

Abstract
Sepsis is the leading cause of acute kidney injury (AKI) in the intensive care unit (ICU). Septic AKI is a complex and multifactorial process that is incompletely understood. During sepsis, the disruption of the mucus membrane barrier, a shift in intestinal microbial flora, and microbial translocation may lead to systemic inflammation, which further alters host immune and metabolic homeostasis. This altered homeostasis may promote and potentiate the development of AKI. As part of this vicious cycle, when AKI develops, the clearance of inflammatory mediators and metabolic products is decreased. This will lead to further gut injury and breakdown in mucous membrane barriers. Thus, changes in the gut during sepsis can initiate and propagate septic AKI. This deleterious gut–kidney crosstalk may be a potential target for therapeutic maneuvers. This review analyses the underlying mechanisms in gut–kidney crosstalk in septic AKI.
Keywords: Sepsis, AKI, Septic AKI, Gut–kidney crosstalk, Inflammation
Background
Acute kidney injury (AKI) is a serious complication in critically ill patients and its development is associated with a high rate of mortality and morbidity as well as increased costs [1]. Sepsis is the leading cause of AKI in the intensive care unit (ICU), and 45 to 70% of all AKI is associated with sepsis [2]. Dialysis requiring AKI, when associated with distant organ dysfunction such as cardiac or respiratory failure, is associated with mortality rates as high as 60 to 80% [3, 4]. Septic AKI is a complex and multifactorial process, and our understanding of its pathogenesis remains incomplete [5]. The current understanding of septic AKI involves: microcirculatory abnormalities, renal tubular epithelial cell metabolic dysfunction and injury, and inflammatory changes [6].
Inflammation is a prominent component of septic AKI. Sepsis and inflammation at the tissue and cellular levels are associated with decreased levels of intracellular adenosine triphosphate (ATP) and with mitochondrial injury in the kidney [7]. It has been well described that higher cytokine concentrations are associated with slower renal recovery from AKI and increased mortality rates [8, 9].
The gastrointestinal tract (gut) is the most common source of secondary infections which occur predominately in the later stages of sepsis. The gut is home to 70–80% of the body’s immune cells and at least hundreds of different microbiome species [10]. In septic patients, inflammation and hypoperfusion play an important role in the pathophysiology of gut injury [11]. Gut injury may lead to impaired gut barrier function, which may result in translocation of bacteria and toxins from the intestinal lumen to the mesenteric lymph and systemic circulation. Subsequently, this bacterial translocation may amplify the systemic inflammatory response and contribute to multiple organ failure and death [12]. This dysregulated crosstalk between the gut’s epithelium, immune system, endogenous microflora, and kidney may lead to worsening of systemic inflammation and potentiation of AKI [13] (Fig. 1).
Fig. 1.
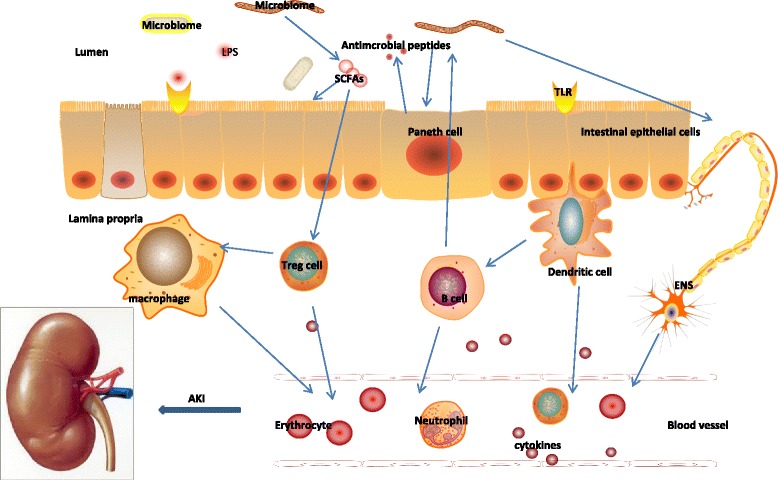
Effect of disruption of the gut mucosal barrier and bacterial translocation on the systemic inflammatory response in septic AKI. During sepsis, the combined effect of disruption of the mucus membrane barrier, a shift in the composition and virulence of intestinal microbes, and microbe translocation in gut lead to expansive inflammation, which will further alter host immune and metabolic homeostasis. The altered immune homeostasis and systemic inflammation can promote AKI in sepsis
The understanding of the gut’s role in initiating septic AKI has led to potential novel therapeutic targets that are currently under investigations. In this review, we summarize the underlying mechanisms of gut–kidney crosstalk in septic AKI.
