

SUMMARY
How mtDNA replication is terminated and the newly formed genomes are separated remain unknown. We here demonstrate that the mitochondrial isoform of topoisomerase 3α (Top3α) fulfills this function, acting independently of its nuclear role as a component of the Holliday junction-resolving BLM-Top3α-RMI1-RMI2 (BTR) complex. Our data indicate that mtDNA replication termination occurs via a hemicatenane formed at the origin of H-strand replication and that Top3α is essential for resolving this structure. Decatenation is a prerequisite for separation of the segregating unit of mtDNA, the nucleoid, within the mitochondrial network. The importance of this process is highlighted in a patient with mitochondrial disease caused by biallelic pathogenic variants in TOP3A, characterized by muscle-restricted mtDNA deletions and chronic progressive external ophthalmoplegia (CPEO) plus syndrome. Our work establishes Top3α as an essential component of the mtDNA replication machinery and as the first component of the mtDNA separation machinery.
Graphical abstract
In Brief: Nicholls et al. identify a role for topoisomerase 3α in the separation of mtDNA following replication. Loss of Top3α activity impairs mtDNA segregation and, consequently, segregation of the mtDNA nucleoid within the mitochondrial network. Mutations in TOP3A cause human mitochondrial disease associated with mtDNA deletions and impaired mtDNA separation.
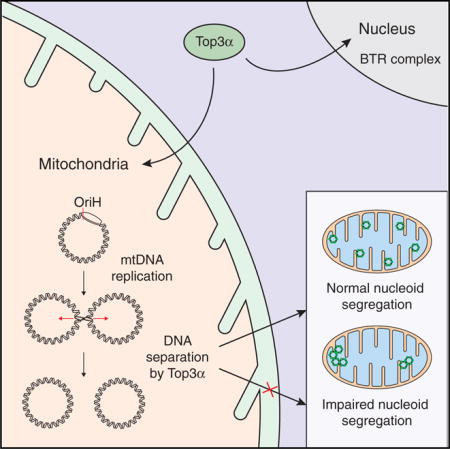
INTRODUCTION
The mammalian mitochondrial genome (mtDNA) is maintained at thousands of copies per cell, and it is packaged into nucleoprotein complexes termed nucleoids. The majority of nucleoids contain a single mtDNA molecule compacted by mitochondrial transcription factor A (TFAM) via cross-strand binding (Kukat et al., 2011, 2015). Nucleoids are readily visible by fluorescence microscopy, where they appear as punctate foci distributed throughout the mitochondrial network (Satoh and Kuroiwa, 1991). The mitochondrial network is itself spread across the cytosol and is highly dynamic, with its morphology modulated by the opposing processes of fission and fusion. Recent data suggest that the segregation of newly replicated nucleoids is coupled to mitochondrial fission at endoplasmic reticulum contact sites, which serves to distribute nucleoids throughout the mitochondrial network (Lewis et al., 2016). The protein products of mtDNA perform essential roles in the respiratory chain, and defects of mitochondrial fission and fusion are associated with improper nucleoid distribution and heterogeneity of membrane potential, as well as mtDNA instability (Friedman and Nunnari, 2014).
The importance of these processes is highlighted by the fact that pathogenic variants in nuclear genes involved in the maintenance and replication of mtDNA are associated with an extensive spectrum of clinical phenotypes, ranging from severe encephalopathy in childhood to late-onset progressive external ophthalmoplegia (PEO), ataxia, and myopathy. Such disorders of mtDNA maintenance manifest as either mtDNA depletion (quantitative) syndromes or multiple mtDNA deletion (qualitative) disorders. PEO and ptosis, a common manifestation of adult mitochondrial disease, are delineated by extraocular muscle paresis that is biochemically defined by focal cytochrome c oxidase (COX)-deficient fibers in skeletal muscle, each harboring high levels of a clonally expanded mtDNA deletion. While at least 17 nuclear-encoded genes have been reported to be associated with adult-onset PEO and muscle-restricted mtDNA deletions (Viscomi and Zeviani, 2017), the underlying nuclear genetic defect remains undetermined in a sizeable number of cases.
The mitochondrial genome is highly compact and has only one major non-coding region (NCR), which contains the origin of heavy (leading) strand DNA synthesis, OriH, and the two transcription promoters LSP and HSP (Gustafsson et al., 2016). Although the mechanisms of mtDNA replication have been studied in detail, little is known about how newly synthesized mtDNA molecules are separated and segregated within the mitochondrial network after the completion of replication. Early pioneering electron microscopy (EM) studies of human mtDNA preparations identified catenanes linked together by X-type branches, which were postulated to have been formed during the completion of mtDNA replication (Hudson and Vinograd, 1967), but the relevant protein factors have remained unidentified.
Catenated DNA forms can be unlinked by certain classes of topoisomerases, which contribute crucial activities during replication, transcription, chromosome segregation, and recombination by creating transient breaks in the DNA backbone to alter DNA topology. Type I topoisomerases cleave one DNA strand to allow strand passage, whereas type II enzymes cleave both strands. Type II topoisomerases are thought to be the major factors that remove catenation links during DNA replication in the eukaryotic nucleus (Pommier et al., 2016). Mammalian mitochondria possess two type I topoisomerases: a dedicated type IB enzyme, Top1mt (Zhang et al., 2001), and Top3α, a type IA enzyme that shows dual nuclear and mitochondrial localization (Wang et al., 2002). Top1mt is thought to act as a DNA swivel to relieve torsional strain during DNA replication and/or transcription, consistent with the mechanism of type IB enzymes (Stewart et al., 1998). A mouse knockout of the Top1mt gene is viable, although it displays signs of mitochondrial dysfunction and shows altered mtDNA supercoiling (Douarre et al., 2012; Zhang et al., 2014). Type IA topoisomerases, in contrast, operate by an enzyme-bridged strand-passage mechanism that permits distinct topological transformations depending on the substrate: if the two strands are part of the same, hypernegatively super-coiled DNA duplex, then the result is relaxation (Plank et al., 2005), whereas if the substrate is interlinked single-stranded DNA (ssDNA) circles, then the result is decatenation (Yang et al., 2010). Top3α contains an N-terminal mitochondrial targeting sequence (MTS) that directs a proportion of the protein pool to mitochondria (Wang et al., 2002), but the functional role of this isoform in mtDNA metabolism has not been addressed. The nuclear isoform of Top3α forms a complex with the helicase BLM and the two OB-fold proteins RMI1 and RMI2 (known as the BLM-Top3α-RMI1-RMI2 [BTR] complex), which together catalyze the dissolution of double-Holliday junctions to produce noncrossover products, in a reaction that is stimulated by RMI1 and RMI2 (Sarbajna and West, 2014).
Previous reports have also identified two type II topoisomerases in mitochondria: Top2α (Zhang et al., 2014) and Top2β, either in a truncated form (Low et al., 2003) or in full-length form (Zhang et al., 2014). Neither Top2α nor Top2β possesses a detectable MTS, and it is unclear how their mitochondrial localization would be achieved. In contrast, the mitochondrial localization of Top1mt and Top3α is supported by proteomic and bioinformatic studies (Calvo et al., 2016; Rhee et al., 2013), in addition to the presence of targeting sequences.
In the present work, we set out to identify enzymes required for the separation of mtDNA molecules after the completion of DNA replication. We identify the mitochondrial isoform of Top3α as being essential for mtDNA separation, and we demonstrate that the loss of this enzyme leads to the formation of catenated mtDNA species, reminiscent of the mtDNA dimers described by the Vinograd laboratory 50 years ago (Hudson and Vinograd, 1967). We characterize the termination structure as a hemicatenane, consisting of two double-stranded DNA (dsDNA) molecules associated through a single-stranded linkage, consistent with the use of a type IA topoisomerase for separation of newly replicated mtDNA molecules. We believe that our work provides a starting point for future investigations into the molecular mechanisms linking mtDNA separation to mtDNA segregation within the dynamic mitochondrial network.
RESULTS
Top3α Is Required for mtDNA Maintenance
After completion of DNA replication in bacteria, topoisomerases are required for the removal of catenation links to separate sister chromosomes. It is not known whether topoisomerases are involved in genome separation in mammalian mitochondria. To address this point, we investigated the functional role of three topoisomerases with reported dual nuclear and mitochondrial localization: Top2α, Top2β, and Top3α. A fourth dedicated mitochondrial topoisomerase, Top1mt, has been extensively studied in the past (Pommier et al., 2016). We first assessed the mitochondrial localization of Top2α, Top2β, and Top3α by preparing highly purified mitochondrial and mitoplast fractions using differential centrifugation and sucrose gradient centrifugation (Figure 1A). Blotting of marker proteins histone H3 (nucleus), TFAM (mitochondrial matrix), and AIF (apoptosis inducing factor; intermembrane space) confirmed the purity of fractions. Consistent with a previous report (Wang et al., 2002), our analysis demonstrated that a proportion of the cellular pool of Top3α localizes to the mitochondrial matrix. However, in contrast to previous reports (Zhang et al., 2014), we were unable to detect either Top2α or Top2β in our mitochondrial preparations.
Figure 1. Loss of Top3α Affects mtDNA Maintenance.
