR-mutated) patients, and n = 19 healthy controls were analyzed by western blotting and densitometry (below). Representative blots are shown. Patient characteristics are given in supplemental Table 6. The Kruskal-Wallis showed that groups did not originate from the same distribution (P < .0001). Groups were therefore compared by using the Wilcoxon signed-rank test. *P < .05; **P < .001. (B) Global H3K9me3, H3K9me2, H3K9me1 levels in PV patients and healthy controls. Peripheral blood granulocytes from n = 10 PV patients and n = 9 healthy controls were analyzed by western blotting. One representative blot of 3 and the densitometric analysis of n = 10 and n = 9, respectively, are shown. ***P < .001; *P < .05. (C) JMJD1C binding in the NFE2 locus in PV patients and healthy controls. Peripheral blood granulocytes of n = 4 PV patients and n = 3 healthy controls were chromatin immunoprecipitated with an antibody against JMJD1C or an IgG control. Top: chromatin immunoprecipitated DNA was amplified with primers covering the -1A and -1F promoter as well as the −5-kb region in the NFE2 locus, illustrated in the schematic representation. Bottom: densitometric analysis of ChIP results. Graphs represent mean ± SEM. *P < .05 by the Student t test. (D) H3K9me2 levels in the NFE2 locus in PV patients and healthy controls. Peripheral blood granulocytes of n = 6 PV patients and n = 7 healthy controls were chromatin immunoprecipitated with an antibody against H3K9me2 or an IgG control. Chromatin immunoprecipitated DNA was amplified as in panel C. One representative PV patient and healthy control each are depicted. Bottom: densitometric analysis. Graphs represent mean ± SEM. ***P < .001; *P < .05 by the Student t test. (E) HP1α binding in the NFE2 locus in PV patients and healthy controls. Peripheral blood granulocytes of n = 4 PV patients and n = 3 healthy controls were chromatin immunoprecipitated with an antibody against HP1α or an IgG control. Chromatin immunoprecipitated DNA was amplified as in panel C. Right, Densitometric analysis. Graphs represent mean ± SEM. *P < .05 by the Student t test. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
JMJD1C has been postulated to demethylate monomethylated and dimethylated histone 3 at lysine 9.19 We therefore investigated whether the increased levels of JMJD1C present in PV patients altered the amount of monomethylated, dimethylated, or trimethylated H3K9 in primary patient cells. Indeed, PV patients showed a statistically significant global reduction in K9 monomethylated and dimethylated histone 3 compared with healthy controls (Figure 3B). The H3K9me3 mark, which is not affected by JMJD1C, was similar in patients and control cells (Figure 3B). Our data thus demonstrate that expression of the histone demethylase JMJD1C is elevated in PV and PMF patients and that, concomitantly, global H3K9methylation levels are altered.
NFE2 and JMJD1C participate in an autoregulatory feedback loop
Because global protein levels and H3K9 methylation do not inform about regulatory events at specific loci, we examined H3K9 methylation in more detail. Because altered gene expression sometimes leads to the formation of self-sustaining autoregulatory loops, we directly assayed JMJD1C binding as well as H3K9me2 at the 2 NFE2 promoters as well as at the −5-kb enhancer. In primary PV cells, there was a significant increase in JMJD1C binding to the 1A promoter as well as a strong trend, which did not quite reach statistical significance, to increased binding at the 1F promoter (Figure 3C). Moreover, in PV patients, all 3 sites within the NFE2 locus carried statistically significantly less H3K9me2 than healthy controls (Figure 3D). The low level of immunoprecipitation with the immunoglobulin G (IgG) control represents nonspecific background binding to either immunoglobulin proteins or to the beads used. Increased binding of the demethylase with a concomitant decrease in the amount of repressive H3K9me2 marks are thus likely to contribute to the overexpression of NFE2 observed in PV patients. As NFE2 directly regulates JMJD1C expression (Figures 1-2), this constitutes a positive feedback loop that continually augments NFE2 expression.
