

Abstract
Cells cope with diverse intrinsic and extrinsic stimuli in order to make adaptations for survival. The epigenetic landscape plays a crucial role in cellular adaptation, as it integrates the information generated from stimuli. Signaling pathways induced by stimuli communicate with chromatin to change the epigenetic landscape through regulation of epigenetic modifiers. Metabolic dynamics altered by these stimuli also affect the activity of epigenetic modifiers. Here, I review the current understanding of epigenetic regulation via signaling and metabolic pathways. In addition, I will discuss possible ways to achieve specificity of epigenetic modifications through the cooperation of stimuli-induced signal transduction and metabolic reprogramming.
Keywords: cancer, differentiation, epigenetics, metabolism, signaling
INTRODUCTION
The epigenetic landscape is crucial to determine the state, behavior and identity of cells through modulation of gene expression. There are numerous epigenetic modifiers, including DNA/histone modifying and de-modifying enzymes (known as writers and erasers, respectively), proteins that recognize specific chromatin structure (readers), and nucleo-some remodeling enzymes. Accumulating evidence shows that distinctive epigenetic alterations are related to cellular differentiation and development (Boland et al., 2014). In addition, aberrant epigenetic changes are functionally associated with many human diseases (Portela et al., 2010). Therefore, understanding the mechanisms of change for the epigenetic landscape is pivotal for both basic and clinical biology.
Signal transduction induced by cell-intrinsic and -extrinsic stimuli is an important way to modify the epigenetic landscape (Badeaux et al., 2013). Signal-mediated posttranslational modifications (PTMs) such as phosphorylation, acetylation, and/or methylation on epigenetic modifiers can modulate the catalytic activity of proteins. Given that many epigenetic modifiers form protein complexes with multiple subunits, including transcription factors, signal-dependent PTMs on the associating proteins can also affect the activity of epigenetic regulators.
Metabolism is a tightly regulated cellular activity to generate energy source, reducing powers, and anabolic building blocks for cell viability, growth, and proliferation. Multiple inputs, such as nutrient availability, the engagement of signaling pathways, and environmental stress, exert influence on cell metabolism. Recently, the cellular metabolism has been implicated in the control of diverse non-metabolic processes, including cell-to-cell communication and cell differentiation (Pavlova et al., 2016). Modifying the epigenetic landscape is one of the emerging non-metabolic effects of metabolism on cell biology. Most, if not all, epigenetic enzymes utilize intermediary metabolites as substrates or co-factors for catalytic reactions. Moreover, unlike kinases, for which the Km values are much lower than intracellular concentration of ATP, the epigenetic enzymes that require metabolites other than ATP have Km values within the range of physiological concentrations of the metabolites (Reid et al., 2017). Therefore, changes in metabolic activity characterized by the concentration and the flux of metabolites can affect the activities of epigenetic modifiers and thereby cause the reconstruction of the epigenetic landscape.
Here, I briefly review the current understanding of how epigenetic modifiers integrate information from dynamic metabolic activities, mainly focusing on DNA/histone methylation, and histone acetylation. Furthermore, to provide insight into the specificity of metabolic activity on epigenetic modifications, I will discuss the functional cooperation between signal transduction and metabolic reprogramming in selecting target genes in different cellular contexts.
EPIGENETIC LANDSCAPE RECONFIGURED THROUGH COOPERATION OF SIGNALING PATHWAYS AND THE ACCOMPANIED ELEVATION IN ACETYL-COA PRODUCTION
One of the critical signaling cascades activated in response to growth factors is the phosphoinositide 3-kinae (PI3K)/AKT pathway, which is highly conserved throughout metazoan. Upon binding of extracellular growth factors to cell membrane receptors, including receptor tyrosine kinases (RTKs), PI3K phosphorylates membrane phosphatidylinositol lipids, which recruit and activate downstream effector kinase AKT. Subsequently, expression of the glucose transporter GLUT1 (glucose transporter 1) is upregulated, and the cellular uptake of glucose is enhanced. In addition, AKT phosphorylates and, in turn, activates several enzymes involved in glycolysis, such as HK (hexokinase) and PFK1/2 (phosphofructokinase 1/2), promoting glucose catabolism (Hay, 2016). Pyruvate, the product generated from glucose by glycolysis enters the mitochondria and is converted to acetyl-CoA. The acetyl-CoA is then condensed with oxaloacetate (OAA) to form citrate, some of which is transported to the cytosol rather than being oxidized via the tricarboxylic acid (TCA) cycle in the mitochondrial matrix. Cytosolic citrate is then converted back to acetyl-CoA by ACL (ATP-citrate lyase). AKT-mediated phosphorylation of ACL promotes cytosolic production of acetyl-CoA, which serves as the acetyl-donor for the acetylation reaction (Fig. 1).
