

Abstract
High mobility group box 2 (HMGB2) is an abundant, chromatin-associated, non-histone protein involved in transcription, chromatin remodeling, and recombination. Recently, the HMGB2 gene was found to be significantly downregulated during senescence and shown to regulate the expression of senescent-associated secretory proteins. Here, we demonstrate that HMGB2 transcription is repressed by p21 during radiation-induced senescence through the ATM-p53-p21 DNA damage signaling cascade. The loss of p21 abolished the downregulation of HMGB2 caused by ionizing radiation, and the conditional induction of p21 was sufficient to repress the transcription of HMGB2. We also showed that the p21 protein binds to the HMGB2 promoter region, leading to sequestration of RNA polymerase and transcription factors E2F1, Sp1, and p300. In contrast, NF-Y, a CCAAT box-binding protein complex, is required for the expression of HMGB2, but NF-Y binding to the HMGB2 promoter was unaffected by either radiation or p21 induction. A proximity ligation assay results confirmed that the chromosome binding of E2F1 and Sp1 was inhibited by p21 induction. As HMGB2 have been shown to regulate premature senescence by IR, targeting the p21-mediated repression of HMGB2 could be a strategy to overcome the detrimental effects of radiation-induced senescence.
Keywords: HMGB2, p21, radiation, senescence, transcription repression
INTRODUCTION
High mobility group box (HMGB) proteins are members of the HMG protein family that contains the characteristic HMG box motif. HMGs compose a group of non-histone chromatin proteins that regulate gene transcription by altering chromatin architecture (Bianchi and Agresti, 2005). These proteins bind to DNA without sequence specificity to increase chromatin accessibility to transcription factors (Thomas, 2001), which generally promotes transcription (Malarkey and Churchill, 2012).
Cellular senescence was first identified as a type of irreversible cell cycle arrest that occurs when cells can no longer replicate (Hayflick and Moorhead, 1961). Many other stresses, such as irradiation, oxidative stress, DNA damage, and oncogenic activation, also trigger premature senescence (Kuilman et al., 2010). Senescence is generally considered a protective response: It has evolved to restrict tumor progression and limits the extent of fibrosis. However, the accumulation of senescent cells can have detrimental consequences, such as in age-related diseases and cancer (Lopez-Otin et al., 2013).
Growing evidence supports the pivotal role of HMGB2 in the regulation of cellular senescence and aging. Aging was associated with the loss of HMGB2 expression and reduced cellularity, which together contribute to the development of osteoarthritis (Taniguchi et al., 2009). Recently, Aird et al. (2016) identified a key role for HMGB2 in the shaping of the chromatin landscape associated with the senescence-associated secretory phenotype (SASP); heterochromatin spreading was prevented, allowing for the exclusion of SASP gene loci from SAHF-mediated gene silencing. We also observed that miRNA-mediated downregulation of HMGB2 contributes the aging of microvascular endothelial cells (unpublished).
In our previous study, HMGB2 was identified as a differentially expressed gene in chemoradiotherapy-resistant colorectal cancers, which modulates the response to radiation therapy (Shin et al., 2013). Although direct killing of tumor cells is the major purpose of radiotherapy, ionizing radiation (IR) inevitably induces premature senescence of some cancer and normal cells within the irradiated field. To preserve the beneficial effects of senescence while minimizing the detrimental effects of radiotherapy-induced senescence, we examined the contribution and downregulation mechanism of HMGB2 in radiation-induced senescence (RIS).
MATERIALS AND METHODS
Cells
Human lung adenocarcinoma cell lines A549 and H1299, lung fibroblast cell line CCD18-Lu, and colorectal adenocarcinoma cell line HT-29 were purchased from the American Type Culture Collection (USA). The cells were maintained in RPMI-1640 medium (A549, H1299, and HT-29) or minimal essential medium (CCD18-Lu), supplemented with 10% FBS and 100 U/mL penicillin-streptomycin. Cells were cultured at 37°C in a humidified incubator with 5% CO2. Ectopic over-expression of HMGB2 and shRNA-expressing pLKO system targeting HMGB2 was described previously (Shin et al., 2013).
Senescence-associated β-galactosidase (SA-β-gal) staining
Cellular SA-β-gal activity was measured as previously described (Dimri et al., 1995). A549 and CCD18-Lu cells were irradiated and incubated further for 3 days. Cells were washed with PBS and fixed with 3.7% (v/v) paraformaldehyde for 15 min at room temperature. The cells were incubated with staining solution containing 1 mg/ml X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactoside), 40 mM citric acid-sodium phosphate (pH 6.0), 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 150 mM NaCl, and 2 mM MgCl2 for 16 h at 37°C. SA-β-gal-stained cells were washed with PBS, counterstained with a 1% eosin solution for five min, and then washed twice with ethanol. The percentage of blue cells per every 400 cells observed under a light microscope was calculated.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated from cells using the Hybrid-R Total RNA Purification kit (GeneAll, Korea). One μg RNA was reverse transcribed using the PrimeScript RT Master Mix (Takara, Japan). Two μl of a 1:10 diluted cDNA were used as templates in 20-μl reactions. qRT-PCR was performed using the qPCR SYBR Green Master Mix (MBiotech, Korea) on a CFX96real-time PCR system (Bio-Rad). Gene expression was normalized to two reference genes (PPIA and RPL13A), and the relative gene expression values were calculated based on the threshold cycle (Ct) value using the 2-ΔΔCt method (Livak and Schmittgen, 2001). PCR conditions were an initial preincubation step at 95°C for 10 min followed by 45 cycles of 95°C for 5 s and 60°C for 30 s. The last amplification cycle was followed by a melting curve analysis to confirm the specificity of the PCR amplification. The primers used for qRT-PCR are listed in Supplementary Table S1.
