

Abstract
The molecular mechanisms by which TGF-β and interleukin 4 (IL-4) signaling control the differentiation of IL-9-producing CD4+ T (TH9) cells remain incompletely understood. We show here that the DNA-binding inhibitor Id3 regulated Th9 cell differentiation, as deletion of Id3 increased IL-9 production from CD4+ T cells. Mechanistically, TGF-β1 and IL-4 down-regulated Id3 expression and this process required the kinase TAK1. Reduction of Id3 expression enhanced the binding of the transcription factors E2A and GATA-3 in the Il9 promoter region, which promoted Il9 gene transcription. Importantly, Id3 control of TH9 cells differentiation regulated anti-tumor immunity in an experimental melanoma-bearing model in vivo, and also in human CD4+ T cells in vitro. Thus, this study reveals a previously unrecognized TAK1-Id3-E2A-GATA-3 pathway that regulates TH9 cell differentiation.
Naïve CD4+ T cells differentiate into distinct subsets upon activation, including T helper type 1 (TH1), TH2, TH17 or regulatory T cells (Treg cells), after encountering antigen in different cytokine milieu microenvironments. IL-9-producing CD4+ T (TH9) cells1 are induced from naïve CD4+ T cells by transforming growth factor-β (TGF-β) together with interleukin 4 (IL-4) during T cell receptor (TCR) stimulation in vitro2, 3. TH9 cells have been shown to play roles in allergic inflammation4, 5, 6, autoimmune diseases3, 7, 8 and cancer9, 10. Although the transcription factors STAT6 2, 11, GATA-33, PU.112, and IRF4 6 are all reported to participate in the differentiation of TH9 cells, the transcriptional networks that control TH9 differentiation have not been fully understood. In addition, the downstream molecular mechanisms by which TGF-β1 and IL-4 signals co-operatively drive the differentiation of TH9 cells are not fully understood.
TGF-β1 mediates the differentiation of Treg cells or TH17 cells depending on the absence or presence of IL-6 respectively13, 14, 15. Id3, a transcription factor that inhibits DNA-binding of basic helix-loop-helix (bHLH) proteins, such as E2A, can control the TGF-β1-mediated reciprocal differentiation of Treg cells and TH17 cells in mice16. Moreover, Id3 deficiency increases IL-4 production from T cells concomitant with high expression of GATA-3. Because TH9 cell differentiation requires both TGF-β and IL-4 signaling, we investigated if Id3 plays a role in TH9 cell differentiation.
Here, we show that Id3 regulated the differentiation of TH9 cells; deletion of Id3 increased the differentiation of TH9 cell from naïve CD4+ T cells. Id3 expression in CD4+ T cells was down-regulated by TGF-β1 and IL-4 stimulation, a process that required the kinase TAK1. Reduction of Id3 expression led to increased binding of transcription factors E2A and GATA-3 at the Il9 promoter region, resulting in increased Il9 gene transcription in naïve CD4+ T cells in response to TGF-β1 and IL-4. Importantly, Id3 controlled the anti-tumor immunity of TH9 cells in an experimental tumor-bearing model in vivo and, IL-9 production was also regulated by Id3 in human CD4+ T cells as well. Collectively, our data revealed a previously unrecognized TAK1-Id3-E2A-GATA-3 pathway in regulation of TH9 cell differentiation.
Results
Id3 deficiency increases IL-9 production in T cells in vitro
The null mutation of Id3 (Id3−/−) results in defective Treg generation from naïve CD4+ T cells, largely due to uncontrolled IL-4 production by the Id3−/− T cells16. As such, we investigated whether the Id3 deficiency also caused increased IL-9 production in CD4+ T cells. To this end, we cultured naïve CD4+CD25−CD62L+ T cells from wild-type or Id3−/− mice17 with TGF-β1 and/or IL-4. We found that Id3-deficient T cells had enhanced expression of Il9 mRNA and IL-9 protein in response to TGF-β1 plus IL-4 compared to wilt-type T cells (Fig. 1a–c). As expected, wild-type naive CD4+ T cells did not produce IL-9 with TGF-β1 alone (Fig. 1b,d). However, naïve Id3−/− T cells readily produced substantial amounts of IL-9 in response to TGF-β1 alone, without exogenous IL-4 (Fig. 1b,d). This was due to the rapid and large induction of endogenous IL-4 in the Id3−/− T cells16, as neutralization of IL-4 with anti-IL-4 antibody completely abrogated IL-9 production in these cells (Fig. 1b,d). In addition to these observations, we used Id3f/f Cd4-Cre+ mice, in which Id3 is specifically deleted in T cells at the CD4+CD8+ (DP) thymocyte stage of differentiation18. Naïve CD4+ T cells from Id3f/fCd4-Cre+ mice had similar expression of the activation-associated markers CD25 and CD69, Il2 mRNA, similar apoptosis rates and T cell proliferation upon TCR stimulation compared to naïve CD4+ T cells form control Id3+/+ Cd4-Cre+ mice (Supplementary Fig. 1). However, Id3f/f Cd4-Cre+ naïve CD4+CD25− T cells differentiated substantially more into TH9 cells compared to CD4+ T cells from Id3+/+ Cd4-Cre+ mice in response to TCR stimulation together with TGF-β1 alone or TGF-β1 plus IL-4 (Fig. 1e). Importantly, we also acutely deleted Id3 in wild-type naive CD4+ T cells using siRNA and found enhanced expression of Il9 gene (Fig. 1f) and IL-9 protein (Fig. 1g) after stimulation with TGF-β1 plus IL-4 in Id3-knocked down naïve T cells compared to wild-type T cells. These data demonstrate that loss of Id3 affects differentiation of TH9 cells.
Figure 1. Id3 deficiency increases TH9 cell differentiation in vitro.
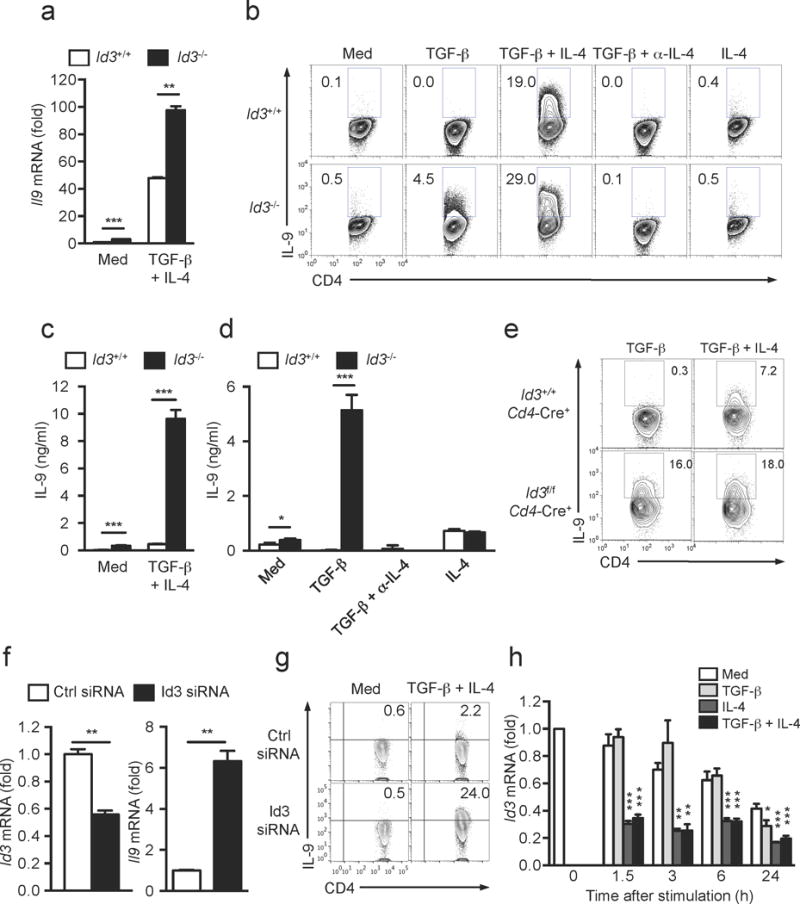
(a) mRNA expression of Il9 in naive CD4+ T cells from Id3+/+ or Id3−/− mice cultured with anti-CD3+CD28, with various combination of TGF-β1, IL-4 and anti-IL-4 for 24h. (b) Intracellular staining of IL-9 protein in CD4+ T cells differentiated as described in a for 72h. (c,d) IL-9 production in TH9-polarizing condition (c) and Treg (TGF-β)- or TH2 (IL-4)-polarizing conditions (d) was determined by ELISA from culture media as described in b. (e) Flow cytometry of intracellular IL-9 expression in CD4+ T cells differentiated from naïve CD4+CD25− T cells isolated from the spleen and lymph nodes of Id3f/fCd4-Cre+ or Id3+/+Cd4-Cre+ mice and cultured with anti-CD3+CD28 together with TGF-β1 alone or TGF-β1 plus IL-4 for 3 days. Data shown are representative of two independent experiments. (f) Il9 and Id3 mRNA expression in naive CD4+CD25− T cells isolated from wild-type mice, transfected with Id3-specific or control siRNA and stimulated with anti-CD3+CD28 with or without TGF-β1 plus IL-4 and analyzed 48h post-stimulation. Expression is relative to Gapdh expression. (g) Flow cytometry analysis of intracellular IL-9 protein in cells differentiated as in f at 72h post-stimulation with anti-CD3+CD28 with or without TGF-β1 plus IL-4. (h) Time course change of Id3 mRNA expression in wild-type naive CD4+ T cells cultured with anti-CD3+CD28 with or without TGF-β1 and/or IL-4. Statistical analysis was shown as comparison to Med of respective time points. Data are representative of two (e-g) or three (a-d) or pooled from five independent experiments (h). Error bars represent mean± SD of duplicate (a,f,h) or triplicate (c,d) well measurements. *p<0.05, **p<0.01, ***p<0.001 (Student’s t-test (a,c,d,f) or one-way ANOVA with post-hoc Bonferroni’s test (h)).