The effect of septic AKI on gastrointestinal function
During septic AKI, increased inflammatory cytokines as well as impairment in the clearance of water and metabolic products (urea in particular) can cause gut injury (Fig. 2). Specifically, increased inflammatory cytokines can damage gut barrier function and increase permeability. The gut’s barrier function is due to apical tight junctions and junctional adhesion molecules (JAM), which prevent luminal contents from escaping into the local extra-luminal environment. During septic AKI, increased levels of cytokines can act on these junctional complexes to modulate permeability [14]. Intestinal hyper-permeability may result when sepsis alters the expression of zonulaoccludens 1 (ZO-1), any one of multiple claudin isoforms, or occludin in the tight junction complex. Alternatively, hyper-permeability may be induced by altering expression of components of JAM [14]. Activation of myosin light chain kinase (MLCK) by cytokines can also worsen para-cellular permeability. MLCK phosphorylates myosin light chain, which results in contraction or opening of the apical tight junction [15]. MLCK activation is associated with an increase in interleukin (IL)-6, tumor necrosis factor α (TNF-α), and IL-1β. The net result is an increase in intestinal permeability. The increased intestinal permeability leads to an amplification of the systemic inflammatory response in a positive feedback response. The increased systemic inflammation further promotes kidney injury.
Fig. 2.
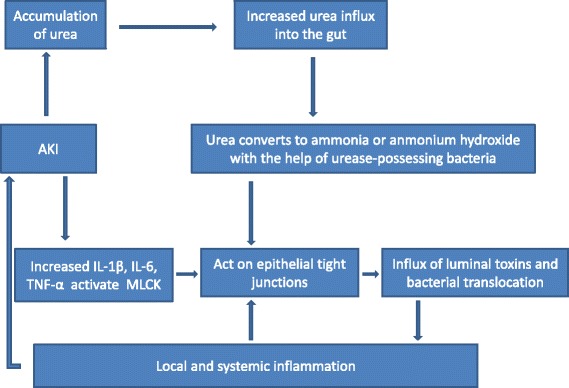
Effect of increased inflammatory cytokines and decreased urea clearance in septic AKI on the gut
During septic AKI, the dysfunction in clearance of metabolic products and water can also directly weaken the gut barrier and increase gut permeability. In septic AKI, kidney dysfunction (with resulting retention of uremic solutes, urea, sodium, and water) as well as aggressive fluid resuscitation can cause a dramatic increase in gut wall edema. This can cause disruption of the colonic epithelial tight junction apparatus [16, 17]. Urea diffuses from the blood into the gut lumen and is metabolized by gut bacterial urease to ammonia (CO(NH2)2 + H2O → CO2 + 2NH3). The ammonia is converted into caustic ammonium hydroxide (NH3 + H2O → NH4OH), which is capable of disrupting tight junction proteins that seal the gap between epithelial cells [17]. Breakdown of this protein triggers influx of luminal toxins as well as bacterial translocation, thus promoting local and systemic inflammation and further damaging the gut’s barrier function.
Increased cytokine levels in septic AKI can lead to dysregulation of gut stem cell proliferation/apoptosis
Toll-like receptors (TLRs) which act as pathogen receptors are expressed on various types of epithelial cells, including in the kidney and gut [13, 18]. Intestinal stem cells express TLR4, which regulates whether they proliferate or die by apoptosis [19]. On a cellular level, crypt proliferation is markedly decreased and both crypt and villus apoptosis are simultaneously increased following sepsis [20]. Although epithelial migration is slowed in critical illness in a TLR4-dependent manner, changes in proliferation and apoptosis overwhelm this slowed migration of cells, resulting in a marked diminution of villus length during sepsis [21]. Simultaneously, critical illness induces global alterations in the mucus layer (reduced thickness, diminished luminal coverage, and poor adherence) and altered gut barrier function [22]. Furthermore, increased cytokine concentrations seen in septic AKI can impair gut cell regeneration and stimulate apoptosis in a TLR4-dependent manner [23]. The increased inflammatory cytokine levels can also cause gut cell apoptosis directly. The net result is that gut apoptosis can enhance hyper-permeability, bacterial translocation, and expansion of the inflammatory response [24].
To summarize, during septic AKI, increased inflammatory cytokine concentrations and retention of urea as well as other metabolites and water can damage the gut barrier, leading to increased permeability. Impaired gut barrier function results in translocation of bacteria and toxins from the intestinal lumen to the mesenteric lymph system and to the systemic circulation. Subsequently, this bacterial translocation may amplify the systemic inflammatory response and contribute to multiple organ failure and death [12].
