

Abstract
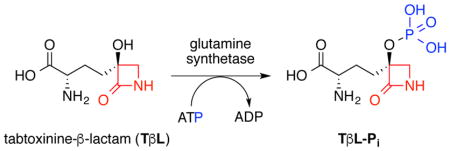
Tabtoxinine-β-lactam (TβL), also known as wildfire toxin, is a time- and ATP-dependent inhibitor of glutamine synthetase produced by plant pathogenic strains of Pseudomonas syringae. Here we demonstrate that recombinant glutamine synthetase from E. coli phosphorylates the C3-hydroxyl group of the TβL 3-(S)-hydroxy-β-lactam (3-HβL) warhead. Phosphorylation of TβL generates a stable, non-covalent enzyme-ADP-inhibitor complex that resembles the glutamine synthetase tetrahedral transition state. The TβL β-lactam ring remains intact during enzyme inhibition making TβL mechanistically distinct from traditional β-lactam antibiotics such as penicillin. Our findings could enable the design of new 3-HβL transition state inhibitors targeting enzymes in the ATP-dependent carboxylate-amine ligase superfamily with broad therapeutic potential in many disease areas.
Keywords: tabtoxin, tabtoxinine-β-lactam, methionine sulfoximine, glufosinate, phosphinothricin, bialaphos, glutamine synthetase, glutamate, glutamine, transition state inhibitor, antibiotic, mechanism-based inhibitor, enzyme inhibitor, penicillin, β-lactam, antibiotic, antibiotic resistance, antimetabolite
Graphical Abstract
Introduction
β-Lactams are the most successful class of clinical antibiotics ever discovered1. Canonical β-lactam antibiotics, such as penicillin G (Fig. 1), are covalent inhibitors of enzymes in the serine hydrolase superfamily, including bacterial transpeptidases (TPases) involved in cell wall assembly2. The mechanistic basis for TPase inhibition is acylation of the active site, catalytic serine residue3. Antibacterial β-lactams are structural mimics of the C-terminal D-Ala- D-Ala peptidyl stem of bacterial peptidoglycan. Formation of the TPase-β-lactam collision complex orients the active site serine residue for nucleophilic attack on the electrophilic β-lactam carbonyl leading to formation of an unstable tetrahedral intermediate. Breakdown of the tetrahedral intermediate opens the β-lactam ring and generates a stable, covalent acyl enzyme adduct. Without functional TPases, bacteria are left unable to crosslink newly formed cell wall polysaccharides resulting in the recruitment of lytic transglycosylases and initiation of a futile cycle ending in autolysis4. To fight back, resistant bacteria express β-lactamases that exploit the hydrolytic liability of β-lactams5. All major classes of β-lactam antibiotics including penicillins, carbapenems, cephalosporins, and monobactams share this common reactivity, driven by the inherent ring strain (~25 kcal/mol) of the β-lactam warhead, that is the source of both clinical success and failure6.
Figure 1.
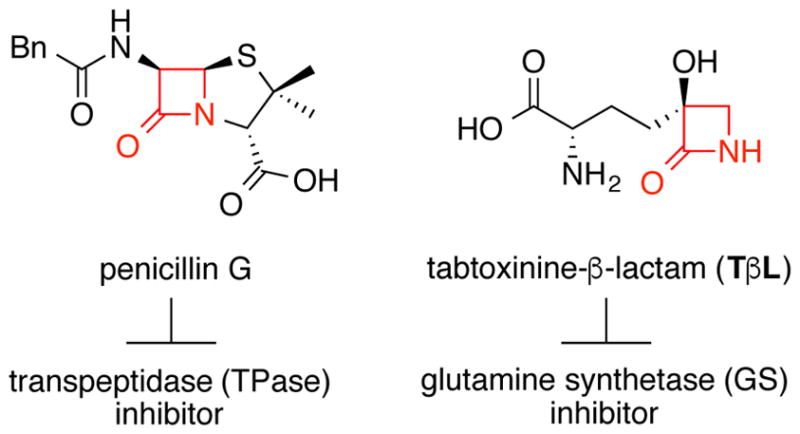
Two classes of β-lactam antibiotics: pencillin G is a transpeptidase (TPase) inhibitor and tabtoxinine-β-lactam (TβL) is a glutamine synthetase (GS) inhibitor.
Tabtoxinine-β-lactam (TβL) is a monocyclic 3-hydroxy-β-lactam (3-HβL) produced by plant pathogenic strains of Pseudomonas syringae and some soil Streptomyces (Fig. 1)7, 8. Unlike canonical β-lactam antibiotics, TβL does not inhibit bacterial TPases but instead is a potent, mechanism-based inhibitor of glutamine synthetase (GS)9, 10. P. syringae pathovars including tabaci, coronofaciens, phaseolicola, tomato and garcae excrete the active phytotoxin TβL as a L-Thr dipeptide pro-drug known as tabtoxin (TβL-Thr)11–13. Unlike traditional β-lactams, both TβL and TβL-Thr are stable towards all major classes of β-lactamase enzymes14. Bacterial resistance to TβL and TβL-Thr can be achieved through acquisition of the ttr gene, which encodes for a cytoplasmic acetyltransferase that deactivates TβL through regioselective acetylation of the α-amino group using acetyl-CoA as the acyl donor11, 15. TβL and TβL-Thr undergo spontaneous rearrangement to the more thermodynamically stable δ-lactam isomers, tabtoxinine-δ-lactam (TδL) and tabtoxin-δ-lactam (TδL-Thr), respectively11. The δ-lactam isomers and hydrolyzed β-amino acids are biologically inactive indicating that the 3-HβL warhead of TβL plays a central role in GS inhibition14. The mechanistic basis for GS inhibition by TβL is unknown. Given the clear differences in enzyme structure and function of bacterial TPases (serine hydrolase family16) and GS (ATP-dependent amine-carboxylate ligase family17), TβL represents a unique opportunity to understand how nature adapted the β-lactam ring to inhibit enzymes other than serine hydrolases18.
Glutamine synthetase is critical for nitrogen metabolism in all forms of life19. GS catalyzes the ATP-dependent conversion of L-Glu and NH3 to L-Gln (Fig. 2a). Bacterial GSs are functional as dodecamers formed by the face-to-face assembly of two hexameric rings20. Bacterial GSs contain 12 active sites formed between each monomer with binding sites for two divalent cations, typically Mg2+ or Mn2+ 21. Active site residues are conserved amongst GS isoforms from different species, but the quaternary structure can vary. For example, human GSs are typically functional as decamers composed of two pentameric rings in a face-to-face arrangement22. The binding of substrates, formation of intermediates, and release of products during the GS biosynthetic reaction is ordered23, 24. First, ATP binds in a site adjacent to the cation-binding sites. ATP binding increases the affinity for L-Glu binding, which takes place adjacent to the ATP site. L-Glu binding causes the active site to close via movement of a mobile loop with subsequent formation of the ammonium-binding site25. Closing of the active site prevents water entry and promotes phosphoryl transfer from the γ-phosphate group of ATP to the γ-carboxylate of L-Glu producing the reactive γ-glutamyl acyl phosphate intermediate (L-Glu-Pi) and ADP that stays bound through the remainder of the biosynthetic reaction. The Asp-50 residue deprotonates the bound ammonium and ammonia attacks the electrophilic δ-carbonyl carbon of the γ-glutamyl acyl phosphate to form a GS-stabilized tetrahedral intermediate that resembles the late transition states for nucleophilic acyl substitution reactions26. Breakdown of the tetrahedral intermediate releases Pi and generates L-Gln27.
Figure 2.

Reactions catalyzed by glutamine synthetase (GS): (A) canonical biosynthetic reaction and (B) phosphorylation of inhibitors generating tight-binding transition state analogs.
Inhibition of GS leads to accumulation of L-Glu and NH3 and blocks downstream production of amino acids, cofactors, nucleotides, and amino sugars28, 29. GS inhibitors have been explored as herbicides30, antimicrobial agents31, and treatments for neurological diseases32. The two main types of GS inhibitors include molecules that bind competitively to either the ATP or L-Glu binding site31. Achieving selectivity for GS isoforms is challenging for both inhibitor types. ATP-competitive inhibitors incorporate hydrophobic heterocycles such as purines, aminoimidazopyridines, and imidazoles. L-Glu-competitive inhibitors, including TβL, are polar α-amino acids that structurally mimic L-Glu. Methionine sulfoximine (MetSox) and glufosinate (Glufos) are L-Glu competitive inhibitors that have been mechanistically and structurally characterized as inhibitors of plant, animal, and bacterial GS. Glufos is sold commercially by BayerCropSciences as an herbicide that is paired with resistant transgenic crops under the tradename LibertyLink®30. GS inhibition by both MetSox and Glufos requires ATP and C2-(S) stereochemistry matching L-Glu substrate20, 33, 34. GS inhibition by MetSox and Glufos is time-dependent, irreversible, and non-covalent. MetSox is phosphorylated on the sulfoximine nitrogen to produce MetSox-Pi that stabilizes GS in a closed, inactive conformation with bound ADP22, 27, 35. Similarly, Glufos is phosphorylated on the phosphinate oxygen to produce Glufos-Pi that stabilizes GS in a closed, inactive conformation with bound ADP (Fig. 2b)20. MetSox-Pi and Glufos-Pi are both tetrahedral transition state analogs36. The phosphorylated inhibitors resemble the predicted structures of the late GS transition state leading to formation of the tetrahedral intermediate after nucleophilic attack of ammonia on the electrophilic acyl phosphate carbonyl and/or the early transition state leading to collapse of the tetrahedral intermediate with release of Pi. The methyl group of MetSox-Pi and Glufos-Pi occupy the GS ammonium-binding site while the N-Pi-sulfoxamine and O-Pi-phosphinate groups interact with the active site metals and cationic amino acid side chains involved in stabilizing the GS tetrahedral intermediate20, 27. Although MetSox and Glufos are non-covalent GS inhibitors, the inhibition is essentially irreversible since MetSox-Pi and Glufos- Pi are tightly bound transition state mimics37.
Since GS inhibition by TβL is time-dependent and requires ATP, we hypothesized that TβL is phosphorylated in the GS active site generating a stable, non-covalent GS-ADP-TβL-Pi inhibitory complex (Fig. 2b). To test our hypothesis we directly detected TβL-Pi released from denatured GS-ADP-TβL-Pi complex using high-resolution LC-mass spectrometry and we characterized the site of TβL phosphorylation within the intact GS-ADP-TβL-Pi inhibition complex using 31P{15N} full echo and rotational-echo double resonance (REDOR) solid state NMR with magic angle-spinning. We used computational modeling to show that TβL-Pi with an intact β-lactam ring resembles the GS tetrahedral intermediate suggesting that TβL-Pi is a GS transition state inhibitor. We also quantitatively compared MetSox, Glufos, and TβL in kinetic assays for inhibition of recombinant GS from Homo sapiens, E. coli, and S. aureus using the Kitz-Wilson model for mechanism-based inhibition. Our results show that TβL is mechanistically distinct from traditional β-lactam antibiotics such as penicillin. Our findings enable the design of new 3-HβLs for inhibiting enzymes in the ATP-dependent carboxylate-amine ligase superfamily with broad therapeutic applications.
Materials and Methods
Strains, Materials, and Instrumentation
E. coli ATCC 29522 and P. syringae pv. tabaci ATCC 11528 were purchased from ATCC (Supplementary Table 1). E. coli BL21-Gold(DE3) was purchased from Agilent. E. coli TOP10 cells were purchased from Invitrogen. Both E. coli BL21-Gold(DE3) and E. coli TOP10 cells were made electrocompetent by standard procedures. Electroporation was accomplished using a MicroPulser electroporator and 0.2 cm gap sterile electroporation cuvettes from Bio-Rad. All bacteria were stored as frozen glycerol stocks at −80 °C. GlnA coding sequences from E. coli, S. aureus, and Homo sapiens were purchased codon-optimized for expression in E. coli BL21 from GenScript in a pET28a vector with a thrombin cleavable N-His6 tag (Supplementary Tables 2,3). DNA purification was performed with kits purchased from Qiagen. All plasmid sequences were confirmed by sequencing performed by Genewiz. Nickel-nitriloacetic acid (Ni-NTA) agarose was purchased from Invitrogen. Any kD SDS-PAGE gels were purchased from Bio-Rad. Proteins were dialyzed using 10K MWCO SnakeSkin dialysis tubing purchased from Thermo Fisher Scientific. Centrifugal filters (30K MWCO) used for protein concentration were purchased from Millipore. Crude phosphodiesterase from Crotalus adamanteus and Crotalus atrox was purchased from Sigma Aldrich as a lyophilized powder. A mixture of pyruvate kinase (PK) and lactate dehydrogenase (LDH) enzymes from rabbit muscle was purchased from Sigma Aldrich as a buffered glycerol solution (900–1400 units/mL LDH; 600–1000 units/mL PK). All aqueous solutions were prepared with water purified using a Milli-Q system. All buffer and salt solutions were sterilized by sterile filtration through a 0.2 μm filter. Luria broth and Difco Nutrient Broth were sterilized in an autoclave. Woolley’s medium was filter sterilized. All pH measurements were recorded using an Orion Star A111 pH meter and a PerpHecT ROSS micro combination pH electrode from Thermo Fisher Scientific. All buffers, salts, media, solvents, and chemical reagents were purchased from Sigma Aldrich unless otherwise stated. D2O and K15NO3 were purchased from Cambridge Isotope Laboratories. Racemic Glufos was purchased from Oakwood Chemical. Fmoc-protected L-Ala-L-Ala dipeptide was purchased from ChemImpex. Glufos-L-Ala-L-Ala tripeptide (AAG), also known as bialaphos, was purchased as a 1 mg/mL aqueous solution from PhytoTechnology Laboratories. L-Ala-L-Ala-Glufos tripeptide (AAG) was synthesized as described in the Supplementary Information (Supplementary Scheme 1; Supplementary Fig. 7–10).