TGF-β1 and IL-4 down-regulate Id3
We next investigated whether TGF-β1 and IL-4 signaling affected Id3 expression. Id3 mRNA expression can be regulated by TGF-β1 signaling16; treatment of naïve CD4+ T cells with TGF-β1 resulted in more Id3 mRNA during the first 1–3 h, followed by less Id3 mRNA by 12-24h compared to cells with TCR stimulation alone (Fig. 1h). The aforementioned regulation of Id3 expression by TGF-β was abolished by using T cells that were deficient either TGF-β receptor I or II (TGFβRI or TGFβRII) (data not shown). When we quantified Id3 expression in naïve CD4+ T cells, we found that Id3 expression was rapidly and significantly decreased at 1.5 h after stimulation with TGF-β1 plus IL-4 compared to TCR stimulation alone, and this reduction lasted for at least 48 h (Fig. 1h and data not shown). Furthermore, the same degree of Id3 down-regulation was observed when cells were treated with IL-4 alone (Fig. 1h). Thus, Id3 expression is down-regulated by cytokine conditions that favour TH9 cell differentiation.
TAK1 is required for Id3 down-regulation downstream TGF-β1
We then studied the molecular mechanisms underlying TGF-β1 and/or IL-4-mediated Id3 down-regulation in CD4+ T cells. We used Tgfbr1f/f Cd4-Cre+ mice and tamoxifen-treated Tgfbr2f/f ER-Cre+ mice that lack either TGFβRI or TGFβRII expression in CD4+ T cells, respectively19 to investigate the requirement for TGF-β signaling in for TH9 cells differentiation. T cells that lacked either TGFβRI or TGFβRII failed to differentiate into TH9 cells following stimulation with TGF-β1 plus IL-4 (Fig. 2a–f), but developed normally or even showed increased differentiation into IFN-γ– or IL-4-producing T cells compared to wild-type (Supplementary Fig. 2a and data not shown). As expected, these TGFβR-deficient T cells failed to differentiate into Foxp3+ Treg cells and TH17 cells (Supplementary Fig. 2a and data not shown), confirming that TGF-β signaling is vital for TH9 cell differentiation.
Figure 2. Non-Smad TGF-β signaling is dominant during TH9 cell differentiation.
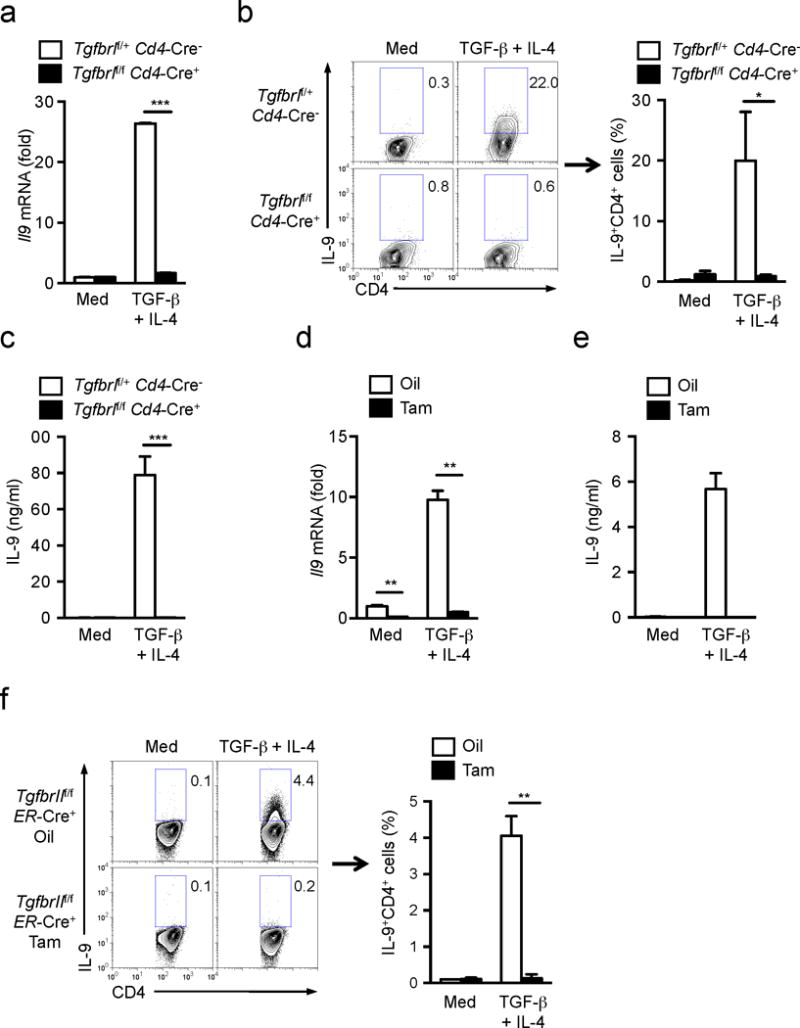
(a) mRNA expression of Il9 in naïve T cells from Tgfbr1f/fCd4-Cre+ and Tgfbr1f/+Cd4-Cre− mice cultured with anti-CD3+CD28 with or without TGF-β1 plus IL-4 for 24h. (b) Intracellular staining of IL-9 protein in CD4+ T cells differentiated as described in a for 72h. Left; Representative of three indepednent experiments. Right; Frequency of IL-9+ TH9 cells from three independent experiments. (c) IL-9 production in culture media of b was determined by ELISA. (d) mRNA expression of Il9 in naïve T cells from TgfbrIIf/fER-Cre+ mice that had been injected with oil or tamoxifen i.p. for consecutive 5 days cultured with anti-CD3+CD28 with or without TGF-β1 plus IL-4 for 24h. (e) IL-9 production in culture media of CD4+ T cells differentiated as described in d was determined by ELISA. (f) Intracellular staining of IL-9 protein in CD4+ T cells differentiated as in e for 72h. Left; Representative of two experiments. Right; Frequency of IL-9+ TH9 cells from two experiments. Data are representative of two (d, e, f(left)) or three (a, b(left), c) independent experiments or are pooled from two (f(right)) or three (b(right)) experiments. Error bars represent mean ± SD. *p<0.05, **p<0.01, ***p<0.001 (Student’s t-test,).
TGF-β1 engagement of its cognate receptors activates signaling through Smad (Smad2, 3 and 4) and non-Smad pathways20, 21, 22. The canonical Smad-dependent signaling pathway is required for the generation of Foxp3+ Treg cells and TH17 cell 19, 23. Smad-dependent signaling is also important for TH9 differentiation24, 25. Consistent with these notions, deletion of Smad2 (Smad2f/fLck-Cre), Smad3 (Smad3−/−), Smad4 (Smad4f/fCd4-Cre+) or Smad3 and Smad4 (Smad3−/−Smad4f/fCd4-Cre+) in T cells resulted in a partial decrease in TH9 cell differentiation compared to their respective wild-type T cells (Supplementary Fig. 2b–e). However, Smad3−/−Smad4f/fCd4-Cre+ T cells did not downregulate Id3 following stimulation with TGF-β1 and IL-4 (Supplementary Fig. 2f), suggesting that although the Smad-dependent pathway might play a role in IL-9 production in CD4+ T cells, it might not be directly associated with Id3.