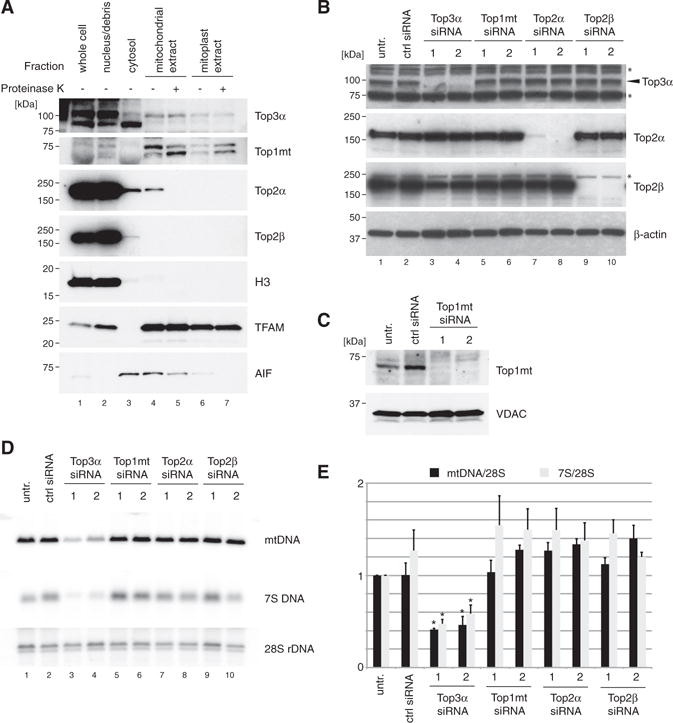
(A) Mitochondrial localization of topoisomerases by western blotting of isolated cell fractions. HeLa cells were fractionated into nuclei/unbroken cells, cytosol, whole mitochondria, and mitoplasts. Mitochondria and mitoplast fractions were additionally treated with proteinase K to remove externally bound proteins. Marker proteins used were histone H3 (nucleus), TFAM (mitochondrial matrix) and apoptosis-inducing factor (AIF; intermembrane space).
(B) Efficiency of depletion of Top3α, Top2α, and Top2β assessed by western blotting of HeLa cell lysates. β-actin is used as a loading control. Asterisk (*) denotes non-specific species.
(C) Efficiency of depletion of Top1mt assessed by western blotting of mitochondrial lysate of HeLa cells. Voltage-dependent anion channel (VDAC) is used as a loading control.
(D) Southern blot analysis of mtDNA copy number and 7S DNA levels following siRNA depletion of topoisomerases as in (B) and (C). mtDNA was linearized using BamHI and detected using probe (a). 28S rDNA is used as a loading control.
(E) Quantification of mean mtDNA copy number and 7S DNA levels following siRNA depletion of topoisomerases (n = 3). Error bars represent SEM.
We next studied how the loss of topoisomerase activity affects mtDNA levels using small interfering RNA (siRNA) in cultured cells. Efficient depletion of protein levels was confirmed using western blotting of cell lysates (Figure 1B). The depletion efficiency of Top1mt was assessed in mitochondrial lysates (Figure 1C) due to antibody limitations. Knockdown of Top3α, but not Top1mt, Top2α, or Top2β, caused an apparent depletion of mtDNA copy number (Figures 1D and 1E), indicating a defect in mtDNA maintenance. In Top3α-depleted cells, we also observed a reduction in the level of 7S DNA, the linear strand of the mitochondrial D-loop (Figure 1D, lanes 3 and 4).
Pathogenic TOP3A Variants Are Associated with mtDNA Instability in Human Pathology
In parallel to the molecular analysis of mitochondrial Top3α, we additionally identified an individual with adult-onset mitochondrial disease characterized by a complex PEO syndrome with prominence of cerebellar features, including upper and lower limb ataxia and dysarthria (Figures S1A–S1D). A detailed summary of the clinical presentation is provided in the STAR Methods. To determine the precise genetic etiology, whole-exome sequencing was undertaken with informed consent. The mean depth per consensus coding sequence was 52-fold, with 74.91% bases at 20-fold coverage (Figure S1E). Analysis of autosomal recessive variants in nuclear genes encoding proteins involved in DNA transcription, replication, and maintenance (Figure S1F) revealed two heterozygous variants in TOP3A (GenBank: NM_004618.3): a c.298A > G (p.Met100Val) missense change and a c.403C > T (p.Arg135*) nonsense change. In the Exome Aggregation Consortium (ExAC) database, the p.Met100Val missense variant was present in 7/120,600 alleles (minor allele frequency [MAF] = 5.804 × 10−5) and the p.Arg135* variant was present in 24/121,054 alleles (MAF = 1.983 × 10−4), all in the heterozygous state. Both variants occurred in the topoisomerase/primase (Toprim) domain (Figure 2A), which contributes to substrate binding and metal ion coordination (Aravind et al., 1998).
Figure 2. Pathological Top3α Variants Are Associated with Altered mtDNA Structure.
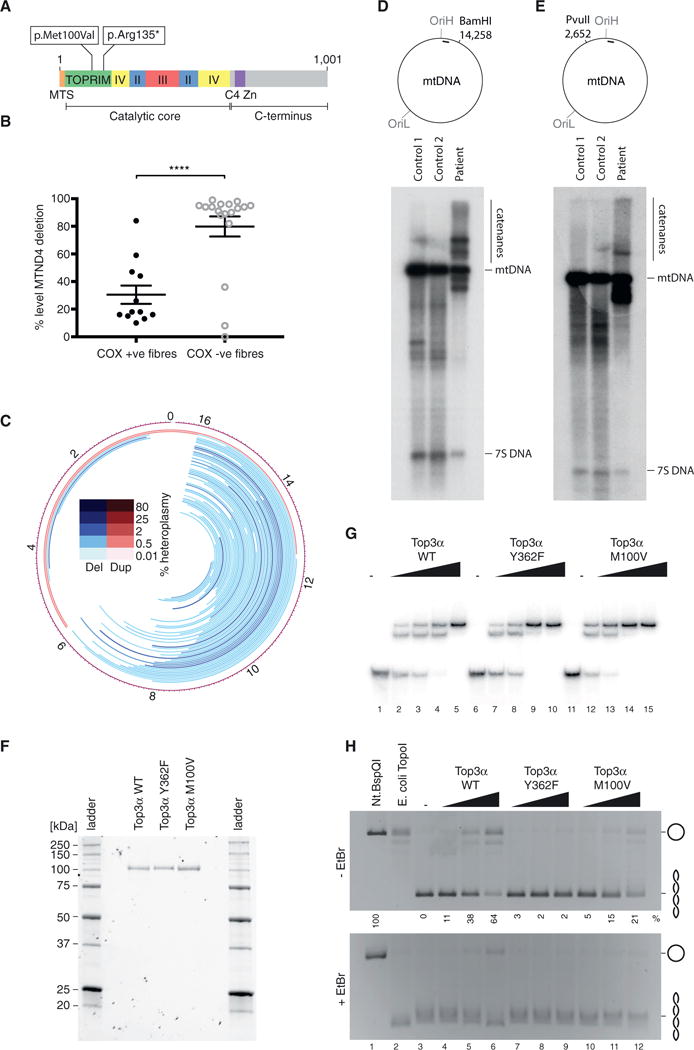
(A) Domain architecture of Top3α, indicating the sites of the identified compound heterozygous variants. MTS indicates the mitochondrial targeting sequence, and roman numerals indicate conserved domains.
(B) Quantitative single-fiber real-time PCR reveals that the majority, but not all, of the COX-deficient fibers exhibit high levels of a clonally expanded mtDNA deletion involving the MTND4 gene, a signature of multiple mtDNA deletions. Data represent median values ± SEM.
(C) DNA rearrangements in Top3α patient. Lines represent predicted mtDNA deleted (blue) or duplicated (red) regions.
(D and E) Southern blot analysis of Top3α patient skeletal muscle DNA indicating the presence of high-molecular-weight mtDNA structures. DNA was cleaved in the major arc with with BamHI (D) or in the minor arc with PvuII (E), separated on agarose, and blotted using probe (a) (indicated by black bar).
(F) Purified Top3α variants used for biochemical characterization. In total, 8 pmol of each protein was separated and detected on Mini-PROTEAN TGX Stain-Free gels (Bio-Rad).
(G) EMSA to assess binding of Top3α variants to a 5′ radiolabeled 80-nt ssDNA oligonucleotide. In total, 10 fmol ssDNA was incubated with increasing amounts of Top3α (0, 107.5, 215, 430, and 860 fmol) for 15 min at 37°C, then separated by 8% PAGE.
(H) DNA relaxation assay. In total, 200 ng negatively supercoiled pBluescript II SK(+) plasmid DNA was incubated with increasing amounts of Top3α protein (1.75, 5, and 10 pmol) for 45 min at 37°C, then separated on 1% agarose and imaged under UV light. Nt.BspQI (a nicking endonuclease) treatment represents the migration of nicked (open circle) DNA and E. coli TopoI treatment represents the migration of relaxed, covalently closed DNA. Numbers beneath lanes for the –EtBr gel indicate the percentage of fully relaxed substrate, with Nt.BspQI-treated sample (lane 1) set at 100%.
The variants were confirmed in the patient by Sanger sequencing, while her unaffected mother and daughter were shown to be heterozygous carriers of the p.Arg135* nonsense change. A diagnostic skeletal muscle biopsy revealed abundant COX-deficient and ragged-red fibers, pathological hallmarks of mtDNA-related disease (Figure S1G). Real-time PCR analysis of mtDNA from individual muscle fibers showed the presence of clonally expanded mtDNA deletions at very high levels (>80% mutated mtDNA) in the majority of COX-deficient, but not COX-positive, fibers (Figure 2B). Patient muscle mtDNA was further subjected to deep sequencing and mapping of breakpoints, which revealed extensive variable mtDNA rearrangements (Figure 2C), while no rearrangements were found in an unaffected control using the same methodology. Most rearrangements of patient mtDNA were predicted deletions within the major arc, with a cluster of breakpoints at the 3′ end of the D-loop, a recognized hotspot for mtDNA deletions (Zeviani et al., 1989). Interestingly, southern blotting of linearized mtDNA from patient skeletal muscle additionally revealed the presence of high-molecular-weight mtDNA species (Figures 2D and 2E), potentially representing catenated mtDNA structures caused by problems in mtDNA separation.