Fig. 1. Cooperation of AKT signaling pathway and the accompanied upregulation of acetyl-CoA metabolism to promote histone acetylation.
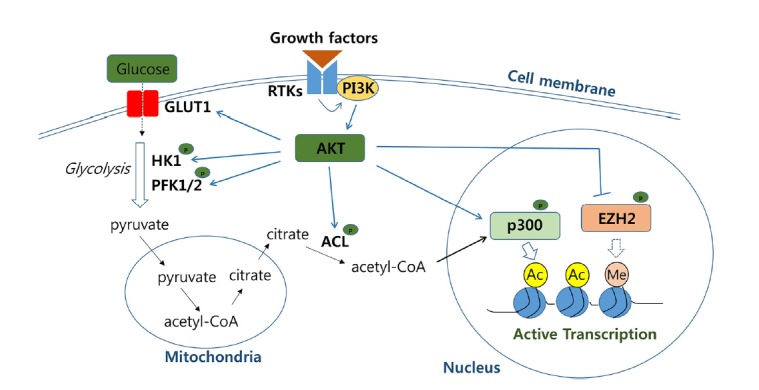
Growth factor-induced AKT signaling pathway upregulates acetyl-CoA production and phosphorylates histone modifying enzymes such as p300 and EZH2 to enhance histone acetylation on chromatin. The enzymes involved in the pathways are in bold. P in green dots denotes for phosphorylation-mediated regulation. Ac and Me refer to acetylation and methylation, respectively. GLUT1; glucose transporter 1, HK1; hexose kinase 1, PFK1/2; phosphofructokinase 1/2, RTKs; receptor tyrosine kinases, PI3K; phosphoinositide 3-kinae, ACL; ATP-citrate lyase, EZH2; enhancer of zeste homolog 2.
PI3K/AKT signaling contributes to aerobic glycolysis, consuming a large amount of glucose to generate lactate (Hay, 2016). Highly proliferating cells, such as cancer and embryonic stem (ES) cells, are characterized by aerobic glycolysis. Production of cytosolic acetyl-CoA, which is dependent on the AKT activity as well as the glucose availability in culture media, determines the histone acetylation level in the highly proliferating cancer cells (Lee et al., 2014). Interestingly, transcriptomic profiling showed that the effect of enhanced acetyl-CoA production was specific rather than global: expression of the genes involved in cell growth and proliferation were selectively increased. However, it remains unclear how a global change in the acetyl-CoA concentration mediates the epigenetic regulation specific for promoting cell growth.
There is perhaps a hint as to mechanism of this specificity from an analogous yeast study showing that when intracellular acetyl-CoA level was elevated, histone acetylation in growth-promoting genes was upregulated by the SAGA complex carrying Gcn5 as the catalytic subunit, with a significant increase in the gene expression (Cai et al., 2011). This study demonstrated that auto-acetylation of the SAGA complex, which was dependent on acetyl-CoA level, facilitated the histone acetylation activity of SAGA toward the target genes. These findings imply that PTMs such as acetylation on specific epigenetic modifiers are involved in selecting chromatin regions to integrate information from metabolic activity. AKT, which enhances acetyl-CoA production, has been shown to phosphorylate the histone acetyltransferase p300 and thereby enhance the co-transcriptional activity of the enzyme (Huang et al., 2005). By contrast, AKT-mediated phosphorylation of the histone lysine methyl-transferase EZH2 suppresses the ability of EZH2 to methylate lysine 27 residue on histone H3, which silences gene expression (Cha et al., 2005). These results suggest that the acetyl-CoA produced by the AKT signaling pathway may be preferentially used for p300-dependent histone acetylation. The AKT-induced inactivation of EZH2 may aid in selecting target genes where histone residues are available for acetylation rather than methylation. Collectively, these findings suggest that the growth-factor stimulated PI3K/AKT pathway serves as a major player in configuring growth promotion in the epigenetic landscape both by reprogramming acetyl-CoA metabolism and by regulating the activities of epigenetic enzymes in a phosphorylation-dependent manner.