1H-NMR spectra were obtained using a Varian Unity Plus 300 MHz NMR spectrometer. LC-MS samples were prepared in 0.45 μ PTFE mini-UniPrep vials from Agilent. LC-MS was performed using an Agilent 6130 quadrupole with G1313 autosampler, G1315 diode array detector, and 1200 series solvent module. LC-MS separations were achieved using a 5 μ Gemini C18 column (50 × 2 mm) from Phenomenex fit with a guard column. LC-MS mobile phases were 0.1% formic acid in (A) water and (B) acetonitrile. Samples were loaded in 10% B holding for 2 min and a linear gradient was then formed to 100% B over 15 min followed by ramping to 10% B over 3 min at a flow rate of 0.5 mL/min. LC-MS data were processed using G2710 ChemStation software. Preparative HPLC was performed using a Beckman Coulter SYSTEM GOLD 127P solvent module and 168 diode array detector using a Luna 10 μ C18(2) 100 A column (250 × 21.2 mm) from Phenomenex fit with a guard column (15 × 21.2 mm). Mobile phases for RP-C18 prep-HPLC were 0.1% TFA in (A) water and (B) acetonitrile. Samples were loaded and eluted in 100% A (held for 15 min) at a flow rate of 10 mL/min. The column was washed with 50% B for 10 min and re-equilibrated with 100% A prior to the next run. For preparative HILIC chromatography the column was a Luna 5 μ HILIC 200 A fit with a guard column (15 × 21.2 mm). HILIC HPLC mobile phases were 5 mM ammonium formate pH 3.2 (pH adjusted with aq. HCl) in (A) 90:10 acetonitrile:water and (B) 50:50 acetonitrile:water. Samples dissolved in 1:1 EtOH:water were loaded in 20% B holding for 10 min and a linear gradient was formed to 60% B over 20 min followed by a re-equilibration to 20% B at a flow rate of 12 mL/min. HPLC data were processed using 32 Karat software, version 7.0. Protein purification was performed on an AKTA Explorer 100 FPLC with UV–vis detection using a Sephadex 75 26/60 HiLoad prep grade gel filtration column purchased from GE Healthcare. DNA and protein concentrations were determined using a NanoDrop 2000 UV–vis spectrophotometer from Thermo Fisher Scientific. Protein extinction coefficients were determined using the ExPasy ProtParam tool. UV–vis spectrophotometry was performed in 1 cm quartz cuvettes on an Agilent Cary 50 fit with an autosampler and water Peltier thermostat system. Bacterial growth studies were performed using polystyrene 96-well plates with polystyrene lids with OD600 measurements were made using a SpectraMax Plus 384 plate reader from Molecular Devices. High-resolution LC-MS/MS spectra were collected using a Q-Exactive (Thermo-Fisher Scientific) equipped with a custom built Eksigent microLC at the Donald Danforth Plant Science Center, St. Louis, MO. The solvents were 0.1% formic acid in (A) water and (B) acetonitrile. The column was a 0.5 × 150 mm Supelco C8. A flow rate of 15 μL/min was held constant while a solvent gradient (2% B held for 3 min, then ramped to 100% B over 11 min, then held at 100% B for 4 min, then ramped to 2% B over 1 min, and re-equilibrated at 2% B for 6 min) was formed. The mass spectrometer was operated in polarity switching mode and scanned from m/z 200–500 at a resolution setting of 70,000 (at m/z 200) for MS1 and a resolution of 17,500 for MS2.
Expression and Purification of GS
Constructs of codon-optimized GlnA in a pET28a vector encoding for N-His6-tagged GS from E. coli, S. aureus, and Homo sapiens were transformed into electrocompetent E. coli BL21- Gold(DE3) via electroporation (Supplementary Tables 2,3). Overnight cultures of E. coli BL21 grown at 37 °C harboring the appropriate plasmid were grown in LB broth containing 50 μg/mL kanamycin and 200 μL was used to inoculate 1L batches of sterile Terrific Broth (12 g/L tryptone, 24 g/L yeast extract, 5 g/L glycerol, 17 mM KH2PO4, and 72 mM K2HPO4) containing 50 μg/mL kanamycin. The cultures were grown at 37 °C with agitation to an OD600 of ~0.9 and then cooled to 16 °C prior to induction with 1 mL of 0.5 M IPTG (0.5 mM final concentration). Cultures were then grown for ~18 h at 16 °C with agitation. Cells were harvested by centrifugation (5,000 r.p.m. for 20 min at 4 °C). Cells/protein solutions were kept at ~4 °C for all remaining steps. Cell pellets were suspended in 40 mL of lysis buffer (50 mM K2HPO4, 500 mM NaCl, 5 mM β-mercaptoethanol, 20 mM imidazole, 10% glycerol, adjusted to pH 8.0) and flash frozen in liquid nitrogen. After thawing, cells were mechanically lysed using an Avestin EmulsiFlex-C5 high-pressure homogenizer. Cell lysate was clarified via ultracentrifugation (45,000 r.p.m. for 35 min) and incubated for 30 min with Ni-NTA resin preconditioned with lysis buffer. The Ni-NTA resin was washed twice with 40 mL aliquots of lysis buffer. For human enzyme the resin was eluted in five separate washes with 10 mL of elution buffer (50 mM K2HPO4, 500 mM NaCl, 5 mM β-mercaptoethanol, 300 mM imidazole, 10% glycerol, adjusted to pH 8.0). Fractions containing pure N-His6-GS as judged by SDS-PAGE analysis (Supplementary Fig. 2) were combined, dialyzed into 100 mM HEPES pH 7.4, concentrated via centrifugal filtration, flash frozen in liquid nitrogen, and stored at −80 °C to give stocks ready for in vitro biochemical assays. For E. coli and S. aureus enzymes the resin was washed several times with snake venom buffer (20 mM Tris, 500 mM NaCl, 20 mM imidazole, 10 mM MgCl2, adjusted to pH 8.0) and then treated with 20 mL of snake venom buffer containing 1 mg/mL of crude phosphodiesterase from Crotalus adamanteus with gentle rocking at 20 °C for 12 h. The resin was washed thoroughly with lysis buffer and then eluted in five separate washes with 10 mL of elution buffer. Fractions containing pure N-His6-GS as judged by SDS-PAGE analysis (Supplementary Fig. 2) were combined, dialyzed into 100 mM HEPES pH 7.4, concentrated via centrifugal filtration, flash frozen in liquid nitrogen, and stored at −80 °C. Typical protein preparations yielded 3–5 mL of E. coli, S. aureus, and human N-His6-GS at concentrations of ~45–150 μM (~6–30 mg/L of induced E. coli BL21 culture). 10
Purification of TβL and TβL-Thr
Safety note: TβL, MetSox, Glufos, and formulations thereof are potentially toxic if ingested. Personal protective equipment and caution should be used when handling solutions containing these compounds. P. syringae pv. tabaci ATCC 11528 is a known plant pathogen and requires a USDA permit for laboratory use. Live cultures should be sterilized using a calibrated autoclave prior to disposal.
A colony selection protocol was followed prior to starting TβL-Thr production cultures. A glycerol stock of P. syringae pv. tabaci ATCC 11528 was used to inoculate a 5 mL culture of Difco Nutrient Broth grown overnight at 26 °C. The overnight culture was diluted 1000x– 100,000x onto Difco nutrient agar plates that were grown overnight at 26 °C. Single colonies (~20) were selected and grown in fresh Difco Nutrient Broth overnight at 26 °C. A portion of each culture was used to prepare a fresh glycerol stock stored at −80 °C and to inoculate 5 mL cultures in Woolley’s medium (10 g/L sucrose, 5 g/L KNO3, 0.8 g/L K2HPO4, 0.8 g/L NaH2PO4•H2O, 0.2 g/L MgSO4•7H2O, 0.1 g/L CaCl2•2H2O, 20 mg/L FeSO4•7H2O) that were grown at 26 °C for 96 h. Each 5 mL culture was analyzed for OD600, pH, activity of filter sterilized supernatant against E. coli in an agar diffusion antibacterial susceptibility assay, presence of TβL-Thr in supernatant detected by LC-MS, and DNA precipitation upon 1:1 dilution in EtOH (more DNA precipitation correlated with more TβL-Thr production). The glycerol stock of the most promising P. syringae culture was used to inoculate a fresh overnight culture in Nutrient Broth at 26 °C. The Nutrient Broth culture was used as inoculum (200 μL) for 0.5 L cultures in Woolley’s medium in 2.8 L baffled flasks grown for 96 h with shaking (225 rpm) at 26 °C. Cells were pelleted by centrifugation for 20 min at 5000 rpm, 4 °C. Supernatant was diluted 1:1 with EtOH to precipitate DNA and 1 M HCl was added to reach pH ~4 to slow TβL-Thr isomerization. The remainder of the TβL-Thr purification was carried out as described in our previous publication (ion exchange chromatography using Dowex 50WX8-200 cation exchange, then prep-HPLC using a HILIC column (typically elutes at retention time of 14–17 min), and finally prep-HPLC using a RP-C18 column (typically elutes at retention time of 7–9 min))11. Stock solutions of TβL-Thr were quantified by 1H-NMR using an acetonitrile internal standard and stored frozen at −80 °C as lyophilized solids or as an aqueous solution at pH <4 (Supplementary Fig. 3). 15N-TβL- Thr was prepared exactly as described for TβL-Thr using Woolley’s medium prepared with K15NO3. Purification of 15N-TβL-Thr was only taken through prep-HPLC using a HILIC column (typically elutes at retention time of 12–14 min) prior to conversion to 15N-TβL. Both 15N-TβL-Thr and TβL-Thr were converted to 15N-TβL and TβL, respectively, by treatment with lysate from P. syringae pv. tabaci ATCC 11528 grown in Woolley’s medium supplemented with 10 μM ZnCl2, as described in our previous publication11. 15N-TβL and TβL were purified by prep-HPLC using a HILIC column and stock solutions were quantified by titration with L-Thr and the amino acid ligase TblF from the TβL-Thr biosynthetic gene cluster, as described in our previous publication11. Standardized solutions of 15N-TβL and TβL were stored at −80 °C at pH <4.
GS Michaelis–Menten Kinetics
All experiments were performed at 37 °C in quartz cuvettes under steady state conditions with continuous monitoring at 350 nm in an Agilent Carey 50 UV–vis spectrophotometer. Glufos was racemic and reported concentrations reflect only the L-enantiomer34. For all experiments the total reaction volume was 500 μL and contained 100 mM HEPES (pH 7.4), 100 mM KCl, 25 mM MgCl2, 10 mM NH4Cl, 0.5 mM PEP, 0.2 mM NADH, 0.2 units PK, 0.3 units LDH, and 10 nM GS. For determination of the apparent Km for ATP, reactions contained 50 mM L-Glu and variable concentrations of ATP ranging from 0.5–5.0 mM. For determination of the apparent Km L-Glu, reactions contained 10 mM ATP and variable concentrations of L-Glu ranging from 1–55 mM. GS was added last to initiate the reaction. Reaction velocities (kobs in absorbance/min) were determined by calculating the slope of the linear region of the 350 nm absorbance vs time plot with background correction for control reactions lacking GS38. Kinetic constants were determined from kobs versus substrate concentration data using a nonlinear, least-squares fitting method with GraphPad Prism, version 7.0a fit to the Michaelis–Menten equation (eq 1),
| (1) |
where kcat is the maximal velocity, [S] is the substrate concentration, and Km is the substrate concentration corresponding to kobs = 1/2kcat. All reactions were performed in triplicate as independent trials (Supplementary Fig. 4).
GS Kitz–Wilson Kinetics
All experiments were performed at 37 °C in quartz cuvettes under steady state conditions with continuous monitoring at 350 nm in an Agilent Carey 50 UV–vis spectrophotometer. MetSox was purchased from Sigma Aldrich as the L-enantiomer. TβL isolated from P. syringae cultures was the L-enantiomer. The total reaction volume for experiments with MetSox and Glufos was 500 μL. To conserve sample, the total reaction volume for experiments with TβL was 400 μL. Each reaction contained 100 mM HEPES (pH 7.4), 100 mM KCl, 25 mM MgCl2, 10 mM NH4Cl, 0.5 mM PEP, 0.2 mM NADH, 0.2 units PK, 0.3 units LDH, 50 mM L-Glu, and enough GS to give a starting rate close to −0.05 absorbance units/min at 350 nm yielding a straight line for ~10 min without consuming all NADH (GS stocks are prepared in advance from frozen concentrated stocks the day of inhibition assay). Control reactions with either no L-Glu or no inhibitor were used to correct for non-specific ATPase activity and NADH degradation. Appropriate pre-incubation time of enzyme and inhibitor was established by investigating the time-dependence of GS inhibition by TβL, MetSox, and racemic Glufos. Percent GS activity relative to a no inhibitor control was measured for inhibitor concentrations ranging from 1–100 μM using pre-incubation times of 0–60 min. No ATP controls were included to demonstrate the requirement of ATP for GS inhibition. Kinetic inhibition constants were determined from kapp versus inhibitor concentration data using a linear fitting method with GraphPad Prism, version 7.0a fit to the Kitz–Wilson equation for irreversible enzyme inhibition (eq 2)39,
| (2) |
where kinact is the rate constant for conversion of the reversible enzyme-inhibitor complex to the irreversible complex, [I] is the inhibitor concentration, and KI is the dissociation constant for the initial reversible enzyme-inhibitor complex. The parameter kapp is defined by equation 3,
| (3) |
where t is the pre-incubation time, vI is the rate with inhibitor, and vo is the rate without inhibitor. Pre-incubation times for Kitz–Wilson kinetic studies were chosen to give linear plots of 1/kapp vs 1/[I] with inhibitor concentrations above and below the KI value. Inhibitor concentrations were varied between 0.33–100 μM and pre-incubation times were typically between 2–10 min. All reactions were performed in triplicate as independent trials.