The role of the microbiome in gut–kidney crosstalk
The intestinal microbiome is made up of more than 100 trillion microorganisms, and is continuously changing over the life of the host, based on diet, age, drug intake, and presence or absence of disease [25]. Increasingly, it is recognized that the microbiome plays a crucial role in the maintenance of health and that alterations in the type, number, and function of microorganisms in the microbiome can have a critical role on survival in critical illness [26]. During septic AKI, elevation in inflammatory cytokine levels as well as ischemia can induce changes in the constituent organisms that make up the microbiome of the gut. Importantly, the severity of inflammation can be modulated by the microbiome [25]. For instance, microbes or microbial products can actively change TLR expression in most cellular compartments of the gut tract, and this alters the host’s ability to sense and respond to the microbiota. Several other changes occur in gut physiology in septic patients, due to either extrinsic factors (antibiotics and parenteral nutrition) or intrinsic factors (systemic inflammation and gut leakage). These changes, in turn, influence the composition of the enteric flora [27]. A massive loss of microbe diversity occurs in patients with severe sepsis, particularly loss of anaerobic diversity [28]. For instance, within 6 h after the onset of sepsis, 90% of the normal anaerobic flora is lost in the gut [29]. In addition, critically ill septic patients relying on parenteral nutrition commonly have thinning of the protective mucus layer. This leads to a decrease in barrier integrity and the availability of immunomodulatory short-chain fatty acids (SCFAs) in the gut [13, 30].
Commensal bacteria (which are lost in sepsis) catabolize polysaccharides to generate SCFAs, and SCFAs play an important role in maintaining immune homoeostasis [31]. SCFAs can enhance the intestinal epithelial barrier function and activate the development of regulatory T (Treg) cells [31]. Monocytes are modulated by microbiota-derived products to prime natural killer (NK) cells via the interferon signaling pathway, which is pivotal in the immune response against other viral infections [32]. Interestingly and as a proof of the importance of the microbiome, therapy with three microbiota-derived SCFAs (acetate, propionate, and butyrate) can improve kidney function in a septic AKI model [32]. This protection was associated with low levels of local and systemic inflammation, oxidative cellular stress, cell infiltration/activation, and apoptosis. In addition, SCFAs ameliorated the effects of hypoxia in kidney epithelial cells by improving mitochondrial biogenesis.
Effect of gut injury on the kidney in septic AKI
The impaired gut barrier function and bacterial translocation in septic AKI increase systemic inflammation, which are associated with slow renal recovery and mortality [8, 9]. After gut injury, the increased permeability results in translocation of bacteria and toxins from the intestinal lumen to the mesenteric lymph and systemic circulation [33, 34]. The influx of toxin contents can directly enter the circulation [35, 36] and/or educate circulating immune cells [37–39], which will cause an amplified inflammation in septic AKI that shifts metabolism towards aerobic glycolysis [40]. These changes are associated with decreased levels of intracellular adenosine triphosphate (ATP) and with mitochondrial injury in the kidney [7]. The decreased ATP synthesis and mitochondrial injury are the main causes of kidney dysfunction and injury [9]. The increased inflammatory cytokines (especially TNF-α) can induce kidney apoptosis through the extrinsic pathway in septic AKI [41, 42]. Up-regulation of Bcl-2 consistently blocks Bax and Bak activation, resulting in the preservation of mitochondrial integrity and cell viability and further supports the intrinsic pathway of apoptosis in septic AKI [41] (Fig. 3).
Fig. 3.