To directly assess the impact of the missense variant upon the enzymatic activity of Top3α, we purified recombinant Top3α from Sf9 insect cells. In addition to the wild-type and M100V variants, we also purified a Y362F mutant, which represents mutation of the catalytic tyrosine residue responsible for covalent linkage to the DNA substrate (Hanai et al., 1996). This variant is capable of binding, but not cleaving, ssDNA substrates (Goulaouic et al., 1999). The p.Arg135* variant lacked the C-terminal 867 amino acids, including the catalytic tyrosine residue, and was not analyzed further. The enzyme preparations were judged to be of high purity by staining of SDS-PAGE gels (Figure 2F). Gel shift analysis of DNA-binding activity confirmed that all three protein variants were capable of binding ssDNA, and it did not show any apparent differences between the binding activities (Figure 2G). We next assessed the effect of the M100V variant upon Top3α-mediated DNA relaxation. When incubated with negatively supercoiled dsDNA substrates, wild-type Top3α produces distinct topological isomers, which can be visualized on ethidium bromide-free agarose gels. The addition of ethidium bromide to the gels induces supercoiling of covalently closed plasmids, which allows nicked substrate to be separated from fully relaxed closed substrate. Nicking activity has been associated with the open, decatenating form of the enzyme (Cejka et al., 2012). Under these conditions, the wild-type enzyme was capable of relaxing the majority of the plasmid substrate, while the M100V variant was substantially less efficient (Figure 2H). The M100V variant also produced less nicked template compared to the wild-type protein. This observed impaired catalytic activity provides a molecular basis for the observed cellular phenotypes.
Loss of Top3α Affects the Topology of mtDNA
To further assess the effect of Top3α upon mtDNA topology, we again used siRNA depletion of Top3α in cultured HeLa cells, due to the tractability of this system. Intact mtDNA was separated on agarose gels and detected by southern blotting to assess the different topological forms of mtDNA. In wild-type cells, mtDNA was present as relaxed open circles, as well as variously supercoiled forms (Figure 3A, lanes 3 and 4). Loss of Top3α, but not Top1mt, Top2α, or Top2β, resulted in a profound loss of monomeric mtDNA forms (Figure 3A, lanes 5 and 6). However, when mtDNA from Top3α-depleted cells was treated with either S1 nuclease (an ssDNA-specific nuclease) or the branch-cutting enzyme T7 endonuclease I, forms of mtDNA migrating as the full-length, linearized mitochondrial genome were released (Figure 3B). We were unable to release the high-molecular-weight mtDNA species by treatment with purified human or prokaryotic type II topoisomerases (Figures S2A and S2B), suggesting that the structures did not represent fully double-stranded, catenated circular mtDNA molecules, but rather contained junctions with ssDNA character.
Figure 3. The M100V Variant of Top3α Shows Impaired Catalytic Activity.
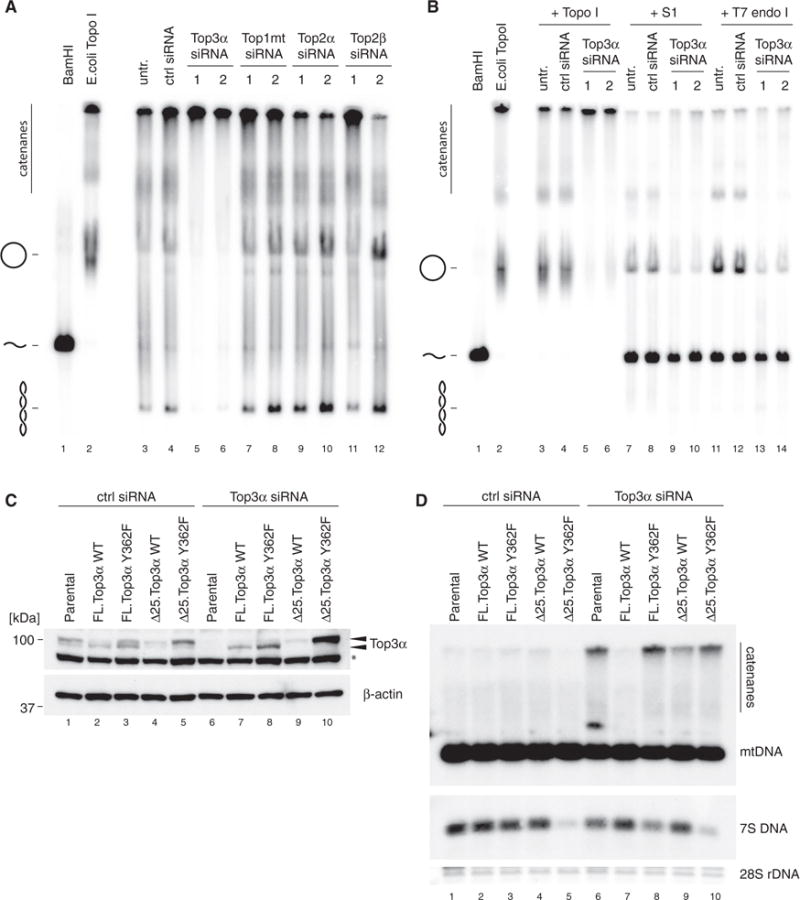
(A) mtDNA topology following siRNA depletion of topoisomerases. Uncut DNA (3 μg) was separated on low-percentage agarose, blotted, and detected using probe (a). BamHI-treated DNA marks the migration of linear mtDNA and E. coli TopoI-treated DNA marks the migration of relaxed, open circular form mtDNA.
(B) Enzymatic resolution of high-molecular-weight mtDNA species from Top3α depleted cells. Where indicated, DNA was treated with E. coli Topo I, S1 nuclease, or T7 endonuclease I prior to separation.
(C and D) Rescue of Top3α siRNA-induced mtDNA catenation phenotype. Flp-In T-REx 293 cells were stably transfected with recoded wild-type (WT) or catalytically inactive (Y362F) TOP3A cDNA in either full-length (FL) or N-terminally truncated (Δ25) form. Expression was induced using 2 ng/mL tetracycline in control siRNA-treated cells (lanes 1–5) or Top3α siRNA-treated cells (lanes 6–10).
(C) Western blot of Top3α depletion and expression of siRNA-resistant constructs. β-actin is used as a loading control. Arrows indicate the two forms or Top3α (nuclear and mitochondrial). Asterisk (*) denotes non-specific species.
(D) Southern blot of mtDNA from samples as in (C). Total DNA samples (3 μg) were restricted using BamHI, separated on agarose, blotted, and detected using probe (a). 28S rDNA is used as a loading control.
To verify that the loss of Top3α was directly responsible for the accumulation of high-molecular-weight mtDNA species, we expressed an siRNA-resistant version of Top3α, and we investigated if it could rescue the effect of endogenous Top3α knockdown in HEK293 cells (Figures 3C and 3D). Dual-targeted proteins typically produce two separable protein products, in which the shorter isoform represents the mitochondrial protein following cleavage of the MTS by the mitochondrial protein peptidase. Accordingly, expression of the full-length Top3α generated a shorter band corresponding to the processed isoform, whereas removal of the N-terminal 25 amino acids prevented mitochondrial targeting and resulted in a larger, unprocessed form of the protein (Figure 3C). The full-length, wild-type, siRNA-resistant Top3α rescued the accumulation of high-molecular-weight mtDNA species following the depletion of endogenous Top3α (Figure 3D). This capacity was lost when either the catalytic tyrosine residue was mutated (Y362F) or the mitochondrial targeting signal was eliminated, confirming that the observed effects are due to the mitochondrial action of Top3α.
Loss of Top3α Induces mtDNA Catenane Formation
We next directly visualized the high-molecular-weight mtDNA species resulting from Top3α depletion using EM. As expected, mtDNA isolated from cells treated with control siRNA was predominantly in monomeric relaxed circular (Figure 4A) or monomeric supercoiled (Figure 4B) forms. In contrast, mtDNA from Top3α-depleted cells was present as large catenated networks. The catenated species appeared as assemblies of circular DNA molecules linked together at a single central point, with only a small proportion of monomeric mtDNA remaining (Figures 4C and 4D).
Figure 4. Loss of Top3α Induces mtDNA Catenane Formation.