Bacterial Growth Assays
A filter sterilized, chemically defined minimal medium (7 g/L K2HPO4, 3 g/L KH2PO4, 0.47 g/L Na citrate dehydrate, 0.1 g/L MgSO4•7H2O, 1 g/L (NH4)2SO4, 4 g/L glucose, 20 mg/L thymine, 0.1 mg/L biotin, 2 mg/L thiamine, 2 mg/L nicotinic acid, 2 mg/L Ca pantothenate, 10 mg MnSO4•7H2O, pH ~7.2) was used for all growth assays with E. coli ATCC 29522. Minimum inhibitory concentrations (MICs) were determined by the broth microdilution method following guidelines outlined by the Clinical and Laboratory Standards Institute (CLSI)40. Each well of a 96-well plate was filled with 50 μL of sterile minimal medium. 50 μL of TβL-Thr or AAG solution in minimal medium was added to the first well of the 96-well plate and diluted 2-fold down each row. 50 μL of E. coli ATCC 25922 inoculum (5 × 105 cfu/mL in minimal medium) was added to each well giving a final volume of 100 μL/well. The final concentration gradient of TβL-Thr and AAG was 128–0.0625 μM. The same procedure was used for studies with L-Glu or L-Gln supplementation with the appropriate amount of amino acid added to the minimal medium. Controls with no antibiotic or kanamycin (MIC = 8 μM) were included on every plate. Plates were covered with lids and incubated at 37 °C for 18–24 h until the MIC could be judged as the lowest concentration of antibiotic required to inhibit visible bacterial growth relative to the no antibiotic control. All MICs were measured in triplicate from independent rows on the same 96-well plate. 14 The same chemically defined minimal medium was used to measure growth curves of E. coli ATCC 29522 in the presence of 5 mM L-Glu or variable concentrations of L-Gln (0.1–5 mM). TβL-Thr was evaluated at final concentrations of 2 μM and 100 μM. AAG was evaluated at a final concentration of 2 μM. The final working volume in each well of the 96- well plate was 100 μL/well. Covered plates were incubated at 37 °C for up to 70 h with OD600 measurements taken every 2 h with plate agitation prior to each measurement. All OD600 measurements were taken in triplicate from independent wells on the same 96-well plate.
Detection of TβL-Pi by LC-MS
GS-inhibitor complexes were formed by incubating purified N-His6-GS from E. coli (84 μM) with 100 μM inhibitor (TβL, 15N-TβL, MetSox, and racemic Glufos), 100 mM HEPES (pH 7.4), 10 mM ATP, 100 mM KCl, and 25 mM MgCl2 at a final volume of 500 μL for 1 h at 37 °C. Full inhibition of GS was confirmed using the PK/LDH coupled spectrophotometric kinetic assay. Control experiments without ATP in the reaction mixture were also performed for each inhibitor. The solution was then applied to a 30K MWCO spin column and concentrated to a final volume 200 μL. The solution was then diluted with 300 μL water and again concentrated to 200 μL. This washing was repeated 10 times to remove all unbound inhibitor and co-substrates. A 200 μL solution of the GS-inhibitor complex was transferred to a microcentrifuge tube and treated with 300 μL MeOH and 20 mM KCl (final concentration) to induce protein precipitation. The mixture was incubated at 60 °C for 1 h during which the solution became cloudy. Solid particulates were removed via centrifugation and the clear, colorless supernatant was transferred to a fresh 30K MWCO spin column. After spinning for 30 min at 5K r.p.m. the flow through (~500 μL) was collected and treated with 50 μL pH 8 sodium borate buffer, 105 μL acetonitrile, and 20 μL of 20 mM FmocCl. After 30 min at room temperature, the sample was analyzed by low-resolution LC-MS in positive ion mode to detect the presence of the phosphorylated and Fmoc-tagged inhibitors (Fmoc-TβL-Pi, Fmoc-15N-TβL-Pi, Fmoc-MetSox-Pi, and Fmoc- Glufos-Pi). During our analysis we found that Fmoc-Glufos-Pi was unstable and we only observed peaks corresponding to the m/z value for Fmoc-Glufos as the corresponding [M+H]+ ion. For low-resolution LC-MS experiments we observed the following m/z values corresponding to [M+H]+ ions: Fmoc-TβL-Pi (retention time = 12.5 min, MS (ESI) calculated for C22H24N2O9P: 491.1 [M+H]+, found 491.1); Fmoc-15N-TβL-Pi (retention time = 12.7 min, MS (ESI) calculated for C22H2415N2O9P: 493.1 [M+H]+, found 493.1); Fmoc- MetSox-Pi (retention time = 9.7 min, MS (ESI) calculated for C20H24N2O8PS: 483.1 [M+H]+, found 483.1); Fmoc-Glufos (retention time = 9.3 min, MS (ESI) calculated for C20H23NO6P: 404.1 [M+H]+, found 404.1). The Fmoc-TβL-Pi sample was further analyzed by high-resolution LC-MS/MS in positive and negative ion modes. In negative ion mode we observed the following retention time and m/z values corresponding to [M−H]− ions for Fmoc-TβL-Pi: HRMS1 (ESI) calculated for C22H22N2O9P: 489.1068 [M−H]−, found 489.1064; HRMS2 (ESI) calculated for loss of Fmoc C7H12N2O7P: 267.0387 [M−H]−, found 267.0381. In positive ion mode we observed m/z values corresponding to [M+H]+ ions for Fmoc-TβL: HRMS1 (ESI) calculated for C22H24N2O9P: 491.1214 [M+H]+, found 491.1230; HRMS2 (ESI) calculated for loss of Fmoc C7H13N2O: 189.0870 [M+H]+, found 189.0877 (Supplementary Fig. 5).
Sample Preparation for Solid-State NMR
GS from E. coli was over expressed in E. coli BL21 as described previously. During large-scale production of GS, the crude phosphodiesterase from C. adamanteus (eastern diamondback rattlesnake) was backordered from the supplier so crude phosphodiesterase from Crotalus atrox (western diamondback rattlesnake) was substituted at this step. Treatment of N-His6-GS with C. atrox phosphodiesterase cleaved the adenylyl groups along with the N-His6 tag at the thrombin-cleavage site. GS was dialyzed into SEC buffer (50 mM potassium phosphate, 150 mM NaCl, 1 mM DTT, 5% glycerol, pH 8) and purified on an AKTA Explorer 100 FPLC with UV–vis detection using a Sephadex 75 26/60 HiLoad prep grade gel filtration column eluting with SEC buffer at a flow rate of 2 mL/min. Fractions containing pure GS as judged by SDS-PAGE analysis and activity in the biosynthetic assay were combined, dialyzed into 100 mM HEPES buffer at pH 7.4, and concentrated via centrifugal filtration to ~6–8 mg/mL. A 10 mL aliquot of 62.8 mg GS in 100 mM HEPES buffer at pH 7.4 was treated with excess 15N-TβL in the presence of 10 mM ATP, 100 mM KCl, 25 mM MgCl2, and 10 mM NH4Cl at 37 °C for 2 h (final volume was 20 mL). Full GS inhibition was confirmed using the coupled PK/LDH assay under biosynthetic reaction conditions. The solution was transferred to a 30K MWCO spin filter and diluted with trehalose buffer (5 mM HEPES, 15 mM trehalose, pH 7.4) prior to centrifugation at 5K r.p.m. and 4 °C. To fully desalt the sample, the dilution and centrifugation process was repeated 10 times until the sample was ultimately concentrated to ~9 mL in trehalose buffer. The sample was treated with PEG 8000 (28.1 mg), 100 kD dextran (6 mg), and 500 kD dextran (6 mg) prior to controlled freezing. The sample was frozen by chilling in a −10 °C ice/CaCl2 bath, freezing in a −70 °C dry ice/acetone bath, and further freezing in a liquid nitrogen bath41. The frozen sample was lyophilized for 72 h to provide a fluffy white powder that was used in solid-state NMR experiments. The final sample components were GS-ADP-15N-TβL-Pi inhibition complex (62.8 mg), trehalose (46.2 mg), PEG 8000 (28.1 mg), 100 kD dextran (6 mg), 500 kD dextran (6 mg), and HEPES (10.3 mg). A control sample was prepared under identical conditions with omission of 15N-TβL and replacement of ATP with ADP. The control sample contained GS-ADP complex (71.5 mg), trehalose (46.2 mg), PEG 8000 (28.1 mg), 100 kD dextran (6 mg), 500 kD dextran (6 mg), and HEPES (10.3 mg).
Solid-State NMR Parameters
Experiments were performed at 12 Tesla with a six-frequency transmission-line probe having a 12-mm long, 6-mm inner-diameter analytical coil, and a Chemagnetics/Varian ceramic spinning module41. Samples were spun using a thin-wall Chemagnetics/Varian (Fort Collins, CO/Palo Alto, CA) 5-mm outer diameter-zirconia rotor at 7143 Hz, with the speed under active control and maintained to within ±2 Hz. A Tecmag Libra pulse programmer (Houston, TX) controlled the spectrometer. Two-kW American Microwave Technology (AMT) power amplifiers were used to produce radio-frequency pulses for 31P (202.3 MHz) and 15N (50.7 MHz). The 1H (500 MHz) radio-frequency pulses were generated by a 2-kW Creative Electronics tube amplifier driven by a 50-W AMT amplifier. All final-stage amplifiers were under active control. The π-pulse lengths were 6 μs for 31P and 1H, and 9 μs for 15N. Proton-phosphorous (or nitrogen) matched cross-polarization transfers were made in 2 ms at 56 kHz. Proton dipolar decoupling was 100 kHz during data acquisition. The S and S0 alternate-scan strategy compensated for short- and long-term drifts in REDOR experiments. Standard XY-8 phase cycling was used for all refocusing observe-channel π pulses (inserted at the end of each rotor period during dipolar evolution) and dephasing π pulses (inserted in the middle of each rotor period) to compensate for finite pulse imperfections. Typically, CPMAS spectra from 100-mg samples were the result of the accumulation of 20,000 to 40,000 scans at room temperature. The P{N}experiment involved the accumulation of 300,000 scans (two weeks) for each spectrum.
Computational Modeling of the GS-ADP-TβL-Pi Complex
Avogadro was used to construct the phosphorylated GS tetrahedral intermediate Gln-Pi and TβL-Pi42. The GS active site was defined using the crystal structure of GS from Salmonella typhimurium (PDB 1FPY)20. Based on sequence alignments from Phyre243, this structure has 98% sequence identity with E. coli GS and is a suitable model for studying receptor-ligand interactions. Autodock Vina44 was used for docking each ligand to the GS active site with ADP and both Mn2+ ions included in the active site. The ligand search space was defined using a box with dimensions 22×22×22A centered on the Mn2+ ion furthest from ADP. To confirm the usability of this search space, the Glufos ligand from the crystal structure (PDB 1FPY) was docked giving the original binding pose. Docking results were visualized using The PyMOL Molecular Graphics System v1.7, Schrodinger, LLC.
Results and Discussion
Purification of TβL-Thr and TβL
The dipeptide TβL-Thr and the corresponding δ-lactam isomer, TδL-Thr, can be isolated from culture supernatants of producing P. syringae strains grown in Woolley’s minimal medium (Supplementary Fig. 1)11, 45. The corresponding GS inhibitor TβL and inactive δ-lactam isomer, TδL, can be obtained if small amounts of ZnCl2 are added to the P. syringae fermentations. Presumably the Zn2+ activates a periplasmic metallopeptidase that hydrolyzes the TβL-Thr dipeptide bond resulting in the efflux of TβL to the extracellular space46. The non-enzymatic isomerization of TβL-Thr and TβL to TδL-Thr and TδL, respectively, is difficult to suppress and most attempts to isolate active phytotoxin result in a majority of the biologically inactive δ-lactam isomers11. Limited access to pure, quantified amounts of TβL-Thr and TβL has restricted the measurement of quantitative GS inhibition parameters and biological activity10, 47, 48. We solved this problem by rigorously purifying TβL-Thr from cultures of P. syringae pv. tabaci ATCC 11528 grown in Woolley’s medium11. Glycerol freezer stocks of P. syringae ATCC 11528 stored at −80 °C stopped producing TβL-Thr at various times during our work. Loss of TβL-Thr production in P. syringae pathovars has been reported previously and is thought to be associated with spontaneous loss of genetic material from mobile genetic islands containing portions of the tabtoxin biosynthetic gene cluster13, 49, 50. To recover TβL-Thr production we used a colony picking strategy and inoculated 5 mL cultures of Woolley’s medium. We monitored for TβL-Thr production using LC-MS and L-Gln-dependent antibacterial activity against E. coli ATCC 25922 in an agar diffusion assay on chemically defined minimal medium (7 g/L K2HPO4, 3 g/L KH2PO4, 0.47 g/L Na citrate dehydrate, 0.1 g/L MgSO4•7H2O, 1 g/L (NH4)2SO4, 4 g/L glucose, 20 mg/L thymine, 0.1 mg/L biotin, 2 mg/L thiamine, 2 mg/L nicotinic acid, 2 mg/L Ca pantothenate, 10 mg MnSO4•7H2O, 30 g/L Bacto-agar, pH ~7.2). The most promising cultures were used to inoculate larger shake flask cultures (500 mL Woolley’s medium per flask). TβL-Thr was purified from P. syringae supernatants by sequential cation exchange chromatography, preparative HILIC HPLC chromatography, and preparative RP-C18 HPLC. TβL-Thr stock solutions were quantified by 1H-NMR and stored as the TFA salt at −80 °C to prevent isomerization to TδL-Thr (Supplementary Fig. 3).
Pure TβL was obtained by treating TβL-Thr with P. syringae ATCC 11528 cell lysate buffered to pH 6.511. TβL was purified from the cell lysate by preparative HILIC HPLC chromatography with fraction collection guided by GS inhibitory activity. Final TβL concentrations were measured by titration with L-Thr and the ATP-dependent amino acid ligase TblF from the tabtoxin biosynthetic gene cluster. TblF activates the carboxyl group of TβL as an acyl phosphate and catalyzes amide bond formation with the μ-amino group of L-Thr to give TβL-Thr with release of ADP and Pi11, 51. We coupled the TblF reaction to a double enzyme reaction with pyruvate kinase (PK) and lactate dehydrogenase (LDH) to convert ADP and PEP to L-lactic acid with stoichiometric consumption of NADH52. The decrease in optical absorbance at 350 nm is proportional to the conversion of NADH to NAD+, which allowed us to determine ADP concentrations and by direct inference TβL concentrations. Quantified stock solutions of TβL were stored in pH 3.2 ammonium formate buffer at −80 °C to prevent isomerization to TδL.