As such, we investigated the Smad-independent pathways that regulate Id3 expression and IL-9 production. The TGF-β-activated kinase TAK1 has been shown to be an important mediator for TGF-β-triggered Smad-independent pathway22, 26. We treated normal naïve CD4+ T cells with the TAK1 inhibitor 5z-7-oxozeaenol under TH9 cell differentiation conditions and found that inhibition of TAK1 almost completely blocked the production of IL-9 in CD4+ T cells (Fig. 3a–c, Supplementary Fig. 2b–e). However, TAK1 inhibition failed to significantly change TH1, TH17 and iTreg cell differentiation from naïve CD4+ T cells (Supplementary Fig. 3a), suggesting a specific function for TAK1 in TH9 cell differentiation. Because the TAK1 inhibitor we used can also inhibit other MAP kinases, we knocked down TAK1 with small interfering RNA (siRNA) in naïve CD4+CD25−CD62L+ T cells from wild-type mouse or by treating naïve CD4+ T cells from Tak1f/f ER-Cre+ mice with 4-hyroxytamoxifen (4-OHT) in vitro (Supplementary Fig. 3b). Reduction of TAK1 expression through these methods resulted in decreased IL-9 expression compared to control siRNA-treated naïve CD4+ T cells or oil treated Tak1f/f ER-Cre+ naïve CD4+ T cells (Fig. 3b) without changes in Foxp3 or IL-17 expression (Supplementary Fig. 3b), indicating a specific role for TAK1 in the differentiation of TH9 cells. Furthermore, treatment of naïve Smad-deficient CD4+ T cells with TAK1 inhibitor abolished the differentiation of TH9 cells (Supplementary Fig. 2b–e). OX40 signaling was reported to amplify the differentiation of TH9 cells27. We cultured naive CD4+ T cells from wild-type mice with antigen-presenting cells (APCs) from OX40L transgenic (OX40L Tg) mice and found that OX40-OX40L signaling indeed promoted the differentiation of TH9 cells (Supplementary Fig. 3c). Importantly, addition of the TAK1 inhibitor to the co-cultures significantly suppressed the frequency of IL-9+ cells after stimulation with TGF-β1 plus IL-4 compared to the cells without TAK1 inhibitor (Supplementary Fig. 3c). This indicates that TAK1 mediated signals play a key role in TH9 cell differentiation.
Figure 3. TAK1 regulates Id3 expression during TH9 cell differentiation.
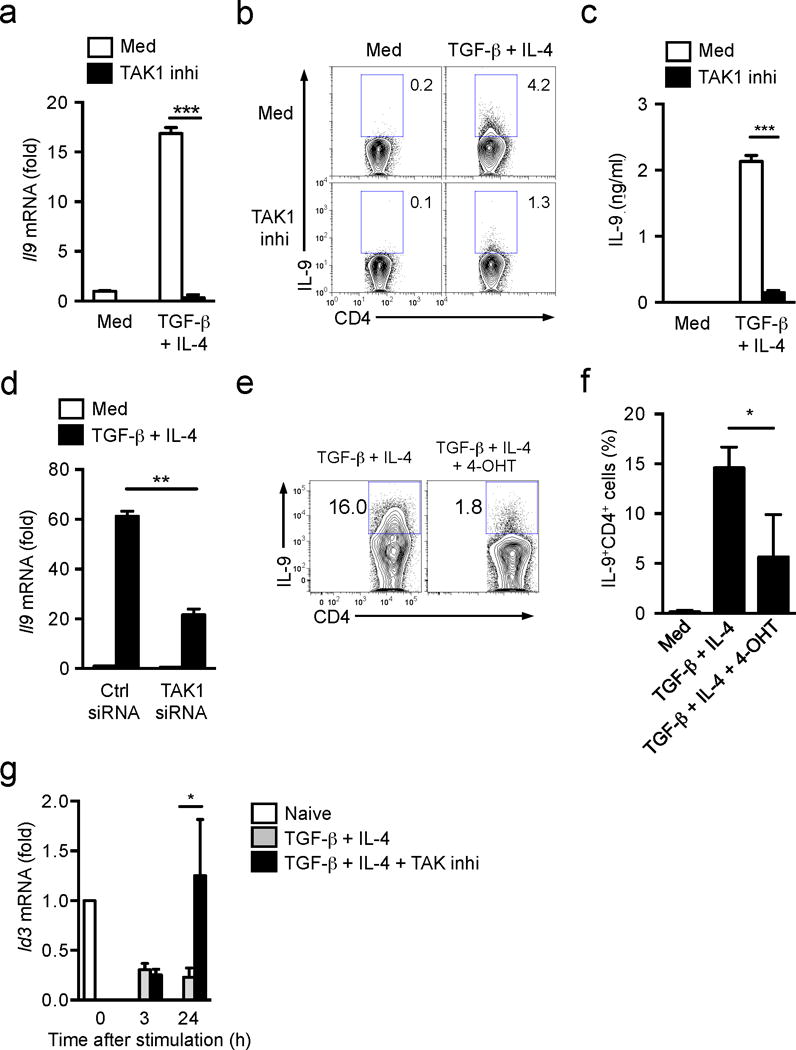
(a) mRNA expression of Il9 in naïve T cells from wild-type mice cultured with anti-CD3+CD28 with or without TGF-β1 plus IL-4 in the absence and presence of TAK1 inhibitor for 24h. (b) Intracellular staining of IL-9 protein in CD4+ T cells differentiated as described in a for 72h. (c) ELISA determination of IL-9 production in culture media of b. (d) mRNA expression of Il9 in wild-type naïve T cells transfected with TAK1-specific or control siRNA followed by the stimulation with anti-CD3+CD28 with or without TGF-β1 plus IL-4 for 24h. (e,f) Flow cytometry of intracellular IL-9 protein in naive CD4+ T cells from Tak1f/f-ERCre+ mice cultured with anti-CD3+CD28 in the absence or presence of 4-hydroxytamoxifen (4-OHT) for overnight to delete TAK1 expression, followed by stimulation with TGF-β1 plus IL-4 for additional 2.5 days. (e) Representative flow cytometry profile from one of three independent experiments; (f) Combined results of three individual experiments. (g) mRNA expression of Id3 in wild-type naive CD4+ T cells cultured with anti-CD3+CD28 and TGF-β1 plus IL-4 in the absence or presence of TAK1 inhibitor for 3-24h. Data are representative of two (d) three (a-c,e) and four (g) independent experiments or are pooled from three experiments (f). Error bars represent mean ± SD. *p<0.05, **p<0.01, ***p<0.001 (Student’s t-test).
We then studied whether TAK1 was involved in TGF-β1- and/or IL-4-mediated inhibition of Id3 expression. The TAK1 inhibitor did not change Id3 suppression in CD4+ T cells stimulated TGF-β1 plus IL-4 during the first 3–6 h in culture compared to the cells without TAK1 inhibitor treatment, but it completely reversed the suppression of Id3 expression by 24 h (Fig. 3g). Treatment with the TAK1 inhibitor did not affect the expression of Irf4 and Sfpi1 (the gene encoding PU.1), two transcription factors known to induce IL-9 expression (Supplementary Fig. 3d)6, 12, suggesting these two pathways are not downstream of TAK1 during induction of IL-9. These data suggest that TAK1 mediates Id3 inhibition during TH9 cell differentiation.
Loss of Id3 expression promotes Il9 transcription
We next investigated the molecular mechanisms by which loss of Id3 expression promoted differentiation of TH9 cells. We focused on the bHLH protein E2A, because Id3 forms a complex with E2A and prevents E2A binding to its target genes28, thus acting as an inhibitor of E2A, As such, we tested if the rapid and persistent reduction of Id3 induced by TGF-β1 and IL-4 promotes E2A binding and transcription at the Il9 promoter. We knocked down E2A expression by siRNA in naïve CD4+ T cells and stimulated them with TGF-β1 plus IL-4. We noticed significantly suppressed Il9 mRNA expression in naïve CD4+ T cells transfected with E2A siRNA compared to control siRNA treated cells (Fig. 4a). Conversely, the ectopic expression of E47 (a splice variant of E2A) enhanced TH9 differentiation (Supplementary Fig. 4a). We measured Il9 promoter activity in response to TGF-β plus IL-4 by luciferase assay in CD4+ T cells transfected with E47-expressing or control vector and found that overexpression of E47 increased the Il9 promoter activity compared to the cells transfected the control vector (Supplementary Fig. 4a). These results suggest that E2A had an important role in the differentiation of TH9 cells. However, there was no considerable change in E2A mRNA in naïve CD4+ T cells stimulated with TGF-β1 plus IL-4 compared with TCR alone (Supplementary Fig. 4b). Consistently, TGF-β1 plus IL-4 did not significantly affect E2A protein in naïve CD4+ T cells (Supplementary Fig. 4c), suggesting the amount of E2A protein is not regulated by TGF-β1 plus IL-4 for IL-9 induction. We have also investigated whether other E-protein family members, E2-2 and HEB, are involved in the TH9 differentiation. Knock-down of E2-2 or HEB did not significantly affect Il9 mRNA induction in CD4+ T cells (data not shown), suggesting a specific role for E2A during TH9 differentiation.