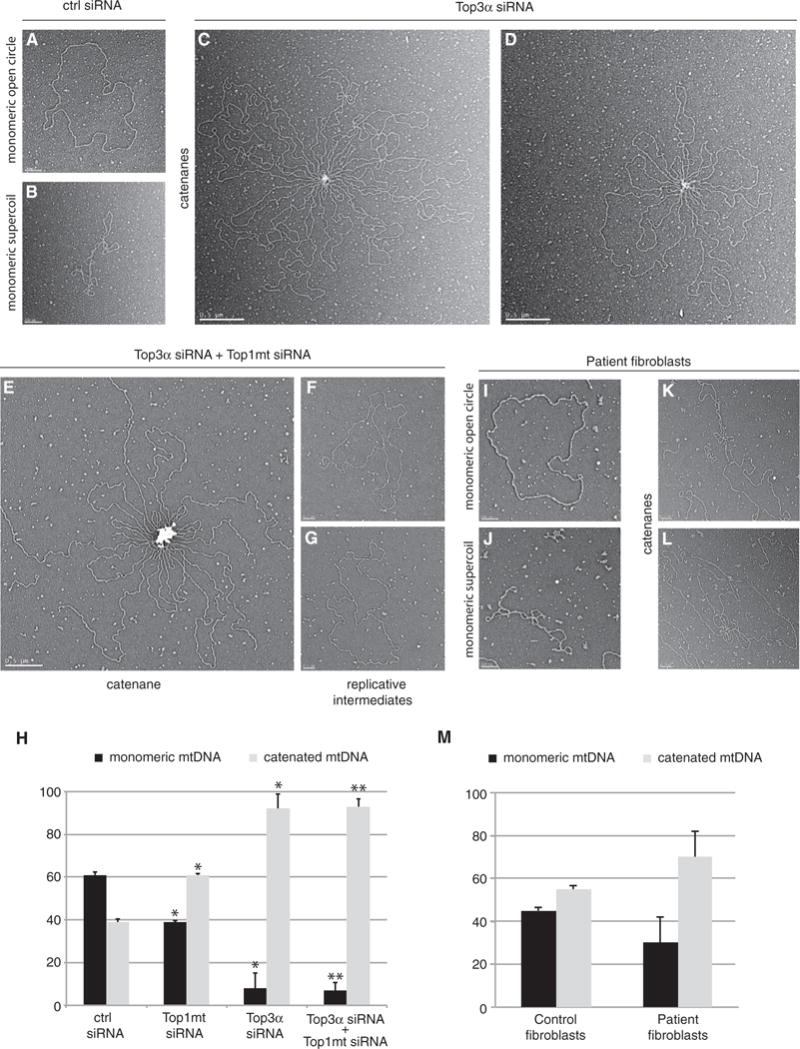
(A and B) Representive EM images of monomeric, open circular form mtDNA (A) and monomeric, supercoiled mtDNA (B) from control siRNA-treated cells. Scale bars, 200 nm.
(C and D) Representative EM images of catenated mtDNA from Top3α-depleted cells, example image 1 (C) and example image 2 (D). Scale bars, 0.5 mm.
(E) Representative EM images of catenated mtDNA from Top3α/Top1mt-depleted cells. Scale bar, 0.5 mm.
(F and G) Representative EM images of late-replicating mtDNA molecules from cells depleted of both Top3α and Top1mt, example image 1 (F) and example image 2 (G). Scale bars, 200 nm.
(H) Quantification of mtDNA molecules found in monomeric (relaxed and supercoiled) or catenated topological forms from EM analysis following the indicated siRNA treatments. Data are expressed as a percentage representing mean values from three independent experiments made in duplicate for each study group. Error bars represent SEM.
(I–L) Representative EM images of mtDNA from patient fibroblasts.
(I) Monomeric open circle (1n).
(J) Supercoiled monomeric mtDNA (1nsc).
(K) Catenane consisting of dimeric circles.
(L) Complex catenane consisting of trimeric circles (2n+1nsc). All scale bars, 200 nm.
(M) EM quantification of mtDNA in monomeric (open circle and supercoiled) and catenated forms from control and patient fibroblasts. Error bars represent SEM. Data are represented as a percentage representing mean values from two independent experiments made in duplicate for each study group.
In EM preparations, longer tracts of ssDNA can be revealed by incubation with a single-strand binding protein (SSB), which visibly coats ssDNA. Incubation of mtDNA with E. coli SSB did not reveal significant tracts of ssDNA (Figures S3A and S3B), further suggesting that the catenated mtDNA species are predominantly composed of fully replicated dsDNA and only contain short ssDNA stretches, rendering them sensitive to S1 nuclease.
In parallel, we studied the effect of Top1mt depletion upon mtDNA structure. Under normal conditions, loss of Top1mt has only mild effects on mtDNA maintenance. Depletion of Top1mt only led to a small increase in the level of catenated species, which were smaller than those seen in Top3α cells and not of a consistent structure (Figures S3C–S3E). We simultaneously depleted Top3α and Top1mt to assess whether these two proteins may cooperate in mtDNA maintenance. Indeed, when both topoisomerases were depleted, we observed a profound and synergistic depletion of mtDNA copy number and an apparent total loss of 7S DNA (Figures S4A and S4B). Visualization of mtDNA from Top3α/Top1mt-depleted cells by EM found the presence of catenated structures very similar to those seen in Top3α-depleted cells (Figure 4E). In addition, we also observed an increased number of late replication intermediates, suggesting a strong replication stalling phenotype (Figures 4F and 4G), consistent with the loss of mtDNA copy number. Quantification of monomeric and catenated mtDNA forms found no difference between the proportion of catenated mtDNA in Top3α/Top1mt-depleted cells compared with Top3α-depleted cells (Figure 4H). This suggests that the loss of Top1mt in combination with the loss of Top3α has an additive inhibitory effect upon replication progression, but not upon mtDNA decatenation.
We additionally assessed the degree of mtDNA catenation in patient fibroblasts carrying the Top3α M100V variant using EM. While mtDNA isolated from control human fibroblasts (Figures S3F–S3H) was equally distributed between free and catenated states (45% free and 55% catenated), in the patient the degree of mtDNA catenation was increased (Figures 4I–4L). In these cells, up to 70% of the mtDNA was assembled in complex structures with only 30% in monomeric form (Figure 4M). Taken together, these features support the direct involvement of Top3α in mtDNA maintenance and segregation, since its deficiency severely affects the topology and stability of human mtDNA in vivo.
Catenane Formation Is Centered around OriH
Many of the catenanes observed in Top3α-depleted cells (Figure 4) were joined through a single central point. This observation suggested that Top3α-mediated mtDNA decatenation is centered at a specific genomic region. Since mtDNA replication is initiated in the NCR and is unidirectional, we hypothesized that separation of fully replicated genomes would also occur in this region.
EM analysis of different NCR-containing restriction fragments of mtDNA from Top3α/Top1mt-depleted cells revealed a large proportion of X-shaped DNA molecules (Figures 5A and 5B). These molecules corresponded to two mtDNA genomes that remained held together through a single junction following digestion, consistent withthe large number of dimeric late replication intermediates found in this study group. By measuring the length of the DNA arms from overlapping restriction fragments, we found that the junction site frequently mapped to the OriH region, establishing this as a major site of Top3α-mediated mtDNA separation. X-shaped molecules were also visible in OriH-containing restriction fragments of mtDNA from Top3α-depleted cells using 2D agarose gel electrophoresis (Figure 5C). Analysis of restriction fragments from regions of the mtDNA excluding the NCR also revealed that depletion of Top3α produced a pronounced and genome-wide replication-stalling phenotype (Figures S4C–S4E), consistent with the loss of mtDNA copy number in these cells.
Figure 5. Catenane Formation Is Centered around OriH.
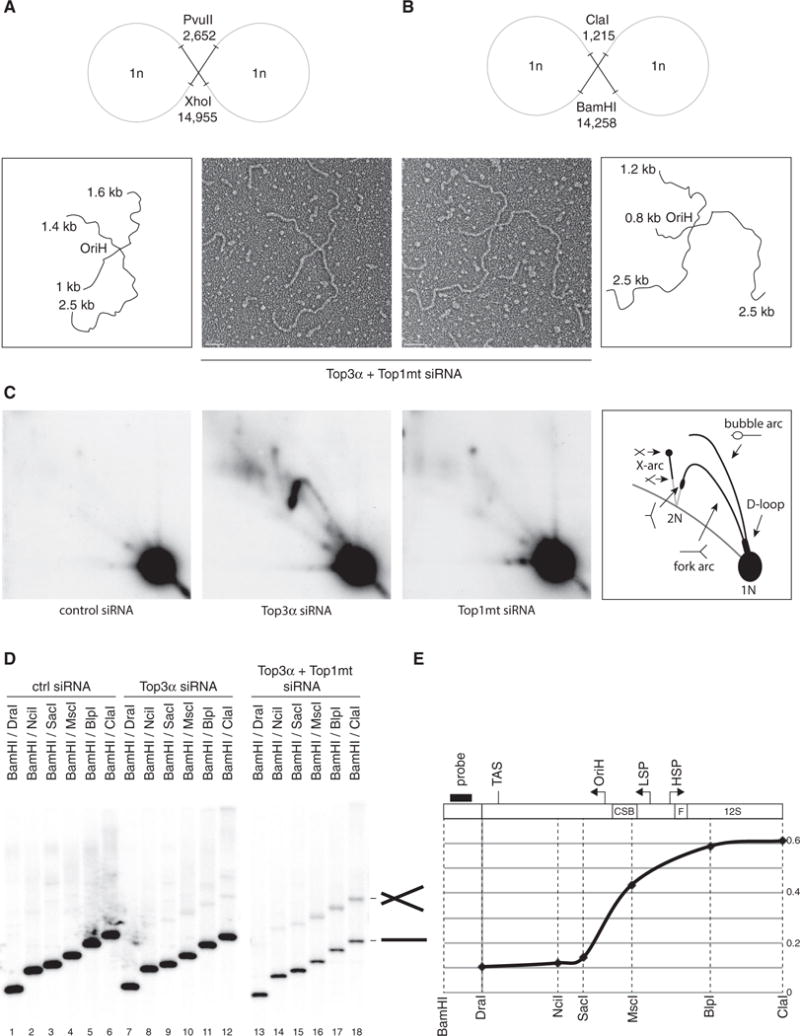
(A and B) Representative electron micrographs of X-shaped, NCR-containing mtDNA molecules, with interpretations. Restriction enzyme digestion with PvuII/XhoI (A) or BamHI/ClaI (B) generates species with arm lengths that map the junction to the OriH region: 1.8 kb/2.5 kb for PvuII/XhoI and 1 kb/2.5 kb for BamHI/ClaI. Scale bars, 200 nm.
(C) mtDNA from control, Top3α, or Top1mt-depleted cells was restricted using HincII, separated by 2D agarose gel electrophoresis, and detected using probe (a). X-shaped molecules are visible as an X-arc in Top3α-depleted cells.