TβL is a Mechanism-Based Inhibitor of GS
TβL inhibits GS from plants10, 47, animals53, and bacteria48. TβL inhibition of cytosolic and chloroplast forms of GS isolated from pea was shown to be time-dependent, ATP-dependent, irreversible, and competitive with respect to L-Glu10, 47. The use of semi-purified TβL and unknown working TβL concentrations prevented the measurement of quantitative inhibition parameters. Another technical challenge for studying GS inhibitors is the specific activity of GS in vitro. GS activity in bacteria is tightly regulated through post-translational modification (Tyrosine adenylylation) making the preparation of homogeneous enzyme difficult54, 55. To overcome this challenge, we codon optimized the glnA genes encoding for GS from Escherichia coli, Staphylococcus aureus, and Homo sapiens in a pET28b expression vector encoding an N-terminal hexahistidine tag with a thrombin cleavage site (Supplementary Table 1,2). We recombinantly expressed the GS homologs in E. coli BL21 and purified the N-His6-tagged proteins by Ni-NTA affinity chromatography (Supplementary Fig. 2). The N-His6-tagged human GS was eluted from the Ni-NTA column, dialyzed, concentrated, flash frozen in liquid nitrogen, and stored at −80 °C. Human GS was used in all subsequent steps with an intact N-His6-tag. The E. coli and S. aureus GSs are adenylylated on a conserved Tyrosine (residue 398 for E. coli GS; residue 375 for S. aureus GS) when expressed in E. coli54. The adenylylated forms of the E. coli and S. aureus GSs show low activity for catalyzing the biosynthetic reaction56. While adhered to the Ni-NTA column, E. coli GS and S. aureus GS were treated with a buffered solution of phosphodiesterase from crude Crotalus adamanteus snake venom to cleave adenylyl groups. This resulted in highly active unadenylylated preparations of N-His6-tagged E. coli and S. aureus GS after elution from the Ni-NTA column. The N-His6-tagged E. coli and S. aureus GS elutions were dialyzed, concentrated, flash frozen in liquid nitrogen, and stored at −80 °C. The flash frozen stocks of N-His6-tagged GS from E. coli, S. aureus, and Homo sapiens were used for all kinetic studies.
With highly pure, quantified solutions of TβL and active, homogenous preparations of GS enzymes in hand we measured in vitro kinetic parameters for the GS inhibition reaction under steady state conditions. A coupled PK/LDH spectrophotometric assay measuring for ADP production was used to analyze GS inhibition kinetics52, 57. GS produces ADP and Pi as byproducts, which enables real-time quantification of ADP with PK/LDH as a measure of GS activity for the biosynthetic reaction. We measured the Michaelis-Menten parameters (apparent Km and kcat for ATP and L-Glu) for the biosynthetic reaction (conversion of L-Glu, ATP, and NH3 to L-Gln, ADP, and Pi) catalyzed by recombinant E. coli, S. aureus, and human GS (Table 1; Supplementary Fig. 4). All three enzymes gave a similar apparent Km for ATP (0.7 ± 0.1 mM for E. coli and human GS; 1.8 ± 0.1 mM for S. aureus GS) under saturating L-Glu (50 mM) and NH3 (10 mM). Apparent Km values for ATP reported in the literature vary from 0.1–0.7 mM for E. coli GS24, 26, 33, 58 and 1.8–2.8 mM for human GS59, both within our measured range. To our knowledge, no kinetic parameters for S. aureus have been reported in the literature. GS from E. coli was faster (apparent kcat = 7300 ± 300 min−1) and more efficient (kcat/Km = 10,400 ± 1500) than both the human (kcat = 2600 ± 100 min−1; kcat/Km = 3700 ± 500 min−1mM−1) and S. aureus (kcat = 720 ± 20 min−1; kcat/Km = 400 ± 30 min−1mM−1) enzymes. The apparent Km values for L-Glu were measured in the presence of saturating ATP (10 mM) and NH3 (10 mM). The final ATP concentration was always kept lower than MgCl2 (25 mM) to avoid the inhibition by ATP that was previously reported for human59, rat liver60, and Chinese hamster liver GS61. Literature values for the E. coli GS apparent Km for L-Glu range from 0.8–6.6 mM24, 26, 33, 58. We measured an apparent Km value of 7.2 ± 0.8 mM for the E. coli enzyme. The apparent L-Glu Km values for the S. aureus (52 ± 7 mM) and human (37 ± 5 mM) enzymes were significantly higher. We found two literature values for the human L-Glu Km, 3.0 ± 1.2 mM and 3.5 ± 0.7 mM, that were measured on the same batch of recombinant human enzyme expressed in E. coli using a radiometric and colorimetric assay, respectively59. The relatively high apparent Km values for L-Glu are consistent with reported in vivo concentrations of L-Glu on the order of 25–100 mM in certain tissues. Recently, the apparent Km for L-Glu was reported to be 26.3 ± 0.4 mM for GS isolated from Leishmania donovani, a protozoan parasite that causes leishmaniasis in tropical regions62. Under saturating ATP and NH3 conditions, E. coli GS (kcat/Km = 1100 ± 100 min−1 mM−1) was more efficient than H. sapiens (kcat/Km = 140 ± 20 min−1 mM−1) and S. aureus (kcat/Km =27± 4 min−1 mM−1) variants.
Table 1.
Apparent Michaelis-Menten parameters for E. coli, S. aureus, and human GS.
| GS | ATP | L-Glu | ||||
|---|---|---|---|---|---|---|
| Km (mM) | kcat (min−1) | kcat/Km (min−1mM−1) | Km (mM) | kcat (min−1) | kcat/Km (min−1mM−1) | |
| E. coli | 0.7 ± 0.1a | 7300 ± 300 | 10400 ± 1500 | 7.2 ± 0.8 | 7900 ± 300 | 1100 ± 100 |
| S. aureus | 1.8 ± 0.1 | 720 ± 20 | 400 ± 30 | 52 ± 7 | 1400 ± 100 | 27 ± 4 |
| Homo sapien | 0.7 ± 0.1 | 2600 ± 100 | 3700 ± 500 | 37 ± 5 | 5100 ± 300 | 140 ± 20 |
All standard deviations were determined for three independent trials.
Toxicity is a concern when developing GS inhibitors into commercial products such as herbicides or antibiotics53. Toxicity arises due to the lack of selectivity of GS inhibitors for a specific GS isoform as well as broad off-target effects associated with the polypharmacology of ATP mimics and non-proteinogenic amino acids63, 64. We measured inhibition parameters for TβL, MetSox, and Glufos against recombinant N-His6-tagged GS from E. coli, S. aureus, and Homo sapien (Table 2; Fig. 3). The GS inhibition was time- and ATP-dependent for all combinations of inhibitors and enzymes. Longer pre-incubation times of enzyme and inhibitor led to increased loss of GS activity consistent with a mechanism-based inhibition model39. GS inhibition was irreversible for all three inhibitors. Treatment of E. coli GS with TβL under biosynthetic reaction conditions provided a stable, soluble enzyme-inhibitor complex. GS activity could not be recovered even after several days of dialysis of the GS-TβL inhibition complex at 4 °C and 37 °C. We used the Kitz- Wilson model for mechanism-based, irreversible enzyme inhibition to compare the potency of TβL, MetSox, and Glufos39. Plots of 1/kapp (min) vs 1/[inhibitor] (μM−1) gave straight lines with R2 values ≥0.9 for all enzyme and inhibitor combinations in support of a mechanism-based inhibition model (Fig. 3cfi). The parameters KI (μM) and kinact (min−1) were interpreted as a metric for the apparent binding affinity and rate of enzyme inactivation, respectively, and the ratio of kinact/KI (min−1μM−1) was used to compare inhibitor efficiency.
Table 2.
Apparent inactivation parameters for TβL, MetSox, and Glufos against E. coli, S. aureus, and human GS.
| GS | TβL | MetSox | Glufos | ||||||
|---|---|---|---|---|---|---|---|---|---|
| KI (μM) | kinact (min−1) | kinact/KI (min−1μM−1 ×103) | KI (μM) | kinact (min−1) | kinact/KI (min−1μM−1 ×103) | KI (μM) | kinact (min−1) | kinact/KI (min−1μM−1 ×103) | |
| EC | 1.7 ± 0.4a | 0.3 ± 0.1 | 180 ± 70 | 3.9 ± 1.1 | 0.4 ± 0.1 | 100 ± 40 | 1.0 ± 0.2 | 0.7 ± 0.1 | 700 ± 200 |
| SA | 50 ± 10 | 0.4 ± 0.1 | 8 ± 3 | 13 ± 3 | 0.4 ± 0.1 | 28 ± 9 | 13 ± 3 | 0.4 ± 0.1 | 30 ± 10 |
| H | 130 ± 40 | 0.3 ± 0.1 | 2 ± 1 | 1000 ± 500 | 0.4 ± 0.2 | 0.4 ± 0.3 | 150 ± 60 | 0.9 ± 0.3 | 6 ± 3 |
All standard deviations were determined for three independent trials.
Figure 3.

Inhibition kinetics for TβL, MetSox, and Glufos against recombinant GS from (A–C) E. coli, (D–F) S. aureus, and (G–I) homo sapiens show time- and ATP-dependence and fit the Kitz-Wilson model for mechanism-based enzyme inhibition. Error bars represent standard deviations for three independent trials.
TβL, MetSox, and Glufos strongly inhibited E. coli, S. aureus, and human GS (Table 2; Fig. 3cfi). Inhibitor efficiency (kinact/KI) towards GS isoforms followed the general trend of E. coli > S. aureus > human. The inhibitor binding (KI) and rate of inactivation (kcat) also followed the same general trend. Inhibitor potency did indicate a promising therapeutic window between E. coli and human GS enzymes. TβL inhibited E. coli GS with a KI = 1.7 ± 0.4 μM and kinact = 0.3 ± 0.1 min−1 resulting in an inhibitor efficiency of kinact/KI = 180 ± 70 min−1μM−1. The inactivation parameters for TβL against human GS were KI = 130 ± 40 μM, kinact = 0.3 ± 0.1 min−1, and kinact/KI = 2 ± 1 min−1μM−1 corresponding to a 90-fold decrease of inhibitor efficiency. The inactivation parameters for TβL against S. aureus GS (KI = 50 ± 10 μM, kinact = 0.4 ± 0.1 min−1, and kinact/KI = 8 ± 3 min−1μM−1) were intermediary compared to E. coli and human GS. The inactivation parameters for Glufos and MetSox were similar to TβL. Literature values of KI reported for Glufos and MetSox against unadenylylated E. coli GS are 1 μM and 2 μM, respectively65, 66. We determined the KI values against E. coli GS to be 1.0 ± 0.2 μM and 3.9 ± 1.1 μM for Glufos and MetSox, respectively. The closeness to literature KI values supports our use of the GS-PK-LDH coupled enzyme assay for measuring inhibition kinetics. Glufos was the most effective inhibitor against all three GS enzymes and gave a promising 120-fold difference in inhibitor efficiency for E. coli and human GS. MetSox gave the largest therapeutic window in terms of inhibitor efficiency for E. coli and human GS (250-fold difference). Although kinact/KI values for E. coli, S. aureus, and human GS enzymes showed good separation, the kinact values are essentially equal and low selectivity is predicted if therapeutic concentrations are high. E. coli, S. aureus, and human GS enzymes show low overall primary sequence homology, but do share highly conserved active site residues (Supplementary Tables 3,4; Supplementary Fig. 6)19. Further computational, structural, and functional studies will be required to fully rationalize the observed binding affinities, inactivation rates, and overall inhibitor efficiencies for TβL, MetSox, and Glufos representing three chemically distinct classes of L-Glu antimetabolites; 3-HβLs, sulfoximines, and phosphinates, respectively.
GS is the Primary Cellular Target for TβL in E. coli
Inhibition of central metabolic pathways with so-called antimetabolites is a classic method to perturb cellular metabolism and is gaining traction in therapeutic and commercial applications67. Antimetabolites such as TβL and other GS inhibitors dysregulate metabolic pathways through enzyme inactivation causing changes in metabolic flux that activate and/or inhibit connected pathways both upstream and downstream of the target metabolite68. MetSox is known to inhibit both glutamine synthetase and γ-glutamylcysteine synthetase69. To support our in vitro GS inactivation measurements, we sought to validate that GS is the primary cellular target for TβL and Glufos in E. coli. To facilitate membrane transport we used pro-drug forms of the GS inhibitors, TβL-Thr and synthetic Ala-Ala- Glufos (AAG) (Supplementary Fig. 1). We synthesized AAG via amide coupling of Fmoc-L-Ala- L-Ala dipeptide with racemic Glufos followed by Fmoc deprotection with 20% piperidine in DMF (Supplementary Scheme 1). Presumably, TβL-Thr and AAG are transported to the periplasm of E. coli cells via outer membrane di- and tri-peptide permeases70–72. Peptidases in the periplasm and/or cytoplasm then cleave the dipeptide bonds releasing the active GS inhibitors that reach cytoplasmic GS46, 73. In order to control for metabolite concentrations we used a chemically defined minimal medium (7 g/L K2HPO4, 3 g/L KH2PO4, 0.47 g/L Na citrate dehydrate, 0.1 g/L MgSO4•7H2O, 1 g/L (NH4)2SO4, 4 g/L glucose, 20 mg/L thymine, 0.1 mg/L biotin, 2 mg/L thiamine, 2 mg/L nicotinic acid, 2 mg/L Ca pantothenate, 10 mg MnSO4•7H2O, pH ~7.2) for growth studies with E. coli ATCC 25922. TβL-Thr and AAG both gave end point MIC90 values of 0.5 μM in broth microdilution assays. Addition of L-Glu had no effect on the antibacterial activity of TβL-Thr and AAG. When the minimal medium was supplemented with 5 mM L-Glu the MIC90 values for TβL-Thr and AAG were 1 μM. Addition of 5 mM L-Gln to the minimal medium completely abolished the antibacterial activity of TβL-Thr and AAG (MIC90 values were >256 μM). If cytoplasmic TβL and Glufos has a polypharmacological antibacterial effect on E. coli, then L-Gln supplementation should not wholly recover bacterial growth. Treatment of E. coli with TβL-Thr and AAG results in glutamine auxotrophy consistent with GS being the primary cellular target for TβL and Glufos in E. coli. The same Gln- auxotroph phenotype has been reported in M. tuberculosis and E. coli treated with MetSox and bialaphos, the naturally occurring Glufos-Ala-Ala (GAA) tripeptide, respectively74, 75. We confirmed this result in E. coli using a commercial source of bialaphos sodium salt. The order of Glufos tripeptide, synthetic AAG or natural GAA (Supplementary Fig. 1), does not appear to influence cellular transport or peptidase cleavage to release the active GS inhibitor in E. coli.