Western blot analysis
Cells were lysed in RIPA buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, and protease inhibitors] and briefly sonicated. Protein content was measured using the Coomassie (Bradford) Protein Assay kit (Thermo Fisher Scientific, Inc., USA), and equal amounts of cell lysate were separated by SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane (Bio-Rad). Membranes were immunoblotted with antibodies against HMGB2 (Abcam, UK), p53, p21, β-actin (Santa Cruz Biotechnology, USA), and ATM (Epitomics, USA), and detected by chemiluminescence using ECL detection reagents.
Chromatin immunoprecipitation (ChIP)
A total of 1 × 107 cells were fixed in 1% formaldehyde for 10 min at room temperature, and the reaction was stopped with the addition of 0.125 M glycine. Fixed cells were washed and incubated in swelling buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.5% NP-40, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and a cocktail of protease inhibitors). Nuclei were pelleted and resuspended in 1 ml SDS lysis buffer (0.2% SDS, 1 mM EDTA, 50 mM Tris-HCl, pH 8, and a cocktail of protease inhibitors). Nuclei were then disrupted by sonication (Q500, Qsonica, LLC, USA) and cell debris was cleared. The supernatants were diluted with dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8, and 167 mM NaCl). One percent of the chromatin from the supernatant was used as the [input] for ChIP normalization. Immunoprecipitation was performed overnight at 4°C with 2 μg antibodies (p53, RNA polymerase II, p21, E2F1, Sp1, and NF-YB, Santa Cruz Biotechnology), followed by incubation with 30 μl protein G magnetic beads (Invitrogen) supplemented with 0.2 mg/ml salmon sperm DNA, 0.1% BSA, and 0.05% NaN3 for 2 h. Beads were washed sequentially as follow: twice with low-salt RIPA buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8, and 150 mM NaCl), twice with high-salt RIPA buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8, and 500 mM NaCl), once with LiCl buffer (0.25 M LiCl, 1% deoxycholic acid, 0.5% NP-40, 1 mM EDTA, and 10 mM Tris-HCl, pH 8), and twice with TE (10 mM Tris-HCl, pH 8, and 1 mM EDTA). The chromatin immunoprecipitates were eluted in elution buffer (EB, 1% SDS and 0.1 M NaHCO3) and reverses the cross-linking. Chromatin was recovered using an Expin PCR SV kit (Gene-All, Korea) and analyzed with a CFX96 real-time PCR system (Bio-Rad). Relative occupancy values were calculated as the percent of input DNA. The primers used for quantitative PCR are listed in Supplementary Table S2.
Transfection of siRNAs
Cells (2.5 × 105) were transfected with 20 nM siRNA using Lipofectamine RNAiMAX transfection reagent (Invitrogen) in a 35-mm dish, according to the manufacturer’s instructions. Two days after transfection, 5 Gy IR was applied, if necessary, and cells were harvested on the next day. The siRNAs (Genolution, KOREA) used in this study are listed in Supplementary Table 3.
Tetracycline inducible gene expression system
The RetroX Tet-OnR advanced inducible expression system (Clontech Laboratories, Inc., USA) was used, according to the manufacturer’s instructions. To generate rtTA-expressing cells, HT-29 or H1299 cells were infected with retrovirus generated from a pRetroX-Tet-On Advanced vector and selected with 500 μg/mL G418 after 7 days. To make the Tet-inducible p21 construct, the pMT5-FLAG-p21 (a gift from Mien-Chie Hung, Addgene plasmid # 16240) was amplified using primers (5′-AAACTCGGATCCATGGACTACAAA GACGATGA-3′ and 5′-AATGCCGAATTCTTAGGGCTTCCTCTT GGAGA-3′), and a BamHI/EcoRI-digested amplified fragment was subcloned into the pRetroX-Tight-pur vector. For generation of the Tet-inducible E2F1 plasmid, HA-E2F-1 wt-pRcCMV (a gift from William Kaelin, Addgene plasmid # 21667) was digested with BamHI and EcoRI, and then sub-cloned into the pRetroX-Tight-pur vector. Retroviruses were introduced to rtTA-expressing cells, and selecting transformants with puromycin (1 μg/ml) for 7 days. For induction of gene expression, cells were treated with 500 μg/mL doxycycline (Sigma) for 24 h.
Luciferase reporter assay
Construction of the HMGB2 luciferase reporter plasmid was previously described (Shin et al., 2013). To assay HMGB2 promoter activity, 5 × 104 293T cells were co-transfected with 100 ng pGL3-basic or pGL3-HMGB2p, and 2 ng pRL-SV40 as a control, for transfection efficiency. To examine the effects of E2F1 on HMGB2 promoter activity, cells were co-transfected with 20 or 100 ng HA-E2F-1 wt-pRcCMV (a gift from William Kaelin, Addgene plasmid # 21667). Cells were allowed to recover for 24 h at 37°C and were then harvested and analyzed using the Dual LuciferaseR Reporter Assay system (Promega, USA). Luminescence was measured using a GloMax 20/20 Single Tube luminometer (Promega). Firefly luciferase activity was normalized to Renilla luciferase activity. Each transfection was performed in triplicate and repeated three times.