Figure 4. E2A and GATA-3 are enriched at Il9 promoter region to promote IL-9 expression.
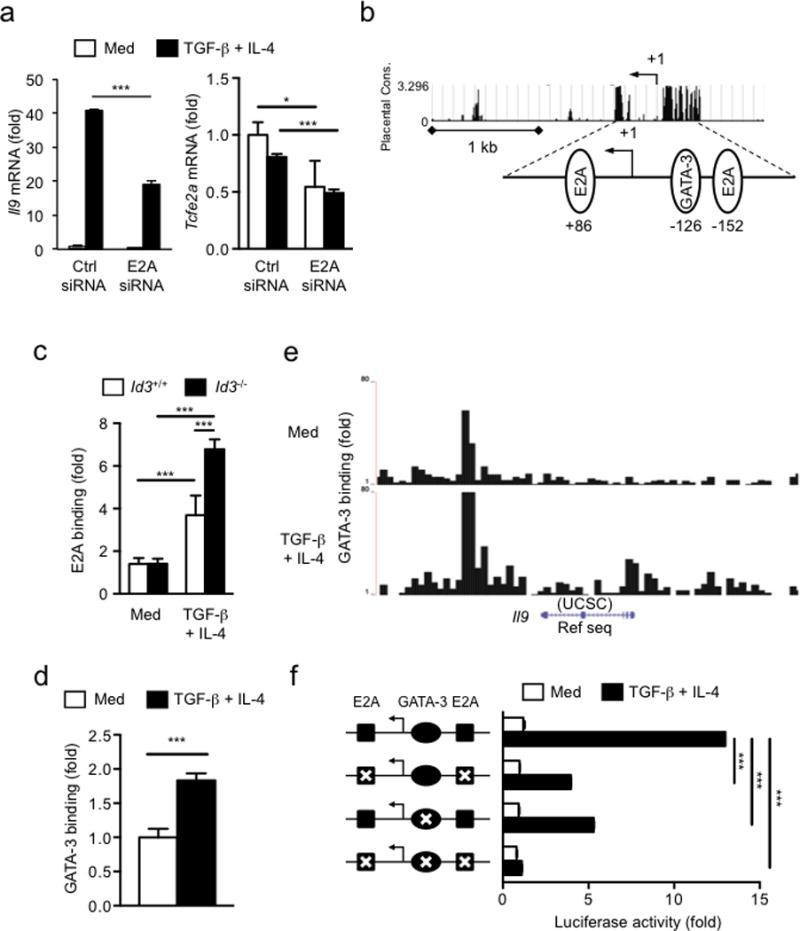
(a), Il9 (left panel) and Tcfe2a (E2A) (right panel) mRNA expression in wild-type naive CD4+ T cells treated with E2A-specific or control siRNA, assessed after stimulation with anti-CD3+CD28 with or without TGF-β1 plus IL-4 for 24h and presented relative to Hprt expression. (b) Genomatix Matinspector analysis of E-protein binding sites (E-boxes) and GATA-3 binding site at the Il9 promoter. (c) ChIP-coupled quantitative PCR analysis of the enrichment of E2A at Il9 promoter region in naive CD4+ T cells isolated from Id3+/+ or Id3−/− mice and cultured for 24 h after stimulation with anti-CD3+CD28 with or without TGF-β1 plus IL-4 and presented relative to results obtained with control IgG, set as 1. (d,e) ChIP-coupled quantitative PCR analysis (d) and ChIP-sequencing analysis (e) of the enrichment of GATA-3 at Il9 promoter region in wild-type naive CD4+ T cells after stimulation with anti-CD3+CD28 with or without TGF-β1 plus IL-4 for 24h. (f) Luciferase assay of TGF-β1 and IL-4-induced Il9 activity (right) in naive CD4+ T cells with wild-type or E-boxes and/or GATA-3 binding site mutated Il9 constructs (left), assessed after stimulation with anti-CD3+CD28 with or without TGF-β1 plus IL-4 for 24h. Data are representative of two (c,d), three (a) and four (f) independent experiments. Error bars represent mean ± SD. * p<0.05, ***p<0.001 (Student’s t-test).
We next investigated whether IL-9 production induced by TGF-β1 plus IL-4 is due to enrichment of E2A binding to the Il9 promoter. We also tested GATA-3 binding to the Il9 promoter since the mechanisms by which GATA-3 controls IL-9 production remained elusive, although GATA-3 has been reported to have a role for TH9 differentiation3 and Id3 deficiency increases GATA-3 expression by an IL-4-dependent manner16. TGF-β1 plus IL-4 significantly up-regulated Gata3 expression in naïve CD4+ T cells, compared to cells treated with TCR alone, 24h post-stimulation, corresponding to induction of Il9 mRNA expression (Supplementary Fig. 5a). Almost all IL-9+ CD4+ T cells co-expressed GATA-3 protein (Supplementary Fig. 5b). Analysis of Il9 promoter with the Genomatix Matinspector software revealed that the Il9 promoter contained at least two canonical (CANNTG) E-protein binding elements (E-boxes) 28, 29, located at +86 and −152 from the transcriptional starting site (TSS) and at least one GATA-3 binding site at −126 bp from the TSS (Fig. 4b). Chromatin immunoprecipitation (ChIP) showed that the binding of E2A protein was significantly enriched onto the Il9 promoter region in response to TGF-β1 plus IL-4 in wild-type T cells compared to the cells with TCR stimulation alone (Fig. 4c). GATA-3 was also enriched at Il9 promoter region in response to TGF-β1 plus IL-4 compared to TCR stimulation alone, as shown by ChIP (Fig. 4d) and ChIP-Sequencing (Fig. 4e). Deletion of Id3 in CD4+ T cells from Id3−/− mice further enhanced E2A binding at the Il9 promoter compared to wild-type CD4+ T cells (Fig. 4c). To assess the direct function of E2A and GATA-3 binding, we measured promoter activity by luciferase assay. Wild-type or mutant Il9 constructs containing mutated E-boxes and/or GATA-3 binding site were transfected into naive CD4+ T cells16 and the cells were cultured with or without TGF-β1 plus IL-4 under TCR stimulation. As expected, the activity of the wild-type Il9 promoter construct increased following stimulation with TGF-β1 plus IL-4 (Fig. 4f), while the Il9 promoter containing mutated E-boxes or GATA-3 binding site showed decreased activity compared to the cells transfected with wild-type Il9 promoter construct in response to TGF-β1 plus IL-4 (Fig. 4f), suggesting that E2A and GATA-3 binding in the Il9 promoter directly promotes transcription. Mutation points in the E-boxes and the GATA-3-binding sites together suppressed most Il9 promoter activation compared to the cells transfected with wild-type Il9 promoter construct (Fig. 4f). These data indicate that the binding of E2A and GATA-3 at the Il9 promoter promote Il9 transcription independently but synergistically, and both regulatory activities are regulated by Id3.
Id3 deficiency increases anti-tumor responses in mice
We next investigated the regulatory function of Id3 in TH9 differentiation in vivo. The frequency of TH9 cells in the spleen, peripheral lymph nodes and lungs of 8–12 weeks-old Id3−/− mice was almost two times greater than in wild-type mice, in which the frequency of TH9 cells is generally low (Supplementary Fig. 6a). Id3−/− mice had higher IL-9 expression in the serum compared to wild-type mice (Supplementary Fig. 6b). We did not detect any increase in the frequency of IL-9-producing type 2 innate lymphoid cells (ILC2s) 30 in Id3−/− mice compared to wild-type mice (data not shown).
