

Abstract
Purpose
Although the role of the angiotensin II type 2 (AT2) receptor in acute lung injury is not yet completely understood, a protective role of this receptor subtype has been suggested. We hypothesized that, in a rodent model of acute lung injury, stimulation of the AT2 receptor with the direct agonist Compound 21 (C21) might have a beneficial effect on pulmonary inflammation and might improve pulmonary gas exchange.
Materials and methods
Male adult rats were divided into a treatment group that received pulmonary lavage followed by mechanical ventilation (LAV, n=9), a group receiving pulmonary lavage, mechanical ventilation, and direct stimulation of the AT2 receptor with C21 (LAV+C21, n=9), and a control group that received mechanical ventilation only (control, n=9). Arterial blood gas analysis was performed every 30 min throughout the 240-min observation period. Lung tissue and plasma samples were obtained at 240 min after the start of mechanical ventilation. Protein content and surface activity of bronchoalveolar lavage fluid were assessed and the wet/dry-weight ratio of lungs was determined. Transcriptional and translational regulation of pro- and antiinflammatory cytokines IL-1β, tumor necrosis factor-alpha, IL-6, IL-10, and IL-4 was determined in lungs and in plasma.
Results
Pulmonary lavage led to a significant impairment of gas exchange, the formation of lung edema, and the induction of pulmonary inflammation. Protein content of lavage fluid was increased and contained washed-out surfactant. Direct AT2 receptor stimulation with C21 led to a significant inhibition of tumor necrosis factor-alpha and IL-6 expressions in the lungs, whereas the expressions of IL-1, IL-10, and IL-4 remained unchanged. During the 240-min observation period, AT2 receptor stimulation did not improve pulmonary gas exchange or lung edema.
Conclusion
In this rodent model of acute lung injury after repeated pulmonary lavage, AT2 receptor stimulation attenuates pulmonary inflammation but does not improve gas exchange.
Keywords: AT2 receptor, lung failure, ARDS, acute lung injury, Compound 21 (C21)
Introduction
Treatment of patients with the acute respiratory distress syndrome (ARDS) remains a challenge. Mortality rates are still high, ranging from 29% to (up to) 60%.1 Despite major advances in the understanding of the disease, acute lung failure is still a leading cause of morbidity and mortality in intensive care units worldwide.2,3
The main pathophysiological feature of ARDS is diffuse damage of the alveolar-epithelial barrier and the subsequent formation of a protein-rich alveolar edema.4,5 The accompanying inflammatory response is a hallmark and mainly characterized by a massive release of proinflammatory cytokines, such as IL-6 or tumor necrosis factor-alpha (TNF-α).5–9 Clinically, the leading symptom is a rapid onset of arterial hypoxemia and/or hypercarbia which, in severe cases, can be life-threatening. Successful treatment strategies include mechanical ventilation with low tidal volumes, fluid-restrictive resuscitation strategies, and prone positioning of the patients.10–12 However, to date, no specific pharmacological treatment is available that targets the underlying pulmonary inflammation during the development or the clinical course of ARDS.
Animal studies have shown that pharmacological inhibition of pulmonary inflammation can lead to beneficial effects in acute lung failure. In different experimental models, the attenuation of pulmonary inflammation mitigated the alveolar edema and subsequently improved arterial oxygenation.13,14 Also, in a rodent model, inhalation of the antiinflammatory cytokine IL-10 not only lowered pulmonary inflammation, but also attenuated lung damage and reduced mortality.15 In humans, however, none of the pharmacological interventions tested so far has demonstrated any beneficial effects in ARDS. Therapies found to be ineffective include inhalation of vasodilators, N-acetyl-cysteine, β2 agonists, surfactant, statins, and others.16–20
The renin–angiotensin–aldosterone system (RAS) plays a major role in human physiology and is involved not only in cardiovascular disease, but also in the regulation of inflammation.21 The two main receptors of the RAS, namely the AT1 receptor and the AT2 receptor, play an intricate and dual role in inflammation. The AT1 receptor contributes to tissue damage caused by uncontrolled inflammation, whereas the AT2 receptor is thought to have beneficial and protective effects.21,22 Overall, the AT2 receptor is regarded as a counterpart to the AT1 receptor, antagonizing the effects of AT1 receptor signaling. There is evidence that the angiotensin-AT2-receptor-axis acts as an endogenous, tissue-protective system involved in the downregulation of inflammation and the promotion of tissue repair.23 Studies with the nonpeptide, specific, and selective AT2 receptor agonist Compound 21 (C21) demonstrated a marked inhibition of inflammatory signaling pathways by direct AT2 receptor activation.24 Based on its antiinflammatory properties, C21 could evoke beneficial effects in animal models of myocardial infarction and stroke.25,26 In a model of pulmonary fibrosis, direct AT2 receptor stimulation significantly attenuated pulmonary and myocardial fibrosis.27 Further, in models of acute lung injury, the survival of mice lacking the AT2 receptor (i.e., AT2 receptor -/y knock-out) was significantly worse.28,29 All these studies provide indirect evidence for the potentially beneficial effects exerted by the AT2 receptor.