We further probed the glutamine dependence of GS inhibitor antibiotics by measuring growth curves for E. coli ATCC 29522 in liquid minimal medium in the presence of varying concentrations of L-Gln (Fig. 4). With no L-Gln present, 2 μM TβL-Thr had a bacteriostatic effect on E. coli ATCC 29522 with full growth recovery after ~24 h (Fig. 4a). E. coli growth was recovered to near control levels when the medium was supplemented with 0.1 mM L-Gln. When L-Gln supplementation reached 2.5 mM the growth curve matched the control with no TβL-Thr. When the concentration of TβL-Thr was increased to 100 μM E. coli growth was suppressed for >50 h (Fig. 4b). Addition of L-Gln antagonized the bacteriostatic activity, but to a lesser extent than the antagonism observed when using 2 μM TβL-Thr (Fig. 4a). The growth of E. coli ATCC 29522 treated with 100 μM TβL-Thr improved with increasing L-Gln concentrations, but never reach the full level of the control (Fig. 4b). AAG had a similar bacteriostatic effect on the growth of E. coli ATCC 29522 with increased potency and decreased susceptibility to L-Gln antagonism (Fig. 4c). The growth of E. coli was fully suppressed for >66 h when treated with 2 μM AAG. Bacterial growth was recovered in the presence of 2 μM AAG with L-Gln supplementation, but higher concentrations of L-Gln were required compared to growth rescue in the presence of 2 μM TβL-Thr. The increased potency of AAG compared to TβL-Thr might be due to spontaneous isomerization of TβL-Thr to TδL-Thr in the growth medium. The isomerization half-life of TβL-Thr is 37 h at 23 °C in pH 7.2 phosphate buffer11. We anticipate that the isomerization rate will increase at 37 °C in pH 7.2 phosphate buffered minimal medium used for bacterial growth studies. E. coli growth started to recover in minimal medium after 26 h in the presence of 2 μM TβL-Thr. No growth recovery was observed after 48 h in the presence of 100 μM TβL-Thr. The concentration dependence and time to growth recovery are consistent with the isomerization of TβL-Thr to TδL-Thr in the growth medium. Since TβL and Glufos are both irreversible GS inhibitors we do not believe that the growth recovery or L-Gln dependence is due to the inhibitor off-rate. Intracellular GS turnover and GS overexpression might play a role in growth recovery. GS overexpression has been shown to be an effective resistance mechanism to GS inhibitors in Mycobacterium tuberculosis76. Some bacteria carry extra copies of glnA that encode for GS homologs with active site mutations that can reduce the inhibitory activity of MetSox and Glufos and potentially play a role in resistance to GS inhibitors74, 77. Spontaneous mutations in glnA coding sequences in Salmonella typhimurium, Bacillus subtilis, Exiguobacterium sp., soybean, and rice have been reported to arise under GS inhibitor selective78–82. The mutations were tracked to active site residues that reduce susceptibility to MetSox and/or Glufos inhibition in vitro. Careful consideration must be given to these potential resistance mechanisms along with endogenous L-Gln concentrations for applications of GS inhibitors as antibiotics and herbicides.
Figure 4.

TβL-Thr and AAG show L-Gln-dependent bacteriostatic activity towards E. coli ATCC 25922. Panels show growth curves of E. coli ATCC 25922 in chemically defined minimal medium supplemented with (A) 2 μM TβL-Thr, (B) 100 μM TβL-Thr, and (C) 2 μM AAG and variable amounts of L-Gln and L-Glu. Error bars represent standard deviations for three independent trials.
GS Catalyzes Phosphorylation of TβL
The mechanistic basis for GS inhibition by TβL is unknown (Fig. 2). We have showed that TβL inhibition is irreversible, ATP-dependent, time-dependent, and fits the Kitz-Wilson kinetic model for mechanism-based enzyme inhibition suggesting that TβL is phosphorylated in the GS active site similar to the related inhibitors MetSox and Glufos (Fig. 3). We used LC-MS to detect the phosphorylated inhibitors TβL-Pi, MetSox-Pi, and Glufos-Pi released after denaturation of inhibition complexes with GS from E. coli (Fig. 5). Recombinant N-His6 GS from E. coli was treated with TβL (Fig. 5a), MetSox (Fig. 5c), and Glufos (Fig. 5d) in the presence and absence of ATP. The initial activity and inhibition of GS was confirmed by the coupled PK/LDH assay monitoring optical absorbance for consumption of NADH that was used for kinetic experiments. The GS-inhibitor complex was washed with buffer via centrifugal filtration to remove excess inhibitor prior to mild heat denaturation and protein precipitation with addition of KCl and MeOH. The clarified methanolic solution was treated with FmocCl to acylate the α-amino groups of the inhibitors to give Fmoc-TβL-Pi, Fmoc-MetSox-Pi, and Fmoc-Glufos-Pi. The Fmoc-tagging step stabilized the phosphorylated inhibitors, increased retention time during LC-MS analysis, and prevented the spontaneous isomerization of TβL or TβL-Pi to TδL or TδL-Pi, respectively11. We also prepared a sample of isotopically labeled 15N-TβL by fermenting P. syringae ATCC 11528 in Woolley’s medium with K15NO3 as the sole nitrogen source. 15N-TβL- Thr and 15N-TβL were purified and quantified as described previously for unlabeled TβL-Thr and TβL. 15N-TβL was used as a positive control isotopomer to confirm the retention time and isotope distribution of TβL-Pi (Fig. 5b).
Figure 5.

GS phosphorylates TβL, MetSox, and Glufos. Panels show extracted ion chromatograms for LC-MS analysis of denatured GS-inhibitor complexes, with (+) or without (−) ATP added, and after treatment with Fmoc-chloride.
Treatment of the supernatant from the agglutinated TβL-treated GS solution with FmocCl followed by LC-MS analysis resulted in a clear peak in the extracted ion chromatogram (EIC) for Fmoc-TβL-Pi (m/z = 491 for [M+H]+) only when ATP was present during incubation of GS and TβL (Fig. 5a). The molecular formula of Fmoc-TβL-Pi was confirmed by high-resolution MS analysis in negative ion mode (HRMS1 (ESI) calculated for C22H22N2O9P: 489.1068 [M−H]−, found 489.1064) (Supplementary Fig. 5). As expected, MS/MS analysis of the Fmoc-TβL-Pi parent ion (observed m/z = 489.1068 for [M−H]−) showed loss of neutral Fmoc giving the TβL-Pi fragment ion (observed m/z = 267.0387 for [M−H]−) and a neutral loss of TβL with formation of the inorganic phosphate (observed m/z = 96.9695 for [M−H]−) and phosphite (observed m/z = 78.9589 for [M−H]−) fragment ions83. The MS/MS fragmentation pattern Fmoc-TβL-Pi is consistent with phosphorylation of the C3-OH group of the 3-HβL group, but does not definitively establish the site of phosphorylation. The MS spectra implied that the β-lactam ring was intact, but it is not possible to distinguish between Fmoc-TβL-Pi and the corresponding phosphorylated δ-lactam isomer without a standard sample. Fmoc-tagging blocks isomerization of TβL to TδL and we suspected that Fmoc-tagging also blocked the spontaneous isomerization of TβL-Pi to the phosphorylated δ-lactam isomer. The same experiments conducted using 15N-TβL produced the expected ion for Fmoc-15N-TβL-Pi (m/z = 493 for [M+H]+) with identical retention time as Fmoc-TβL-Pi (Fig. 5b). We also confirmed that N-His6-tagged GS from E. coli catalyzes the ATP-dependent conversion of MetSox to MetSox-Pi (Fig. 5c). LC-MS analysis of the denatured GS-MetSox-Pi solution after treatment with FmocCl produced a strong peak for Fmoc-MetSox-Pi in the EIC (m/z = 483 for [M+H]+) only when ATP was included during enzyme incubation. Glufos-Pi is an unstable phosphoanhydride that hydrolyzes rapidly upon denaturing the GS-Glufos-Pi complex84. We observed a large enhancement in ion counts for Fmoc-Glufos (m/z = 483 for [M+H]+) when ATP was present in the incubation of GS with Glufos (Fig. 5d). Small amounts of Fmoc-Glufos were also present in the no ATP control reaction that might be a result of insufficient washing prior to denaturing, weak binding of Glufos to the GS active site, or co-purification of ATP in the GS active site resulting in a small amount of Glufos-Pi production.
In order to gain insight on the mechanism of GS inhibition by TβL we needed to establish the site of phosphorylation on TβL-Pi. We initially attempted to obtain an X-ray crystal structure of TβL-Pi bound to GS from E. coli without success. Although we could regularly obtain stable crystals of the GS- TβL-Pi inhibition complex, the quality of X-ray diffraction was not sufficient to solve the structure. During these crystallization attempts we turned to solid-state NMR as a proven method to provide structural information on enzyme-inhibitor complexes in heterogeneous mixtures41. We hypothesized that rotational-echo double resonance (REDOR) could be used to restore dipolar couplings between 15N and 31P spin pairs of GS-bound TβL-Pi that are removed by magic angle spinning85–87. We scaled up production of GS from E. coli using the same recombinant expression system as described previously. During our scale up the phosphodiesterase from crude Crotalus adamanteus (eastern diamondback rattlesnake) snake venom, used to cleave adenylyl PTMs, was backordered from the supplier. Thus, we turned to a new source of phosphodiesterase from crude Crotalus atrox (western diamondback rattlesnake) to cleave the adenylyl groups. Treatment of N-His6-tagged GS bound to Ni-NTA resin with C. atrox phosphodiesterase cleaved the adenylyl groups as well as the N-His6-tag resulting in elution of highly active GS. C. atrox venom is known to contain thrombin-like proteases capable of cleaving the N-His6 tag at the thrombin cleavage site (Supplementary Table 3)88. Untagged GS was further purified by size-exclusion chromatography (SEC) and catalytic activity towards the L-Glu to L-Gln biosynthetic reaction was confirmed using the PK/LDH coupled spectrophotometric assay. TβL inhibited the untagged, SEC-purified GS with time- and ATP-dependence with the same efficiency as the N-His6-GS. SDS-PAGE and MALDI analysis confirmed the anticipated molecular weight of the GS monomer (51.9 kDa). Samples were prepared by treating E. coli GS (~63 mg) with excess 15N-TβL in the presence of ATP followed by extensive washing of the resulting GS-ADP-15N-TβL-Pi complex with trehalose/HEPES buffer using centrifugal filtration. The sample was cryoprotected using trehalose, PEG-8000, and dextran prior to controlled freezing at −10 °C (ice/CaCl2 bath) with gradual cooling to −78 °C (dry ice/acetone) and final freezing at liquid nitrogen temperatures. Lyophilization for 72 h gave ~160 mg of a fluffy white powder that was used for solid-state NMR experiments41. A control sample lacking 15N-TβL inhibitor and replacing ATP with ADP was prepared in the same manner.
A control sample of GS, ADP, and buffer was used to establish a zero parts per million (ppm) chemical shift (δp) in the 202-MHz 31P cross-polarization magic-angle spinning NMR spectrum (Fig. 6a). No signals associated with free ADP were observed. Next we tried a 31P{15N} REDOR experiment on GS-ADP-15N-TβL-Pi. This experiment is always done in two parts41. The first is a so-called “full echo”, which is obtained with 31P π pulses on the completion of each rotor period. This establishes a reference signal that takes account of homogeneous decay (T2). Both free phosphate (Pi) and ADP peaks are observed, as well as a bound phosphate peak at -2 ppm (Fig. 6b). The second part of the REDOR experiment inserts 15N π pulses in the middle of each rotor period. These are so-called “dephasing” pulses. Their function is to defeat the spatial averaging of magic-angle spinning and allow 31P–15N dipolar coupling to appear as a diminution of the full echo resulting in S, a “dephased echo” (not shown). The REDOR difference is ΔS=S0-S (Fig. 6c), which has a simple interpretation in terms of the heteronuclear dipolar coupling between 31P and 15N. This coupling yields rPN, the internuclear separation. Because of the massive Pi peak near 0 ppm in S0, we chose to use the first spinning sideband of the bound phosphate peak to estimate S0 and ΔS (Fig. 6b,c; dotted line). Interference from the small (and broad) Pi spinning sideband could be ignored. Integrals of both ΔS and S0 first sidebands improved sensitivity and led to rPN = 4.1 A (± 0.2 A).
Figure 6.
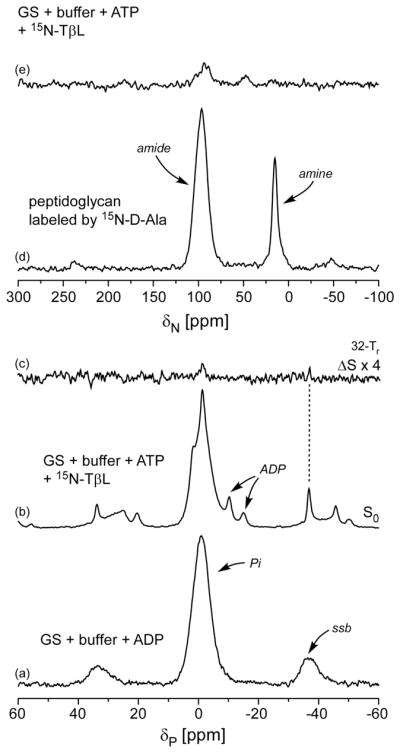
(a) 202-MHz 31P cross-polarization magic-angle spinning NMR spectrum of a mixture of glutamine synthetase, buffer, and ADP. The resulting phosphate peak is assigned a zero ppm chemical shift. (b) 31P{15N} full echo and (c) rotational-echo double resonance (REDOR) difference for a mixture of glutamine synthetase, buffer, ATP, and 15N-labeled TβL after dipolar evolution for 32 rotor periods (4.48 ms). (d) 50.7-MHz 15N cross-polarization magic-angle spinning NMR spectrum of intact whole cells of Staphylococcus aureus whose cell walls have been labeled by D-[15N]alanine. The chemical-shift reference is solid ammonium sulfate. (To switch to a liquid ammonia nitrogen chemical-shift scale, add 20 ppm.) (e) 50.3-MHz 15N cross-polarization magic-angle spinning NMR spectrum of the sample of panel (b).