We then investigated the role of Id3 in the TH9 cell-related pathology in vivo. TH9 cells have been reported to inhibit tumor growth9, 10. To test if Id3 deficiency increased the anti-tumor response of T cells we used an experimental melanoma mouse model9, 10. To avoid potential non-specific effects in Id3−/− mice, which have defects in thymic development of CD4+ T cells16, we used a T cell adoptive transfer-system. Because naïve Id3−/− CD4+ T cells preferentially differentiate into TH9 cells, but not Treg cells when stimulated with TCR in the presence of TGF-β1 alone, we cultured Id3−/− and wild-type naïve CD4+CD25− T cells with anti-CD3 and anti-CD28 antibodies together with TGF-β1 for three days and then injected them i.v. into Rag1−/− mice, which were then subcutaneously injected with B16 melanoma cells. Mice that have received Id3−/− CD4+ T cells consistently showed slower tumor growth compared to mice that have received wild-type CD4+ T cells; the tumor size was almost reduced half at day 15 in the mice received Id3−/− CD4+ T cells compared to the mice received wild-type CD4+ T cells (Fig. 5 and data not shown). Anti-IL-9 neutralizing antibody treatment had no effect on the tumor growth in tumor-bearing mice transferred with wild-type CD4+ T cells compared to isotype control antibody treatment (Fig. 5a,b), consistent with the absence of TH9 cells in wild-type T cells treated with TGF-β1 alone (Fig. 1b,d), while anti-IL-9 antibody treatment significantly promoted the melanoma growth in mice that received Id3−/− CD4+ T cells compared to isotype control antibody treated mice (Fig. 5a,b). Analysis of the CD4+ T cells in the tumor and in the draining lymph nodes or spleens of mice receiving Id3−/−CD4+ T cells indicated that the anti-IL-9 treatment did not significantly change the frequency of IL-17+ TH17 and Foxp3+ Treg cells and did not decrease (actually increased) the frequency of IFN-γ+TH1 cells compared to the mice without anti-IL-9 treatment (Fig. 5c and data not shown), suggesting that the increased tumor growth in these mice was not due to a decrease in IFN-γ+ T cells and/or an increase in Treg cells.
Figure 5. IL-9 production by Id3−/− CD4+ T cells is involved in anti-tumor immunity.
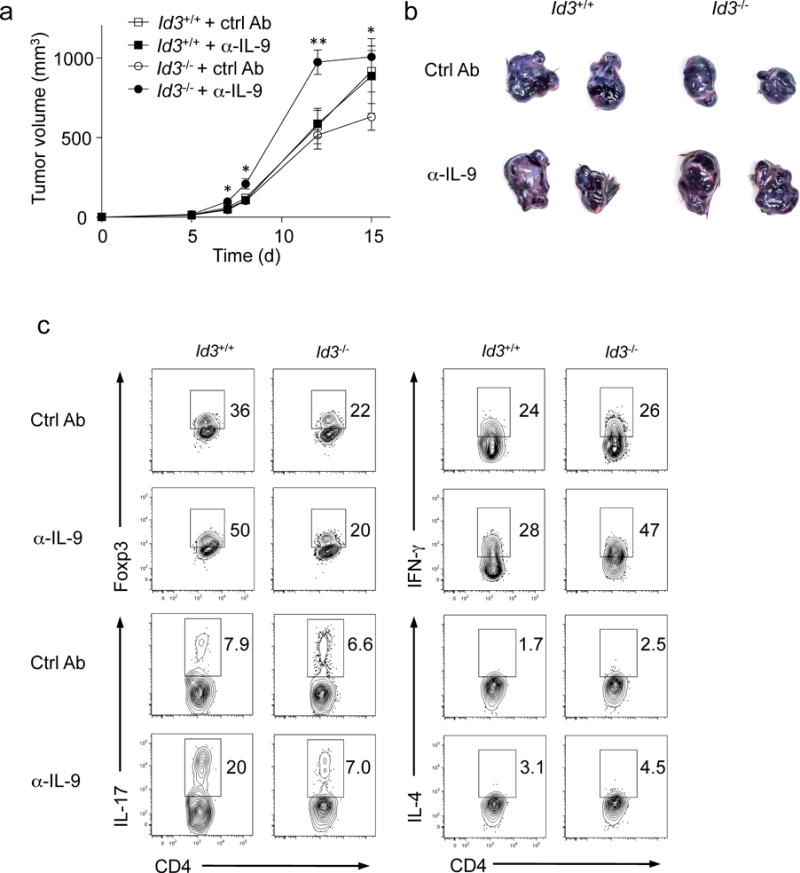
(a) Tumor growth was monitored over time of Rag1−/− mice transferred intravenously with TCR plus TGF-β1-treated CD4+ CD25− T cells from Id3−/− or Id3+/+ mice simultaneously injected with B16 melanoma cells subcutaneously on day 0. The mice were treated with anti-IL-9 or isotype control antibodies every three days from day 0. (b) Representative tumors in each group harvested at the end of experiments as in a. (c) Intracellular staining of Foxp3, IFN-γ, IL-17, and IL-4 in intratumor CD4+ T cells as in a. Error bars represent mean ± SEM (n=4, Id3+/++ CtrlAb and Id3+/++α-IL-9; n=5, Id3−/−+ CtrlAb and Id3−/−+α-IL-9). *p<0.05, **p<0.01 (Student’s t-test). Data are representative of three independent experiments.
We also investigated the anti-tumor effect of Id3-deficient TH9 cells in Id3f/f Cd4-Cre+ T cells to exclude the possible effect of Id3 deficiency on thymic development in systemic Id3−/− mice. We adoptively transferred TGFβ1-treated naïve CD4+CD25− T cells from Id3f/fCd4−Cre+ or Id3+/+ Cd4−Cre+ mice into Rag1−/− mice followed by challenged with melanoma cancer cells. Mice that received TGF-β1-treated naive Id3f/fCd4-Cre+ T cells significantly suppressed the development and growth of tumors compared to the mice that received TGF-β1-treated naive Id3+/+Cd4-Cre+ T cells (Supplementary Fig. 7a). Administration of an anti-IL-9 antibody completely abolished the anti-tumor effect of Id3f/f Cd4-Cre+ T cells, while having no effect on the growth of tumors in mice transferred with Id3+/+Cd4-Cre+ T cells (Supplementary Fig. 7a). These data indicate that differentiated TH9 cells from Id3-deficient CD4+ T cells have an important role in the suppression of melanoma tumor growth.
Because TAK1 was required in the Id3-mediated TH9 cell differentiation, we next investigated whether TAK1 deficiency affected TH9 cell-mediated anti-tumor immunity in vivo. We adoptively transferred TAK1-deficient and wild-type CD4+ T cells that were pre-stimulated with TGF-β1 plus IL-4 in cultures into Rag1−/− mice (Supplementary Fig. 7b,d). Rag1−/− mice transferred with TAK1-deficient CD4+ T cells had substantially faster tumor growth than mice transferred with wild-type CD4+ T cells (Supplementary Fig. 7b). Anti-IL-9 antibody were administered every three days from day 0 to assess whether the anti-tumor immunity was indeed mediated by IL-9 in this setting. Blockade of IL-9 with anti-IL-9 antibody promoted melanoma tumor cell growth in the Rag1−/− host mice transferred with wild-type T cells compared to control antibody-treated Rag1−/− mice transferred with wild-type T cells (Supplementary Fig. 7b), while it had no effect in the mice transferred with TAK1-deficient CD4+ T cells (Supplementary Fig. 7b). Analysis of the intratumor T cells showed no significant changes in the frequencies of IFN-γ+ and Foxp3+ T cells between TAK1-difficient and wild-type T cells (Supplementary Fig. 7d). These data suggest that Id3 and TAK1 regulate TH9 cell differentiation and their anti-tumor activity.
Id3 controls IL-9 production in human CD4+ T cells
Finally, we investigated whether Id3 also has a regulatory role in the differentiation of human Th9 cells. We isolated naïve CD4+CD25−CD45RA+ T cells from peripheral blood mononuclear cells of healthy human subjects and stimulated them with anti-CD3 alone or plus anti-CD28 in the presence of indicated various stimuli. Similarly to mouse cells, TGF-β1 plus IL-4 induced the highest amounts of Il9 mRNA (Fig. 6a) and protein (Fig. 6b) under anti-CD3 plus anti-CD28 stimulation compared to the cells with all other culture conditions,, although TGF-β1 plus IL-4 also induced IL-9 under anti-CD3 stimulation alone (Supplementary Fig. 8a). As seen in mouse T cells, the TAK1 inhibitor (5z-7-oxozeaenol) completely blocked the Il9 mRNA and IL-9 protein expression induced by TGF-β1 plus IL-4 in human CD4+ T cells (Fig. 6a,b), suggesting a critical role for TAK1 in human Th9 cell differentiation.