Although AT2 receptor signaling in acute lung injury is not yet fully elucidated, it is suggested that this receptor subtype has a protective role. To the best of our knowledge, no study has used the AT2 receptor agonist C21 in experimental acute lung injury. Based on the data described above, we hypothesized that direct stimulation of the AT2 receptor with C21 could attenuate pulmonary inflammation and might improve pulmonary edema and gas exchange in an experimental model of acute lung injury.
Materials and methods
Animal preparation and acute lung injury with repeated pulmonary lavage
This animal study was approved by the Committee on Use and Care of Animals at the Charité-Universitätsmedizin Berlin, Germany, by the state animal committee (approval No. G0076/11; LAGeSo, Berlin, Germany). All procedures were conducted in accordance with institutional guidelines.
Adult male Sprague-Dawley rats (300–350 g) underwent general anesthesia with isoflurane and pentobarbital sodium (60 mg/kg/h, intraperitoneally [i.p.]); muscle relaxation was attained with pancuronium bromide (2 mg/kg/h, i.p.). Animals were placed supine on a heating pad. A tracheotomy was performed and a catheter was inserted into the left carotid artery. Rats were ventilated with an Evita XL mechanical ventilator (Drägerwerk, Lübeck, Germany) with the following settings: pressure-controlled ventilation; fraction of inspired oxygen (FiO2): 1,0; ventilation rate: 30/min; peak inspiratory pressure (PIP): 14 cm H2O; positive end-expiratory pressure (PEEP): 2 cm H2O; and inspiratory/expiratory ratio: 1:2. After arterial partial pressure of oxygen reached a steady state (PaO2 >500 mmHg), respiratory failure was induced by repeated bronchopulmonary lavage with warm saline (37°C; 30 mL/kg) until a PaO2 <150 mmHg was reached, as described previously.30 Rats that were ventilated without pulmonary lavage served as controls. A recruitment maneuver was conducted after the cycles of lavage by increasing the PIP to 34 cm H2O for three breathing cycles; this same recruitment procedure was also performed in the controls. Afterward, in all groups, ventilator settings were set to PIP=26 mmHg and PEEP=6 mmHg and remained unchanged throughout the entire observation period of 240 min.
On completion of the experiments, animals were sacrificed by an overdose of pentobarbital. Lung samples were harvested, snap-frozen in liquid nitrogen, and stored at −80°C until further investigation. Whole blood was drawn into tubes containing EDTA and centrifuged at 1000×g for 2 min at 4°C. The plasma was frozen at −80°C until further processing.
C21 treatment
The selective AT2-receptor agonist C21 (a kind gift from Vicore Pharma, Mölndal, Sweden) was dissolved in sterile-filtered phosphate-buffered saline and administered intraperitoneally at a dosage of 0.03 mg/kg body weight 1 h before induction of lung failure.
In vivo measurements
Arterial blood samples were taken before lavage and every 30 min until 240 min posttreatment for analysis of PaO2, PaCO2, pH, and base excess (ABL 800 Basic; Radiometer, Copenhagen, Denmark). Mean arterial blood pressure and heart rate were continuously recorded with a digital monitoring system (Picco; PULSION Medical Systems, Feldkirchen, Germany).
Bronchoalveolar lavage fluid measurements
Protein concentrations of the bronchoalveolar lavage fluid (BALF) samples were determined using the bicinchoninic acid assay (Pierce, Rockford, IL, USA). Also, surface activity of recovered BALF was assessed using a modified Wilhelmy balance (E. Biegler GmbH, Mauerbach, Austria). In this method, a tight-fitting teflon barrier reduces the surface area of a teflon trough from 100% to 20% at a cycle speed of 0.33/min. Saline is used as subphase and is kept at 37°C. The force on a platinum slide (1×1 cm), dipped into the subphase, is measured by a force transducer and expressed as surface tension. Maximal surface tension is measured at 100% surface area and minimal surface tension at 80% surface compression and expressed as millinewtons per meter (mN/m). Surface tension characteristics of a sample are measured after application on the surface of the saline-filled trough. In the present study, 200 µL of recovered lavage fluid was applied on the surface of the trough; maximal and minimal surface tensions were measured after three cycles.
Wet/dry ratio
To evaluate the formation of lung edema in the different treatment groups, the wet/dry-weight ratio of lungs was assessed. Lungs were extracted and weighted, and the wet/dry ratio was calculated using the method described by Peterson et al.31
Quantitative real-time polymerase chain reaction
Total RNA was isolated from left lungs using the Absolutely RNA kit (Stratagene, La Jolla, CA, USA) according to the manufacturer’s recommendations including a DNase digest. cDNA synthesis was performed using random hexamer primers and M-MLV reverse transcriptase (Promega, Mannheim, Germany); no template controls and reactions without addition of reverse transcriptase (RT-) served as negative controls. cDNA was quantified by real-time polymerase chain reaction (RT-PCR) Master Mix (Applied Biosystems, Darmstadt, Germany) using FAM-5′⁄TAMRA-3′ labeled probes for IL-1β, TNF-α, IL-10, IL-6, IL-4, IFN-γ, TGF-β, heme oxygenase-1, and GAPDH (Metabion, Munich, Germany). Data represent the mean expression level±SD, standardized to GAPDH expression, calculated according to the 2−ΔΔCT method of at least three independent measurements per cDNA (technical triplicates). The sequences of primers used are listed in Table 1.