(D) Total DNA from cells depleted of Top3α and Top1mt was restricted with the indicated enzymes, separated on agarose, and blotted using probe (b). X-shaped molecules are only visible when the probed fragment includes the molecule junction.
(E) Quantification of the proportion of X-shaped species, normalized to the level of linear species for the same digestion. Black bar indicates the probe. Dotted lines indicate the sites of restriction enzymes used. Gene loci and key sequence elements are indicated above the graph.
We next sought to determine the location of the catenane junction using restriction mapping. When mtDNA from cells depleted of Top3α was cleaved to create a series of fragments of different lengths (between 3,526 and 12,728 bp) containing the replication origin OriH, we consistently observed an additional band that migrated at approximately twice the expected fragment size, corresponding to the X-shaped molecules seen by EM (Figures S5A and S5B). Ethidium bromide was included in the restriction digests to inhibit thermal branch migration of cleaved interlinked molecules (Harmon et al., 1999). These species were specific for combinations of restriction enzymes that spanned the NCR, and they were particularly prominent in cells depleted of both Top3α and Top1mt (Figure S5C). X-shaped molecules could also be generated from high-level expression of catalytically inactive Top3α in HEK cells (Figures S5D–S5F), presumably as a dominant-negative effect. Consistent with previous observations that junction-containing mtDNA molecules are sensitive to ssDNA endonucleases (Figure 3B), S1 nuclease treatment caused specific depletion of the X-shaped molecules without affecting the linear restriction fragments (Figure S5C).
To more precisely map the location of the catenane junction, we created a series of mtDNA restriction fragments from cells depleted of Top3α and Top1mt to produce DNA fragments either excluding or including OriH. The probe was then placed outside of the NCR, on the basis that catenated species would only be observed when the restriction digest extends beyond the catenane junction. High levels of X-shaped species were limited to the region including OriH and immediately upstream (Figures 5D and 5E), suggesting that this is the primary site of post-replicative mtDNA separation.
To better characterize the structure of the X-shaped species, a junction-containing restriction fragment was subjected to treatment with a series of enzymes with characterized activities. The X-shaped molecules were found to be susceptible to cleavage by the DNA branch-cutting enzymes T7 endonuclease I and T4 endonuclease VII, but resistant to the E. coli Holliday junction resolvase RuvC (Figures S6A and S6B). In addition, X-shaped molecules were susceptible to thermal branch migration in a buffer containing magnesium, which inhibits the migration of Holliday junctions (Panyutin and Hsieh, 1994) (Figure S6C). Taken together, these data establish that the mtDNA resolution structure consists of a branched molecule with ssDNA character that does not represent a Holliday junction. We therefore propose that this structure consists of a hemicatenane formed during the termination of mtDNA replication.
Mitochondrial Top3α Functions Independently of the BTR Complex
In the nucleus, Top3α is a subunit of the BTR complex together with the helicase BLM and the OB-fold proteins RMI1 and RMI2, which together dissolve double-Holliday junctions to produce noncrossover products. Of these proteins, only Top3α was detectable in our mitochondrial preparations, whereas BLM, RMI1, and RMI2 were not (Figure 6A). To address if the observed mtDNA maintenance defect associated with loss of Top3α was secondary to its role in the nuclear BTR complex, we used siRNA to deplete BLM, RMI1, and RMI2. Efficient depletion of protein levels was confirmed using western blotting of cell lysates (Figure 6B). Interestingly, whereas knockdown of RMI1 caused a destabilization of the longer nuclear isoform of Top3α, the processed, shorter mitochondrial isoform was unaffected, supporting the notion that mitochondrial Top3α functions independently of the nuclear BTR complex (Figure 6B, lanes 5 and 6). Furthermore, knockdown of BLM, RMI1, or RMI2 did not cause depletion of mtDNA levels (Figure 6C) or alteration of mtDNA topology (Figure 6D), demonstrating that loss of mtDNA was not secondary to nuclear effects caused by loss of the BTR complex.
Figure 6. Mitochondrial Top3α Is Independent of the BTR Complex.
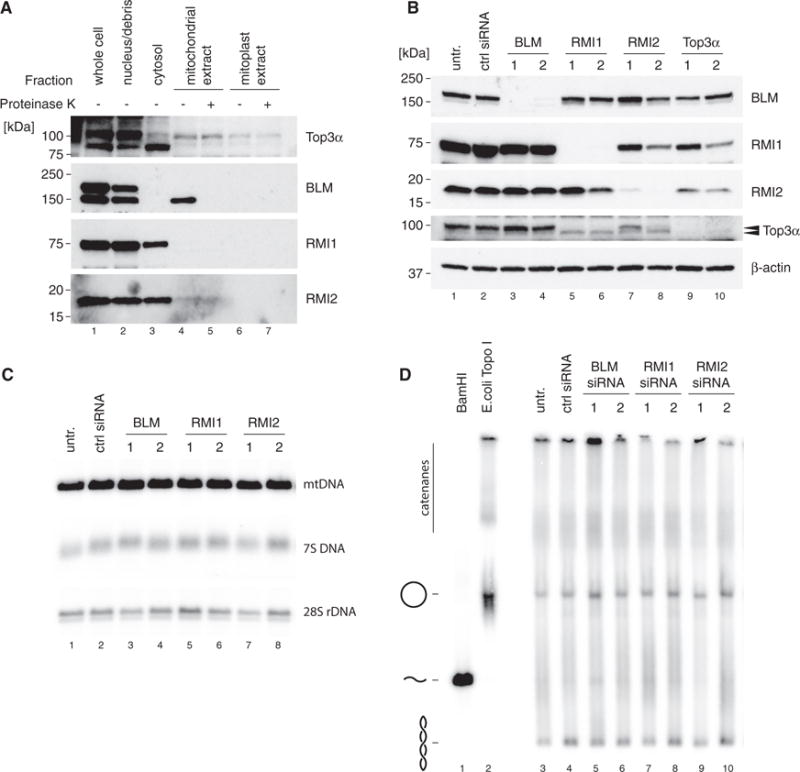
(A) Mitochondrial localization of protein components of the BTR complex by western blotting of cell fractions as in Figure 1A. Top3α blot is reproduced from Figure 1A for reference.
(B) Efficiency of depletion of BTR complex components assessed by western blotting of HeLa cell lysates. β-actin is used as a loading control. Arrows indicate the two forms or Top3α (nuclear and mitochondrial).
(C) Southern blot analysis of mtDNA copy number and 7S DNA levels following siRNA depletion of BLM, RMI1, and RMI2 as in (B). 28S rDNA is used as a loading control.
(D) mtDNA topology following siRNA depletion of BTR complex proteins. Uncut DNA (3 mg) was separated on low-percentage agarose, blotted, and detected using probe (a). BamHI-treated DNA marks the migration of linear mtDNA and E. coli TopoI-treated DNA marks the migration of relaxed, open circular form mtDNA.
Loss of Top3α Perturbs Nucleoid Separation and Distribution within the Mitochondrial Network
We used confocal microscopy to visualize the effects of Top3α depletion upon mtDNA separation and segregation. Cells were stained with anti-DNA antibodies to detect the mtDNA nucleoid and with antibodies against the outer mitochondrial membrane protein TOM20 to stain the mitochondrial network. In control siRNA-treated cells, nucleoids appeared as a punctate pattern spread throughout the cytoplasm, which co-localized with the mitochondrial network (Figure 7A). In cells depleted of Top3α, there was a noticeable change in nucleoid size and distribution, with cells containing fewer and larger nucleoids. This loss of nucleoid number was exacerbated in cells depleted of both Top3α and Top1mt in combination, consistent with the loss of mtDNA copy number in these cells. Quantification of nucleoid numbers found that Top3α depletion induced a decrease of approximately 70% in the number of nucleoids per cell, and depletion of both Top3α and Top1mt caused an 83% decrease (Figure 7C). Depletion of RMI1 did not appear to affect either nucleoid size or number, confirming that the observed effect of Top3α depletion was due to the mitochondrial isoform of Top3α.
Figure 7. Loss of Top3α Perturbs Nucleoid Separation and Distribution within the Mitochondrial Network.
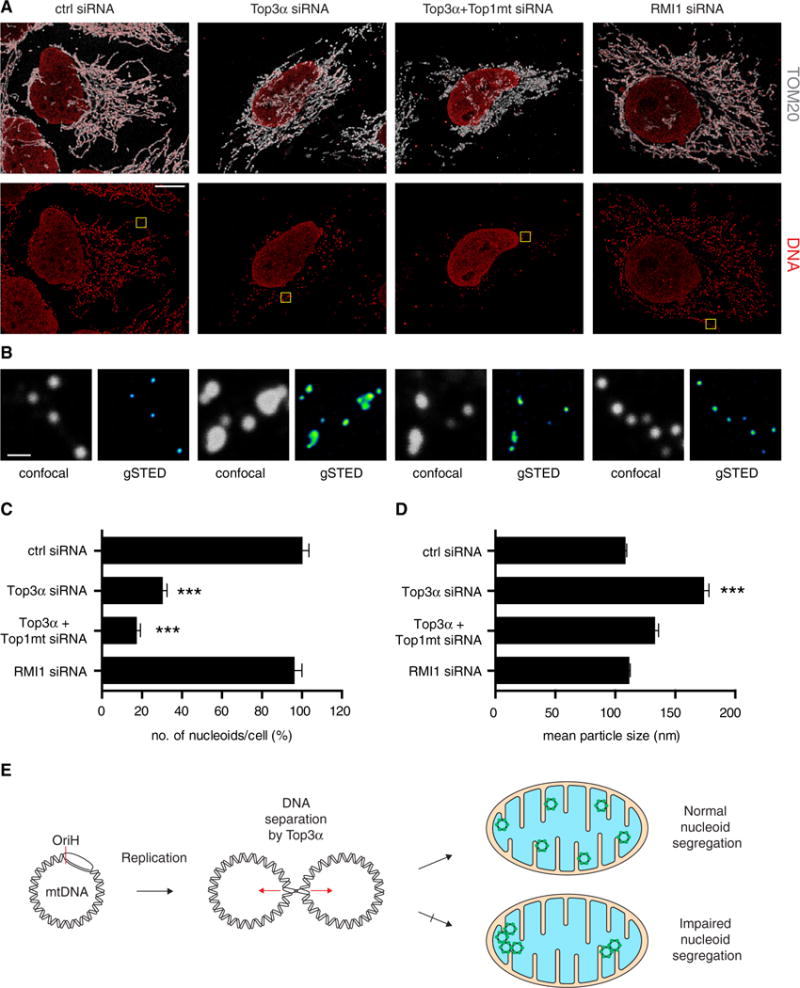
(A) Representative confocal microscopy images of HeLa cells following the indicated siRNA treatments, showing merged channels of DNA and TOM20 (upper panels) and single channels of DNA (lower panels). Scale bar, 10 μm.