Binding of HP1 at the NFE2 locus is decreased in PV patients
Methylation of H3K9 regulates binding of heterochromatin protein 1 (HP1), which can mediate gene silencing by facilitating heterochromatin formation.20 We therefore investigated HP1α binding to the NFE2 locus in PV patients and healthy controls. HP1α binding to the NFE2-1A locus was significantly decreased in PV patients (Figure 3E). Increased binding of JMJD1C (Figure 3C) and decreased H3K9 dimethylation at the NFE2-1A promoter (Figure 3D) thus lead to a decrease in HP1α binding (Figure 3E). A decrease in binding of the repressive HP1α protein will contribute to an open chromatin structure with consequentially increased NFE2 expression as observed in MPN patients.
Decitabine treatment restores HP1α binding and reduces NFE2 expression
We sought to reduce the elevated NFE2 levels and thereby lower JMJD1C expression, and restore the physiological H3K9me2 amounts and HP1α binding at the NFE2 locus by pharmacological intervention. Treatment of HEL cells with the epigenetic modifier decitabine significantly reduced both NFE2 and JMJD1C levels (Figure 4A). Concomitantly, H3K9me2 levels at the NFE2-1A and -1F promoter increased significantly (Figure 4B). Fitting with this model, decitabine treatment augmented HP1α binding on the NFE2 locus (Figure 4C). Importantly, global H3K9me2 levels were not affected by decitabine treatment, indicating some site specificity (Figure 4A). Our data thus demonstrate that decitabine treatment reverses aberrant histone methylation at the NFE2 locus, reestablishes physiological HP1α binding, and thereby reduces NFE2 expression.
Figure 4.

Decitabine treatment reduces NFE2 expression by reversing H3K9 hypomethylation and H3Y41 phosphorylation. (A) Effect of decitabine treatment on the expression of NFE2, of JMJD1C, and on total H3K9me2 levels in HEL cells. HEL cells were treated with 400 ng/μL decitabine or dimethyl sulfoxide (DMSO). Lysates were interrogated by western blot. (B-D) Effect of decitabine treatment on H3K9me2 levels, HP1α binding, and H3Y41 phosphorylation at the NFE2 locus in HEL cells. Top panels: HEL cells were treated as in panel A and chromatin immunoprecipitated with an antibody against H3K9me2 (B), HP1α (C), H3Y41ph (D), or an IgG control as indicated. Chromatin immunoprecipitated DNA was amplified with the primers illustrated in Figure 3C. One representative result of at least 3 independent experiments is shown. Bottom panels: densitometric analysis of the ChIP results. Graphs represent mean ± SEM. *P < .05, **P < .01 by the Student t test. DAC, decitabine.
NFE2 is regulated via JAK2-mediated histone tyrosine phosphorylation
HP1α binding has recently also been shown to be regulated by a novel function of JAK2.21 JAK2 phosphorylates Tyr41 on histone 3 (H3Y41), thereby reducing HP1α binding.21 We therefore investigated whether Tyr41 is phosphorylated in the NFE2 locus and whether this is altered by decitabine treatment. Indeed, the NFE2-1A promoter carries phospho-H3Y41 and this is significantly reduced following decitabine treatment (Figure 4D).
HEL cells are homozygous for the constitutively activating JAK2V617F mutation found in the majority of MPN patients. We therefore investigated the effect of decitabine in a JAK2wt cell line, K562. In K562 cells, decitabine treatment did not alter NFE2 levels (Figure 5A), nor did it change H3K9me2 levels at the NFE2-1A and -1F promoters (Figure 5B). We confirmed the difference between JAK2V617F mutant and JAK2wt cells using 3 additional lines: decitabine treatment reduced NFE2 levels in JAK2V617F mutant HEL, UKE-1, and SET-2 cells (Figure 5C), but not, or only slightly in JAK2wt K562 or KMOE-2 cells (Figure 5D). The small reduction observed in KMOE-2 cells is much slighter than that observed in the 3 JAK2V617F mutant cell lines. Therefore, NFE2 silencing by decitabine may occur selectively in JAK2V617F mutant cells.
Figure 5.