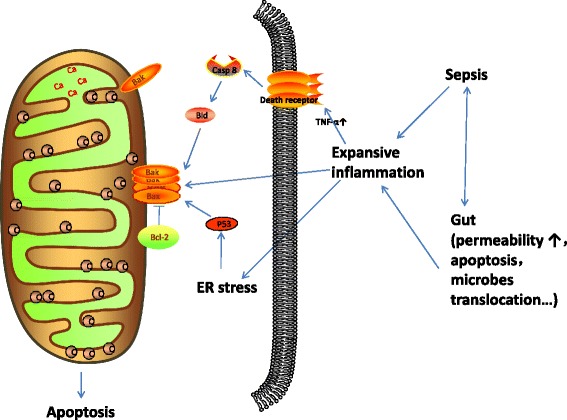
The different pathways (intrinsic and extrinsic) involved in renal apoptosis in septic AKI. ER endoplasmic reticulum
The role of monocytes/macrophages in gut–kidney crosstalk
Monocytes/macrophages play an important role in the initiation or progression of inflammatory diseases [43]. Inflammation is closely related to macrophage activation: M1 macrophages exert pro-inflammatory activities, whereas M2 macrophages are involved in resolving inflammation [44] and in facilitating tissue repair [45]. Infection causes monocytes to migrate and infiltrate into organs where they differentiate via M1 or M2 pathways into pathogen-killing or tissue-repair phenotypes, respectively. Therefore, strategies that limit early macrophage infiltration or activation may represent a novel approach in the prevention or treatment of AKI in septic patients. However, the signaling pathway involved in the repair mechanism of M2 macrophages needs further investigation [46]. During sepsis, as the gut barrier function is impaired, bacteria translocation and expanding inflammation change the immune microenvironment, which may determine the fate of macrophage differentiation in the gut, kidney, or other distant organs into a more pro-inflammatory phenotype.
Targeting the microbiome for therapeutic gain in gut–kidney crosstalk
As the gut plays an important role in the progression of inflammation and septic AKI, efforts to modulate the gut microbiome and barrier function to improve outcomes make great sense. Strategies include intake of live microbiota, addition of the necessary nutrients for microbiota regeneration, and administration of exogenous supplements such as SCFAs that are the products of microbiota. Selective decontamination of the digestive tract, probiotics, phosphate, and SCFAs are the most studied interventions in this regard and targeting the microbiome for therapeutic gain to regulate the immune function is a promising strategy to improve outcomes in sepsis. However, human data are still evolving in this therapeutic area.
Selective decontamination of the digestive tract
Selective decontamination of the digestive tract (SDD) uses non-absorbable microbials, which are applied daily in the oropharynx and the gastrointestinal tract. The aim of this intervention is to prevent secondary colonization and overgrowth of potential bacterial pathogens while preserving the anaerobic microbiota, thereby preventing excess infectious disease in critically ill patients [47, 48]. As 90% of the normal anaerobic flora is lost after a sudden insult such as sepsis [29], the addition of SDD to a septic AKI patient is promising to rebuild the gut’s barrier, microbiome, and immune function. The aim of this intervention is to break down the continuous cycle of injury followed by amplification of inflammation that occurs in the gut–kidney crosstalk pathway [32]. A comprehensive systematic review and network meta-analysis [48], which encompasses more than 60 clinical studies and ten meta-analyses, suggested that SDD can prevent noscomial infections in critically ill patients and decreases overall mortality rates. However, randomized controlled trials are still needed in this area.
Probiotics
Probiotics are live bacteria and yeasts that, when administered in adequate amounts, confer a health benefit on the host. Although their clinical use is rising, data on efficacy are still emerging. In a murine cecal ligation and perforation model of sepsis, the administration of the probiotics Lactobacillus rhamnosus GG (LGG) and Bifidobacterium longum (BL) improved mortality following sepsis and prevented sepsis-induced changes in gut epithelial apoptosis and proliferation [49]. Additionally, probiotics can attenuate growth of pathogenic intestinal bacteria, potentially limiting endotoxin production and preventing bacteremia [50]. As a corollary to probiotic administration, there is also increasing evidence that fecal microbiota transplantation is significantly more effective in the treatment of recurrent Clostridium difficile infection than standard antibiotic therapy by increasing fecal bacterial diversity in recipients [51].
Short-chain fatty acids
SCFAs are microbial metabolic products of dietary fibers, and the most studied SCFAs are butyrate, propionate, and acetate. These metabolites are sensed by the G-protein-coupled receptors (GPR) on intestinal epithelium and can also diffuse across the epithelium [13, 30] to affect enteric nervous and immune systems. SCFAs can directly affect Treg cells [52, 53], neutrophils [54], monocytes [55], and mast cell [56] through the GPRs expressed on them, which will modulate the immune homeostasis of the body. It has been shown in animal experiments that therapy with three microbiota-derived SCFAs (acetate, propionate, and butyrate) improved renal dysfunction in a sepsis model, largely through epigenetic modulation of the inflammatory process [32]. SCFAs were also sufficient to up-regulate serotonin (5-HT) [57] . 5-HT can promote immune function of leukocytes by either enhancing dendritic cell-mediated T-cell activation or affecting macrophage polarization and phagocytosis [58] (Fig. 4). This promising avenue of research requires future study.
Fig. 4.