(B) Magnifications of the yellow boxed areas in (A) showing the imaged nucleoids in confocal (gray, left) and g-STED (Green Fire LUT, right). Scale bar, 600 nm.
(C) Quantification of nucleoid number per cell following the indicated siRNA treatments. Nucleoid number is expressed as a percentage of control siRNA-treated cells (from 4 independent experiments, n ≥ 9 cells analyzed). Error bars represent SEM.
(D) Mean size of nucleoids determined by g-STED under the indicated siRNA conditions. Number of nucleoids analyzed from three independent experiments: control siRNA = 244; Top3α siRNA = 357; Top3α + Top1mt siRNA = 361; RMI1 siRNA = 393. Error bars represent SEM.
(E) Model for the mechanism of action of Top3α in human mitochondria. Following mtDNA replication, Top3α is required to resolve mtDNA hemicatenanes and separate the newly synthesized mtDNAs. Loss of mitochondrial Top3α activity leads to mtDNA catenation and impaired nucleoid segregation.
We next used gated stimulated emission depletion (g-STED) super-resolution microscopy to make quantitative measurements of nucleoid size. STED is preferable for such measurements because the diameter of the nucleoid, at approximately 100 nm, is below the diffraction barrier of conventional confocal microscopy. Indeed, application of g-STED to DNA-stained cells depleted of Top3α permitted the resolution of apparently single DNA structures in confocal images into multiple juxtaposed structures (Figure 7B). Calibration of the microscope using fluorescent DNA origamis confirmed that the resolution was sufficient for quantitative nucleoid measurement under the conditions used (Figures S7A and S7B). Measurement of nucleoid diameter in control siRNA-treated cells confirmed the previously determined size of approximately 100 nm (Figure 7D), corresponding to one mtDNA molecule compacted with proteins (Kukat et al., 2011). In contrast, cells depleted of Top3α showed a greatly enlarged mean nucleoid diameter of approximately 170 nm. This effect was also apparent, albeit at lower resolution, from confocal images of the same cells (Figure S7C). Top3α-depleted cells also showed a greatly enlarged range of nucleoid sizes, extending from around 100 nm up to approximately 350–400 nm (Figure S7D). The fact that these structures could not be resolved at the resolution permitted by g-STED implies that they represent multiple physically conjoined mtDNA genomes, consistent with the structures observed using EM (Figure 4). Top3α-mediated DNA separation is therefore required for nucleoid segregation within the mitochondrial network.
DISCUSSION
At the completion of mtDNA replication, nucleoids must not only be physically separated but also distributed throughout the mitochondrial network. The co-localization of the mitochondrial genome with its protein products of the oxidative phosphorylation (OXPHOS) system at the inner mitochondrial membrane means that proper mtDNA distribution is crucial to maintain proper cellular energy homeostasis. In this work, we demonstrate that Top3α is required for mtDNA separation in vivo (Figure 7E). Top3α has been characterized as an ssDNA decatenase (Yang et al., 2010), but it is also capable of resolving hemicatenanes, which are structures in which only one strand of a dsDNA molecule is interlinked with one strand of another dsDNA molecule. These types of structures can form as a final intermediate from the unwinding of converging replication forks, and E. coli topoisomerase III (together with the RecQ helicase) is capable of resolving late-replicating plasmid intermediates in vitro (Suski and Marians, 2008). Resolution of fully replicated mtDNA molecules through a hemicatenane intermediate as outlined here would presumably obviate the requirement for a type II topoisomerase activity in mitochondria, consistent with the absence of either type II topoisomerase in our mitochondrial preparations. The function of Top3α may be evolutionarily conserved, in as much as Drosophila mutants specifically lacking the mitochondrial isoform of Top3α show loss of mtDNA copy number and an increased rate of mtDNA deletions (Tsai et al., 2016; Wu et al., 2010), even if the molecular basis for this effect in flies remains unknown.
The role of nuclear Top3α in the dissolution of double-Holliday junctions during homologous recombination requires the RecQ family helicase BLM, as well as the OB-fold proteins RMI1 and RMI2 (the BTR complex). However, whereas the loss of Top3α was found to affect mtDNA maintenance, topology, and separation, knockdown of the other BTR complex components (BLM, RMI1, or RMI2) had no discernible effect, and no mitochondrial isoforms of these proteins were detectable (Figure 6). These data demonstrate that mitochondrial Top3α functions independently of its nuclear cofactors. Since mitochondria are not known to possess a machinery for homologous recombination, it could be expected that the physiological substrates of Top3α in mitochondria differ from those in the nucleus. It is possible that mitochondrial Top3α is capable of performing its role independently of other cofactors or, alternatively, that a separate set of cofactors exists that helps Top3α to fulfil its mitochondrial function.
We have mapped the location of the junction between nonseparated daughter mtDNA molecules to a region near OriH. Our data suggest that replication has been completed but that the newly replicated mtDNA molecules remains interlocked at this site. Available models of mtDNA replication stipulate that replication initiates in the region of OriH and is essentially unidirectional, and so this region would logically also be the site of leading strand replication completion. However, as lagging strand replication initiates at a separate site, the steps that lead to the completion of lagging strand replication, and to the formation of the resolution structure, remain unclear.
mtDNA is anchored to the mitochondrial inner membrane, with the OriH region being the probable attachment site (Albring et al., 1977). It therefore seems plausible that the unlinking of replicated mtDNA molecules at the origin of replication would be coupled to the separation of nucleoids through mitochondrial dynamics. Mitochondrial division sites coincide with regions in which contacts are formed between mitochondria and the endoplasmic reticulum (ER). These contact sites are in turn spatially linked to nucleoids involved in active mtDNA synthesis, a process that occurs before mitochondrial division (Lewis et al., 2016). The data suggest that contacts between the ER and mitochondria may help to coordinate licensing of mtDNA synthesis with division to distribute newly replicated nucleoids to daughter mitochondria. This process could be disrupted by either a failure of mtDNA separation (leading to mtDNA catenation) or a failure of mitochondrial fission (leading to nucleoid clustering). In support of this notion, loss of the fission GTPase DRP1 in mice causes severe mtDNA nucleoid clustering and a mosaic deficiency of mitochondrial respiration (Ishihara et al., 2015).
Defects in the mitochondrial fusion machinery are also associated with mtDNA instability and OXPHOS deficiency in both animal models (Chen et al., 2010) and in human pathology (Amati-Bonneau et al., 2008; Hudson et al., 2008; Rouzier et al., 2012). For instance, a proteolytic fragment of the OPA1 protein has been implicated in anchoring of mtDNA to the membrane, with loss of this fragment leading to nucleoid clustering (Elachouri et al., 2011). Pathogenic variants in the OPA1 gene cause changes in the distribution of mtDNA molecules, with some regions within the mitochondrial network being devoid of mtDNA and other regions containing larger nucleoid assemblies. Pathological sequence variants in RNASEHI have also been associated with defective mtDNA segregation (Akman et al., 2016); in both examples, these are associated with adult-onset PEO+ phenotypes (Amati-Bonneau et al., 2008; Hudson et al., 2008; Reyes et al., 2015). In this respect, the identification of pathogenic TOP3A variants in a patient with the clinicopathological hallmarks of a disturbance in mtDNA maintenance is significant, and as such pathogenic variants in this gene should be considered in patients with PEO and multiple mtDNA deletions in muscle but without a firm genetic diagnosis. The precise mechanisms by which pathogenic TOP3A variants may cause mtDNA deletions remain to be addressed. However, we propose that deletion formation is secondary to changes in mtDNA topology, caused by reduced Top3α activity and leading to widespread replication stalling. In support of this notion, TWINKLE mutations that cause replication stalling predispose to deletion formation (Goffart et al., 2009).