Decitabine-dependent downregulation of NFE2 occurs selectively in the presence of JAK2V617F. (A) Effect of decitabine treatment on NFE2 expression and total H3K9me2 levels in K562 cells. K562 cells were treated with 400 ng/μL decitabine or DMSO. Lysates were interrogated by western blot. (B) Effect of decitabine treatment on H3K9me2 levels in the NFE2 locus in K562 cells. Cells were treated as in panel A. Cell lysates were chromatin immunoprecipitated with an antibody against H3K9me2 or an IgG control. Chromatin immunoprecipitated DNA was amplified with primers covering the NFE2 locus as illustrated in Figure 3C. One representative result of 3 independent experiments is shown. Bottom panel; densitometric analysis. Graphs represent mean ± SEM. (C-D) NFE2 expression after decitabine treatment in JAK2V617F-positive and JAK2wt cell lines. Whole-cell extracts of the indicated cell lines were treated as noted and analyzed as described in panel A. (C) JAK2V617F-positive; (D) JAK2wt. (E) Effect of decitabine treatment on SHP1, phosphorylated JAK2 (p-JAK2), JAK2, and HP1α expression. HEL cells were treated as in panel A. Total cell lysates were interrogated by western blotting. One representative result of 3 independent experiments is shown. (F) Effect of JAK2 inhibitor treatment on NFE2 and JMJD1C expression. HEL cells were treated with the indicated concentrations of the JAK2 inhibitor TG101348 (fedratinib)26 for 48 hours as indicated. Whole-cell lysates were analyzed by western blotting for JMJD1C, NFE2, PU.1, and actin. One representative result of at least 2 independent experiments is shown. (G) H3Y41P ChIPseq density profiles at the NFE2 gene locus in HEL cell ± JAK2 inhibition. Visualization of H3K4me3, H3Y41P before and after inhibition of JAK2 by TG101209 and IgG control. ChIPseq data from Dawson et al.27 (H) Effect of Jmjd1c knockdown on cytokine-independent cell growth. Baf/3-Jak2V617F cells expressing Jak2V617F were transduced with a lentivirus encoding an shRNA against Jmjd1c or a scrambled control. Western blot analysis showed successful knockdown of JMJD1C (left, top). The cells were cultured without (left) or with (right) IL3. Cells were counted every 24 hours. Three independent experiments were performed in triplicates. Data points represent mean ± SEM. ***P < .001; **P < .01, *P < .05 by 2-way ANOVA.
Decitabine treatment has been shown to inhibit JAK2 activity by upregulating expression of the phosphatase Src homology region 2 domain-containing phosphatase-1 (SHP1), which dephosphorylates and deactivates JAK2.22 We confirmed the effect of decitabine on SHP1 expression and JAK2 phosphorylation by western blot (Figure 5E), supporting our hypothesis that the decrease of Y41 phosphorylation of histone 3 observed upon decitabine treatment is indeed due to decreased JAK2 activity. Because the decrease in JAK2 phosphorylation by decitabine does not directly prove that this kinase modulates NFE2 levels, we treated HEL cells with the JAK2 inhibitor TG101348 (fedratinib) and assessed NFE2 expression by western blot. Treatment with the JAK2 inhibitor reduced NFE2 protein expression in a dose-dependent mechanism (Figure 5F, top), but not expression of a second myeloid transcription factor, PU.1 (Figure 5F, bottom). Likewise, inhibition of JAK2 activity by TG101348 strongly reduced JMJD1C expression (Figure 5F, bottom). ChIPseq analysis demonstrates that the NFE2 locus is bound by H3 phosphorylated at Y41 in vivo and that inhibition of JAK2 activity strongly decreases the presence of this mark at the NFE2 locus (Figure 5G). In summary, these data strongly argue that NFE2 expression is regulated both via the novel epigenetic STAT-independent activity of JAK2 as well as by JMJD1C in a positive feedback mechanism.
Depletion of Jmjd1c selectively reduces JAK2V617F-mediated cytokine-independent proliferation
Therapeutic JAK2 inhibition, while ameliorating symptoms and decreasing splenomegaly in MPN patients, has only a modest effect on the JAK2V617F allele burden. These observations suggest that the MPN clone does not rely solely on activated JAK2 for its growth advantage. We therefore investigated whether decreasing aberrantly elevated JMJD1C expression (Figure 3A) would affect cell proliferation despite the presence of constitutively active JAK2. Erythroid cells rendered cytokine independent by expression of JAK2V617F (Baf/3-Jak2V617F) were left untreated, transduced with a vector expressing a scrambled control shRNA, or transduced with a vector expressing an shRNA against JMJD1C. In the latter cells, expression of the demethylase was reduced by ∼60% (Figure 5H, top left). The 3 cell lines were subsequently assessed for proliferation both in the presence of IL3 and in absence of the cytokine. Decreased JMJD1C expression caused a highly significant decrease specifically in JAK2V617F-driven, cytokine-independent proliferation, whereas cytokine-dependent growth was affected only slightly (Figure 5H right). These data suggest that inhibiting JMJD1C expression or activity in MPN patients would retard expansion of the malignant clone.