Growth factor-induced signaling pathways can mediate nuclear localization of acetyl-CoA generating enzymes. Upon stimuli by serum or epidermal growth factor, pyruvate dehydrogenase complex (PDC), which produces acetyl-CoA, translocates from mitochondria to nucleus (Sutendra et al., 2014). The increase of nuclear PDC is dependent on cell cycle as well. Inhibition of nuclear PDC leads to a decrease in histone acetylation in genes involved in cell cycle progression, resulting in the reduced expression of these genes. Cell differentiation signals can also drive nuclear localization of acetyl-CoA producing enzymes ACL and ACSS2 (Mews et al., 2017; Wellen et al., 2009). Strikingly, the physical association of ACSS2 to upregulated neuronal genes displaying elevated histone acetylation has been observed in differentiated mouse neurons. An ACSS2-enriched motif analysis identified the transcription factor YY1 (Yin Yang1), which interacts with the histone acetyltransferase CBP, as a potential interacting partner for chromatin association. Supporting the possibility of ACSS2/YY1/CBP complex formation, ACSS2 physically associates with the histone acetyltransferase CBP in the neuron. These findings raise the possibility that differentiation-related stimuli drive epigenetic regulation through a signal-induced “on-site” acetyl-CoA generation for specific sets of genes associated with cell differentiation.
REGULATION OF THE EPIGENETIC LANDSCAPE BY ENERGY STRESS-RESPONSIVE AMPK SIGNALING AND THE SUBSEQUENT CHANGE IN NAD+ METABOLISM
AMPK (AMP-activated protein kinase) is an AMP-sensitive protein kinase that functions as an energy stress sensor in cells (Mihaylova et al., 2011). AMPK signaling activated by ATP depletion and/or an increase in AMP/ATP ratio mediates cellular responses affecting cellular proliferation, apoptosis, and metabolism. A major downstream event of AMPK activation is the retention and restoration of the cellular ATP pool by rewiring genetic and metabolic programs, including an increase in intracellular NAD+ level. When AMPK is activated by glucose-restriction, which decreases the cellular ATP pool or by the treatment of AICAR (5-aminoimidazole-4-carboxamide- 1-b-D-ribofuranoside), a direct AMPK activator, the expression of NAMPT (nicotinamide phosphoribosyltransferase), which is involved in the NAD biosynthesis salvage pathway, is upregulated to promote NAD production (Canto et al., 2009). At the same time, AMPK increases the ratio of NAD+/NADH in a mitochondrial oxidation-dependent manner.
Sirtuins are a family of histone lysine deacetylases requiring NAD+ as the essential catalytic cofactor. Of the seven different sirtuins (SIRT1-7), SIRT1 is a well-characterized effector of AMPK signaling. Enhanced production of NAD+ upon AMPK activation promotes the activity of SIRT1 (Canto et al., 2009), which deacetylates lysine residues of histones, including the acetylated lysine 9 of histone H3 (H3K9ac), H3K14ac, H4K16ac, and H1K26ac (Chang and Guarente, 2014). In addition, SIRT1 removes acetylation from non-histone proteins such as PGC-1α, the activity of which is positively regulated by AMPK-mediated phosphorylation. By contrast, acetylation of PGC-1α suppresses the transcriptional activity of the protein, even when it is phosphorylated. Thus, the SIRT1-mediated deacetylation of PGC-1α functions to eliminate a negative mark from the protein (Canto et al., 2009). Consequently, AMPK signaling leads to the transcriptional activation of PGC-1α target regions through direct phosphorylation of PGC-1α and an increase in NAD+ level, which facilitates SIRT1 histone deacetylase activity.