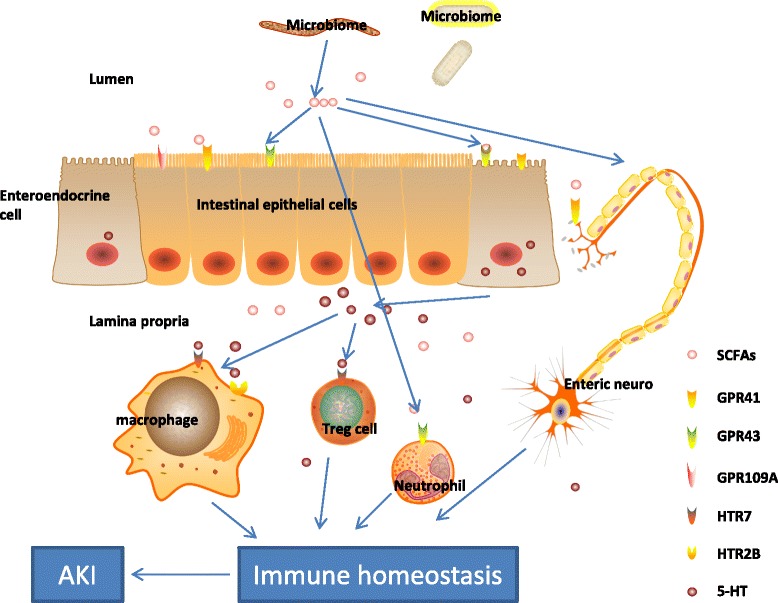
SCFAs, including acetate, propionate, and butyrate, are produced at high concentration by bacteria in the gut and subsequently released in the bloodstream. Receptors for SCFAs are G-protein-coupled receptors, GPR41, GPR43, GPR109A. Propionate is the most potent agonist for both GPR41 and GPR43. Acetate is more selective for GPR43, whereas butyrate and isobutyrate (nicotinic) are more active on GPR41. GPR41 is expressed in a number of tissues. GPR43 is selectively expressed in leukocytes and recruited toward sites of bacterial infection. Nicotinic acid can be anti-inflammatory in monocytes. Butyrate can also inhibit Jun NH(2)-terminal kinase activation and cytokine transcription in mast cells. SCFAs promote biosynthesis of 5-HT, enhancing T-cell activation by signaling through the 5-HT7 receptor, skews human macrophage polarization through HTR2B and HTR7, and activates immune responses and inflammation
Other therapies
The most common treatment for septic AKI is continuous renal replacement therapy (CRRT). CRRT can aid in removal of uremic toxins, sodium, and excessive volume. Thus, CRRT might positively impact gut wall edema and lessen the risk of bacterial translocation; future studies on CRRT should explore this possibility.
Conclusions
Many aspects of sepsis remain undefined, and the interplay between the gut and kidney during septic AKI remains an interesting avenue for investigation. During sepsis, the combined effect of erosion of the mucus barrier, a shift in the composition and virulence of intestinal microbes, and the inability of the host epithelium to regulate its proliferative and apoptotic responses may lead to a tipping point in gut function where cascading inflammation drives AKI. During AKI, the clearance of inflammatory mediators is decreased, and metabolic products accumulate that can increase systemic inflammation. The continuous cycle of injury/amplification of inflammation can lead to devastating consequences. In theory, rational therapies aimed at restoring gut integrity, the microbiome, and the homeostatic balance between the two systems represents an exciting avenue in the battle against critical illness.
Abbreviations
- 5-HT
Serotonin
- AKI
Acute kidney injury
- ATP
Adenosine triphosphate
- Bcl-2
B-cell lymphoma-2
- BL
Bifidobacterium longum
- CRRT
Continuous renal replacement therapy
- ENS
Enteric nervous system
- GPR
G-protein-coupled receptors
- ICU
Intensive care unit
- IECs
Intestinal epithelial cells
- IL
Interleukin
- JAM
Junctional adhesion molecules
- LGG
Lactobacillus rhamnosus GG
- LPS
Lipopolysaccharide
- MCP-1
Monocyte chemo attractant protein-1
- MLCK
Myosin light chain kinase
- mTOR
Mechanistic target of rapamycin
- NK
Natural killer
- PRR
Pattern recognition receptor
- SCFA
Short-chain fatty acid
- SDD
Selective decontamination of the digestive tract
- TLRs
Toll-like receptors
- TNF-α
Tumor necrosis factor α
- Treg
Regulatory T
- ZO-1
Zonulaoccludens 1
Authors’ contributions
JZ, CR, and KD contributed to manuscript writing and editing. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jingxiao Zhang, Email: zjingxiao1101@163.com.