Discussion
In our previous work, we have demonstrated the influence of NFE2 on MPN pathophysiology. Elevated NFE2 levels induce a MPN phenotype in several murine models and suffice to cause spontaneous transformation to acute myeloid leukemia.4,5 However, the upstream pathways by which NFE2 overexpression is effected in MPN patients, as well as the downstream pathways through which it exerts its effect, remain incompletely understood. As a transcription factor, NFE2 itself constitutes a difficult drug target because pharmacological modulation of transcription factor activity has not been widely achieved. However, a deeper understanding of the NFE2 upstream and downstream effectors may point the way to novel targets for MPN therapy, as these proteins may prove more amenable to pharmacological inhibition. As our murine models have shown that elevated NFE2 levels predispose to leukemic transformation, our data argue that early intervention to normalize NFE2 levels in patients at risk may prevent transformation to acute leukemia.
Here, we demonstrate that the histone demethylase JMJD1C constitutes a novel NFE2 target gene. Moreover, JMJD1C participates in a positive feedback loop, as the protein itself binds the NFE2 promoter, thereby epigenetically regulating the expression of its own transcriptional activator. In addition, the NFE2 locus is regulated by the novel epigenetic function of JAK2 through histone phosphorylation. Our data show that NFE2 expression is regulated by compounding epigenetic mechanisms and that these are perturbed in MPN patients.
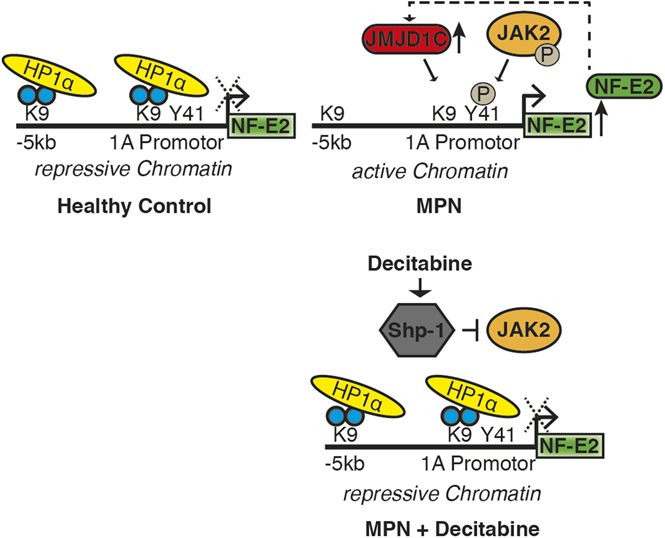
Our data suggest that in PV patients, the NFE2 locus is found in an open chromatin formation (Figure 6A). Open chromatin at the NFE2 locus is the result of 2 interlocking epigenetic mechanisms: for 1, constitutive JAK2 activity causes phosphorylation of H3Y41 in the NFE2 promoter. At the same time, increased JMJD1C binding to the NFE2 locus causes demethylation of H3K9me2. Both modifications prevent binding of the repressive HP1α protein, leading to open, active chromatin, allowing increased NFE2 expression. NFE2, in turn, activates transcription of JMJD1C, completing an autoregulatory loop (Figure 6B).
Figure 6.

JAK2 and JMJD1C epigenetically regulate NF-E2. (A) Schematic representation of NFE2 regulation by JAK2 and JMJD1C and the effect of decitabine. (B) Illustration of the regulation of NF-E2 by JMJD1C in a positive feedback loop. P, phosphoryl group.
In our study, decitabine treatment, among the pleiotropic effects it exhibits on cells, counteracts this mechanism by inhibiting JAK2 activity, via the induction of SHP1, leading to decreased H3Y41ph levels. Consequently, HP1α binding is increased, decreasing NFE2 expression and subsequently lowering JMJD1C expression, which augments the levels of H3K9me2, further increasing HP1α binding and thereby decreasing NFE2 transcription. Hence, decitabine acts at several points to restore the inactive chromatin configuration at the NFE2 locus, found in healthy controls (Figure 5G). It is important to note that the observations stem from data obtained in cell lines. We cannot exclude that decitabine has additional effects, for example by altering DNA methylation, but we did not observe any changes in DNA methylation of the NFE2 gene following decitabine treatment.