Cross-polarization 15N NMR of a labeled bacterial cell-wall sample established a chemical-shift (δN) reference spectrum for known amide (NH) and primary amine (NH2) nitrogens (Fig. 6d)89. The same experiment performed on GS-ADP-15N-TβL-Pi produced an amide-nitrogen peak near 100 ppm and an isopropyl primary amine nitrogen peak near 50 ppm (Fig. 6e), but no peaks associated with sp2 nitrogens. Again spectral integration improved signal-to-noise ratios. The E. coli GS dodecamer has 12 functional active sites. Based on peak integrations we estimate that ~6 of the GS active sites are occupied by15N-TβL-Pi. With only 50% active site occupancy, our samples of GS-ADP-15N-TβL-Pi might be heterogeneous, which might explain difficulty obtaining quality X-ray diffraction data on stable crystals. Previous studies showed that 11 of the 12 active sites of E. coli GS are occupied by Glufos-Pi and MetSox-Pi produced ~80% active site occupancy35, 84. Sub-stoichiometric inhibitor occupancy of GS active sights might be a result of communication between active sites, which is consistent with previous observations of progressively slower inhibitor binding and phosphorylation90.
Based on structural models of GS bound to MetSox-Pi and Glufos-Pi there are three possible sites of phosphorylation on TβL: (1) β-lactam nitrogen, (2) β-lactam oxygen, (3) C3-hydroxyl group. Amide O-phosphorylation is catalyzed by aminoimidazole ribonucleotide synthetase in the purine biosynthetic pathway. Recently, a glutamine kinase was discovered that catalyzes N-phosphorylation of the L-Gln amide in a pathway for phosphoramidite capsular polysaccharide biosynthesis in Campylobacter jejuni, a common foodborne pathogen91. The 15N cross-polarization experiment rules out phosphorylation of the β-lactam nitrogen and β-lactam oxygen, which would leave the 15N without a direct bond to a proton or with δN corresponding to full sp2 hybridization, respectively. Furthermore, a 31P–15N through space distance of 4.1 A rules out direct phosphorylation of nitrogen. The observed distance measurement and 15N cross-polarization experiments are consistent with phosphorylation of the TβL C3-hydroxyl group with an intact β-lactam ring. Phosphorylation at this site makes TβL-Pi structurally related to MetSox-Pi and Glufos-Pi that mimic the tetrahedral GS transition state (Fig. 2).
TβL-Pi is a GS Transition State Analog
Transition states for nucleophilic acyl substitution reactions resemble the tetrahedral intermediate36. MetSox-Pi, Glufos-Pi, and TβL-Pi show structural similarity to the high-energy GS tetrahedral intermediate, Gln-Pi, and are predicted to bind with abnormally high-affinity (MetSox-Pi Ki < 10−19 M against GS37) and stabilize the normally short-lived GS transition state geometry. We built computational models for the GS tetrahedral intermediate, Gln-Pi (Fig. 7a), and TβL-Pi (Fig. 7b) with phosphorylation of the C3- hydroxyl group bound to dimetallic (Mn2+) GS from E. coli. We used Avogadro to build structures of Gln-Pi and TβL-Pi42. We used a crystal structure of GS from Salmonella typhimurium (PDB 1FPY) as template to build a homology model of E. coli GS (98% sequence identity) using the Phyre2 software20, 43. We used Autodock Vina to identify low energy ligand docked poses in the GS active site with bound ADP44. The ligand search space was validated by docking Glufos, which recovered the original binding pose from the crystal structure (PDB 1FPY). Both Gln-Pi (Fig. 7c) and TβL-Pi (Fig. 7d) were found to bind as expected in the L-Glu binding site with the α-amino acid group interacting with Glu212A, Asn264A, Glu327A, and His269A27. Some key differences were observed when comparing interaction networks for the phosphorylated head groups of Gln-Pi and TβL-Pi. The 34 phosphate of Gln-Pi interacts with Mn2+, His210A, and Tyr179A. The Gln-Pi oxyanion and nitrogen cation are predicted to interact with Tyr179A. The stereochemistry and conformation of TβL-Pi resembles that of Gln-Pi with the phosphate groups occupying similar chemical space. The β-lactam oxygen and nitrogen aligned closely with the oxyanion and nitrogen cation, respectively, of Gln-Pi suggesting that the 3-HβL warhead mimics the structure, polarity, and chirality of the GS transition state. TβL-Pi showed more extensive interactions with amino acid side chains including residues from chain B. The phosphate of TβL-Pi interacted with Mn2+, His210A, and Tyr179A similar to Gln-Pi. The β-lactam oxygen was stabilized by hydrogen bonding to Arg359A and the β-lactam NH group donated a hydrogen bond to Asp50B. Interaction of TβL-Pi with Asp50B is an interesting observation for several reasons. Asp50B is highly conserved on the central, mobile flap of GS chain B that is involved in active site closure upon L-Glu binding25. Asp50B also forms part of the ammonium-binding site and is thought to deprotonate NH4+ prior to nucleophilic attack on the δ-carbonyl carbon of the γ-glutamyl acyl phosphate leading to Gln-Pi formation20, 26. Asp50B is not a strong enough base to deprotonate the β-lactam NH group (pKa ~25) so a stable H-bond might result holding GS in a closed conformation resembling the transition state structure. The strained β-lactam imparts amine-like character on the β-lactam nitrogen (pKa of protonated twisted amides ~4.592–94), which might also play a role in stabilization of the Asp50 interaction and overall stereoelectronic resemblance of the GS transition state. The stable H-bond between Asp50B and the β-lactam NH might be a distinguishing feature for TβL compared to MetSox and Glufos, which both place a less polar CH3 group in this chemical space20. Efforts to substitute the Glufos CH3 group with more polar groups such as amines resulting in slower binding and decreased inactivation rates65, 66, 95–97. The tetrahedral γ-phosphinate structure of Glufos increases the rate of phosphoryl transfer due to similarity in structure to the GS transition state and tetrahedral intermediate, but slows the rate of initial GS binding due to differences in structure to the trigonal planar substrate L-Glu γ-carboxylate33. Detailed kinetic and structural studies of TβL, similar to those reported for Glufos and MetSox, are needed to fully appreciate the contribution of GS binding and rate of phosporyl transfer to the observed KI and kinact.
Figure 7.

Model for glutamine synthetase complexes with the reaction tetrahedral intermediate Gln-Pi (A,C) and TβL-Pi (B,D). Panels A and B depicts the glutamine synthetase active site formed between chain A and chain B with the top scoring docked poses of ADP, Gln-Pi, and TβL-Pi shown as sticks with nitrogens in blue, oxygens in red, phosphorous in orange, and carbon in green (ADP) or teal (Gln-Pi and TβL-Pi). The two active site Mn2+ metals are shown as spheres colored sea green. The GS cartoon structure is shown in grey with selected amino acid side chains shown as navy blue line structures. The P–N through-space distance for Gln-Pi (2.9 A) and TβL-Pi (4.2 A) are highlighted as yellow dashed lines. Panels C and D show the 2D-interaction network between glutamine synthetase amino acid side chains and Gln-Pi or TβL-Pi for the top scoring docked poses. The images in panels A and B were generated using PyMOL v1.7.
The (S)-C3-hydroxyl group of TβL is positioned in the GS active site to attack the ATP γ-phosphate group to achieve phosphoryl transfer. The C3-hydroxyl is predicted to be protonated at physiological pH, but α-orientation to the β-lactam carbonyl might lower the pKa by at least an order of magnitude, via hydrogen bonding and inductive effects, making it a better mimic of the L-Glu carboxylate and increasing the nucleophilicity98. Thus, lowering the pKa of the C3-hydroxyl group might increase the rate of GS binding and phosphoryl transfer. Interestingly, a TβL analog that is chlorinated on the γ-carbon (α to the C3-OH) was isolated from a strain of Streptomyces species 372A8. The purpose for chlorination at this position is unknown – enhanced binding through favorable polarity effects99 in the GS active site or increased rate of phosphoryl transfer to the C3-hydroxyl group? Chlorination likely lowers the pKa of the C3-hydroxyl proton through inductive effects, making deprotonation more thermodynamically favorable and kinetically faster. Reactivation of GS inhibited by MetSox-Pi was shown to have a strong pH dependence, implying that pKa of active site amino acid residues and the inhibitor are important for the formation and stability of the inhibition complex37. Furthermore, a γ-hydroxylated variant of Glufos was reported to increase the rate of GS activation by 50%, which might be the result of faster binding and/or phosphoryl transfer66. Inhibitor potency towards GS isoforms seems to correlate with Km values for L-Glu (Table 1,2). A higher apparent Km value for L-Glu correlated with an increase in KI for TβL, MetSox, and Glufos, which is consistent with a competitive inhibition model. Both KI and kinact are important and physiologically relevant metrics for mechanism-based inhibitors. Lessons learned from tetrahedral transition state analogs36 including phosphinates (ATP-dependent ligase inhibitors100) and tertiary alcohols (protease inhibitors101) might prove useful for the synthetic optimization of 3-HβLs as inhibitors of ATP-dependent ligases102, 103.
Conclusions
We have established the mechanistic basis for the ATP-dependent inhibition of GS by TβL using enzyme kinetics, mass spectrometry, solid-state NMR, and computational modeling. TβL is competitive with L-Glu and GS catalyzes phosphoryl transfer from ATP to the C3- hydroxyl group of the β-lactam warhead. Phosphorylated TβL-Pi resembles the GS tetrahedral transition state and forms a stable inhibition complex with bound ADP that is non-covalent and essentially irreversible. TβL is mechanistically distinct from traditional β-lactam antibiotics, such as penicillin, that covalently inhibit transpeptidases in the serine hydrolase superfamily by a strain-driven, β-lactam ring-opening acylation mechanism. The TβL β-lactam ring remains intact during GS inhibition and serves as a tetrahedral template that matches the conformation, polarity, and chirality of the GS transition state. The 3-HβL warhead of TβL might be broadly applicable as a tetrahedral scaffold for designing transition state analogs of enzymes that stabilize tetrahedral intermediates including proteases and enzymes in the ATP-dependent carboxylate-amine ligase superfamily102, 103. The phosphinate warhead of the related GS inhibitor Glufos has been repurposed many times for such targeted applications100. Targeting the active site of ATP-dependent ligases might be advantageous over the commonly exploited ATP site, which can generate off-target toxicity. Dipeptide pro-drugs of TβL show broad-spectrum bacteriostatic antibacterial activity. GS is the primary cellular for TβL in E. coli and treatment induces L-Gln auxotrophy. TβL and the related inhibitors MetSox and Glufos show moderate selectivity for inhibiting bacterial GS over human GS. Toxicity associated with inhibition of human GS, competition with endogenous L-Gln, cell permeability, and emerging resistance mechanisms are challenges for developing GS inhibitors as antimicrobial agents31. Applications of GS inhibitors for treating infectious diseases might be limited to scenarios where endogenous L-Gln levels are low, such as L-Gln depletion associated with sepsis104, since L-Gln strongly antagonizes antimicrobial activity67, 68, 105. MetSox was shown to be effective and synergistic with isoniazid in a guinea pig lung infection model of M. tuberculosis and long exposure to low doses of MetSox was well tolerated106. Prospects for synergistic antibiotics combinations with GS inhibitors are attractive since GS plays a central role in nitrogen metabolism feeding important downstream pathways targeted by established clinical antibiotics. Glutamine synthetase activity has been associated with bacterial virulence and is required for some multi-drug resistant bacterial phenotypes107, 108. Improved structural analogs of TβL, MetSox, and Glufos structures and new pro-drug formulations might enhance the therapeutic window and cellular uptake of GS inhibitors. Methods reported here will be useful for establishing SAR for GS inhibitors in vitro and in whole cell antibacterial assays. Target binding (KI) and rate of enzyme inactivation (kinact) are important metrics for mechanism-based inhibitors that expand dimensions for structural optimization. TβL, MetSox, and Glufos represent three chemically distinct classes (3-HβL, sulfoximine, and phosphinate, respectively) of mechanism-based GS inhibitors with the potential to offer differential selectivity towards GS isoforms in applications as herbicides, pesticides, antimicrobials, and therapeutics for treating human diseases.
Supplementary Material
Acknowledgments
Funding
Research was supported by start-up funds from Washington University in St. Louis.
We thank A. D’Avignon (formerly WUSTL, Dept. of Chemistry; currently Sanford Burnham Medical Research Institute, Orlando, FL), J. Kao (WUSTL, Dept. of Chemistry), and B. Marsden (WUSTL, Dept. of Chemistry) for assistance in the acquisition of solution NMR spectra. We thank Dr. Brad Evans at the Proteomics & Mass Spectrometry Facility at the Donald Danforth Plant Science Center, St. Louis, MO for assistance with the acquisition of the QTRAP LC-MS/MS spectra (supported by the National Science Foundation under Grant No. DBI-0521250). We thank Margaret Reck (WUSTL, Dept. of Chemistry) for assistance purifying tabtoxin from P. syringae cultures. We thank Dr. Joe Jez and Cynthia Holland (WUSTL, Dept. of Biology) for diligent efforts crystallizing GS and analyzing diffraction patterns at the advanced photon source at Argonne National Laboratory.
Abbreviations
- AAG
L-Ala-L-Ala-Glufos
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- BME
β-mercaptoethanol
- DMF
dimethylformamide
- DTT
dithiothreitol
- ESI
electrospray ionization
- Fmoc
fluorenylmethyloxycarbonyl
- Gln-Pi
glutamine synthetase tetrahedral intermediate
- Glufos
glufosinate
- Glufos-Pi
phospho-glufosinate
- GS
glutamine synthetase
- 3-HβL
3-hydroxy-β-lactam
- LC
liquid chromatography
- LDH
lactate dehydrogenase
- MS
mass spectrometry
- MetSox
methionine sulfoximine
- MetSox-Pi
phospho-methionine sulfoximine
- MIC
minimum inhibitory concentration
- NAD+
nicotinamide adenine dinucleotide oxidized form
- NADH
nicotinamide adenine dinucleotide reduced form
- NMR
nuclear magnetic resonance
- NTA
nitriloacetic acid
- OD600
optical density at 600 nm
- PEP
phosphoenolpyruvate
- Pi
inorganic phosphate
- PK
pyruvate kinase
- pv
pathovar
- REDOR
rotational-echo double resonance
- r.p.m.
rotations per minute
- SAR
structure-activity relationship
- SEC
size exclusion chromatography
- TβL
tabtoxinine-β-lactam
- TβL-Pi
phospho-tabtoxinine-β-lactam
- TβL-Thr
β-tabtoxin
- TδL
tabtoxinine-δ-lactam
- TδL-Thr
δ-tabtoxin
- TδL-Pi
phospho-tabtoxinine-δ-lactam
- TPase
transpeptidase
Footnotes
Notes
The authors declare no competing financial interest.