Table 1.
Sequences of oligonucleotides used for qRT-PCR
| Gene | Forward primer 5′–3′ reverse primer 5′–3′ | Probe 5′ 6-FAM–TAMRA-3′ |
|---|---|---|
| IL-1b | aacaaaaatgcctcgtgctgtct tgttggcttatgttctgtccattg |
acc cat gtgagctgaaagctctccacc |
| IL-6 | aactccatctgcccttcagga ggcagtggctgtcaacaa cat |
tttctctccgcaagagacttc cag cca |
| IL-10 | gaagaccctctg gat acagctgc tgctcc act gccttgctttt |
cgctgt cat cgatttctc ccc tgtga |
| TNF-α | tcg agt gac aag ccc gta gc ctcagccactccagctgctc |
cgtcgtagcaaaccaccaagcaga |
| IL-4 | cggtga act gag gaa act ctg tag a tcagtgttgtgagcgtgg act c |
tcagtgttgtgagcgtgg act c |
| TGF-β | ccc tgc ccc tacatttgg a ccc tgc ccc tacatttgg a |
cacacagta cag caaggtccttgccct |
| GAPDH | ccc taaggccaaccgtgaaaa gat g tcaggt gag ccc cag cct |
ccc taaggccaaccgtgaaaa gat g |
Abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; qRT-PCR, quantitative real-time reverse transcriptase polymerase chain reaction; TGF-β, transforming growth factor-beta; TNF-α, tumor necrosis factor-alpha.
Determination of cytokines
After the experimental procedure, lungs were removed (without prior lavage). Therefore, the samples contained both cytokines of the alveolar compartment and cytokines of the interstitial tissue. For measurement of cytokine levels, right lungs were thawed, minced, and homogenized in cold lysis buffer containing 10 mM Tris/HCl, pH 7.5, 300 mM NaCl, 1% Triton X-100, 2 mM MgCl2, 5 M EDTA, and the protease inhibitor cocktail, CompleteMini (Roche Diagnostics, Mannheim, Germany). Homogenates were centrifuged at 6000×g for 10 min at 4°C. For analysis of cytokine levels in plasma, respective blood samples were centrifuged at 1000×g for 2 min at 4°C and plasma was separated. Protein concentration was determined using the bicinchoninic acid assay (Pierce, Rockford, IL, USA). The protein levels of IL-1β, TNF-α, and IL-10 were evaluated using the cytometric bead array mouse/rat inflammation kit and fluorescence-activated cell sorting analysis according to the manufacturer’s instructions (BD Biosciences, San Diego, CA, USA). A total of 50 µL of lung protein lysate or plasma were used per measurement. The sensitivity of this system was between 1.9 and 7.2 pg per mL for each of the cytokines.
Immunoblotting
Sodium dodecyl sulfate polyacrylamide gel electrophoresis, electrotransfer to a nitrocellulose membrane, and antibody detection were carried out as described previously.32 Polyclonal rabbit anti-rat IL-6, rabbit anti-rat AT2 receptor, rabbit anti-rat TNF antibody (Cell Signaling, Cambridge, UK), and rabbit anti-rat β-actin antibody (Sigma-Aldrich, St. Louis, MO, USA) were used. Depicted blots represent a Western blot series (technical triplicates; n=3). Quantification of respective band density was performed using the image analysis program ImageJ 1.42q (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data in diagrams are presented as mean±standard error of the mean (SEM). For nonparametric samples, statistical analysis was performed with the Mann–Whitney U-test. All other statistical analyses were performed with IBM SPSS Statistics version 20 (SPSS, Chicago, IL, USA) and GraphPad PRISM version 7 (San Diego, CA, USA). Differences were considered significant at p<0.05.
Results
Pulmonary gas exchange and systemic hemodynamics
PaO2, PaCO2, heart rate, and mean blood pressure at baseline and during the 240-min observation period are presented in Figure 1. Initially, PaO2 and PaCO2 levels showed no significant differences between the control group, the lavaged group, and the lavaged group receiving C21. Immediately after pulmonary lavage, in all lavaged groups, the arterial oxygen levels decreased significantly and did not fully recover throughout the observation period (control vs. LAV, p<0.001; control vs. LAV+C21; p<0.001). Also, after lavage, PaCO2 levels increased significantly and remained highly elevated, indicating severe impairment and dysfunction of gas exchange (control vs. LAV, p<0.0001; control vs. LAV+C21; p<0.0001; Figure 1A and B). Treatment with the selective AT2 receptor agonist C21 had no significant effect on PaO2 and PaCO2 levels during the 240-min observation period. Further, parameters of systemic hemodynamics (e.g., heart rate and mean arterial blood pressure) showed no significant difference between the groups (Figure 1C and D). Repeated pulmonary lavage led to the formation of lung edema, as shown by a significant increase in the wet/dry-weight ratio of the lungs in lavaged animals (Figure 2). In this model of acute lung failure, treatment with C21 had no significant effect on the wet/dry ratio of the lungs.
Figure 1.