AMPK was also observed to phosphorylate SIRT1 (Lau et al., 2014). Physical association of SIRT1 with DBC1 (deleted in breast cancer 1) is inhibitory of the lysine deacetylase activity of the protein (Kim et al., 2008; Zhao et al., 2008). When AMPK is activated by different compounds, including AICAR, SIRT1 becomes phosphorylated and is released from DBC1 (Lau et al., 2014; Nin et al., 2012). In turn, inactivation of p53 by SIRT1-dependent deacetylation is accompanied by AMPK activation. These studies imply that AMPK leads to transcriptional inactivation of the p53 target regions. Altogether, energy stress sensed by the AMPK pathway affects changes in the epigenetic landscape by changing NAD+ metabolism and by mediating PTMs of SIRT1 and the interacting transcription factors (Fig. 2).
Fig. 2. Energy stress induced AMPK signaling to promote NAD+-dependent SIRT1 activity.

Upon energy stress-stimuli, the AMPK signaling pathway is upregulated, resulting in the increase of intracellular NAD+ level. At the same time, histone deacetylase SIRT1 and/or transcription factors such as PGC-1α are phosphorylated in AMPK-dependent manner. The enzymes involved in the pathways are in bold. P in green dots denotes for phosphorylation-mediated regulation. De-Ac in blue circles indicates deacetylation-mediated regulation. AMPK; AMP-activated kinase, NAMPT; nicotinamide phosphoribosyltransferase, SIRT1; sirtuin 1, PGC-1α; peroxisome proliferator-activated receptor gamma coactivator 1-alpha, AICAR; 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside.
SAM METABOLISM, WHICH IS RESPONSIVE TO NUTRIENT STATUS AND THE ACTIVITIES OF SIGNALING PATHWAYS, IS LINKED TO A DNA/HISTONE METHYLATION-DEPENDENT EPIGENETIC LANDSCAPE
Methyltransferases targeting DNA and lysine or arginine residues of histones catalyze the transfer of methyl groups from S-adenosylmethionine (SAM) to specific substrates. Thus, changes in cellular SAM levels can influence the enzymatic activities of methylation writers. One-carbon metabolism that couples the folate cycle and the methionine cycle is essential for SAM production (Fig. 3). When SAM production is restricted in cancer cells or in mouse models by methionine depletion, histone methylation prominently decreases, with trimethylation at lysine 4 of H3 (H3K4me3) exhibiting the largest change (Mentch et al., 2015). In the same study, analyzing significantly depleted regions of H3K4me3 in cancer cells showed that cancer-associated genes such as AKT1, MYC and MAPK were enriched for loss of H3K4me3 at promoters, with resulting decreased expression. Similarly, when threonine metabolism was attenuated in mouse embryonic stem cells by threonine restriction in culture medium or by genetic depletion of TDH (threonine dehydrogenase), the levels of cellular SAM and H3K4me3 decreased, affecting cell growth and differentiation (Shyh-Chang et al., 2013). These studies suggest that the status of nutrients such as methionine and threonine fueling one-carbon metabolism has a dramatic effect on the epigenetic landscape through histone methylation.
Fig. 3. Cooperation of SAM metabolism and cell signaling to alter DNA/histone methylation.

DNA/histone methylation is tightly linked with one-carbon metabolism. The oncogenic JAK2 signaling pathway regulates arginine methyltransferase PRMT5 activity and subsequently modulates methylation of histones and non-histone proteins, including transcription factors, NF-κB, p53 and E2F-1. The enzymes involved in the pathways are in bold. Metabolism pathways are marked in italic. P in green dots denotes for phosphorylation-mediated regulation. TDH; threonine dehydrogenase, MTAP; methylthioadenosine phosphorylase, PRMT5; protein arginine N-methyltransferase 5, JAK2; Janus kinase 2, DNMT1/3; DNA methyltransferase 1/3, LKB1; liver kinase B1, mTOR; mechanistic target of rapamycin, SAM; S-adenosylmethionine, SAH; S-adenosylhomocysteine, Hcy; homocysteine, MTA; methylthioadenosine.