Figure 6. Id3 controls IL-9 production in human CD4+ T cells.
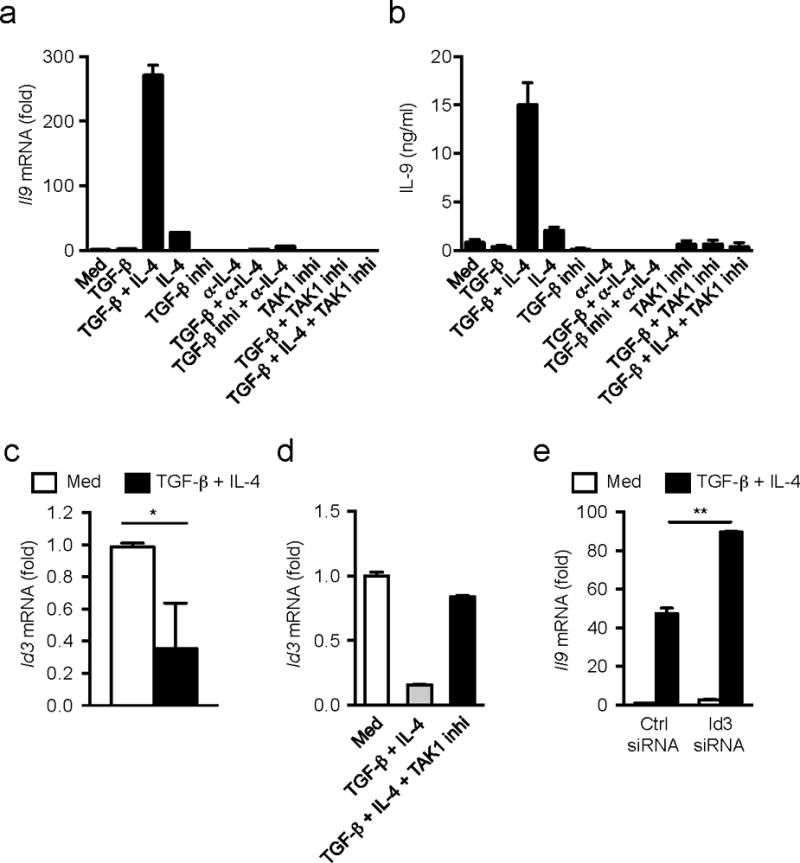
(a) mRNA expression of Il9 in naïve CD4+CD25−CD45RA+ cells isolated from human peripheral blood mononuclear cells after stimulation with anti-CD3+CD28 with various combination of TGF-β1, IL-4, TGF-β receptor inhibitor, anti-IL-4, or TAK1 inhibitor for 48h. (b) ELISA determination of IL-9 production in culture media of a. (c,d) mRNA expression of Id3 in human naïve CD4+ T cells after stimulation with anti-CD3+CD28 with or without TGF-β1 plus IL-4 in the absence (c) or presence (d) of TAK1 inhibitor for 48h. (e) mRNA expression of Il9 in human naïve CD4+ T cells transfected with Id3-specific or control siRNA followed by the stimulation with anti-CD3+CD28 with or without TGF-β1 plus IL-4 for 48h. mRNA expressions are relative to Gapdh expression. Data representative of two (d,b) or three (a,e) experiments or combined four separate experiments (c). Error bars represent mean ± SD. *p<0.05, **p<0.01 (Student’s t-test).
Consistent with mouse cells, Id3 mRNA expression was substantially decreased in human naïve CD4+ T cells in response to TGF-β1 plus IL-4 compared to TCR stimulation alone (Fig. 6c), and this effect was reversed by treatment with the TAK1 inhibitor (Fig. 6d). Knock down of Id3 expression by siRNA (Supplementary Fig. 8b) increased Il9 mRNA expression in human CD4+ T cells stimulated by TGF-β1 plus IL-4 (Fig. 6e). Thus, TAK1 is required for the differentiation of human TH9 cell differentiation by inhibiting Id3 expression. Overall, these data show that a TAK1-Id3-E2A-GATA-3 pathway modulates Il9 transcription in naïve CD4+ T cells in response to TGF-β1 and IL-4 stimulation (Supplementary Fig. 9).
Discussion
The molecular pathways responsible for TH9 cell differentiation remain incompletely understood. In this paper, we show that Id3 negatively regulates IL-9 production in CD4+ T cells. First, deletion of Id3 resulted in significantly larger amounts of IL-9 in CD4+ T cells and Id3−/− naïve CD4+ T cells readily produced IL-9 even with TGF-β1 alone, due largely to the endogenous IL-4 secretion in Id3−/− T cells16. Second, the TGF-β1 and IL-4-induced down-regulation of Id3 was key in promoting IL-9 production in T cells, and this required TAK1.
Id3 was previously shown to be critical for TGF-β1 signaling in T cells16. Id3 mRNA expression was reported to increase at early time points (<3h) after TGF-β1 stimulation, followed by a decrease at later time points 16. Here, TGF-β1 plus IL-4 down-regulated Id3 expression at early during T cell stimulation in culture and this down-regulation lasted at least 48 h. Further analysis revealed that IL-4 alone also inhibited Id3 expression at early time points after stimulation (3h), and that TGF-β1 signaling acted in a synergistic manner to down-regulate Id3 at later time points (24h). The molecular mechanism underlying the IL-4-mediated early down-regulation of Id3 in CD4+ T cells remains unknown, but it appears that this early decrease in Id3 expression alone is necessary, but insufficient, to promote TH9 differentiation.
We showed that blockade of TAK1 activity with an inhibitor completely reversed the down-regulation of Id3 induced by TGF-β plus IL-4, and importantly, blocked TH9 cell differentiation in TAK1 inhibitor- or TAK1 siRNA-treated wild-type CD4+ T cells as well as in 4-OHT-treated Tak1f/f ER-Cre+ CD4+ T cells. However, TAK1 inhibition did not affect the expression of the transcription factors IRF46 and PU.112, which have been reported to control TH9 cell differentiation. Smad2 and Smad4 have been shown to have a role in TH9 cell differentiation24, 25. Here we expand these findings and suggest Smad2, 3 and 4 play a partial function in TH9 differentiation. However, TAK1 inhibition almost completely abrogated TH9 cell differentiation, suggesting a dominant role for the TAK1-dependent pathway in this process. Notably, blockade of TAK1 activity with a specific inhibitor also reduced IL-9 expression in Id3−/− T cells (data not shown), suggesting that TAK1 might modulate IL-9 expression though additional Id3-independent pathways, such as NF-κB27 and/or NFATs31. These possibilities remain to be investigated. Nevertheless, down-regulation of Id3 represents an important effector downstream of TAK1 during TH9 cell differentiation, The binding of E2A and GATA-3 in the Il9 promoter had a key role downstream the reduction of Id3 expression during TH9 differentiation. Down-regulation of Id3 led to enhanced binding of E2A in the Il9 promoter and multiple experimental approaches showed that E2A and GATA-3 binding promoted Il9 transcription in CD4+ T cells. Although both GATA-3 expression and binding of E2A are known to be regulated by Id316, 28, these two factors seemed to have independent, yet additive functions for Il9 gene activation, because mutation of both of them resulted in the synergetic and maximal reduction of IL-9 production. In contrast to E2A, other E-proteins E2-2 and HEB seemed dispersible for Id3-mediated IL-9 production in T cells.
The biological relevance of Id3 regulation of TH9 cells was demonstrated in an experimental tumor model. Although TH9 cells have been reported to be important in allergic inflammation4, 5, 6, autoimmune diseases 3, 7, 8 and tumor immunity9, 10, the detection of TH9 cells in vivo is rather difficult as the generation and differentiation of TH9 cells is transient30, 32. However, we showed that TH9 cells differentiated from Id3 deficient CD4+ T cells play an important role in the suppression of melanoma tumor growth in mice. This finding may have important implication for considering Id3 as potential target to develop anti-tumor immunotherapy, as reduction or deletion of Id3 in T cells could drive them to differentiate into anti-tumor TH9 cells rather than Treg cells in response to relevant tumor antigens in the TGFβ-rich environment of tumors.