(A) Arterial PO2 (mmHg), (B) arterial PCO2 (mmHg), (C) heart rate (min−1), and (D) mean arterial pressure (mmHg) at baseline (0 min), after pulmonary lavage (5 min), and over the entire observation period (240 min) in rats with mechanical ventilation only (control; n=9), surfactant washout by repeated pulmonary lavage and mechanical ventilation (LAV; n=9), or surfactant washout by repeated pulmonary lavage, mechanical ventilation, and direct stimulation of the AT2 receptor with C21 (0.03 mg/kg body weight; LAV+C21; n=9).
Notes: Data are presented as mean±SD; multivariate nonparametric analysis for longitudinal data over the course of time (two-way MANOVA), p>0.05 for comparison between the LAV and LAV+C21 groups.
Abbreviations: AT2, angiotensin II type 2; C21, Compound 21; MANOVA, multivariate analysis of variance; MAP, mean arterial pressure.
Figure 2.
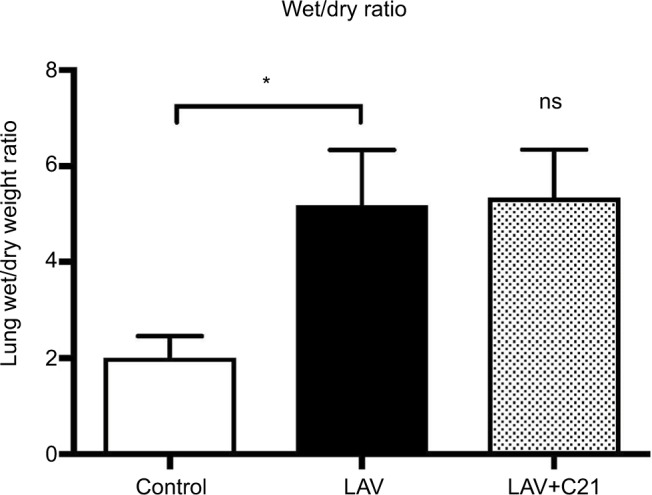
Wet/dry-weight ratio of lungs of rats with mechanical ventilation only (control; n=9), repeated pulmonary lavage and mechanical ventilation (LAV; n=9), and repeated pulmonary lavage, mechanical ventilation, and direct stimulation of the AT2 receptor with C21 (0.03 mg/kg body weight; LAV+C21; n=9).
Notes: Data are presented as mean±SD, Mann–Whitney U-test (n=9). n.s., not significantly different compared with lavaged group. *p<0.05.
Abbreviations: AT2, angiotensin II type 2; C21, Compound 21.
Bronchoalveolar lavage fluid
Protein content of the recovered BALF was significantly increased in all consecutive lavage samples as compared with controls, indicating increased permeability of the alveolar-capillary barrier caused by the lavage procedure (Figure 3A). With increasing numbers of lavage cycles, the protein content of recovered BALF increased significantly. Assessment of the surface activity of recovered BALF showed a significant reduction of surface tension, indicating effective removal of surfactant from the brochoalveolar compartment (Figure 3B). Recovery rate of lavage fluid was 85%±5%.
Figure 3.
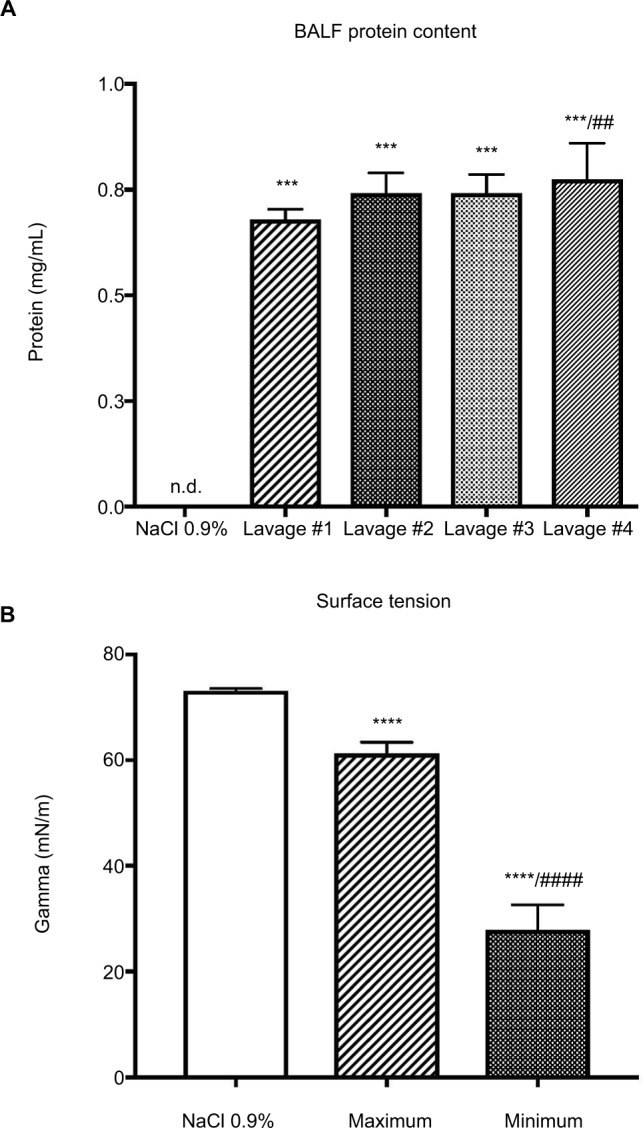
(A) Protein content of recovered BALF of the first consecutive lavage samples; ***p<0.001 when compared with control (lavage fluid, NaCl 0.9%); ##p<0.01 when compared with the first lavage. Data are presented as mean±SD, Mann–Whitney U-test (n=9). (B) Dynamic measurement of surface tension expressed as Gamma (mN/m) of recovered BALF of the first lavage samples; ****p<0.0001 compared with control (lavage fluid, NaCl 0.9%); ####p<0.0001 compared with minimal surface tension of the first lavage; data are presented as mean±SD, Mann–Whitney U-test (n=9).