CRISPR/CAS9-mediated ATM gene knockout
A lentiviral gRNA vector was obtained from transOMIC technologies, Inc. (USA). To produce lentivirus particles, 5 × 106 293FT cells were seeded onto 100-mm dishes and transiently transfected with 5 μg pCLIP-All-EFS-puro containing gRNA targeting ATM, 4 μg pCMV-dR8.2 dvpr (a gift from Bob Weinberg, Addgene plasmid # 8455), and 1 μg pCMV−VSV-G (a gift from Bob Weinberg, Addgene plasmid # 8454) after 16 h using PEI (Polysciences, USA). Supernatants were collected 48 and 72 h post-transfection, and viruses were concentrated using a Centricon 100K (Millipore) and subsequently used for infection in the presence of 8 μg/ml polybrene. One day after infection, 1 μg/ml puromycin (Sigma) was applied for 4 days for selection. Surviving cells were digested with trypsin and diluted in media for colony formation. Single colonies were selected, and each colony was passaged and subjected to western blot analysis using anti-ATM antibodies (Epitomics, USA).
In situ proximity ligation assay (PLA)
In situ PLA was performed according to the manufacturer’s instructions (Olink Bioscience, Sweden). Briefly, cells were cultured on 18 × 18-mm cover glasses and fixed in ice-cold methanol for 15 min, permeabilized with 0.5% Triton X-100 for 10 min, and blocked at 37°C for 30 min. Two primary antibodies generated from rabbits and mice were bound together for 16 h at 4°C. After the removal of unbound primary antibodies, cells were incubated with proximity probes (anti-rabbit PLUS and anti-mouse MINUS) for 1 h at 37°C. Ligation was performed for 30 min at 37°C, followed by polymerization for 2 h at 37°C. Cells were counterstained with DAPI, and immunofluorescence images were captured using a laser-scanning confocal microscope.
Statistical analysis
All results are expressed as means±SE. Student’s t-test was used for statistical comparisons. All tests were two-sided, and p values less than 0.01 considered significant (*).
RESULTS
HMGB2 is downregulated during RIS
Various external stresses can damage cellular components, especially chromosomal DNA, causing the senescence of damaged cells. IR used for therapeutic purpose is a well-known senescence-inducing agent. As predicted, SA-β-gal positive population was increased after IR exposure in A549 (lung adenocarcinoma) and CCD18-Lu (lung fibroblast) cells (Fig. 1A), suggesting that cancer radiotherapy could trigger senescence of adjacent stromal cells as well as cancer cells. Recently, HMGB2 was identified as one of most commonly downregulated genes in replicative senescence and oncogene-induced senescence (Aird et al., 2016). We thus measured HMGB2 protein levels during RIS. In both carcinoma and fibroblast cells, HMGB2 protein expression was decreased following IR in a dose- and time-dependent manner and inversely correlated with canonical senescence markers (Figs. 1B and 1C). A qRT-PCR analysis revealed that mRNA expression of HMGB2 was downregulated by IR in a dose-associated manner, supporting that IR induces the transcriptional inactivation of the gene (Fig. 1D). It was also recognized that both mRNA and protein levels of HMGB2 were markedly decreased by 2 Gy of IR, a commonly applied dose in conventional radiotherapy. These results suggest that transcriptional inactivation of HMGB2 might be involved in IR induction of cellular senescence.
Fig. 1. HMGB2 is downregulated during radiation-induced senescence.
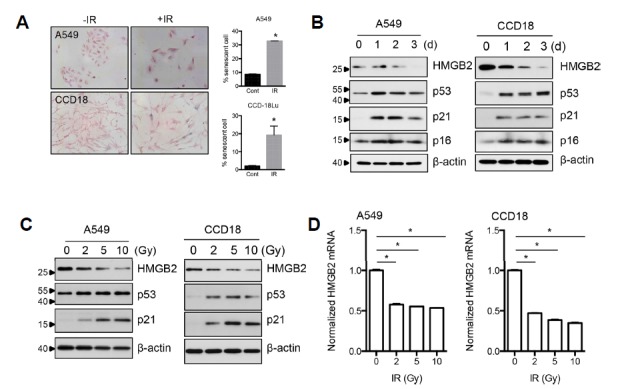
(A) A549 and CCD18-Lu lung cells were exposed to IR, and senescent cells were detected by SA-β-gal staining after 3 days. (B) A549 and CCD18-Lu cells were exposed to radiation and harvested after 1, 2, or 3 d. HMGB2 protein levels were examined by immunoblot analysis. Canonical senescence markers p53, p21, and p16 were also examined. (C) A549 and CCD18-Lu cells were exposed to the indicated doses of radiation and harvested after 1 day. Immunoblot analysis was performed as in (B). (D) A549 and CCD18-Lu cells were exposed to the indicated doses of radiation and harvested after 16 h. HMGB2 mRNA levels were examined by qRT-PCR.
HMGB2 regulates RIS
To determine whether HMGB2 regulates premature senescence induced by irradiation, HMGB2-overexpressing and HMGB2-depleted cell lines were established. As expected, overexpression of HMGB2 delayed RIS in both A549 and CCD18-Lu cells (Fig. 2A). In contrast, depletion of HMGB2 using shRNAs accelerated RIS (Fig. 2B). Expression of HMGB2 was confirmed by immunoblot analysis (Fig. 2C).
Fig. 2. HMGB2 regulates radiation-induced senescence.