Disorders of methionine metabolism can occur through pathological loss of MTAP (methylthioadenosine phosphorylase), which is frequently linked to the deletion of the tumor suppressor gene CDKN2A in sporadic cancer due to physical proximity (Kryukov et al., 2016). MTAP metabolizes MTA (methylthioadenosine) to adenine and methionine in the methionine salvage pathway. Interestingly, MTA selectively inhibits the enzymatic activity of the arginine methyltransferase PRMT5 but not the other histone methyltransferases. Genetic manipulation to modulate the expression of MTAP leads to a change in the level of the symmetric dimethylation of arginine 3 on histone H4 (H4R3me2s), which is associated with transcriptional regulation. In addition, PRMT5 regulates transcription through the methylation of transcription factors such as NF-κB, p53, and E2F-1 (Stopa et al., 2015). Therefore, it is possible that the aberrant methionine metabolism caused by MTAP inactivation influences the epigenetic landscape through the alteration of PRMT5 activity, affecting methylation of histones and transcription factors. In the myeloproliferative neoplasm, oncogenic JAK2 phosphorylates and inhibits PRMT5, preventing histone methylation (Liu et al., 2011). Intriguingly, unlike the sporadic solid cancer associated with MTAP loss, for which PRMT5 depletion was detrimental, inhibition of PRMT5 in hematopoietic cells promotes myeloproliferation and/or erythroid differentiation by altering gene expression profiles. The distinctive consequence of PRMT5 loss in different cellular context implies that the specificity is achieved through the cooperation of different metabolic and signaling activities in a given condition.
Serine biosynthesis is important for one-carbon metabolism by fueling the methionine cycle. A few genetic factors, including NRF2, c-MYC, G9a, and mTOR, regulate serine biosynthesis and in turn SAM metabolism (Mattaini et al., 2016). Recently, the signaling pathway to functionally link serine and subsequent SAM metabolism with DNA methylation has been observed in genetically modified mouse models (Kottakis et al., 2016). When the loss of LKB1 (liver kinase B1), an upstream kinase of AMPK, was combined with KRASG12D, the serine biosynthesis pathway was induced to increase the serine concentration, rendering cells dependent on glycolysis-mediated serine availability. At the same time, the expression of the DNA methyltransferases DNMT1 and DNMT3 was upregulated in an mTOR-dependent manner, resulting in the increase of DNA but not histone methylation in the cells. Given that AMPK suppresses mTOR activity, this study suggests that the activation of mTOR pathway through the LKB1 loss-mediated AMPK inactivation is the key molecular event to trigger the upregulation of serine biosynthesis and DNMTs expression and, in turn, enhance DNA methylation.
Although the evidence of a functional link between SAM metabolism and DNA/histone methylation is accumulating, the specificity of this link remains unclear in many cases. A plausible mechanism for the specificity can be found in the cooperation of mTOR-dependent serine metabolism and DNMTs expression as described above. Alternatively, the specificity may be determined through the differential biochemical characteristics of methyltransferases. Kinetic properties of many histone methyltransferases have been studied (Mentch et al., 2016). Interestingly, there is a range of Km values for SAM across the histone methyltransferases, suggesting that a change in SAM concentration would differentially affect enzymatic activity. For example, MLL1, with a Km for SAM of 10.4 μM, would be more responsive to a decrease in the intracellular SAM level than EZH2, with a Km for SAM of 1.64 μM. Related to this, methionine restriction to reduce SAM level results in a much larger decrease in H3K4me3, which can be generated by MLL1, than EZH2-mediated H3K27me3 (Mentch et al., 2015).