Ghada Ankawi, Email: ghadaankawi@gmail.com.
Jian Sun, Email: sunjiandoctor1987@163.com.
Kumar Digvijay, Email: librandigu@gmail.com.
Yongjie Yin, Email: yinyongjie2003@sina.cn.
Mitchell H. Rosner, Email: MHR9R@hscmail.mcc.virginia.edu
Claudio Ronco, Email: cronco@goldnet.it.
References
- 1.Mandelbaum T, Scott DJ, Lee J, et al. Outcome of critically ill patients with acute kidney injury using the Acute Kidney Injury Network criteria. Crit Care Med. 2011;39(12):2659–2664. doi: 10.1097/CCM.0b013e3182281f1b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uchino S. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294(7):813. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 3.Chao C-T, Hou C-C, Wu V-C, et al. The impact of dialysis-requiring acute kidney injury on long-term prognosis of patients requiring prolonged mechanical ventilation: nationwide population-based study. PLoS One. 2012;7(12):e50675. doi: 10.1371/journal.pone.0050675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bagshaw SM, Laupland KB, Doig CJ, et al. Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: a population-based study. Crit Care. 2005;9(6):R700–R709. doi: 10.1186/cc3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoste EAJ, Bagshaw SM, Bellomo R, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. 2015;41(8):1411–1423. doi: 10.1007/s00134-015-3934-7. [DOI] [PubMed] [Google Scholar]
- 6.Gómez H, Kellum JA. Sepsis-induced acute kidney injury. Curr Opin Crit Care. 2016;22(6):546–553. doi: 10.1097/MCC.0000000000000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol. 2014;25(12):2689–2701. doi: 10.1681/ASN.2014030262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murugan R, Wen X, Shah N, et al. Plasma in fl ammatory and apoptosis markers are associated with dialysis dependence and death among critically ill patients receiving renal replacement therapy. Nephrol Dial Transpl. 2014;8(10):1854–1864. doi: 10.1093/ndt/gfu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gómez H, Kellum JA, Ronco C. Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat Rev Nephrol. 2017;13(3):143–151. doi: 10.1038/nrneph.2016.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell. 2016;164(3):337–340. doi: 10.1016/j.cell.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 11.Habes QLM, van Ede L, Gerretsen J, Kox M, Pickkers P. Norepinephrine contributes to enterocyte damage in septic shock patients; a prospective cohort study. Shock. 2017;75(6):1. doi: 10.1097/SHK.0000000000000955. [DOI] [PubMed] [Google Scholar]
- 12.Dominguez JA, Coopersmith CM. Can we protect the gut in critical illness? The role of growth factors and other novel approaches. Crit Care Clin. 2010;26(3):549–565. doi: 10.1016/j.ccc.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mittal R, Coopersmith CM. Redefining the gut as the motor of critical illness. Trends Mol Med. 2014;20(4):214–223. doi: 10.1016/j.molmed.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9(11):799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 15.Zahs A, Bird MD, Ramirez L, Turner JR, Choudhry MA, Kovacs EJ. Inhibition of long myosin light-chain kinase activation alleviates intestinal damage after binge ethanol exposure and burn injury. Am J Physiol Gastrointest Liver Physiol. 2012;303(6):G705–G712. doi: 10.1152/ajpgi.00157.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaziri ND, Yuan J, Rahimi A, Ni Z, Said H, Subramanian VS. Disintegration of colonic epithelial tight junction in uremia: A likely cause of CKD-associated inflammation. Nephrol Dial Transplant. 2012;27(7):2686–2693. doi: 10.1093/ndt/gfr624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaziri ND, Yuan J, Norris K. Role of urea in intestinal barrier dysfunction and dysruption of epithelial tight junction in CKD. Am J Nephrol. 2014;37(1):1–6. doi: 10.1159/000345969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El-Achkar TM, Huang X, Plotkin Z, Sandoval RM, Rhodes GJ, Dagher PC. Sepsis induces changes in the expression and distribution of Toll-like receptor 4 in the rat kidney. Am J Physiol Ren Physiol. 2006;290(5):F1034–F1043. doi: 10.1152/ajprenal.00414.2005. [DOI] [PubMed] [Google Scholar]