Abbreviations: BALF, bronchoalveolar lavage fluid; n.d., not detected.
AT2 receptor stimulation inhibits pulmonary inflammation
To analyze the impact of selective AT2 receptor stimulation with C21 on the transcriptional regulation of pro and antiinflammatory cytokines, mRNA levels of relevant mediators were measured in the lungs. Repeated pulmonary lavage led to a significant induction of proinflammatory TNF-α, IL-1β, IL-6, and IL-4 mRNA (Figure 4A–F). Also, a significant induction of antiinflammatory IL-10 mRNA was observed. Levels of TGFβ mRNA remained unchanged compared with the control group. Concomitant stimulation of the AT2 receptor with C21 significantly inhibited TNF-a and IL-6 mRNA expressions compared with respective controls. In contrast, IL-1β, IL-10, IL-4, and TGFβ mRNA expressions showed no significant change after AT2 receptor stimulation.
Figure 4.

Relative mRNA expression of (A) TNF-α, (B) IL-6, (C) IL-1β, (D) IL-10, (E) IL-4, and (F) TGF-β in lungs of rats with mechanical ventilation only (control; n=9), repeated pulmonary lavage and mechanical ventilation (LAV; n=9) and repeated pulmonary lavage, mechanical ventilation, and direct stimulation of the AT2 receptor with C21 (0.03 mg/kg body weight; LAV+C21; n=9).
Notes: Lungs were harvested after 240 min of mechanical ventilation and real-time polymerase chain reaction was performed, GAPDH served as housekeeping gene, expression of control was set to 100%. Data are presented as mean±SEM, Mann–Whitney U-test, *p<0.05 compared with LAV group (n=9).
Abbreviations: AT2, angiotensin II type 2; C21, Compound 21; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; n.d., not detected; SEM, standard error of the mean; TGF-β, transforming growth factor-beta; TNF-α, tumor necrosis factor-alpha.
On the protein level, pulmonary lavage significantly induced TNF-α and IL-1β expressions in the lungs (Figure 5A and B). Direct stimulation of the AT2 receptor led to a significant decrease in the expression of TNF-α, but not IL-1β protein. IL-6, IL-10, IL-4, and TGF-β could not be detected in the protein level in the lungs of either the control or lavaged groups.
Figure 5.

Protein expression of (A) TNF-α and (B) IL-1β in lungs of rats with mechanical ventilation only (control; n=9), repeated pulmonary lavage and mechanical ventilation (LAV; n=9), and repeated pulmonary lavage, mechanical ventilation, and direct stimulation of the AT2 receptor with C21 (0.03 mg/kg body weight; LAV+C21; n=9).
Notes: Lungs were harvested after 240 min of mechanical ventilation, and protein levels were analyzed using CBA and FACS analysis. Results are given as pg (of the respective cytokine) per mg protein of lung tissue. Data are presented as mean±SEM, Mann–Whitney U-test, *p<0.05 compared with LAV group (n=9).
Abbreviations: AT2, angiotensin II type 2; C21, Compound 21; CBA, cytometric bead array; FACS, fluorescence-activated cell sorting; SEM, standard error of the mean; TNF-α, tumor necrosis factor-alpha.
Further, we aimed to clarify whether the proinflammatory stimulus induced by the model of acute lung injury resulted in systemic inflammation (i.e., in the blood). However, determination of cytokines in the plasma showed that none of the cytokines listed above were present.
In summary, in this experimental model of acute lung injury, the results suggest that direct stimulation of the AT2 receptor exerts antiinflammatory effects by inhibiting pro-inflammatory cytokine production in the lung.
AT2 receptor expression
Finally, pulmonary AT2 receptor expression was analyzed (Figure 6). AT2 receptor mRNA was significantly induced by treatment with C21 and concomitant pulmonary lavage. This effect translated into significantly increased AT2 receptor protein expression in the lung.
Figure 6.

Relative mRNA and protein expression of the AT2 receptor in the lung of rats with mechanical ventilation (control; n=9), repeated pulmonary lavage and mechanical ventilation (LAV; n=9), and repeated pulmonary lavage, mechanical ventilation, and direct stimulation of the AT2 receptor with C21 (0.03 mg/kg body weight; LAV+C21; n=9).
Notes: Lungs were extracted after 240 min of mechanical ventilation and real-time polymerase chain reaction (left) or immunoblotting (right) was performed. GAPDH served as housekeeping gene and α-actinin served as loading control. Expression of control was set to 100%. Data are presented as mean±SEM, Mann–Whitney U-test, *p<0.05 compared with LAV group (n=9).