The observation that TAK1-difficient T cells did not produce IL-9 further strengthened the idea of a cross-talk between TAK and Id3 in TH9 cell differentiation. Furthermore, we also demonstrated a role for TAK1 and Id3 in human TH9 cell differentiation. Although the mechanistic details of how TAK1 and Id3 signalings interact each other remain to be elucidated, our findings provide further understanding of the molecular mechanisms by which TGF-β1 controls the differentiation of helper T cell subsets.
Online Methods
Mice
C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Smad3−/− (on a C57BL/6 background)33, Smad4f/fCd4-Cre+, Smad4f/+Cd4-Cre+, TgfbrIf/fCd4-Cre+, TgfbrIf/+Cd4-Cre−34_ENREF_18, Tgfbr2f/fER-Cre+, Id3−/−17, Id3f/fCd4-Cre+, Id3+/+Cd4-Cre+, Tak1f/fER-Cre+, Rag1−/− mice were bred in our facility under specific pathogen-free conditions. All animal studies were performed according to National Institutes of Health guidelines for use and care of live animals and approved by the Animal Care and Use Committees of National Institute of Dental and Craniofacial Research.
Antibodies and reagents
The following antibodies were from eBioscience: purified anti-mouse CD3 (no azide and low endotoxin; 145-2C11), purified anti-mouse CD28 (no azide and low endotoxin; 37.51), purified anti-human CD3 (OKT3), purified anti-human CD28 (CD28.2), and following fluorochrome-conjugated anti-mouse antibodies; anti-CD4 (RM4-5), anti-TCR-β (H57-597), anti-Foxp3 (FJK-16a), and anti-GATA3 (TWAJ). The following antibodies were from BD Pharmingen: fluorochrome-conjugated anti-mouse IL-17 (TC11-18H10) and IFN-γ (XMG1.2), purified anti-mouse IL-4 (no azide and low endotoxin; 11B11), purified anti-human IL-4 (no azide and low endotoxin; MP4-25D2), and isotype-matched control antibodies (rat IgG1κ (R3-34), rat IgG2a (R35-95), and rat IgG2b (A95-1)). Fluorochrome-conjugated anti-mouse IL-9 (RM9A4) was purchased from BioLegend. Recombinant mouse IL-4 (404-ML), human IL-4 (204-IL) and TGF-β1 (240-B) were purchased from R&D Systems. Anti-IL-9 antibody (9C1) and isotype control were purchased from BioXCell. 5z-7-Oxozeaenol (TAK1 inhibitor, Sigma) was used at 50 nM, and SB431542 (TGF-βR inhibitor) was used at 5 μM.
Flow cytometry analysis
Intranuclear staining was carried out using the Fixation/Permeabilization buffer solution (eBioscience) according to manufacturer’s protocol. For intracellular cytokine staining, cells were stimulated with PMA (10 ng/ml), Ionomycin (250 ng/ml) with Golgi-Plug (1:1000 dilution, BD pharmingen) at 37°C for 4 hr, using the Fixation/Permeabilization buffer solution (BD bioscience) according to manufacturer’s protocol. Stained cells were analyzed on a FACS-Calibur (BD Bioscience) and Flow Jo software.
Th9 cell differentiation in vitro
Naive CD4+CD25−CD62L+ cells were purified by magnetic cell sorting (Miltenyi Biotec) and culture at 0.5 × 106 cells/well in 24-well plates with plate-bound anti-CD3 (1 μg/mL) and soluble anti-CD28 (1 μg/mL), TGF-β1 (2 ng/mL) with or without IL-4 (10 ng/mL) at 37°C. Three days later, cells were analyzed using FACS.
Real-time RT-PCR
Total RNA was derived from cultured cells with an RNeasy Mini kit (Qiagen) and cDNA was synthesized by using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Quantitative real-time PCR was performed according to the protocol of TaqMan gene expression assay kits (Applied Biosystems) with the following primers: Il9, Mm00434305_m1; Hprt, Mm00446968_m1; Tcfe2a, Mm01175595_m1; Id3, Mm00492575_m1; Gata3, Mm00484683_m1; Sfpi1, Mm00488142_m1; Irf4, Mm00516431_m1. Results were normalized to the expression of Hprt1 mRNA.
RNA-mediated interference
An Amaxa Mouse T cell Nucelofector kit was used for knockdown of E2A by small interfering RNA (siRNA) in naive CD4+ T cells according to the manufacturer’s instructions (Lonza). Wild-type naive CD4+CD25−CD62L+ cells were stimulated for 24h with anti-CD3 and anti-CD28 and were transduced by electroporation with E2A-specific siRNA (5 nM) or AllStars Negative Control siRNA (Qiagen). After overnight culture, cells were restimulated for 24 h with TGF-β1 plus IL-4, followed by real-time PCR analysis of Tcfe2a or Il9 mRNA. Of the E2A-specific siRNAs (SI01444359, SI01444366, SI01444373 and SI01444380) tested (all from Qiagen), results obtained with SI01444373 are presented.
Chromatin Immunoprecipitation (ChIP) assay and Chip-Sequencing
ChIP assay were carried out as described previously16. In brief, CD4+CD25−CD62L+ T cells were purified by magnet cell sorting (Miltenyi Biotec) and cultured at 1 × 106/well in 24-well plates with plate-bound anti-CD3 (1 μg/ml) and soluble anti-CD28 (1 μg/ml) in the presence or absence of 2 ng/ml TGF-β1 and 10 ng/ml IL-4. 24 h later, cells were collected and fixed with 1% formaldehyde at room temperature for 10 min and lysed in the lysis buffer (Diagenode). The lysate were sonicated and precipitate with anti-E2A (V-18, Santacruz), anti-GATA3 (B-10, Santacruz), control rabbit IgG (Abcam) or control mouse IgG (CellSignaling). ChIP DNA was analyzed by qPCR (Bio-Rad) with the following primers: IL-9 promoter-forward: 5′-GGC CCA GCA CAG AAC TGA AGA GC and reverse: 5′-AGG GTT TTT CCC GGT TTG AAG AGC. ChIP-sequencing was performed as described previously35.
Luciferase assay
The mouse IL-9 conserved promoter and several exon regions (−464 to +284) were amplified from genomic DNA by PCR with 5′-GCT GTG AAC ACT GAG Aga agc ttG G and 5′-ACT Gag gta ccA ATA GCC AGA GG. Small letters indicate the authentic Hind III and Kpn I site that was introduced to allow forced cloning into the pGL4 basic luciferase reporter gene vector. Site-directed mutagenesis of the E2A and GATA3 binding site was performed by QuickChange XL kit (Stratagene), according to manufacturer’s instruction. The sequence was changed as follows: E2A1: ATAGCG, E2A2: ATCACG, Gata3: ATTC. Purified naive CD4+ T cells were transfected with reporter construct together with pRL-TK vector (Promega) for internal control by using a mouse T cell Nucreofector kit (LONZA). The Il9 promoter activity was determined by a dual-luciferase assay system (Promega) according to manufacturer’s instructions. All samples were normalized for transfection efficiency by dividing firefly luciferase activity with that of the renilla luciferase activity.
Tumor-bearing model
CD4+CD25− T cells were isolated from Id3−/− and wild-type mice or from Id3f/f CD4-cre+ and Id3+/+CD4-cre+ mice and cultured with anti-CD3+CD28 and TGF-β1 for three days. The cells were collected and transferred i.v. into Rag1−/− mice (5 × 106 cells/mouse) on day 0. On the same day, B16 melanoma cells (5 × 105 cells/mouse) were subcutaneously injected at the side of the abdomen of T-cell-transferred mice. Anti-IL-9 Ab or control mouse IgG2a were treated i.p. every three days from day 0.
Human T cells in vitro
Human peripheral blood mononuclear cells were provided by healthy volunteers and obtained from the NIH Department of Transfusion Medicine (DTM) through their approved protocol number: NCT000001846. These blood samples were provided by the DTM on a de-identified basis. A signed informed consent was obtained from all donors. Naïve CD4+ T cells were purified by naïve T cell isolation kit (Miltenyi Biotec). Naïve T cells were cultured with plate-bound anti-human CD3 (1 μg/mL) and soluble anti-human CD28 (1 μg/mL) with the combination of TGF-β1 (2 ng/mL), IL-4 (10 ng/mL), anti-human IL-4 (10 μg/mL), TGF-βR inhibitor (SB431542, 5 μM), and TAK1 inhibitor (50 nM) for 48 h. mRNA expression were measured according to the protocol of TaqMan gene expression assay kits (Applied Biosystems) with following primers: Gapdh, Hs99999905_m1; Il9, Hs00914237_m1; Id3, Hs00954037_g1; Gata3, Hs00231122_m1. Id3 knockdown were performed by using Amaxa Human T cell Nucleofector kit with Id3-specific siRNAs (SI04431714) or AllStars Negative Control siRNA (Qiagen).