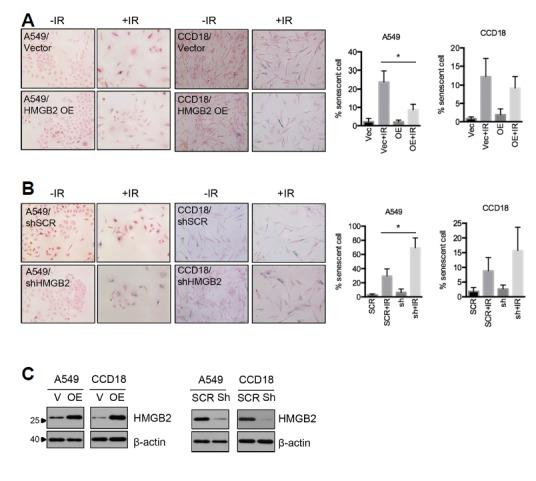
(A) HMGB2 was overexpressed in A549 and CCD18-Lu lung cells, and senescent cells were detected using SA-β-gal staining 3 days after irradiation. (B) HMGB2 was depleted using shRNA in A549 and CCD18-Lu cells, and senescent cells were detected using SA-β-gal staining 3 days after irradiation. (C) HMGB2 protein levels were examined by immunoblot analysis.
p53 is not a direct repressor of HMGB2 transcription
We have shown that IR downregulated HMGB2 expression in a p53-dependent manner in colorectal cancer cells (Shin et al., 2013). We thus tested whether p53 is required for IR-mediated downregulation of HMGB2 in lung carcinoma and fibroblast cells. Knockdown of p53 using siRNAs clearly eliminated IR-mediated HMGB2 downregulation (Fig. 3A). Next, to reveal the mechanism of p53-mediated repression of transcription, we determined whether p53 binds directly to the HMGB2 promoter region. A549 cells were exposed to 10 Gy IR, and a ChIP assay was performed using anti-p53 antibodies. No significant enrichment of HMGB2 promoter DNA was observed, irrespective of exposure to IR (Supplementary Fig. S1). In contrast, p53 was shown to bind to the promoter region of p21, a well-known site of p53 binding (Lagger et al., 2003), and p53 recruitment was dramatically increased after exposure to IR. In addition, a conserved p53-response element was not identified within the 2 kb promoter sequence region of HMGB2. Together, these results show that p53 mediates IR-induced HMGB2 downregulation but does not act as a direct transcriptional repressor, raising the possibility that downstream effector(s) of p53 signaling might be involved in IR-mediated repression of HMGB2 transcription.
Fig. 3. IR-induced HMGB2 downregulation is mediated by the ATM-p53-p21 signaling cascade.
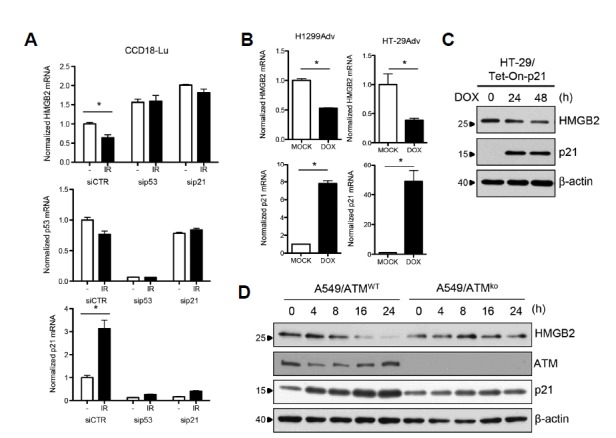
(A) CCD18-Lu cells were transfected with siRNAs to knockdown p53 or p21, and exposed to IR. Sixteen hours after exposure, total RNA was prepared, and expression of HMGB2, p21, or p53 was examined by qRT-PCR. (B) H1299 and HT-29 cells harboring a Tet-inducible p21 expression system were treated with doxycycline, and qRT-PCR analysis was performed. (C) Western blot analysis was performed to verify protein levels. (D) ATM knockout A549 cells were established using the CRISPR-CAS9 system. A549 cells with different ATM background levels were treated with 10 Gy IR, and western blot analyses were performed.
HMGB2 downregulation is mediated by the ATM-p53-p21 DNA damage signaling cascade
The CDKN1A gene, which encodes p21Cip1, is a direct transcriptional target of p53 and was originally identified as a cyclin-dependent kinase (CDK) inhibitor that could induce cell cycle arrest and senescence following p53 activation (Harper et al., 1993; Li et al., 1994; Xiong et al., 1993). In addition, p21 has been implicated in the control of the transcription of several genes, including those involved in S phase and mitosis independent of CDK inhibition (Ferrandiz et al., 2012). To examine whether p21 is involved in HMGB2 downregulation, RNAi-mediated p21 gene knockdown was performed. Similar to results from siRNAs targeting p53, transfection of siRNAs targeting p21 increased basal HMGB2 expression levels and completely abolished the downregulation of HMGB2 by IR (Fig. 3A). The knockdown effects of siRNAs against p53 could be explained by the simultaneous knockdown of p21 in these cells. In accordance with these findings, the p53-activating drug, nutlin-3A, could not block HMGB2 transcription in p53- or p21-deficient HCT116 cells (data not shown). The conditional overexpression of p21 using the Tet-On expression system was sufficient to down-regulate HMGB2 (Figs. 3B and 3C). As the ataxia telangiectasia mutated (ATM) kinase is a master regulator of DNA damage by IR, the effects of ATM on p21-mediated HMGB2 regulation were also examined. ATM-deficient A549 clones were generated using the CRISPR/CAS9 genome editing system, and IR was applied. In contrast to the ATM-wild-type control, HMGB2 levels did not decrease after irradiation in ATM-deficient cells (Fig. 3D).
p21 binds the HMGB2 promoter and inhibits the recruitment of RNA polymerase II
As HMGB2 expression is regulated at the transcriptional level (Fig. 1D), RNA polymerase II binding to HMGB2 promoter DNA was expected to be affected by either IR or p21 induction. Results of a ChIP assay using anti-RNA polymerase II antibodies showed that both IR (Fig. 4B) and p21 induction (Fig. 4C) inhibited the recruitment of RNA polymerase II to the HMGB2 promoter region. To assess whether p21 works as a repressor of transcription by binding to the HMGB2 promoter, a ChIP assay was performed using anti-p21 antibodies. IR promoted the recruitment of p21 to the HMGB2 promoter region (Fig. 4D) near the transcription start site (TSS). A ChIP assay performed in Tet-inducible p21-expressing cells showed similar results (Fig. 4E). The amount of chromatin DNA in the distal promoter region (approximately 10 kb upstream of the TSS) (Fig. 4A, region A) was not enriched.