Supplementary Tables, Figures, and Schemes including protein gels, NMR spectra, Michaelis-Menten kinetic plots, mass spectrometry data, HPLC and LC-MS chromatograms, and synthetic procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Walsh C, Wencewicz T. Antibiotics: Challenges, Mechanisms, Opportunities. ASM Press; 2016. [Google Scholar]
- 2.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev. 2008;32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 3.Lee W, McDonough MA, Kotra LP, Li ZH, Silvaggi NR, Takeda Y, Kelly JA, Mobashery S. A 1.2-Å snapshot of the final step of bacterial cell wall biosynthesis. Proc Natl Acad Sci U S A. 2001;98:1427–1431. doi: 10.1073/pnas.98.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho H, Uehara T, Bernhardt TG. Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell. 2014;159:1300–1311. doi: 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bush K, Jacoby GA. Updated functional classification of beta-lactamases. Antimicrob Agents Chemother. 2010;54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roux MV, Jimenez P, Davalos JZ, Castano O, Molina MT, Notario R, Herreros M, Abboud JLM. The first direct experimental determination of strain in neutral and protonated 2-azetidinone. J Am Chem Soc. 1996;118:12735–12737. [Google Scholar]
- 7.Stewart WW. Isolation and proof of structure of wildfire toxin. Nature. 1971;229:174–178. doi: 10.1038/229174a0. [DOI] [PubMed] [Google Scholar]
- 8.Scannell JP, Pruess D, Blount JF, Ax HA, Kellett M, Weiss F, Demny TC, Williams TH, Stempel A. Antimetabolites produced by microorganisms. XII (S)-alanyl-3-[alpha-(S)-chloro-3-(S)-hydroxy 2-oxo-3- azetidinylmethyl]-(S)-alanine, a new beta-lactam containing natural product. J Antibiot. 1975;28:1–6. doi: 10.7164/antibiotics.28.1. [DOI] [PubMed] [Google Scholar]
- 9.Sinden SL, Durbin RD. Glutamine synthetase inhibition: Possible mode of action of wildfire toxin from Pseudomonas tabaci. Nature. 1968;219:379–380. doi: 10.1038/219379a0. [DOI] [PubMed] [Google Scholar]
- 10.Langston-Unkefer PJ, Macy PA, Durbin RD. Inactivation of glutamine synthetase by tabtoxinine-β-lactam. Plant Physiol. 1984;76:71–74. doi: 10.1104/pp.76.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wencewicz TA, Walsh CT. Pseudomonas syringae self-protection from tabtoxinine-beta-lactam by ligase TblF and acetylase Ttr. Biochemistry. 2012;51:712–7725. doi: 10.1021/bi3011384. [DOI] [PubMed] [Google Scholar]
- 12.Bender CL, Alarcon-Chaidez F, Gross DC. Pseudomonas syringae phytotoxins: Mode of action, regulation, and biosynthesis by peptide and polyketide synthetases. Microbiol Mol Biol Rev. 1999;63:266–292. doi: 10.1128/mmbr.63.2.266-292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Brien HE, Desveaux D, Guttman DS. Next-generation genomics of Pseudomonas syringae. Curr Opin Microbiol. 2011;14:24–30. doi: 10.1016/j.mib.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Hart KM, Reck M, Bowman GR, Wencewicz TA. Tabtoxinine-β-lactam is a “stealth” β-lactam antibiotic that evades β-lactamase-mediated antibiotic resistance. MedChemComm. 2016;7:118–127. [Google Scholar]
- 15.He H, Ding Y, Bartlam M, Sun F, Le Y, Qin X, Tang H, Zhang R, Joachimiak A, Liu J, Zhao N, Rao Z. Crystal structure of tabtoxin resistance protein complexed with acetyl coenzyme A reveals the mechanism for β-lactam acetylation. J Mol Biol. 2003;325:1019–1030. doi: 10.1016/s0022-2836(02)01284-6. [DOI] [PubMed] [Google Scholar]
- 16.Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, Cravatt BF. Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc Natl Acad Sci U S A. 2010;107:20941–20946. doi: 10.1073/pnas.1011663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iyer LM, Abhiman S, Maxwell Burroughs A, Aravind L. Amidoligases with ATP-grasp, glutamine synthetase-like and acetyltransferase-like domains: synthesis of novel metabolites and peptide modifications of proteins. Mol BioSyst. 2009;5:1636–1660. doi: 10.1039/b917682a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galletti P, Giacomini D. Monocyclic beta-lactams: new structures for new biological activities. Curr Med Chem. 2011;18:4265–4283. doi: 10.2174/092986711797200480. [DOI] [PubMed] [Google Scholar]
- 19.Eisenberg D, Gill HS, Pfluegl GM, Rotstein SH. Structure-function relationships of glutamine synthetases. Biochim Biophys Acta. 2000;1477:122–145. doi: 10.1016/s0167-4838(99)00270-8. [DOI] [PubMed] [Google Scholar]
- 20.Gill HS, Eisenberg D. The crystal structure of phosphinothricin in the active site of glutamine synthetase illuminates the mechanism of enzymatic inhibition. Biochemistry. 2001;40:1903–1912. doi: 10.1021/bi002438h. [DOI] [PubMed] [Google Scholar]
- 21.Hunt JB, Smyrniotis PZ, Ginsburg A, Stadtman ER. Metal ion requirement by glutamine synthetase of Escherichia coli in catalysis of γ-glutamyl transfer. Arch Biochem Biophys. 1975;166:102–124. doi: 10.1016/0003-9861(75)90370-7. [DOI] [PubMed] [Google Scholar]
- 22.Krajewski WW, Collins R, Holmberg-Schiavone L, Jones TA, Karlberg T, Mowbray SL. Crystal structures of mammalian glutamine synthetases illustrate substrate-induced conformational changes and provide opportunities for drug and herbicide design. J Mol Biol. 2008;375:217–228. doi: 10.1016/j.jmb.2007.10.029. [DOI] [PubMed] [Google Scholar]
- 23.Meek TD, Johnson KA, Villafranca JJ. Escherichia coli glutamine synthetase. Determination of rate-limiting steps by rapid-quench and isotope partitioning experiments Derivation of equations for isotope partitioning experiments for a terreactant enzyme. Biochemistry. 1982;21:2158–2167. doi: 10.1021/bi00538a027. [DOI] [PubMed] [Google Scholar]
- 24.Meek TD, Villafranca JJ. Kinetic mechanism of Escherichia coli glutamine synthetase. Biochemistry. 1980;19:5513–5519. doi: 10.1021/bi00565a008. [DOI] [PubMed] [Google Scholar]
- 25.Pearson JT, Dabrowski MJ, Kung I, Atkins WM. The central loop of Escherichia coli glutamine synthetase is flexible and functionally passive. Arch Biochem Biophys. 2005;436:397–405. doi: 10.1016/j.abb.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 26.Alibhai M, Villafranca JJ. Kinetic and mutagenic studies of the role of the active site residues Asp-50 and Glu-327 of Escherichia coli glutamine synthetase. Biochemistry. 1994;33:682–686. doi: 10.1021/bi00169a008. [DOI] [PubMed] [Google Scholar]
- 27.Liaw SH, Eisenberg D. Structural model for the reaction mechanism of glutamine synthetase, based on five crystal structures of enzyme-substrate complexes. Biochemistry. 1994;33:675–681. doi: 10.1021/bi00169a007. [DOI] [PubMed] [Google Scholar]
- 28.Trenkamp S, Eckes P, Busch M, Fernie AR. Temporally resolved GC-MS-based metabolic profiling of herbicide treated plants treated reveals that changes in polar primary metabolites alone can distinguish herbicides of differing mode of action. Metabolomics. 2009;5:277–291. doi: 10.1007/s11306-008-0149-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, Miletic H, Sakariassen PO, Weinstock A, Wagner A, Lindsay SL, Hock AK, Barnett SC, Ruppin E, Morkve SH, Lund-Johansen M, Chalmers AJ, Bjerkvig R, Niclou SP, Gottlieb E. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol. 2015;17:1556– 1568. doi: 10.1038/ncb3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donn G, Kocher H. Inhibitors of Glutamine Synthetase. In: Boger P, Wakabayashi K, Hirai K, editors. Herbicide Classes in Development: Mode of Action, Targets, Genetic Engineering, Chemistry. Springer Berlin Heidelberg; Berlin, Heidelberg: 2002. pp. 87–101. [Google Scholar]
- 31.Mowbray SL, Kathiravan MK, Pandey AA, Odell LR. Inhibition of glutamine synthetase: a potential drug target in Mycobacterium tuberculosis. Molecules. 2014;19:13161–13176. doi: 10.3390/molecules190913161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeitner TM, Cooper AJ. Inhibition of human glutamine synthetase by L-methionine- S,R-sulfoximine-relevance to the treatment of neurological diseases. Metab Brain Dis. 2014;29:983–989. doi: 10.1007/s11011-013-9439-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abell LM, Villafranca JJ. Investigation of the mechanism of phosphinothricin inactivation of Escherichia coli glutamine synthetase using rapid quench kinetic techniques. Biochemistry. 1991;30:6135–6141. doi: 10.1021/bi00239a008. [DOI] [PubMed] [Google Scholar]
- 34.Shrake A, Ginsburg A, Wedler FC, Sugiyama Y. On the binding of L-S- and L-R-diastereoisomers of the substrate analog L-methionine sulfoximine to glutamine synthetase from Escherichia coli. J Biol Chem. 1982;257:8238–8243. [PubMed] [Google Scholar]
- 35.Ronzio RA, Rowe WB, Meister A. Mechanism of inhibition of glutamine synthetase by methionine sulfoximine. Biochemistry. 1969;8:1066–1075. doi: 10.1021/bi00831a038. [DOI] [PubMed] [Google Scholar]
- 36.Schramm VL. Enzymatic transition states, transition-state analogs, dynamics, thermodynamics, and lifetimes. Annu Rev Biochem. 2011;80:703–732. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maurizi MR, Ginsburg A. Reactivation of glutamine synthetase from Escherichia coli after auto-inactivation with L-methionine-S-sulfoximine, ATP, and Mn2+ J Biol Chem. 1982;257:4271–4278. [PubMed] [Google Scholar]
- 38.Horecker B, Kornberg A. The extinction coefficients of the reduced band of pyridine nucleotides. J Biol Chem. 1948;175:385–390. [PubMed] [Google Scholar]
- 39.Kitz R, Wilson IB. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J Biol Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- 40.CLSI. CLSI document M07-A9. Clinical and Laboratory Standards Institute; Wayne, PA: 2012. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard-Ninth Edition. [Google Scholar]
- 41.Studelska DR, McDowell LM, Adler M, O’Connor RD, Mehta AK, Guilford WJ, Dallas JL, Arnaiz D, Light DR, Schaefer J. Conformation of a bound inhibitor of blood coagulant factor Xa. Biochemistry. 2003;42:7942–7949. doi: 10.1021/bi027369g. [DOI] [PubMed] [Google Scholar]
- 42.Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminf. 2012;4:17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woolley DW, Pringle RB, Braun AC. Isolation of the phytopathogenic toxin of Pseudomonas tabaci, an antagonist of methionine. J Biol Chem. 1952;197:409– 417. [PubMed] [Google Scholar]
- 46.Levi C, Durbin RD. The isolation and properties of a tabtoxin-hydrolysing aminopeptidase from the periplasm of Pseudomonas syringae pv. tabaci. Physiol Mol Plant Pathol. 1986;28:345–352. [Google Scholar]
- 47.Thomas MD, Langston-Unkefer PJ, Uchytil TF, Durbin RD. Inhibition of glutamine synthetase from pea by tabtoxinine-β-lactam. Plant Physiol. 1983;71:912–915. doi: 10.1104/pp.71.4.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas MD, Durbin RD. Glutamine synthetase from Pseudomonas syringae pv. tabaci: Properties and inhibition by tabtoxinine-β-lactam. J Gen Microbiol. 1985;131:1061–1067. [Google Scholar]
- 49.Kinscherf TG, Coleman RH, Barta TM, Willis DK. Cloning and expression of the tabtoxin biosynthetic region from Pseudomonas syringae. J Bacteriol. 1991;173:4124–4132. doi: 10.1128/jb.173.13.4124-4132.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kinscherf TG, Willis DK. The biosynthetic gene cluster for the β-lactam antibiotic tabtoxin in Pseudomonas syringae. J Antibiot. 2005;58:817–821. doi: 10.1038/ja.2005.109. [DOI] [PubMed] [Google Scholar]
- 51.Arai T, Arimura Y, Ishikura S, Kino K. L-amino acid ligase from Pseudomonas syringae producing tabtoxin can be used for enzymatic synthesis of various functional peptides. Appl Environ Microbiol. 2013;79:5023–5029. doi: 10.1128/AEM.01003-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grassl M. In: Methods of Enzymatic Analysis. 2. Bergmeyer HU, editor. Academic Press, Inc; New York, NY: 1974. [Google Scholar]
- 53.Sinden SL, Durbin RD, Uchytil TF, Lamar C. The production of convulsions by an exotoxin from Pseudomonas tabaci. Toxicol Appl Pharmacol. 1969;14:82–88. doi: 10.1016/0041-008x(69)90167-7. [DOI] [PubMed] [Google Scholar]
- 54.Rhee SG, Chock PB, Stadtman ER. Advances in Enzymology and Related Areas of Molecular Biology. John Wiley & Sons, Inc; 2006. Regulation of Escherichia coli Glutamine Synthetase; pp. 37–92. [DOI] [PubMed] [Google Scholar]
- 55.Jiang P, Mayo AE, Ninfa AJ. Escherichia coli glutamine synthetase adenylyltransferase (ATase, EC 2.7.7.49): Kinetic characterization of regulation by PII, PII-UMP, glutamine, and α-ketoglutarate. Biochemistry. 2007;46:4133–4146. doi: 10.1021/bi0620510. [DOI] [PubMed] [Google Scholar]
- 56.Luo S, Kim G, Levine RL. Mutation of the adenylylated tyrosine of glutamine synthetase alters its catalytic properties. Biochemistry. 2005;44:9441–9446. doi: 10.1021/bi050554k. [DOI] [PubMed] [Google Scholar]