Abbreviations: AT2, angiotensin II type 2; C21, Compound 21; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; SEM, standard error of the mean.
Discussion
This study demonstrates that, in an experimental model of acute lung injury, direct stimulation of the angiotensin AT2 receptor with C21 exerts antiinflammatory effects. AT2 receptor stimulation led to significant inhibition of the expression of proinflammatory cytokines in the lungs on both the transcriptional and translational levels. However, in this model, the antiinflammatory effect did not translate into significant improvement of pulmonary gas exchange or lung edema.
Overall, the model of acute lung injury used in this study sufficiently and adequately reproduced the main features of acute lung failure. First, pulmonary lavage and subsequent mechanical ventilation led to impaired pulmonary gas exchange. On average, PaO2 levels were <100 mmHg during the time course of the model, indicating a marked functional impairment. Second, the experimental procedure induced relevant pulmonary inflammation. This is in line with previous studies using repeated pulmonary lavage as the method of choice for inducing lung failure.30,33,34 In the present study, analysis of the recovered lavage fluid showed a significant protein content and demonstrated that surface tension was significantly reduced by the lavage fluid. These findings confirm that the pulmonary alveolar-capillary barrier was effectively disrupted and pulmonary surfactant was efficiently removed from the alveolar compartment by the procedures applied.
However, pulmonary inflammation is a hallmark and a main pathophysiological feature of acute lung failure.5,8 Early onset proinflammatory cytokines such as TNF-α, IL-6, or IL-1 are the predominant mediators of the disease and are involved in the course of lung injury.9,35 TNF-α is known to alter pulmonary membrane permeability, to trigger leukocyte recruitment, and to initiate downstream effects of inflammation in the lung.6,7,9 In the present study, there was a significant induction of proinflammatory cytokines on both the mRNA and the protein levels. The inflammatory response, however, was restricted to the lungs, as we could not detect any spillover of cytokines into the systemic circulation. Also, the overall amount of TNF-α in the lungs was lower than in other experimental settings that applied much more potent triggers of inflammation, such as lipopolysaccharides (LPS).36
In the present study, AT2 receptor activation with the selective agonist C21 significantly attenuated pulmonary inflammation and lowered expression levels of proinflammatory cytokines on both the transcriptional and translational levels. This is in line with previous findings demonstrating that AT2 receptor signaling can efficiently suppress inflammatory cytokines in vitro and in vivo.37,38 The mechanisms responsible for the antiinflammatory actions of the AT2 receptor are not yet completely understood. However, in vitro studies have shown that AT2 receptor signaling leads to a marked suppression of nuclear factor-kB (NF-kB) activation.38 AT2 receptor-mediated inhibition of NF-kB has been observed in human and mouse dermal fibroblasts, smooth muscle cells, in cultured human coronary artery endothelial cells, and in the brain.39,40,41 Also, the AT2 receptor has been shown to have an antiinflammatory effect by the activation of protein phosphatases and the synthesis of epoxyeicosatrienoic acid.38 We suggest that, in our model also, these mechanisms might be responsible for the antiinflammatory properties of C21 in the lung. However, more studies are required to perform detailed analyses of the signaling pathways involved in AT2 receptor-mediated antiinflammation in acute lung injury.
Pharmacological interventions at the RAS level have been shown to protect from experimental lung injury. For example, Imai et al demonstrated that ACE2, a negative regulator of the RAS, exerts beneficial effects in acute lung injury.28 Klein et al showed that Angiotensin (1–7 (Ang (1–7)), a small peptide and an agonist of the receptor Mas, attenuates the key features of experimental acute lung injury, such as pulmonary inflammation.13Ang (1–7) exerts vasodilatory and antioxidative effects, mainly via its receptor Mas; however, Ang (1–7) also partly activates the AT2 receptor.42 To the best of our knowledge, no other study has examined acute lung injury directly involving the AT2 receptor or its agonist C21. In contrast to the results of Klein et al, in our experimental setting, we could not demonstrate a substantial, clinical improvement. Pulmonary gas exchange (probably the most important clinical parameter) showed no significant improvement with C21. Further, the wet/dry-ratio (regarded as a valid marker of pulmonary edema) showed no significant improvement in the C21 treatment group. Thus, the attenuation of pulmonary inflammation alone did not lead to subsequent clinical benefit. The lavage model reproduces the failure of gas exchange and induces pulmonary inflammation; however, in contrast to other animal models of acute lung injury (e.g., acid aspiration) or models that induce lung failure by systemic injection of oleic acid or LPS, no additional proinflammatory stimulus is applied during the lavage procedure. However, this additional inflammatory stimulus might be a relevant prerequisite. In this regard, it would be worthwhile to test the antiinflammatory properties of C21 in alternative models of acute lung injury. Further, the observation time in the present model is limited and, with longer exposure times, the attenuation of inflammation might eventually lead to some clinical improvement. This possibility has, however, to be addressed in further studies.
Conclusion
Direct stimulation of the AT2 receptor attenuates pulmonary inflammation in a model of acute lung injury. However, relevant beneficial clinical effects of this approach are still to be elucidated.