Fig. 4. p21 binds to the HMGB2 promoter and inhibits RNA polymerase II recruitment.
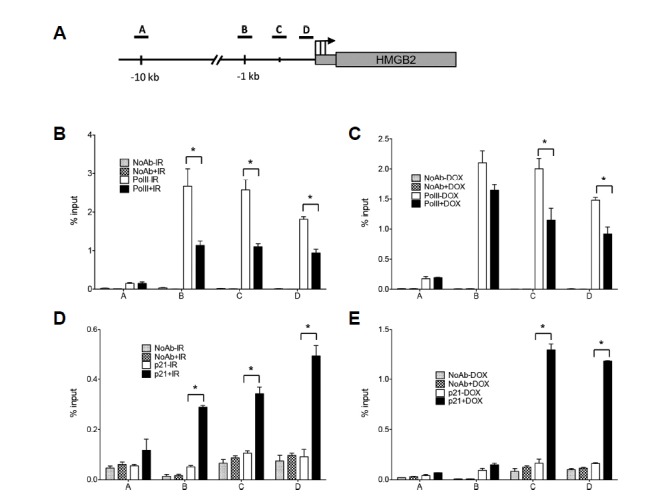
(A) The diagram indicates the location of the amplicons, TSS (arrow), and HMGB2 gene structure. Three regions (B, C, and D) in promoter were analyzed and far upstream sequences (−10 kb, region A) were included as negative controls. A549 cells were exposed to 10 Gy IR (B, D) and Tet-On-p21 HT-29 cells were treated with doxycycline for 24 h to induce p21 expression (C, E). ChIP was performed using anti-RNA polymerase II (B, C) and anti-p21 (D, E) antibodies as indicated. DNA in the immunoprecipitated chromatin was analyzed by real-time PCR. The results are expressed as a relative enrichment of DNA in chromatin immunoprecipitated with antibodies to input DNA.
The Rb-E2F1 pathway regulates HMGB2 expression
It is well known that CDK inhibition by p21 sustains the hypophosphorylation status of the retinoblastoma tumor suppressor protein (Rb) and inhibits the release of the E2F1 transcription factor from Rb-E2F1 complexes (Fagan et al., 1994). Intriguingly, we found that HMGB2 mRNA expression was increased by siRNA-mediated knockdown of Rb while it was decreased by knockdown of E2F1 (Figs. 5A and 5B). Consistently, E2F1 induction using the Tet-On system resulted in a dose-dependent elevation of HMGB2 mRNA level (Fig. 5C). E2F1-mediated transcriptional activation of HMGB2 was confirmed using a luciferase reporter assay (Fig. 5D). The HMGB2 promoter activity was increased by E2F1 in a dose-dependent manner. To assess whether E2F1 directly binds to the HMGB2 promoter region and, if so, whether IR and/or p21 induction affects E2F1 binding, a ChIP assay was performed using anti-E2F1 antibodies. E2F1 bound specifically near the TSS of the HMGB2 promoter region, and its binding was decreased by IR (Fig. 5E) or p21 induction (Fig. 5F).
Fig. 5. Rb-E2F1 regulates HMGB2 expression.
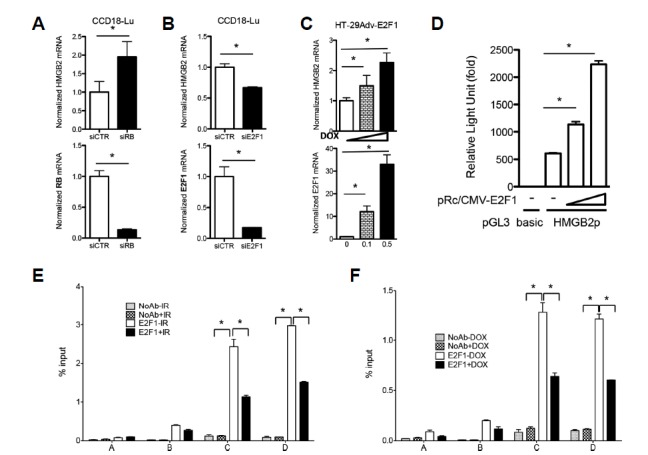
Gene knockdown using siRNAs targeting Rb (A) or E2F1 (B) was performed, and mRNA levels of HMGB2, Rb, and E2F1 were examined by qRT-PCR. (C) Increasing amounts of doxycycline were added in the Tet-inducible E2F1 expression system, and HMGB2 and E2F1 transcription was examined by qRT-PCR. (D) A pGL3 luciferase reporter vector harboring HMGB2 promoter DNA (pGL3-HMGB2p) was co-transfected with increasing amounts of E2F1 expression plasmid (pRc/CMV-E2F1) into 293T cells, and transcriptional activity driven by the HMGB2 promoter was measured by a dual luciferase assay. ChIP was performed using anti-E2F1 antibodies in A549 cells exposed to 10 Gy IR (E) and in p21-induced HT-29 cells (F). DNA in the immunoprecipitated chromatin was analyzed by real-time PCR, as described in Fig. 4.