JMJD1C has recently been shown to be required for establishment of MLL-AF9 driven leukemia in a murine model.23 Moreover, shRNA-mediated knockdown of JMJD1C inhibited both colony formation and growth of murine MLL-AF9 cells as well as primary human leukemic cells ex vivo.23 We could likewise demonstrate that JMJD1C is required for cytokine-independent growth (Figure 5H). These data implicate JMJD1C as a potential therapeutic target in leukemia and MPNs. However, to date, effective JMJD1C inhibitors have not been developed. Decitabine treatment, by lowering NFE2 levels, decreasing JMJD1C expression, and subsequently increasing H3K9me2 levels (Figure 4A-B), may provide a viable alternative.
Treatment with decitabine or the related agent 5-azacitidine has been investigated in small cohorts of selected MPN patients. In a cohort of 34 PMF patients, 5-Aza-C led to clinical improvement in 21% of patients, after a median of 5 months, whereas only 1 patient showed a partial response.24 Likewise, of 6 patients with myelofibrosis in blast phase unable to undergo allogeneic bone marrow transplant, 4 remained alive on decitabine treatment after a median follow-up of 9 months, demonstrating that this is a viable treatment option in this challenging clinical setting.25 As JAK2 inhibition and decitabine both decrease NFE2 levels (Figures 4A and 5F), combination of both drugs may provide synergistic effects and constitute a further promising therapy option. Such a combination (ruxolitinib and decitabine) is currently being investigated in blast-phase MPN and post-MPN AML patients in a phase 1/2 trial by the MPD Research Consortium (MPD-RC, trial 109).
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Franziska Zipfel for expert technical assistance, and Konstanze Doehner and Frank Stegelmann for their valuable contribution.
This work was supported by grants from the National Institutes of Health, National Cancer Institute (1 PO1 CA108671 [H.L.P.]) and the Deutsche Forschungsgemeinschaft (Pa 611/8-1 and SFB 992–TP02 [H.L.P.]). HLP is a member of the MPD Research Consortium (MPD-RC). J.C.P. was funded by the “MOTIVATE” scholarship program supported by the Else-Kroener-Fresenius-Stiftung. This work was supported, in part, by the Excellence Initiative of the German Research Foundation (GSC-4, Spemann Graduate School [J.S.J. and H.L.P.]).
H.L.P. is a member of the MPD Research Consortium (MPD-RC).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.C.P. designed and performed research, analyzed data, and wrote the paper; J.S.J., J.W., C.K., H.F.S., T.S.S., and C.J.O. designed and performed research and analyzed data; H.B. performed research and analyzed data; E.S. designed and performed research; D.H.S. and M.G. performed research; A.G. supervised research and analyzed data; and H.L.P. designed and supervised research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Heike L. Pahl, University Medical Center Freiburg, Klinik fuer Tumorbiologie, Breisacher Str. 117, 79106 Freiburg, Germany; e-mail: heike.pahl@uniklinik-freiburg.de.