- 19.Neal MD, Sodhi CP, Jia H, et al. Toll like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53-upregulated modulator of apoptosis. J Biol Chem. 2012;287(44):37296–37308. doi: 10.1074/jbc.M112.375881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neal MD, Sodhi CP, Dyer M, et al. A critical role for TLR4 induction of autophagy in the regulation of enterocyte migration and the pathogenesis of necrotizing enterocolitis. J Immunol. 2013;190(7):3541–3551. doi: 10.4049/jimmunol.1202264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dominguez JA, Vithayathil PJ, Khailova L, et al. Epidermal growth factor improves survival and prevents intestinal injury in a murine model of pseudomonas aeruginosa pneumonia. Shock. 2011;36(4):381–389. doi: 10.1097/SHK.0b013e31822793c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang M, Alsaigh T, Kistler EB, Schmid-Schönbein GW. Breakdown of Mucin as barrier to digestive enzymes in the ischemic rat small intestine. PLoS One. 2012;7(6):1–12. doi: 10.1371/journal.pone.0040087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazmanian SK, Round JL, Kasper D. A microbial symbiosis factor prevents inflammatory disease. Nature. 2008;453(7195):620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 24.Hotchkiss RS, Swanson PE, Freeman BD, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27(7):1230–1251. doi: 10.1097/00003246-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 25.Cho I, Blaser MJ. Applications of next-generation sequencing: The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13(4):260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ojima M, Motooka D, Shimizu K, et al. Metagenomic analysis reveals dynamic changes of whole gut microbiota in the acute phase of intensive care unit patients. Dig Dis Sci. 2016;61(6):1628–1634. doi: 10.1007/s10620-015-4011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Defazio J, Fleming ID, Shakhsheer B, Zaborina O, Alverdy JC. The opposing forces of the intestinal microbiome and the emerging pathobiome. Surg Clin North Am. 2014;94(6):1151–1161. doi: 10.1016/j.suc.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng Y, Ralls MW, Xiao W, Miyasaka E, Herman RS, Teitelbaum DH. Loss of enteral nutrition in a mouse model results in intestinal epithelial barrier dysfunction. Ann N Y Acad Sci. 2012;1258(1):71–77. doi: 10.1111/j.1749-6632.2012.06572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayakawa M, Asahara T, Henzan N, et al. Dramatic changes of the gut flora immediately after severe and sudden insults. Dig Dis Sci. 2011;56(8):2361–2365. doi: 10.1007/s10620-011-1649-3. [DOI] [PubMed] [Google Scholar]
- 30.Charney AN, Micic L, Egnor RW. Nonionic diffusion of short-chain fatty acids across rat colon. Am J Phys. 1998;274(3):G518–G524. doi: 10.1152/ajpgi.1998.274.3.G518. [DOI] [PubMed] [Google Scholar]
- 31.Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev. 2013;13(5):321–335. doi: 10.1038/nri3430. [DOI] [PubMed] [Google Scholar]
- 32.Andrade-Oliveira V, Amano MT, Correa-Costa M, et al. Gut bacteria products prevent AKI induced by ischemia-reperfusion. J Am Soc Nephrol. 2015;26(8):1877–1888. doi: 10.1681/ASN.2014030288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deitch EA. Gut lymph and lymphatics: a source of factors leading to organ injury and dysfunction. Ann N Y Acad Sci. 2010;1207(Suppl):E103–E111. doi: 10.1111/j.1749-6632.2010.05713.x. [DOI] [PubMed] [Google Scholar]
- 34.Purohit V, Bode JC, Bode C, et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol. 2008;42(5):349–361. doi: 10.1016/j.alcohol.2008.03.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bellot P, Francés R, Such J. Pathological bacterial translocation in cirrhosis: pathophysiology, diagnosis and clinical implications. Liver Int. 2013;33(1):31–39. doi: 10.1111/liv.12021. [DOI] [PubMed] [Google Scholar]
- 36.Mizuno T, Yokoyama Y, Nishio H, et al. Intraoperative bacterial translocation detected by bacterium-specific ribosomal RNA-targeted reverse-transcriptase polymerase chain reaction for the mesenteric lymph node strongly predicts postoperative infectious complications after major hepatectomy for. Ann Surg. 2010;252(6):1013–1019. doi: 10.1097/SLA.0b013e3181f3f355. [DOI] [PubMed] [Google Scholar]
- 37.Sumagin R, Robin AZ, Nusrat A, Parkos CA. Transmigrated neutrophils in the intestinal lumen engage ICAM-1 to regulate the epithelial barrier and neutrophil recruitment. Mucosal Immunol. 2014;7(4):905–915. doi: 10.1038/mi.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fujiu K, Shibata M, Nakayama Y, et al. A heart-brain-kidney network controls adaptation to cardiac stress through tissue macrophage activation. Nat Med. 2017;23(5):611–622. doi: 10.1038/nm.4326. [DOI] [PubMed] [Google Scholar]