Sp1 and p300 are involved in HMGB2 transcription
Through a computer-based search for HMGB2 promoter sequences, binding elements of transcription factors, Sp1, p300, and NF-Y, were identified. Among the candidates, Sp1 was tested first because mithramycin A, a specific inhibitor of Sp1, downregulated HMGB2 (data not shown). A ChIP assay using anti-Sp1 antibodies revealed that Sp1 bound specifically near the TSS of the HMGB2 promoter region and that IR and p21 inhibited the recruitment of Sp1 to this region (Figs. 6A and 6B). Similarly, p300 was shown to bind to the HMGB2 promoter and this binding was inhibited by both IR exposure or p21 induction (Figs. 6C and 6D). Finally, we tested the involvement of NF-Y, a CCAAT box-binding protein complex. A ChIP assay using anti-NF-YB antibodies showed that NF-Y bound to the predicted NF-Y-binding element near the TSS (Figs. 6E and 6F). However, in contrast to E2F1, Sp1 and p300, the NF-Y binding to the HMGB2 promoter was unaffected by either IR exposure or p21 induction, indicating that NF-Y is not associated with p21 downregulation of HMGB2 transcription.
Fig. 6. Analysis of transcription factor binding to the HMGB2 promoter.
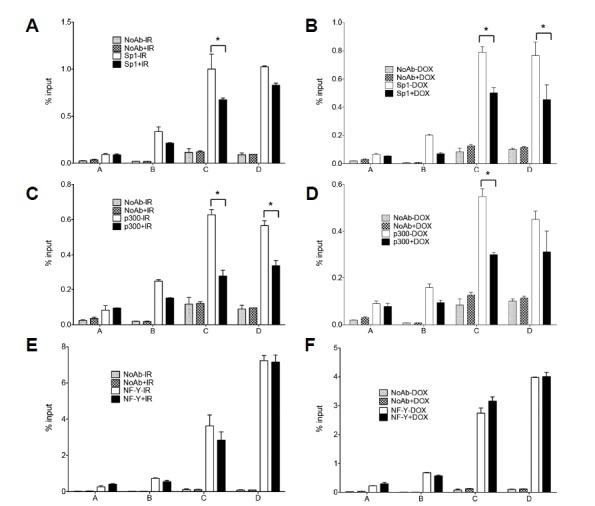
A549 cells were exposed to 10 Gy IR (A, C, and E) or Tet-inducible p21-expressing HT-29 cells were treated with doxycycline for 24 h (B, D, and F). ChIP was performed using anti-Sp1 (A, B), anti-p300 (C, D), and anti-NF-YB (E, F) antibodies as indicated. DNA in the immunoprecipitated chromatin was analyzed by real-time PCR, as described in Fig. 4.
Recruitment of E2F1 and Sp1 to chromosome is inhibited by p21 induction
To examine the effect of p21 on transcription factors binding to chromosome, we performed PLA assay, which enables the detection of intracellular protein-protein interactions in situ. We first examined the interactions with histone H1, one of the five main histone protein families that are components of chromatin (Fig. 7, left panel). After p21 induction, chromosome-bound portion of p21 was increased accordingly and the interactions between histone H1 and E2F1 or Sp1 were dramatically inhibited. However, induction of p21 did not affect on the interaction between histone H1 and NF-YB. Next, as NF-Y generally binds near the TSS of transcriptionally active genes, interactions between NF-Y and other factors were also tested (right panel). Overexpressed p21 proteins were recruited to the vicinity of NF-Y-binding sites. The interactions between NF-YA and E2F1 or Sp1 were dramatically inhibited by p21 induction. Although p21 could interact with soluble form of NF-YA, overall interaction between NF-Y and E2F1 or Sp1 decreased by p21 induction.
Fig. 7. Effects of p21 induction on transcription factor binding to chromosome.
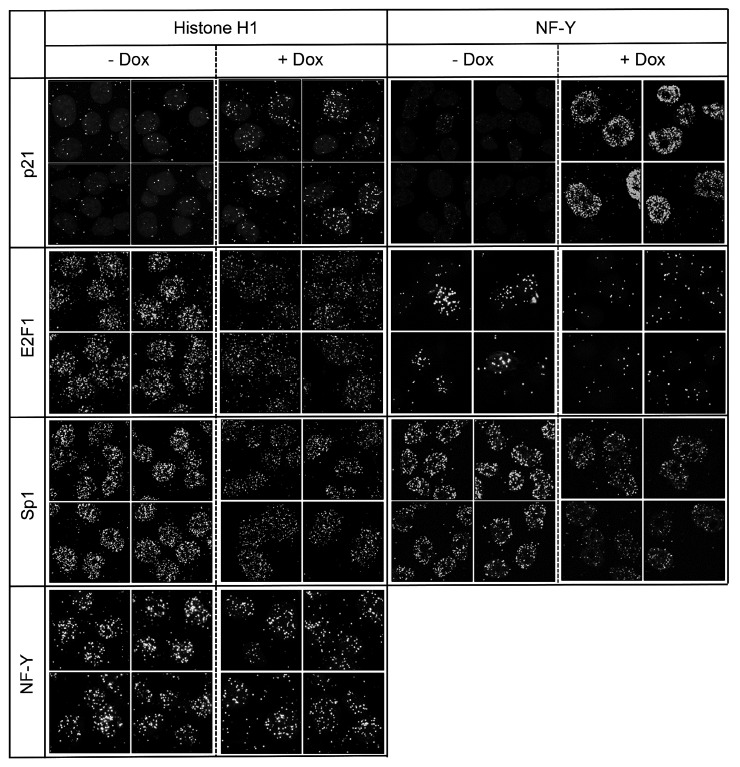
Interactions between p21 and transcription factors were examined by in situ PLA. H1299/Tet-On-p21 cells were treated with 0.1 μM doxycycline for 24 h, and PLA assays were performed using the combination of indicated antibodies from different sources, according to the manufacturer’s instructions.