REFERENCES
- 1.Goerttler PS, Kreutz C, Donauer J, et al. Gene expression profiling in polycythaemia vera: overexpression of transcription factor NF-E2. Br J Haematol. 2005;129(1):138-150. [DOI] [PubMed] [Google Scholar]
- 2.Wang W, Schwemmers S, Hexner EO, Pahl HL. AML1 is overexpressed in patients with myeloproliferative neoplasms and mediates JAK2V617F-independent overexpression of NF-E2. Blood. 2010;116(2):254-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kapralova K, Lanikova L, Lorenzo F, et al. RUNX1 and NF-E2 upregulation is not specific for MPNs, but is seen in polycythemic disorders with augmented HIF signaling. Blood. 2014;123(3):391-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaufmann KB, Gründer A, Hadlich T, et al. A novel murine model of myeloproliferative disorders generated by overexpression of the transcription factor NF-E2. J Exp Med. 2012;209(1):35-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jutzi JS, Bogeska R, Nikoloski G, et al. MPN patients harbor recurrent truncating mutations in transcription factor NF-E2. J Exp Med. 2013;210(5):1003-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wehrle J, Seeger TS, Schwemmers S, Pfeifer D, Bulashevska A, Pahl HL. Transcription factor nuclear factor erythroid-2 mediates expression of the cytokine interleukin 8, a known predictor of inferior outcome in patients with myeloproliferative neoplasms. Haematologica. 2013;98(7):1073-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011;29(10):1356-1363. [DOI] [PubMed] [Google Scholar]
- 8.ENCODE Project Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011;9(4):e1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fathi AT, Abdel-Wahab O. Mutations in epigenetic modifiers in myeloid malignancies and the prospect of novel epigenetic-targeted therapy. Adv Hematol. 2012;2012:469592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mascarenhas J, Roper N, Chaurasia P, Hoffman R. Epigenetic abnormalities in myeloproliferative neoplasms: a target for novel therapeutic strategies. Clin Epigenetics. 2011;2(2):197-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roelz R, Pilz IH, Mutschler M, Pahl HL. Of mice and men: human RNA polymerase III promoter U6 is more efficient than its murine homologue for shRNA expression from a lentiviral vector in both human and murine progenitor cells. Exp Hematol. 2010;38(9):792-797. [DOI] [PubMed] [Google Scholar]
- 12.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402-408. [DOI] [PubMed] [Google Scholar]
- 13.Swerdlow S, Campo E, Harris N. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IRAC Press; 2008. [Google Scholar]
- 14.Temerinac S, Klippel S, Strunck E, et al. Cloning of PRV-1, a novel member of the uPAR receptor superfamily, which is overexpressed in polycythemia rubra vera. Blood. 2000;95(8):2569-2576. [PubMed] [Google Scholar]
- 15.Jin F, Li Y, Dixon JR, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503(7475):290-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet. 2013;14(11):765-780. [DOI] [PubMed] [Google Scholar]
- 17.Gu L, Frommel SC, Oakes CC, et al. ; ICGC Project on Early Onset Prostate Cancer. BAZ2A (TIP5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence. Nat Genet. 2015;47(1):22-30. [DOI] [PubMed] [Google Scholar]
- 18.Kralovics R, Teo SS, Buser AS, et al. Altered gene expression in myeloproliferative disorders correlates with activation of signaling by the V617F mutation of Jak2. Blood. 2005;106(10):3374-3376. [DOI] [PubMed] [Google Scholar]
- 19.Kim SM, Kim JY, Choe NW, et al. Regulation of mouse steroidogenesis by WHISTLE and JMJD1C through histone methylation balance. Nucleic Acids Res. 2010;38(19):6389-6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bannister AJ, Zegerman P, Partridge JF, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410(6824):120-124. [DOI] [PubMed] [Google Scholar]
- 21.Dawson MA, Bannister AJ, Göttgens B, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461(7265):819-822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiong H, Chen ZF, Liang QC, et al. Inhibition of DNA methyltransferase induces G2 cell cycle arrest and apoptosis in human colorectal cancer cells via inhibition of JAK2/STAT3/STAT5 signalling. J Cell Mol Med. 2009;13(9B):3668-3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sroczynska P, Cruickshank VA, Bukowski JP, et al. shRNA screening identifies JMJD1C as being required for leukemia maintenance. Blood. 2014;123(12):1870-1882. [DOI] [PubMed] [Google Scholar]
- 24.Quintás-Cardama A, Tong W, Kantarjian H, et al. A phase II study of 5-azacitidine for patients with primary and post-essential thrombocythemia/polycythemia vera myelofibrosis. Leukemia. 2008;22(5):965-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mascarenhas J, Navada S, Malone A, Rodriguez A, Najfeld V, Hoffman R. Therapeutic options for patients with myelofibrosis in blast phase. Leuk Res. 2010;34(9):1246-1249. [DOI] [PubMed] [Google Scholar]
- 26.Wernig G, Kharas MG, Okabe R, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13(4):311-320. [DOI] [PubMed] [Google Scholar]
- 27.Dawson MA, Foster SD, Bannister AJ, et al. Three distinct patterns of histone H3Y41 phosphorylation mark active genes. Cell Reports. 2012;2(3):470-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.