In addition, the local supply of the methyl donor SAM to specific chromatin regions may be a specificity factor. It has been shown that the enzymes involved in SAM metabolism form nuclear protein complexes with specific DNA/histone methyltransferases and/or transcription factors. The MAT (methionine adenosyltransferase) complex with the transcription repressor MafK, found in mouse cells, and the SESAME (serine-responsive SAM-containing metabolic enzyme) complex purified from yeast cells are recruited to specific chromatin regions and thus mediate the metabolic effect on specific target sites (Katoh et al., 2011; Li et al., 2015). The list of similar complexes containing metabolic enzymes along with epigenetic enzymes may be expanded.
CHANGE IN α-KG PRODUCTION REGULATES DNA/HISTONE DEMETHYLATION
The removal of methylation from DNA and histones requires ten-eleven translocation (TET) enzymes and histone demethylases, respectively. There are two types of histone demethylases. LSD1 and LSD2 are in the flavin adenine dinucleotide (FAD)-dependent amine oxidase family and the others, containing JmjC domain, belong to the alpha-ketoglutarate (α-KG)-dependent hydroxylase family, the activities of which require both Fe2+ and oxygen (Kooistra et al., 2012). α-KG is generated as an intermediate metabolite in the TCA cycle or is produced by the transamination of cytosolic glutamate converted from glutamine. Therefore, the intracellular α-KG level can be influenced by the activity of mitochondrial TCA cycle and glutamine availability. In proliferative cells, glutamine uptake is upregulated and promotes α-KG production (Wise et al., 2010).
Intracellular α-KG level is critical for mouse and human embryonic stem (ES) cell maintenance and differentiation (Carey et al., 2015; Hwang et al., 2016). A decrease in α-KG level either by glutamine starvation or by genetic depletion of PAST1, which participates in α-KG production, results in a failure to maintain the pluripotency of the ES cells due to an attenuation of DNA and histone demethylases activity. Interestingly, for different histone methylation, H3K9me3 and H3K36me3 are prominently affected by a change in α-KG level. By contrast, H3K4me3 level is not responsive to the manipulation of intracellular α-KG concentration. Contrary to these studies, an increase in α-KG accelerates the differentiation of primed human pluripotent cells (PSCs) or mouse epiblast stem cells (EpiSCs) by promoting the activities of TETs and JmjC family histone demethylases (TeSlaa et al., 2016). This opposite outcome in different stages of stem cell differention suggests that despite the same molecular mechanism, the effect of α-KG is context-specific. Nonetheless, physiological changes in α-KG level, which are likely to be modulated by differentiation stimuli, have proven to be crucial for the features of the epigenetic landscape associated with distinctive cellular states.
The influence of glutamine availability on epigenetic regulation has also been observed in tumors. Depending on the intratumoral regions, glutamine levels vary largely (Pan et al., 2016). In the core region of solid tumors with poor vasculature, glutamine is significantly deficient compared with peripheral regions. In turn, glutamine deficiency leads to the depletion of the α-KG level in the core of the tumor, suppressing histone demethylation activities. An increase of H3K27me3 is the most prominent in the α-KG-deficient region. Furthermore, the results of this study demonstrate that lack of glutamine and subsequent increase in H3K27me3 at the core of tumor is involved in cancer cell dedifferentiation and cancer drug resistance by suppressing expression of specific genes.
The mechanism by which specific histone demethylases respond to changes in global α-KG level is still unclear. The differential kinetic properties of enzymes that confer sensitivities to α-KG or other co-factors may be responsible for the specificity. Careful biochemical characterization of histone demethylases remains to be done. PTMs may be an alternative explanation for achieving the specificity. PKA-dependent phosphorylation activates the enzymatic activity of KDM7C by facilitating complex formation with ARID5B (Baba et al., 2011). Similarly, CDK2 phosphorylates and activates KDM7B for demethylation of H3K9me1/2 (Sun et al., 2015). However, whether the signaling pathways are cooperative with α-KG metabolism needs to be addressed.