Acknowledgments
The authors wish to thank Laraine Visser-Isles for English language editing.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8):788–800. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 2.Pham T, Rubenfeld GD. Fifty years of research in ARDS. The epidemiology of acute respiratory distress syndrome. A 50th birthday review. Am J Respir Crit Care Med. 2017;195(7):860–870. doi: 10.1164/rccm.201609-1773CP. [DOI] [PubMed] [Google Scholar]
- 3.Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353(16):1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 4.Albertine KH. Ultrastructural abnormalities in increased-permeability pulmonary edema. Clin Chest Med. 1985;6(3):345–369. [PubMed] [Google Scholar]
- 5.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122(8):2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mukhopadhyay S, Hoidal JR, Mukherjee TK. Role of TNF-alpha in pulmonary pathophysiology. Respir Res. 2006;7(1):125. doi: 10.1186/1465-9921-7-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang G, Hamacher J, Gorshkov B, et al. The dual role of TNF in pulmonary edema. J Cardiovasc Dis Res. 2010;1(1):29–36. doi: 10.4103/0975-3583.59983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson MR, Takata M. Inflammatory mechanisms of ventilator-induced lung injury: a time to stop and think? Anaesthesia. 2013;68(2):175–178. doi: 10.1111/anae.12085. [DOI] [PubMed] [Google Scholar]
- 9.Hegeman MA, Hennus MP, Heijnen CJ, et al. Ventilator-induced endothelial activation and inflammation in the lung and distal organs. Crit Care. 2009;13(6):R182. doi: 10.1186/cc8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT, Wheeler A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 11.Wiedemann HP, Wheeler AP, Bernard GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med. 2006;354(24):2564–2575. doi: 10.1056/NEJMoa062200. [DOI] [PubMed] [Google Scholar]
- 12.Guérin C, Reignier J, Richard JC, et al. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med. 2013;368(23):2159–2168. doi: 10.1056/NEJMoa1214103. [DOI] [PubMed] [Google Scholar]
- 13.Klein N, Gembardt F, Supé S, et al. Angiotensin-(1–7) protects from experimental acute lung injury. Crit Care Med. 2013;41(11):e334–e343. doi: 10.1097/CCM.0b013e31828a6688. [DOI] [PubMed] [Google Scholar]
- 14.Hoegl S, Brodsky KS, Blackburn MR, Karmouty-Quintana H, Zwissler B, Eltzschig HK. Alveolar epithelial A2B adenosine receptors in pulmonary protection during acute lung injury. J Immunol. 2015;195(4):1815–1824. doi: 10.4049/jimmunol.1401957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoegl S, Boost KA, Czerwonka H, et al. Inhaled IL-10 reduces biotrauma and mortality in a model of ventilator-induced lung injury. Respir Med. 2009;103(3):463–470. doi: 10.1016/j.rmed.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 16.Adhikari NK, Dellinger RP, Lundin S, et al. Inhaled nitric oxide does not reduce mortality in patients with acute respiratory distress syndrome regardless of severity: systematic review and meta-analysis. Crit Care Med. 2014;42(2):404–412. doi: 10.1097/CCM.0b013e3182a27909. [DOI] [PubMed] [Google Scholar]
- 17.Bernard GR, Wheeler AP, Arons MM, et al. A trial of antioxidants n-acetylcysteine and procysteine in ARDS. The antioxidant in ARDS study group. Chest. 1997;112(1):164–172. doi: 10.1378/chest.112.1.164. [DOI] [PubMed] [Google Scholar]
- 18.Matthay MA, Brower RG, Carson S, et al. Randomized, placebo-controlled clinical trial of an aerosolized β2-agonist for treatment of acute lung injury. Am J Respir Crit Care Med. 2011;184(5):561–568. doi: 10.1164/rccm.201012-2090OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davidson WJ, Dorscheid D, Spragg R, Schulzer M, Mak E, Ayas NT. Exogenous pulmonary surfactant for the treatment of adult patients with acute respiratory distress syndrome: results of a meta-analysis. Crit Care. 2006;10(2):R41. doi: 10.1186/cc4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McAuley DF, Laffey JG, O’Kane CM, et al. Simvastatin in the acute respiratory distress syndrome. N Engl J Med. 2014;371(18):1695–703. doi: 10.1056/NEJMoa1403285. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. 2003;35(6):881–900. doi: 10.1016/s1357-2725(02)00271-6. [DOI] [PubMed] [Google Scholar]
- 22.Rompe F, Unger T, Steckelings UM. The angiotensin AT2 receptor in inflammation. Drug News Perspect. 2010;23(2):104–111. doi: 10.1358/dnp.2010.23.2.1475901. [DOI] [PubMed] [Google Scholar]