It appears that mitochondrial dynamics and mtDNA replication are closely linked processes, and in this context Top3α may function to coordinate the separation of newly replicated mtDNA daughter molecules with mitochondrial fission. Similar to the separation of sister chromatids in the nucleus during mitosis, mitochondrial fission may apply a physical separating force upon newly replicated mtDNA molecules, which could stimulate Top3α-dependent separation. In future studies, we hope to further address these and related ideas to clarify the molecular machinery underlying proper mtDNA separation and segregation in mammalian cells.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Top3α | Proteintech Group | Cat#14525-1-AP; RRID: AB_2205881 |
| Rabbit polyclonal anti-Top1mt | Sigma-Aldrich | Cat#HPA001915; RRID: AB_1080331 |
| Rabbit monoclonal anti-Top2α | Abcam | Cat#ab52934; RRID: AB_883143 |
| Mouse monoclonal anti-Top2β | Santa Cruz Biotechnology | Cat#sc-25330; RRID: AB_628384 |
| Rabbit polyclonal anti-Histone H3 | Abcam | Cat#ab1791; RRID: AB_302613 |
| Rabbit polyclonal anti-TFAM | Shi et al. (2012) | N/A |
| Rabbit polyclonal anti-AIF | Abcam | Cat#ab1998; RRID: AB_302748 |
| Mouse monoclonal anti-β-actin (clone AC-15) | Abcam | Cat#ab6276; RRID: AB_2223210 |
| Mouse monoclonal anti-VDACI/Porin | Abcam | Cat#ab14734; RRID: AB_443084 |
| Rabbit polyclonal anti-BLM | Novus | Cat#NB100-214; RRID: AB_10001628 |
| Rabbit polyclonal anti-RMI1 | Proteintech Group | Cat#14630-1-AP; RRID: AB_2301203 |
| Rabbit polyclonal anti-RMI2 | Abcam | Cat#ab122685; RRID: AB_11127123 |
| Mouse monoclonal IgM anti-DNA (clone AC-30-10) | Progen | Cat#61014; RRID: AB_1541137 |
| Rabbit polyclonal anti-TOM20 | Santa Cruz Biotechnology | Cat#sc-11415; RRID: AB_2207533 |
| Sheep anti-Mouse HRP | GE Healthcare | Cat#NA931; RRID: AB_772210 |
| Goat anti-Rabbit HRP | Thermo Fisher | Cat#31460; RRID: AB_228341 |
| Goat anti-Rabbit Atto647N | Sigma-Aldrich | Cat#40839; RRID: AB_1137669 |
| Goat anti-Mouse IgM Alexa Fluor 594 | Thermo Fisher | Cat#A-21044; RRID: AB_2535713 |
| Bacterial and Virus Strains | ||
| Autographa californica nuclear polyhedrosis virus | Clontech | Cat# 631401 |
| XL1-Blue Competent Cells | Agilent Technologies | Cat#200249 |
| Biological Samples | ||
| Human skeletal muscle biopsy | Isolated from patient (this paper) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine RNAiMAX | Thermo Fisher | Cat#13778075 |
| Aqua-Poly/Mount | Polysciences | Cat#18606 |
| Top3α-6His - WT, Y326F and M100V | This paper | N/A |
| Proteinase K | Thermo Fisher | Cat#AM2548 |
| T7 endonuclease I | New England Biolabs | Cat#M0302 |
| S1 nuclease | Promega | Cat#M5761 |
| E. coli Topo I | New England Biolabs | Cat#M0301 |
| Human TopIIα | Inspiralis | Cat#HT201 |
| Human TopIIβ | Inspiralis | Cat#HTB201 |
| E. coli Gyrase | Inspiralis | Cat#G1001 |
| E. coli TopoIV | Inspiralis | Cat#T4001 |
| E. coli SSB | Thermo Fisher | Cat#70032Z500UG |
| Exonuclease VII | New England Biolabs | Cat#M0379S |
| T4 endonuclease VII | MCLab | Cat#ENDO7-100 |
| RuvC | Abcam | Cat#ab63828 |
| Lambda exonuclease | New England Biolabs | Cat#M0262S |
| Critical Commercial Assays | ||
| Mitochondria isolation kit, human | Miltenyi Biotec | Cat#130-094-532 |
| Pre-Separation Filters (20 μm) | Miltenyi Biotec | Cat#130-101-812 |
| Prime-It II Random Primer Labeling Kit | Agilent Technologies | Cat#300385 |
| HIS-Select Nickel Affinity Gel | Sigma-Aldrich | Cat#P6611 |
| Superdex 200 16/600 Gel Filtration Columns | GE Healthcare | Cat#28989335 |
| HiTrap SP HP Cation Exchange Columns | GE Healthcare | Cat#17115101 |
| HiTrap Heparin HP Columns | GE Healthcare | Cat#17-0406-01 |
| SureSelectXT Human All Exon V5 bait library 96 | Agilent | Cat#5190-6209 |
| Nextera DNA library preparation kit | Illumina | Cat#FC-121-1030 |
| Deposited Data | ||
| mtDNA sequencing data | This paper | https://www.ebi.ac.uk/ena; ENA: PRJEB23559 |
| Mendeley Data dataset | This paper | https://doi.org/10.17632/43wnnd4tjw.1 |
| ClinVar NCBI | This paper | https://www.ncbi.nlm.nih.gov/clinvar/; ClinVar: SCV000611605 and SCV000611606 |
| Experimental Models: Cell Lines | ||
| Human: HeLa cells | Sahlgrenska Academy Cell Culture Core Facility | N/A |
| Human: Flp-In T-REx 293 cells | Sahlgrenska Academy Cell Culture Core Facility | N/A |
| Human: patient primary fibroblasts passage 5–10 | This paper | N/A |
| Spodoptera frugiperda: Sf9 cells | Clontech | 631402 |
| Oligonucleotides | ||
| Primers for radiolabelled probes, see Table S1 | This paper | N/A |
| Silencer Select siRNAs, see Table S1 | Thermo Fisher | N/A |
| Primers for Sanger sequencing of TOP3A variants, see Table S1 | This paper | N/A |
| EMSA substrate oligo, see Table S1 | This paper | N/A |
| AllStars Negative Control siRNA | QIAGEN | Cat#SI03650318 |
| Recombinant DNA | ||
| Plasmid: pBacPAK9 | Clontech | Cat#631402 |
| Plasmid: pBacPAK9-Top3α16-1001-6His | This paper | N/A |
| Plasmid: pBluescript II SK(+) | Stratagene | Cat#212205 |
| Plasmid: pcDNA5 FRT/TO | Thermo Fisher | Cat#V652020 |
| Plasmid: pcDNA5 FRT/TO-Top3α | This paper | N/A |
| Plasmid: pOG44 | Thermo Fisher | Cat#V600520 |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Adrian’s FWHM plugin for ImageJ | NIH | https://imagej.nih.gov/ij/plugins/fwhm/ |
| MultiGauge V3.0 | Fujifilm | N/A |
| Gatan Digital Micrograph | Gatan | http://www.gatan.com/products/tem-analysis/gatan-microscopy-suite-software |
| Bowtie 2 | Johns Hopkins University | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| LAST | CBRC, AIST | http://last.cbrc.jp/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Claes Gustafsson (claes.gustafsson@medkem.gu.se).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Case Report
A 67-year-old-woman presented with slowly progressive ptosis, intermittent double vision, mild dysphagia and nasal regurgitation, sensorineural hearing loss, rapid cycling mood disorder, cardiac arrhythmias (necessitating a permanent pace maker insertion), reduced exercise tolerance and prominent cerebellar ataxia. Her sister died aged 27 years from complications of systemic lupus erythematosus. Her father was deceased but no history of myopathy and her mother, aged 93 years and five children were otherwise well. Examination revealed bilateral ptosis, moderate to severe asymmetric PEO, frontalis muscle over activity, staccato speech, neck flexor weakness (MRC4+/5), proximal limb weakness (MRC4+/5), upper limb dysmetria, retained tendon reflexes and a broad-based gait. Pyramidal or extrapyramidal features were not found. Serum lactate was 1.3mmol/L (< 2) and serum creatine kinase was 251 U/L (10-160). Nerve conduction studies showed a mild sensory neuropathy and brain MRI (Magnetic Resonance Imaging) revealed bilateral symmetrical changes within the thalami, red nuclei and long tracts within the midbrain, in addition to cerebellar atrophy. Cardiology work up including an echocardiogram revealed initially impaired left ventricular (LV) function (LVEF 55%–60%) and right ventricular outflow tract ectopic beats with symptoms necessitating computerized assisted balloon mapping of right ventricular outflow tract ectopy and ablation. Holter electrocardiogram (ECG) showed non-sustained atrial and ventricular tachycardia necessitating placement of a permanent pace maker. Exome sequencing had local ethical and review board approval (13/YH/0310 Braford-Leeds research ethics committee and Newcastle upon Tyne Hospitals NHS Foundation Trust).
Cell lines
HeLa cells were grown at 37°C, 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM; 4.5 g/l glucose, 2 mM glutamine, 110 mg/ml sodium pyruvate) supplemented with 10% fetal bovine serum (FBS) and 5% penicillin/streptomycin. Fibroblasts were grown as above with the addition of 50 mg/ml uridine. Untransfected (parental) Flp-In T-REx 293 cells were grown in DMEM supplemented with 10% FBS, 5% penicillin/streptomycin, 15 mg/ml blasticidin, and 100 mg/ml Zeocin. Following stable transfection, these cells were grown as above with the addition of 50 mg/ml hygromycin and without Zeocin. Spodoptera frugiperda (Sf9) cells used for recombinant protein expression were grown in suspension in HyClone SFX-Insect medium supplemented with 5% FBS and 5% penicillin/streptomycin at 27°C.
METHOD DETAILS
Cell line manipulation
For siRNA transfections, HeLa cells were reverse transfected with 5 nM of siRNA using Lipofectamine RNAiMAX. After three days cells were re-transfected, for a total of six days of transfection. For siRNA transfections of Flp-In T-REx 293, cells were plated 24 hr before transfection and forward transfected using the same concentrations and conditions as for HeLa cells. Sequences of siRNAs are provided in Table S1.