PERSPECTIVE
For the last couple of decades, our understanding of how epigenetic regulations function in diverse cellular processes has progressed at an astonishing pace. However, determining the molecular details of how different stimuli generate distinctive epigenetic landscapes needs more investigation. Careful biochemical characterization of individual epigenetic enzymes, such as determining the Km values of histone demethylases for α-KG, is necessary. In addition, it will be important to determine the dynamics of intermediary metabolites such as acetyl-CoA and methionine in different physiological states driven by intrinsic and extrinsic stimuli. Further, the mechanistic understanding of how the signaling pathways reprogram metabolic activities will be informative in revealing their specificity. Altogether, understanding the specificity of epigenetic regulation through signaling and metabolism (Fig. 4) will pave the way towards comprehending how cells integrate information from environmental factors such as diet to adapt for survival.
Fig. 4. Stimuli-responsive cellular adaptations through specific epigenetic regulation.
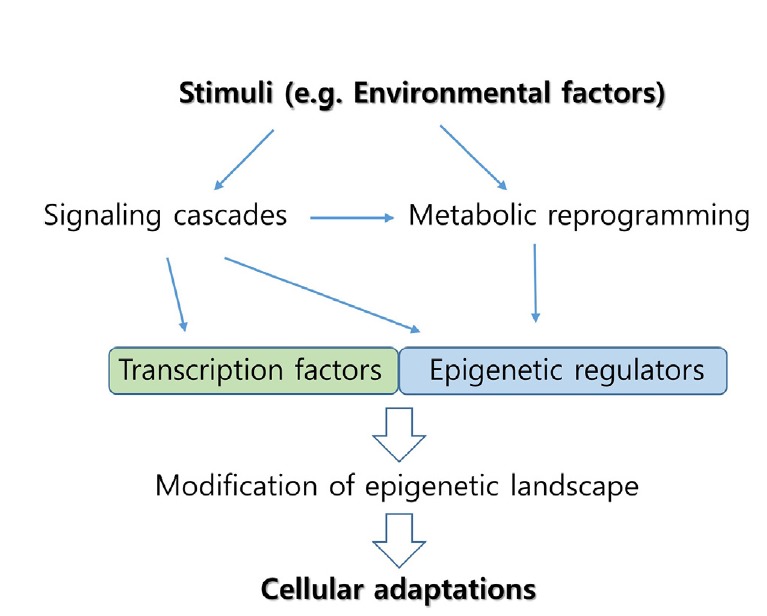
Cellular stimuli induce signaling cascades and metabolic reprogramming, leading to the modification of epigenetic regulators and transcription factors at multiple levels, such as posttranslational modifications or the availability of metabolites. Subsequent epigenetic changes targeted by the epigenetic regulators, along with interacting transcription factors, mediate cellular adaptations to the given stimuli by modulating gene expressions.
ACKNOWLEDGMENTS
This work was supported by the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning (NRF-2013R1A1A 1006638) and by the KRIBB Research Initiative Program.