Supplementary Material
Acknowledgments
We thank Drs. JE. Konkel and C. Chia at NIDCR for their critically reading the manuscript. We thank Dr. Yuan Zhuang at Duke University for Id3f/f Cd4-Cre+ mice; Dr. Xian C. Li, at Harvard Medical School for OX40L transgenic APCs; Dr. Wan Y. at University of North Carolina for Tak1f/f-ERCre mice; Dr. A. Yoshimura. at Keio University of Medicine for Smad2f/f-LckCre+ mice. This work was supported by the Intramural Research Program of the National Institute of Dental and Craniofacial Research (US National Institutes of Health) and the JSPS Research Fellowship for Japanese Biomedical and Behavioral Researchers at NIH (to H.N).
Footnotes
Author contributions: H.N., D.Z., T.M. designed and performed most of the experiments, interpreted the data and drafted paper; K.C. performed ChIp-Seq experiments, H.C., M.I., L.D., P.Z., E.T., B.A., W.J., N.G. performed experiments, Q. C., L.S. provided critical scientific input. K.Z. supervised and designed ChIp-Seq experiments and interpreted data, W.J.C. conceived and supervised the research, designed experiments and wrote the paper.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Schmitt E, Germann T, Goedert S, Hoehn P, Huels C, Koelsch S, et al. IL-9 production of naive CD4+ T cells depends on IL-2, is synergistically enhanced by a combination of TGF-beta and IL-4, and is inhibited by IFN-gamma. J Immunol. 1994;153(9):3989–3996. [PubMed] [Google Scholar]
- 2.Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nature immunology. 2008;9(12):1341–1346. doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]
- 3.Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nature immunology. 2008;9(12):1347–1355. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicolaides NC, Holroyd KJ, Ewart SL, Eleff SM, Kiser MB, Dragwa CR, et al. Interleukin 9: a candidate gene for asthma. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(24):13175–13180. doi: 10.1073/pnas.94.24.13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng G, Arima M, Honda K, Hirata H, Eda F, Yoshida N, et al. Anti-interleukin-9 antibody treatment inhibits airway inflammation and hyperreactivity in mouse asthma model. American journal of respiratory and critical care medicine. 2002;166(3):409–416. doi: 10.1164/rccm.2105079. [DOI] [PubMed] [Google Scholar]
- 6.Staudt V, Bothur E, Klein M, Lingnau K, Reuter S, Grebe N, et al. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity. 2010;33(2):192–202. doi: 10.1016/j.immuni.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 7.Jager A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol. 2009;183(11):7169–7177. doi: 10.4049/jimmunol.0901906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh TP, Schon MP, Wallbrecht K, Gruber-Wackernagel A, Wang XJ, Wolf P. Involvement of IL-9 in Th17-associated inflammation and angiogenesis of psoriasis. PloS one. 2013;8(1):e51752. doi: 10.1371/journal.pone.0051752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Purwar R, Schlapbach C, Xiao S, Kang HS, Elyaman W, Jiang X, et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nature medicine. 2012 doi: 10.1038/nm.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Y, Hong S, Li H, Park J, Hong B, Wang L, et al. Th9 cells promote antitumor immune responses in vivo. The Journal of clinical investigation. 2012;122(11):4160–4171. doi: 10.1172/JCI65459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goswami R, Jabeen R, Yagi R, Pham D, Zhu J, Goenka S, et al. STAT6-dependent regulation of Th9 development. J Immunol. 2012;188(3):968–975. doi: 10.4049/jimmunol.1102840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang HC, Sehra S, Goswami R, Yao W, Yu Q, Stritesky GL, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nature immunology. 2010;11(6):527–534. doi: 10.1038/ni.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. The Journal of experimental medicine. 2003;198(12):1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126(6):1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 15.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 16.Maruyama T, Li J, Vaque JP, Konkel JE, Wang W, Zhang B, et al. Control of the differentiation of regulatory T cells and T(H)17 cells by the DNA-binding inhibitor Id3. Nature immunology. 2011;12(1):86–95. doi: 10.1038/ni.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Dai M, Zhuang Y. A T cell intrinsic role of Id3 in a mouse model for primary Sjogren’s syndrome. Immunity. 2004;21(4):551–560. doi: 10.1016/j.immuni.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 18.Jones-Mason ME, Zhao X, Kappes D, Lasorella A, Iavarone A, Zhuang Y. E protein transcription factors are required for the development of CD4(+) lineage T cells. Immunity. 2012;36(3):348–361. doi: 10.1016/j.immuni.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nature immunology. 2008;9(6):632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 20.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 21.Mu Y, Gudey SK, Landstrom M. Non-Smad signaling pathways. Cell and tissue research. 2012;347(1):11–20. doi: 10.1007/s00441-011-1201-y. [DOI] [PubMed] [Google Scholar]
- 22.Choi ME, Ding Y, Kim SI. TGF-beta signaling via TAK1 pathway: role in kidney fibrosis. Seminars in nephrology. 2012;32(3):244–252. doi: 10.1016/j.semnephrol.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka K, Ichiyama K, Hashimoto M, Yoshida H, Takimoto T, Takaesu G, et al. Loss of suppressor of cytokine signaling 1 in helper T cells leads to defective Th17 differentiation by enhancing antagonistic effects of IFN-gamma on STAT3 and Smads. J Immunol. 2008;180(6):3746–3756. doi: 10.4049/jimmunol.180.6.3746. [DOI] [PubMed] [Google Scholar]
- 24.Tamiya T, Ichiyama K, Kotani H, Fukaya T, Sekiya T, Shichita T, et al. Smad2/3 and IRF4 Play a Cooperative Role in IL-9-Producing T Cell Induction. J Immunol. 2013;191(5):2360–2371. doi: 10.4049/jimmunol.1301276. [DOI] [PubMed] [Google Scholar]
- 25.Wang A, Pan D, Lee YH, Martinez GJ, Feng XH, Dong C. Cutting edge: Smad2 and Smad4 regulate TGF-beta-mediated Il9 gene expression via EZH2 displacement. J Immunol. 2013;191(10):4908–4912. doi: 10.4049/jimmunol.1300433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Molecular cell. 2008;31(6):918–924. doi: 10.1016/j.molcel.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao X, Balasubramanian S, Liu W, Chu X, Wang H, Taparowsky EJ, et al. OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nature immunology. 2012;13(10):981–990. doi: 10.1038/ni.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murre C. Helix-loop-helix proteins and lymphocyte development. Nature immunology. 2005;6(11):1079–1086. doi: 10.1038/ni1260. [DOI] [PubMed] [Google Scholar]
- 29.Murre C, McCaw PS, Vaessin H, Caudy M, Jan LY, Jan YN, et al. Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell. 1989;58(3):537–544. doi: 10.1016/0092-8674(89)90434-0. [DOI] [PubMed] [Google Scholar]
- 30.Wilhelm C, Hirota K, Stieglitz B, Van Snick J, Tolaini M, Lahl K, et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nature immunology. 2011;12(11):1071–1077. doi: 10.1038/ni.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jash A, Sahoo A, Kim GC, Chae CS, Hwang JS, Kim JE, et al. Nuclear factor of activated T cells 1 (NFAT1)-induced permissive chromatin modification facilitates nuclear factor-kappaB (NF-kappaB)-mediated interleukin-9 (IL-9) transactivation. The Journal of biological chemistry. 2012;287(19):15445–15457. doi: 10.1074/jbc.M112.340356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan C, Aziz MK, Lovaas JD, Vistica BP, Shi G, Wawrousek EF, et al. Antigen-specific Th9 cells exhibit uniqueness in their kinetics of cytokine production and short retention at the inflammatory site. J Immunol. 2010;185(11):6795–6801. doi: 10.4049/jimmunol.1001676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. The EMBO journal. 1999;18(5):1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Konkel JE, Maruyama T, Carpenter AC, Xiong Y, Zamarron BF, Hall BE, et al. Control of the development of CD8alphaalpha+ intestinal intraepithelial lymphocytes by TGF-beta. Nature immunology. 2011;12(4):312–319. doi: 10.1038/ni.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei G, Hu G, Cui K, Zhao K. Genome-wide mapping of nucleosome occupancy, histone modifications, and gene expression using next-generation sequencing technology. Methods in enzymology. 2012;513:297–313. doi: 10.1016/B978-0-12-391938-0.00013-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.