- 39.Schuijt TJ, van der Poll T, de Vos WM, Wiersinga WJ. The intestinal microbiota and host immune interactions in the critically ill. Trends Microbiol. 2013;21(5):221–229. doi: 10.1016/j.tim.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 40.Waltz P, Carchman E, Gomez H, Zuckerbraun B. Sepsis results in an altered renal metabolic and osmolyte profile. J Surg Res. 2016;202(1):8–12. doi: 10.1016/j.jss.2015.12.011. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Wei Q, Wang CY, Hill WD, Hess DC, Dong Z. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J Biol Chem. 2004;279(19):19948–19954. doi: 10.1074/jbc.M313629200. [DOI] [PubMed] [Google Scholar]
- 42.wasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16(4):343–53. 10.1038/ni.3123. [DOI] [PMC free article] [PubMed]
- 43.Gordon S, Plüddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunol Rev. 2014;262(1):36–55. doi: 10.1111/imr.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gabanyi I, Muller PA, Feighery L, Oliveira TY, Costa-Pinto FA, Mucida D. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell. 2016;164(3):378–391. doi: 10.1016/j.cell.2015.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sica A, Mantovani A. Macrophage plasticity and polarization: In vivo veritas. J Clin Invest. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xing L, Genhua M, Chunmei S, et al. Role of M2 macrophages in sepsis-induced acute kidney injury. Shock. 2017;1 10.1097/SHK.0000000000001006. [DOI] [PubMed]
- 47.de Smet AMGA, Kluytmans JAJW, Cooper BS, et al. Decontamination of the digestive tract and oropharynx in ICU patients. N Engl J Med. 2009;360(1):20–31. doi: 10.1056/NEJMoa0800394. [DOI] [PubMed] [Google Scholar]
- 48.Price R, MacLennan G, Glen J, SuDDICU Collaboration Selective digestive or oropharyngeal decontamination and topical oropharyngeal chlorhexidine for prevention of death in general intensive care: systematic review and network meta-analysis. BMJ. 2014;348:g2197. doi: 10.1136/bmj.g2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khailova L, Frank DN, Dominguez JA, Wischmeyer PE. Probiotic administration reduces mortality and improves intestinal epithelial homeostasis in experimental sepsis. Anesthesiology. 2013;119(1):166–177. doi: 10.1097/ALN.0b013e318291c2fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shimizu K, Ogura H, Asahara T, et al. Probiotic/synbiotic therapy for treating critically Ill patients from a gut microbiota perspective. Dig Dis Sci. 2013;58(1):23–32. doi: 10.1007/s10620-012-2334-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Nood E, Vrieze A, Nieuwdorp M, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368(5):407–415. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- 52.Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science. 2013;341(6145):569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Atarashi K, Tanoue T, Shima T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331(6015):337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carretta MD, Conejeros I, Hidalgo MA, Burgos RA. Propionate induces the release of granules from bovine neutrophils. J Dairy Sci. 2013;96(4):2507–2520. doi: 10.3168/jds.2012-6111. [DOI] [PubMed] [Google Scholar]
- 55.Digby JE, Martinez F, Jefferson A, et al. Anti-inflammatory effects of nicotinic acid in human monocytes are mediated by GPR109A dependent mechanisms. Arterioscler Thromb Vasc Biol. 2012;32(3):669–676. doi: 10.1161/ATVBAHA.111.241836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diakos C, Prieschl EE, Säemann MD, et al. n-Butyrate inhibits Jun NH(2)-terminal kinase activation and cytokine transcription in mast cells. Biochem Biophys Res Commun. 2006;349(2):863–868. doi: 10.1016/j.bbrc.2006.08.117. [DOI] [PubMed] [Google Scholar]
- 57.Yano JM, Yu K, Donaldson GP, et al. Indigenous bacterica from the gut microbiota regulate host serotonin biosnthesis. Cell. 2015;161(2):264–276. doi: 10.1016/j.cell.2015.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de las Casas-Engel M, Dominguez-Soto A, Sierra-Filardi E, et al. Serotonin skews human macrophage polarization through HTR2B and HTR7. J Immunol. 2013;190(5):2301–2310. doi: 10.4049/jimmunol.1201133. [DOI] [PubMed] [Google Scholar]