REFERENCES
- Baba A, Ohtake F, Okuno Y, Yokota K, Okada M, Imai Y, Ni M, Meyer CA, Igarashi K, Kanno J, et al. PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nat Cell Biol. 2011;13:668–675. doi: 10.1038/ncb2228. [DOI] [PubMed] [Google Scholar]
- Badeaux AI, Shi Y. Emerging roles for chromatin as a signal integration and storage platform. Nat Rev Mol Cell Biol. 2013;14:211–224. doi: 10.1038/nrm3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland MJ, Nazor KL, Loring JF. Epigenetic regulation of pluripotency and differentiation. Circ Res. 2014;115:311–324. doi: 10.1161/CIRCRESAHA.115.301517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. 2011;42:426–437. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413–416. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metabol. 2014;25:138–145. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16:635–649. doi: 10.1038/nrc.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WC, Chen CC. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol Cell Biol. 2005;25:6592–6602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang IY, Kwak S, Lee S, Kim H, Lee SE, Kim JH, Kim YA, Jeon YK, Chung DH, Jin X, et al. Psat1-dependent fluctuations in alpha-Ketoglutarate affect the timing of ESC differentiation. Cell Metabol. 2016;24:494–501. doi: 10.1016/j.cmet.2016.06.014. [DOI] [PubMed] [Google Scholar]
- Katoh Y, Ikura T, Hoshikawa Y, Tashiro S, Ito T, Ohta M, Kera Y, Noda T, Igarashi K. Methionine adenosyltransferase II serves as a transcriptional corepressor of Maf oncoprotein. Mol Cell. 2011;41:554–566. doi: 10.1016/j.molcel.2011.02.018. [DOI] [PubMed] [Google Scholar]
- Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- Kooistra SM, Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13:297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- Kottakis F, Nicolay BN, Roumane A, Karnik R, Gu H, Nagle JM, Boukhali M, Hayward MC, Li YY, Chen T, et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature. 2016;539:390–395. doi: 10.1038/nature20132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. 2016;351:1214–1218. doi: 10.1126/science.aad5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AW, Liu P, Inuzuka H, Gao D. SIRT1 phosphorylation by AMP-activated protein kinase regulates p53 acetylation. Am J Cancer Res. 2014;4:245–255. [PMC free article] [PubMed] [Google Scholar]
- Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metabol. 2014;20:306–319. doi: 10.1016/j.cmet.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Swanson SK, Gogol M, Florens L, Washburn MP, Workman JL, Suganuma T. Serine and SAM responsive complex SESAME regulates histone modification crosstalk by sensing cellular metabolism. Mol Cell. 2015;60:408–421. doi: 10.1016/j.molcel.2015.09.024. [DOI] [PubMed] [Google Scholar]
- Liu F, Zhao X, Perna F, Wang L, Koppikar P, Abdel-Wahab O, Harr MW, Levine RL, Xu H, Tefferi A, et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell. 2011;19:283–294. doi: 10.1016/j.ccr.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattaini KR, Sullivan MR, Vander Heiden MG. The importance of serine metabolism in cancer. J Cell Biol. 2016;214:249–257. doi: 10.1083/jcb.201604085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentch SJ, Locasale JW. One-carbon metabolism and epigenetics: understanding the specificity. Ann New York Acad Sci. 2016;1363:91–98. doi: 10.1111/nyas.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, Gomez Padilla P, Ables G, Bamman MM, Thalacker-Mercer AE, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metabol. 2015;22:861–873. doi: 10.1016/j.cmet.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mews P, Donahue G, Drake AM, Luczak V, Abel T, Berger SL. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature. 2017;546:381–386. doi: 10.1038/nature22405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nin V, Escande C, Chini CC, Giri S, Camacho-Pereira J, Matalonga J, Lou Z, Chini EN. Role of deleted in breast cancer 1 (DBC1). protein in SIRT1 deacetylase activation induced by protein kinase A and AMP-activated protein kinase. J Biol Chem. 2012;287:23489–23501. doi: 10.1074/jbc.M112.365874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, Yang Y, Hernandez-Davies JE, Rosales KK, Li H, et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol. 2016;18:1090–1101. doi: 10.1038/ncb3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metabol. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- Reid MA, Dai Z, Locasale JW. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol. 2017;19:1298–1306. doi: 10.1038/ncb3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013;339:222–226. doi: 10.1126/science.1226603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopa N, Krebs JE, Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci. 2015;72:2041–2059. doi: 10.1007/s00018-015-1847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Huang Y, Wei Q, Tong X, Cai R, Nalepa G, Ye X. Cyclin E-CDK2 protein phosphorylates plant homeodomain finger protein 8 (PHF8). and regulates its function in the cell cycle. J Biol Chem. 2015;290:4075–4085. doi: 10.1074/jbc.M114.602532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell. 2014;158:84–97. doi: 10.1016/j.cell.2014.04.046. [DOI] [PubMed] [Google Scholar]
- TeSlaa T, Chaikovsky AC, Lipchina I, Escobar SL, Hochedlinger K, Huang J, Graeber TG, Braas D, Teitell MA. alpha-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metabol. 2016;24:485–493. doi: 10.1016/j.cmet.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427–433. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]