- 23.Sumners C, de Kloet AD, Krause EG, Unger T, Steckelings UM. Angiotensin type 2 receptors: blood pressure regulation and end organ damage. Curr Opin Pharmacol. 2015;21:115–121. doi: 10.1016/j.coph.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan Y, Wallinder C, Plouffe B, et al. Design, synthesis, and biological evaluation of the first selective nonpeptide AT2 receptor agonist. J Med Chem. 2004;47(24):5995–6008. doi: 10.1021/jm049715t. [DOI] [PubMed] [Google Scholar]
- 25.Kaschina E, Grzesiak A, Li J, et al. Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation. 2008;118(24):2523–2532. doi: 10.1161/CIRCULATIONAHA.108.784868. [DOI] [PubMed] [Google Scholar]
- 26.Schwengel K, Namsolleck P, Lucht K, et al. Angiotensin AT2-receptor stimulation improves survival and neurological outcome after experimental stroke in mice. J Mol Med (Berl) 2016;94(8):957–966. doi: 10.1007/s00109-016-1406-3. [DOI] [PubMed] [Google Scholar]
- 27.Bruce E, Shenoy V, Rathinasabapathy A, et al. Selective activation of angiotensin AT2 receptors attenuates progression of pulmonary hypertension and inhibits cardiopulmonary fibrosis. Br J Pharmacol. 2015;172(9):2219–2231. doi: 10.1111/bph.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imai Y, Kuba K, Rao S, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imai Y, Kuba K, Penninger JM. Angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Cell Mol Life Sci. 2007;64(15):2006–2012. doi: 10.1007/s00018-007-6228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menk M, Graw JA, Steinkraus H, et al. Characterization of inflammation in a rat model of acute lung injury after repeated pulmonary lavage. Exp Lung Res. 2015;41(8):466–476. doi: 10.3109/01902148.2015.1075079. [DOI] [PubMed] [Google Scholar]
- 31.Peterson BT, Brooks JA, Zack AG. Use of microwave oven for determination of postmortem water volume of lungs. J Appl Physiol Respir Environ Exerc Physiol. 1982;52(6):1661–1663. doi: 10.1152/jappl.1982.52.6.1661. [DOI] [PubMed] [Google Scholar]
- 32.Menk M, von Haefen C, Funke-Kaiser H, et al. Ethanol-induced downregulation of the angiotensin AT2 receptor in murine fibroblasts is mediated by PARP-1. Alcohol. 2010;44(6):495–506. doi: 10.1016/j.alcohol.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 33.Lachmann B, Robertson B, Vogel J. In vivo lung lavage as an experimental model of the respiratory distress syndrome. Acta Anaesthesiol Scand. 1980;24(3):231–236. doi: 10.1111/j.1399-6576.1980.tb01541.x. [DOI] [PubMed] [Google Scholar]
- 34.Henzler D, Hochhausen N, Chankalal R, et al. Physiologic and biologic characteristics of three experimental models of acute lung injury in rats. Anesth Analg. 2011;112(5):1139–1146. doi: 10.1213/ANE.0b013e3182104dac. [DOI] [PubMed] [Google Scholar]
- 35.Liu YY, Chiang CH, Chuang CH, Liu SL, Jheng YH, Ryu JH. Spillover of cytokines and reactive oxygen species in ventilator-induced lung injury associated with inflammation and apoptosis in distal organs. Respir Care. 2014;59(9):1422–1432. doi: 10.4187/respcare.02992. [DOI] [PubMed] [Google Scholar]
- 36.Voelker MT, Fichtner F, Kasper M, et al. Characterization of a double-hit murine model of acute respiratory distress syndrome. Clin Exp Pharmacol Physiol. 2014;41(10):844–853. doi: 10.1111/1440-1681.12283. [DOI] [PubMed] [Google Scholar]
- 37.Menk M, Graw JA, von Haefen C, et al. Stimulation of the angiotensin II AT2 receptor is anti-inflammatory in human lipopolysaccharide-activated monocytic cells. Inflammation. 2015;38(4):1690–1699. doi: 10.1007/s10753-015-0146-9. [DOI] [PubMed] [Google Scholar]
- 38.Rompe F, Artuc M, Hallberg A, et al. Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor kappa b. Hypertension. 2010;55(4):924–931. doi: 10.1161/HYPERTENSIONAHA.109.147843. [DOI] [PubMed] [Google Scholar]
- 39.Zhu L, Carretero OA, Xu J, et al. Activation of angiotensin II type 2 receptor suppresses TNF-α-induced ICAM-1 via NF-κB: possible role of ACE2. Am J Physiol Heart Circ Physiol. 2015;309(5):H827–H834. doi: 10.1152/ajpheart.00814.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu L, Iwai M, Li Z, et al. Regulation of inhibitory protein-kappaB and monocyte chemoattractant protein-1 by angiotensin II type 2 receptor-activated src homology protein tyrosine phosphatase-1 in fetal vascular smooth muscle cells. Mol Endocrinol. 2004;18:666–678. doi: 10.1210/me.2003-0053. [DOI] [PubMed] [Google Scholar]
- 41.McCarthy CA, Vinh A, Callaway JK, Widdop RE. Angiotensin AT2 receptor stimulation causes neuroprotection in a conscious rat model of stroke. Stroke. 2009;40(4):1482–1489. doi: 10.1161/STROKEAHA.108.531509. [DOI] [PubMed] [Google Scholar]
- 42.Bader M. ACE2, angiotensin-(1–7), and mas: the other side of the coin. Pflugers Arch. 2013;465(1):79–85. doi: 10.1007/s00424-012-1120-0. [DOI] [PubMed] [Google Scholar]