- 57.Shapiro BM, Stadtman ER. [130] Glutamine synthetase (Escherichia coli) Methods Enzymol. 1970;17:910–922. doi: 10.1016/s0076-6879(85)13032-6. [DOI] [PubMed] [Google Scholar]
- 58.Woolfolk CA, Shapiro B, Stadtman ER. Regulation of glutamine synthetase. Arch Biochem Biophys. 1966;116:177–192. doi: 10.1016/0003-9861(66)90026-9. [DOI] [PubMed] [Google Scholar]
- 59.Listrom DC, Morizono H, Rajagopal SB, McCann TM, Tuchman M, Allewell MN. Expression, purification, and characterization of recombinant human glutamine synthetase. Biochem J. 1997;328:159–163. doi: 10.1042/bj3280159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iqbal K, Wu C. Glutamine synthetase. 8 Purification and some properties of the enzyme from rat liver. Enzyme. 1971;12:553–560. doi: 10.1159/000459585. [DOI] [PubMed] [Google Scholar]
- 61.Tiemeier DC, Milman G. Chinese Hamster Liver Glutamine Synthetase: Purification, physical and biochemical properteis. J Biol Chem. 1972;247:2272–2277. [PubMed] [Google Scholar]
- 62.Kumar V, Yadav S, Soumya N, Kumar R, Babu NK, Singh S. Biochemical and inhibition studies of glutamine synthetase from Leishmania donovani. Microb Pathog. 2017;107:164–174. doi: 10.1016/j.micpath.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 63.Dar AC, Das TK, Shokat KM, Cagan RL. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature. 2012;486:80–84. doi: 10.1038/nature11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nunn PB, Bell EA, Watson AA, Nash RJ. Toxicity of non-protein amino acids to humans and domestic animals. Nat Prod Commun. 2010;5:485–504. [PubMed] [Google Scholar]
- 65.Logusch EW, Walker DM, McDonald JF, Franz JE, Villafranca JJ, DiIanni CL, Colanduoni JA, Li B, Schineller JB. Inhibition of Escherichia coli glutamine synthetase by α- and γ-substituted phosphinothricins. Biochemistry. 1990;29:366–372. doi: 10.1021/bi00454a009. [DOI] [PubMed] [Google Scholar]
- 66.Walker DM, McDonald JF, Logusch EW. Synthesis of D,L-γ-hydroxyphosphinothricin, a potent new inhibitor of glutamine synthetase. J Chem Soc, Chem Commun. 1987:1710–1711. [Google Scholar]
- 67.Murima P, McKinney JD, Pethe K. Targeting bacterial central metabolism for drug development. Chem Biol. 2014;21:1423–1432. doi: 10.1016/j.chembiol.2014.08.020. [DOI] [PubMed] [Google Scholar]
- 68.Zlitni S, Ferruccio LF, Brown ED. Metabolic suppression identifies new antibacterial inhibitors under nutrient limitation. Nat Chem Biol. 2013;9:796–804. doi: 10.1038/nchembio.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meister A. Glutathione biosynthesis and its inhibition. Methods Enzymol. 1995;252:26–30. doi: 10.1016/0076-6879(95)52005-8. [DOI] [PubMed] [Google Scholar]
- 70.Bush DR, Langston-Unkefer PJ. Tabtoxinine-β-lactam transport into cultured corn cells: Uptake via an amino acid transport system. Plant Physiol. 1987;85:845–849. doi: 10.1104/pp.85.3.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smith MW, Tyreman DR, Payne GM, Marshall NJ, Payne JW. Substrate specificity of the periplasmic dipeptide-binding protein from Escherichia coli: experimental basis for the design of peptide prodrugs. Microbiology. 1999;145:2891–2901. doi: 10.1099/00221287-145-10-2891. [DOI] [PubMed] [Google Scholar]
- 72.Zboinska E, Lejczak B, Kafarski P. Organophosphonate utilization by the wild-type strain of Pseudomonas fluorescens. Appl Environ Microbiol. 1992;58:2993–2999. doi: 10.1128/aem.58.9.2993-2999.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Circello BT, Miller CG, Lee JH, van der Donk WA, Metcalf WW. The antibiotic dehydrophos is converted to a toxic pyruvate analog by peptide bond cleavage in Salmonella enterica. Antimicrob Agents Chemother. 2011;55:3357–3362. doi: 10.1128/AAC.01483-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tullius MV, Harth G, Horwitz MA. Glutamine synthetase GlnA1 is essential for growth of Mycobacterium tuberculosis in human THP-1 macrophages and guinea pigs. Infect Immun. 2003;71:3927–3936. doi: 10.1128/IAI.71.7.3927-3936.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bayer E, Gugel KH, Hagele K, Hagenmaier H, Jessipow S, Konig WA, Zahner H. Stoffwechselprodukte von mikroorganismen. 98 Mitteilung Phosphinothricin und phosphinothricyl-alanyl-alanin. Helv Chim Acta. 1972;55:224–239. doi: 10.1002/hlca.19720550126. [DOI] [PubMed] [Google Scholar]
- 76.Carroll P, Waddell SJ, Butcher PD, Parish T. Methionine Sulfoximine resistance in Mycobacterium tuberculosis is due to a single nucleotide deletion resulting in increased expression of the major glutamine synthetase, GlnA1. Microb Drug Resist. 2011;17:351–355. doi: 10.1089/mdr.2010.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Behrmann I, Hillemann D, Puhler A, Strauch E, Wohlleben W. Overexpression of a Streptomyces viridochromogenes gene (glnII) encoding a glutamine synthetase similar to those of eucaryotes confers resistance against the antibiotic phosphinothricyl-alanyl-alanine. J Bacteriol. 1990;172:5326–5334. doi: 10.1128/jb.172.9.5326-5334.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schreier HJ, Rostkowski CA, Kellner EM. Altered regulation of the glnRA operon in a Bacillus subtilis mutant that produces methionine sulfoximine-tolerant glutamine synthetase. J Bacteriol. 1993;175:892–897. doi: 10.1128/jb.175.3.892-897.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miller ES, Brenchley JE. L-Methionine SR-sulfoximine-resistant glutamine synthetase from mutants of Salmonella typhimurium. J Biol Chem. 1981;256:11307–11312. [PubMed] [Google Scholar]
- 80.Pornprom T, Prodmatee N, Chatchawankanphanich O. Glutamine synthetase mutation conferring target-site-based resistance to glufosinate in soybean cell selections. Pest Manage Sci. 2009;65:216–222. doi: 10.1002/ps.1671. [DOI] [PubMed] [Google Scholar]
- 81.Tian YS, Xu J, Zhao W, Xing XJ, Fu XY, Peng RH, Yao QH. Identification of a phosphinothricin-resistant mutant of rice glutamine synthetase using DNA shuffling. Sci Rep. 2015;5:15495. doi: 10.1038/srep15495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang S, Han Y, Kumar A, Gao H, Liu Z, Hu N. Characterization of an L-phosphinothricin resistant glutamine synthetase from Exiguobacterium sp. and its improvement. Appl Microbiol Biotechnol. 2017;101:3653–3661. doi: 10.1007/s00253-017-8103-1. [DOI] [PubMed] [Google Scholar]
- 83.Jiang Y, Sun C, Ding X, Yuan D, Chen K, Gao B, Chen Y, Sun A. Simultaneous determination of adenine nucleotides, creatine phosphate and creatine in rat liver by high performance liquid chromatography–electrospray ionization-tandem mass spectrometry. J Pharm Biomed Anal. 2012;66:258–263. doi: 10.1016/j.jpba.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 84.Colanduoni JA, Villafranca JJ. Inhibition of Escherichia coli glutamine synthetase by phosphinothricin. Bioorg Chem. 1986;14:163–169. [Google Scholar]
- 85.Schaefer J. Development of REDOR rotational-echo double-resonance NMR” by Terry Gullion and Jacob Schaefer. [J. Magn” Reson 81 (1989) 196–200. J Mag Res. 2011;213:421–422. doi: 10.1016/j.jmr.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 86.Gullion T, Schaefer J. Rotational-echo double-resonance NMR. J Mag Res. 1989;81:196–200. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 87.Gullion T, Schaefer J. Detection of weak heteronuclear dipolar coupling by rotational-echo double-resonance nuclear magnetic resonance. Adv Magn Reson. 1989;13:57–83. [Google Scholar]
- 88.Pandya BV, Budzynski AZ. Anticoagulant proteases from western diamondback rattlesnake (Crotalus atrox) venom. Biochemistry. 1984;23:460–470. doi: 10.1021/bi00298a010. [DOI] [PubMed] [Google Scholar]
- 89.Cegelski L, Steuber D, Mehta AK, Kulp DW, Axelsen PH, Schaefer J. Conformational and quantitative characterization of oritavancin–peptidoglycan complexes in whole cells of Staphylococcus aureus by in vivo 13C and 15N labeling. J Mol Biol. 2006;357:1253–1262. doi: 10.1016/j.jmb.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 90.Wedler FC, Sugiyama Y, Fisher KE. Catalytic cooperativity and subunit interactions in E coli glutamine synthetase binding and kinetics with methionine sulfoximine and related inhibitors. Biochemistry. 1982;21:2168–2177. doi: 10.1021/bi00538a028. [DOI] [PubMed] [Google Scholar]
- 91.Taylor ZW, Brown HA, Narindoshvili T, Wenzel CQ, Szymanski CM, Holden HM, Raushel FM. Discovery of a glutamine kinase required for the biosynthesis of the O-methyl phosphoramidate modifications found in the capsular polysaccharides of Campylobacter jejuni. J Am Chem Soc. 2017;139:9463–9466. doi: 10.1021/jacs.7b04824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kirby AJ, Komarov IV, Feeder N. Synthesis, structure and reactions of the most twisted amide. J Chem Soc, Perkin Trans. 2001;2:522–529. [Google Scholar]
- 93.Kirby AJ, Komarov IV, Feeder N. Spontaneous, millisecond formation of a twisted amide from the amino acid, and the crystal structure of a tetrahedral intermediate. J Am Chem Soc. 1998;120:7101–7102. [Google Scholar]
- 94.Greenberg A, Moore DT, DuBois TD. Small and medium-sized bridgehead bicyclic lactams: A systematic ab initio molecular orbital study. J Am Chem Soc. 1996;118:8658–8668. [Google Scholar]
- 95.Logusch EW, Walker DM, McDonald JF, Leo GC, Franz JE. Synthesis of α- and γ-alkyl-substituted phosphinothricins: potent new inhibitors of glutamine synthetase. J Org Chem. 1988;53:4069–4074. [Google Scholar]
- 96.Johnson CR, Boettcher BR, Cherpeck RE, Dolson MG. Design and synthesis of potent inhibitors of glutamine synthetase: 1. Cyclic analogs of phosphonothricin. Bioorg Chem. 1990;18:154–159. [Google Scholar]
- 97.Berlicki Ł, Obojska A, Forlani G, Kafarski P. Design, synthesis, and activity of analogues of phosphinothricin as inhibitors of glutamine synthetase. J Med Chem. 2005;48:6340–6349. doi: 10.1021/jm050474e. [DOI] [PubMed] [Google Scholar]
- 98.Silva AM, Kong X, Hider RC. Determination of the pKa value of the hydroxyl group in the alpha-hydroxycarboxylates citrate, malate and lactate by 13C NMR: implications for metal coordination in biological systems. Biometals. 2009;22:771–778. doi: 10.1007/s10534-009-9224-5. [DOI] [PubMed] [Google Scholar]
- 99.Sirimulla S, Bailey JB, Vegesna R, Narayan M. Halogen interactions in protein–ligand complexes: Implications of halogen bonding for rational drug design. J Chem Inf Model. 2013;53:2781–2791. doi: 10.1021/ci400257k. [DOI] [PubMed] [Google Scholar]
- 100.Parsons WH, Patchett AA, Bull HG, Schoen WR, Taub D, Davidson J, Combs PL, Springer JP, Gadebusch H, Weissberger B, et al. Phosphinic acid inhibitors of D-alanyl-D-alanine ligase. J Med Chem. 1988;31:1772–1778. doi: 10.1021/jm00117a017. [DOI] [PubMed] [Google Scholar]
- 101.Motwani HV, De Rosa M, Odell LR, Hallberg A, Larhed M. Aspartic protease inhibitors containing tertiary alcohol transition-state mimics. Eur J Med Chem. 2015;90:462–490. doi: 10.1016/j.ejmech.2014.11.036. [DOI] [PubMed] [Google Scholar]
- 102.Greenlee WJ, Springer JP, Patchett AA. Synthesis of an analog of tabtoxinine as a potential inhibitor of D-alanine:D-alanine ligase (ADP forming) J Med Chem. 1989;32:165–170. doi: 10.1021/jm00121a030. [DOI] [PubMed] [Google Scholar]
- 103.Dolle RE, McNair D, Hughes MJ, Kruse LI, Eggelston D, Saxty BA, Wells TN, Groot PH. ATP-citrate lyase as a target for hypolipidemic intervention. Sulfoximine and 3-hydroxy-beta-lactam containing analogues of citric acid as potential tight-binding inhibitors. J Med Chem. 1992;35:4875–4884. doi: 10.1021/jm00104a014. [DOI] [PubMed] [Google Scholar]
- 104.Karinch AM, Pan M, Lin CM, Strange R, Souba WW. Glutamine metabolism in sepsis and infection. J Nutr. 2001;131:2535S–2538S. doi: 10.1093/jn/131.9.2535S. [DOI] [PubMed] [Google Scholar]
- 105.Price DRG, Feng H, Baker JD, Bavan S, Luetje CW, Wilson ACC. Aphid amino acid transporter regulates glutamine supply to intracellular bacterial symbionts. Proc Natl Acad Sci U S A. 2014;111:320–325. doi: 10.1073/pnas.1306068111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Harth G, Horwitz MA. Inhibition of Mycobacterium tuberculosis glutamine synthetase as a novel antibiotic strategy against tuberculosis: Demonstration of efficacy in vivo. Infect Immun. 2003;71:456–464. doi: 10.1128/IAI.71.1.456-464.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stranden AM, Roos M, Berger-Bachi B. Glutamine synthetase and heteroresistance in methicillin-resistant Staphylococcus aureus. Microb Drug Resist. 1996;2:201–207. doi: 10.1089/mdr.1996.2.201. [DOI] [PubMed] [Google Scholar]
- 108.Liu X, Pai PJ, Zhang W, Hu Y, Dong X, Qian PY, Chen D, Lam H. Proteomic response of methicillin-resistant S. aureus to a synergistic antibacterial drug combination: a novel erythromycin derivative and oxacillin. Sci Rep. 2016;6:19841. doi: 10.1038/srep19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.