DISCUSSION
Mechanism of transcriptional repression of the HMGB2 gene by p21
In this study, we revealed the mechanism of transcriptional repression of the HMGB2 gene during RIS. A schematic model of HMGB2 regulation is shown in Fig. 8. Several transcription factors, including E2F1, Sp1, p300, and NF-Y, are required for the proper transcription of HMGB2. However, after exposure to IR, DNA damage activates p53, leading to p21 induction (Fig. 3). Overexpressed p21 proteins bind to the HMGB2 promoter region (Figs. 4D and 4E) and sequester RNA polymerase and transcription factors like E2F1, Sp1, and p300 (Figs. 4B and 4C, 5, and 6). Furthermore, PLA results confirm that p21 induction inhibits the binding of E2F1 and Sp1 to chromosome (Fig. 7). Thus, p21-mediated transcriptional regulation can be summarized as follows: First, p21 interferes with basal transcriptional machinery, including RNA polymerase II, likely as a result of the inhibition of TATA-binding protein (TBP) and TBP-associated factors by p21. Second, p21 sequesters sequence-specific transcriptional activators, like E2F1, Sp1, and p300, from the promoter region, which might be because of competition from a common DNA sequence or via conformational and/or epigenetic changes mediated by p21. Third, as p21 does not inhibit the binding of the NF-Y trimeric complex to the CCAAT box, a sequence-specific or transcription factor-specific regulatory mechanism of p21 is likely.
Fig. 8. Diagram of HMGB2 transcriptional regulation by IR.
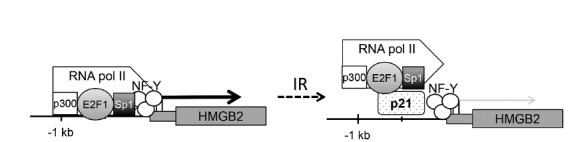
Transcription factors E2F1, Sp1, p300, and NF-Y bind to the HMGB2 promoter region and promote the transcription of the HMGB2 gene. When cells are exposed to IR, DNA damage activates p53, leading to p21 induction. Overexpressed p21 binds to HMGB2 promoter and inhibits HMGB2 transcription.
Recently, it was reported that p21 functions not only as a CDK inhibitor but also as a transcriptional co-repressor in some systems and both the cell cycle-dependent element (CDE) and the cell cycle gene homology region (CHR) are found in most genes under the transcriptional regulation of p21 (Ferrandiz et al., 2012; Fischer et al., 2016). In addition, sequential ChIP/ReChIP using anti-E2F1 and then anti-p21 antibodies support the premise that p21 associates with the E2F1 transcription factor at the Wnt4 promoter and limits the recruitment of c-Myc and p300 (Devgan et al., 2005). However, there is no evidence (except ChIP) that supports p21 binding to DNA yet. Although we also observed p21 binding to the HMGB2 promoter region (Figs. 4D and 4E), oligonucleotides harboring CDE/CHR sequences could not pull down p21, even after conditional overexpression (data not shown), suggesting that p21 might not bind to the CDE/CHR directly but that other factors or physiological conditions are necessary to facilitate p21 binding to DNA. It is therefore necessary to demonstrate the direct binding of p21 to specific DNA elements via ChIP-seq analysis, identify the consensus binding element, and verify the results with either an EMSA or DNA pulldown assay.
Additionally, growing evidence supports that p21 mediates the activation of the DREAM complex, which is composed of DP, Rb-like, E2F4, and MuvB proteins, which leads to the specific binding of the DREAM complex to CHR. Fisher et al. (2016) identified 210 potential target genes regulated by the p53-p21-DREAM-CDE/CHR signaling pathway via a bioinformatics analysis, in which HMGB2 was included as a candidate target gene. As most target genes are essential regulators of G2 phase and mitosis, the p53-p21-DREAM-CDE/CHR pathway appears to be the principal mechanism by which p53 affects G2/M cell cycle arrest. Interestingly, a mutation study performed by Quaas et al. (2012) showed that NF-Y binding was unrelated to CDE/CHR, similar to the effects of radiation or p21 on NF-Y binding (Figs. 6E and 6G). Taken together, transcriptional repression by p21 could work via two independent ways: the p53-p21-DREAM-CDE/CHR signaling pathway by CDK inhibition and by binding to the promoter region. Further studies are required to reveal the details of such p21 regulation.
A number of transcriptional regulators have been shown to alter the chromatin structure of their target genes via the recruitment of histone acetyltransferases, histone deacetylases, and chromatin remodeling complexes. It is plausible that p21 might alter chromatin structure through epigenetic modifications. However, we did not see any changes in histone acetylation or methylation in ChIP experiments using anti-acetyl-histone H3 or anti-methyl histone antibodies (data not shown). In addition, trichostatin A, an inhibitor of HDAC, could not recover the IR-mediated downregulation of HMGB2 (data not shown). These results support that epigenetic changes and chromosome remodeling may not function as major contributors to transcriptional repression by p21.
As p21 is a downstream target of p53, the contribution of p21 in p53-mediated transcriptional regulation was examined. Our previous microarray analysis using a Tet-On p53 and p21 expression system (Supplementary Fig. S2) showed that more than half of the repressed genes by p53 are mediated through p21. This finding suggests that p21 is a major regulator of p53-dependent transcriptional repression. Genes downregulated by p21 are involved in DNA metabolism and replication, chromatin organization and remodeling, and transcriptional regulation. Most are also involved in chromosome organization and DNA metabolism.
Role of HMGB2 in senescence
Cellular senescence is an irreversible arrest of the cell cycle that occurs after extensive serial passages in culture (Hayflick and Moorhead, 1961) due to telomere loss or dysfunction. Later, it was determined that oncogenes and external stresses also trigger premature senescence (Kuilman et al., 2010), playing a favorable role in tumor suppression. However, senescent cells secret proinflammatory cytokines, growth factors, and matrix metalloproteinases (Davalos et al., 2010), which cause chronic inflammation that leads to age-related diseases (van Deursen, 2014). Therefore, understanding and controlling these two aspects of senescence are of great importance. The prime role of HMGB2 in senescence can be determined by using Hmgb2 knockout mice, a relevant model of aging-related diseases, like osteoarthritis (Taniguchi et al., 2009; 2017). We observed that HMGB2 downregulated by miRNAs during RS and depletion of HMGB2 accelerated endothelial cell senescence (unpublished). Recently, HMGB2 was identified as a key regulator of SASP expression during senescence through the modulation of heterochromatin formation (Aird et al., 2016). HMGB2 was identified as the most often repressed protein during RS and OIS, and results of ChIP-seq analysis showed that HMGB2 bound to the SASP gene promoter region, which prevents HP1α protein recruitment. As many detrimental effects of senescence are mediated by SASP, targeting HMGB2 may be a good strategy to preserve the beneficial effects of senescence while minimizing the detrimental effects of RIS induced by chronic inflammation.
Supplementary data
ACKNOWLEDGMENTS
This study was supported by a grant of the Korea Institute of Radiological and Medical Sciences (KIRAMS) (1711045557; 1711045538;1711045554/50531-2017) funded by the Ministry of Science and ICT and by a grant from the Basic Science Research Program through the National Research Foundation of Korea (NRF) (NRF-2015R1D1A1A01060810) funded by the Ministry of Science and ICT, Republic of Korea.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Aird KM, Iwasaki O, Kossenkov AV, Tanizawa H, Fatkhutdinov N, Bitler BG, Le L, Alicea G, Yang TL, Johnson FB, et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol. 2016;215:325–334. doi: 10.1083/jcb.201608026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev. 2005;15:496–506. doi: 10.1016/j.gde.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Davalos AR, Coppe JP, Campisi J, Desprez PY. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273–283. doi: 10.1007/s10555-010-9220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devgan V, Mammucari C, Millar SE, Brisken C, Dotto GP. p21WAF1/Cip1 is a negative transcriptional regulator of Wnt4 expression downstream of Notch1 activation. Genes Dev. 2005;19:1485–1495. doi: 10.1101/gad.341405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan R, Flint KJ, Jones N. Phosphorylation of E2F-1 modulates its interaction with the retinoblastoma gene product and the adenoviral E4 19 kDa protein. Cell. 1994;78:799–811. doi: 10.1016/s0092-8674(94)90522-3. [DOI] [PubMed] [Google Scholar]
- Ferrandiz N, Caraballo JM, Garcia-Gutierrez L, Devgan V, Rodriguez-Paredes M, Lafita MC, Bretones G, Quintanilla A, Munoz-Alonso MJ, Blanco R, et al. p21 as a transcriptional co-repressor of S-phase and mitotic control genes. PloS One. 2012;7:e37759. doi: 10.1371/journal.pone.0037759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Quaas M, Steiner L, Engeland K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016;44:164–174. doi: 10.1093/nar/gkv927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagger G, Doetzlhofer A, Schuettengruber B, Haidweger E, Simboeck E, Tischler J, Chiocca S, Suske G, Rotheneder H, Wintersberger E, et al. The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol Cell Biol. 2003;23:2669–2679. doi: 10.1128/MCB.23.8.2669-2679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jenkins CW, Nichols MA, Xiong Y. Cell cycle expression and p53 regulation of the cyclin-dependent kinase inhibitor p21. Oncogene. 1994;9:2261–2268. [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkey CS, Churchill ME. The high mobility group box: the ultimate utility player of a cell. Trend Biochem Sci. 2012;37:553–562. doi: 10.1016/j.tibs.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaas M, Muller GA, Engeland K. p53 can repress transcription of cell cycle genes through a p21(WAF1/CIP1)-dependent switch from MMB to DREAM protein complex binding at CHR promoter elements. Cell Cycle. 2012;11:4661–4672. doi: 10.4161/cc.22917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin YJ, Kim MS, Kim MS, Lee J, Kang M, Jeong JH. High-mobility group box 2 (HMGB2) modulates radioresponse and is downregulated by p53 in colorectal cancer cell. Cancer Biol Ther. 2013;14:213–221. doi: 10.4161/cbt.23292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi N, Carames B, Ronfani L, Ulmer U, Komiya S, Bianchi ME, Lotz M. Aging-related loss of the chromatin protein HMGB2 in articular cartilage is linked to reduced cellularity and osteoarthritis. Proc Natl Acad Sci USA. 2009;106:1181–1186. doi: 10.1073/pnas.0806062106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi N, Kawakami Y, Maruyama I, Lotz M. HMGB proteins and arthritis. Hum Cell. 2017;31:1–9. doi: 10.1007/s13577-017-0182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JO. HMG1 and 2: architectural DNA-binding proteins. Biochem Soc Trans. 2001;29(Pt 4):395–401. doi: 10.1042/bst0290395. [DOI] [PubMed] [Google Scholar]
- van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.