Cell fractionation
Suspension HeLa cells (approx. 5 × 108) were pelleted by centrifugation at 300 × g for 10 min, washed in PBS and weighed to determine the volume (assuming a cell density of 1.25 g/ml). Pellet was resuspended in 9 volumes hypotonic buffer (20 mM HEPES pH 8.0, 5 mM KCl, 1.5 mM MgCl2, 2 mM DTT, 1 mg/ml BSA, 1 mM PMSF and 1 3 proteinase inhibitors (1 mM PMSF, 2 mM pepstatin A, 0.6 mM leupeptin and 2 mM benzamidine in 100% ethanol)), incubated on ice for 10 minutes then homogenized using 10 strokes of a 15 mL glass Dounce homogenizer with tight-fitting pestle. A two-thirds volume of 2.5 × MSH buffer (525 mM mannitol, 175 mM sucrose, 20 mM HEPES (pH 8.0), 5 mM EDTA, 1 mg/ml BSA, 0.2 mM PMSF, 2 mM DTT, and 1 3 protein inhibitors) was then added and the homogenate was centrifuged twice at 1600 × g for 10 min to pellet nuclei and unbroken cells. The supernatant was collected and centrifuged at 10,000 × g for 10 min, and the supernatant from this step was retained as the cytosolic fraction. The pellet was then resuspended in 1 3 MSH (210 mM mannitol, 70 mM sucrose, 20 mM HEPES (pH 8.0), 2 mM EDTA, 1 mg/ml BSA, 0.2 mM PMSF and 1 × proteinase inhibitors), supplemented with 10 mM MgCl2, to a final concentration of 2 mg/ml (assuming 10 mg of mitochondria per gram of cells). 125 U of benzonase was added and incubated for 30 min on a roller at 4°C. Samples were pelleted at 10,000 × g for 10 min and washed twice with 5 mL of 1 × MSH without BSA. Pellets were resuspended in a minimal volume of 1 × MSH and loaded onto a two-step sucrose gradient (1.5 M/1 M sucrose made up in gradient buffer (10 mM HEPES pH 7.8, 5 mM EDTA, 2 mM DTT)) and centrifuged at 130,000 × g (39,000 rpm in a Beckman Coulter TLS55 rotor) for 1 hour at 4°C. The interface fraction was collected and four volumes gradient buffer added, then pelleted at 10,000 × g for 10 min. This stage represents the mitochondrial fraction. This pellet was divided into two, and half used for making mitoplasts according to the ‘swell-contract’ method (Pallotti and Lenaz, 2007). Mitochondria were resuspended in potassium phosphate buffer (pH 7.2) and incubated on ice for 20 min. ATP and MgCl2 were then added to final concentration of 1 mM each, incubated on ice for a further 5 min, then pelleted by centrifugation at 15,000 × g for 10 min and washed once with 1 × MSH. Half of each of the mitochondrial and mitoplast fractions were treated with proteinase K to remove externally-bound proteins. Fractions were incubated with 50 μg/ml proteinase K for 30 min at 4°C, then the reaction was stopped by the addition of 5 mM PMSF and centrifuged at 15,000 × g for 10 min, and the pellets washed once with 1 × MSH.
DNA preparation
For extraction of total cellular DNA, cells were washed once with PBS before being solubilised in cell lysis buffer (75 mM NaCl, 50 mM EDTA, 20 mM HEPES (pH 7.8), 0.5% SDS, 0.2 mg/ml proteinase K) and added to an equal volume of phenol. DNA was purified by two consecutive rounds of sequential extraction with phenol and chloroform, followed by isopropanol precipitation and resuspension in TE pH 8.0. For preparation of mtDNA for EM analysis, mitochondria were first isolated from 1 × 107 cells using a MACS mitochondria isolation kit (Miltenyi Biotec) according to manufacturer’s instructions. Cells were lysed by 15 passages through a 27 gauge needle fitted to a 1 mL syringe, and nuclei were removed by centrifugation at 300 × g for 10 min at 4°C. The supernatant from this step was applied to the column through a 20 μm pre-separation filter. Isolated mitochondria were lysed by the addition of 0.5% SDS and 0.5 mg/ml proteinase K at 55°C for 2 hr. DNA was then extracted by phenol/chloroform extraction and ethanol precipitation, and pellets were resuspended in TE pH 8.0.
DNA modifying enzyme treatments and Southern blotting
DNA (3 mg) was digested using 10 U of the indicated restriction enzymes (all from New England Biolabs) according to the manufacturer’s instructions. For restriction mapping experiments (Figures 5 and S5), 500 ng/ml ethidium bromide was included in the reactions to inhibit thermal branch migration. Where indicated, samples were treated with DNA modifying enzymes immediately prior to loading on the gel, according to manufacturer’s instructions. Enzymatic treatments were all carried out for 30 min at 37°C, and used 5 U E. coli Topoisomerase I, 25 U S1 nuclease, 5 U T7 endonuclease I, 5 U human Topoisomerase IIα or Topoisomerase IIβ, 2.5 U E. coli gyrase, 5 U E. coli TopoIV, 5 U Exonuclease VII, 5 U T4 exonuclease VII, 20 nM RuvC or 5 U lambda exonuclease. All buffers were provided by the manufacturer except RuvC, for which the buffer used was 12 mM Tris-HCl pH 8, 10 mM MgCl2, 1 mM DTT and 100 μg/ml BSA (Zou and Rothstein, 1997). For branch migration experiments, restricted and precipitated total DNA samples (5 μg) were resuspended in branch migration buffer (50 mM NaCl, 0.1 mM EDTA, 10 mM Tris-Cl pH 8, with or without 10 mM MgCl2) and incubated at 65°C for 2 hours (Panyutin and Hsieh, 1994).
For restricted samples, DNA was separated on 0.6% agarose gels containing 500 ng/ml ethidium bromide at 100 V for 4 hr. Uncut DNA samples were separated on 0.4% agarose without ethidium bromide, at 35 V for 22 hr. After electrophoresis, DNA gels were depurinated by incubation in 0.25 M HCl for 20 min, then incubated in denaturation buffer (0.5 M NaOH, 1.5 M NaCl) twice for 10 min, and neutralisation buffer (0.5 M tris-HCl (pH 7.4), 1.5 M NaCl) twice for 10 min. DNA was blotted onto a charged nylon membrane overnight, then cross-linked by exposure to 254 nm UV, 200 mJ/cm2. Radiolabelled dsDNA probes were made by random labeling of gel-extracted PCR products using a Prime-It II kit (Agilent). For probe primer sequences see Table S1.
Immunoblotting
Whole cell lysates were prepared by incubation in cell lysis buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% (w/v) Triton X-100, 1 × proteinase inhibitors) for 30 min at 4°C, followed by centrifugation at 11,000 × g for 3 min and retaining the supernatant. Protein samples were separated by SDS-PAGE on gradient (4%–20%) polyacrylamide gels, blotted onto nitrocellulose membranes and probed with the indicated antibodies.
EM analysis of mtDNA
After purification as described above, mtDNA was prepared for EM as described in Thresher and Griffith (1992). In brief, mtDNA was suspended in a solution of 0.25 M ammonium acetate pH 7.5 and cytochrome C (7 μg/ml). A 50 μL drop was placed on a sheet of parafilm for 3–10 minutes to allow the DNA to become trapped in the denatured protein film. The film was picked up with a parlodion covered EM grid, dehydrated, air-dried, and shadow cast with 80% platinum: 20% palladium in a high vacuum. At least 100 images for each study group were captured using a Gatan Orius real time CCD camera (Pleasanton, CA, USA) attached to an FEI Tecnai T12 TEM/STEM instrument (Hillsboro, OR, USA) operated at 40 kV. EM analysis was made using Gatan Digital Micrograph software. For publication the contrast was inverted and gray scale levels optimized using Adobe Photoshop.
EM analyses of single stranded mtDNA
mtDNA isolated under different experimental conditions was incubated with E. coli SSB protein (Amersham Biochemicals), at a ratio of 3 μg of SSB per microgram of mtDNA, in 10 mM HEPES pH 7.6, 4 mM MgCl2, and 50 mM NaCl for 20 min at room temperature (RT). After incubation the complexes were fixed with 0.6% glutaraldehyde for 5 min at RT and chromatographed through 2 mL columns of 6% agarose beads (Agarose Bead Technologies, Madrid, Spain) equilibrated in 10 mM Tris HCl pH 7.6 and 0.1 mM EDTA. The samples were adsorbed to thin carbon supports in the presence of 2 mM spermidine, washed and rotary shadow-cast with tungsten. Imaging was done as above.
Immunocytochemistry
Cells previously seeded on coverslips were fixed in 4% PFA in PBS for 10 min at RT, and subsequently permeabilized with 0.1% Triton X-100 in PBS for 5 min at RT. After blocking of unspecific sites, cells were incubated overnight at 4°C with the indicated primary antibodies, prepared in 3% BSA in PBS. Cells were then incubated with the corresponding secondary antibodies in 3% BSA in PBS for 2 hr at RT. Finally, coverslips were extensively washed with PBS, prior to mounting with Aqua/Poly-mount (Polyscience).
Confocal and g-STED microscopy
Imaging was performed by using a Leica TCS SP8 g-STED microscope, equipped with a white light laser and a 100 × objective lens (HC PL APO CS2 100 × oil, 1.40 NA). For confocal images of TOM20 and DNA, Z stacks in accordance with the Nyquist sampling criteria were taken by exciting the fluorophores at 647 nm and 594 nm respectively, and signal was collected by Hybrid detectors. Stimulated emission depletion was performed with a 775 nm depletion laser. 2D confocal and g-STED images were acquired sequentially with the optical zoom set to obtain a voxel size of 22.7 × 22.7 nm. Excitation was provided at 594 nm and signal was collected by Hybrid detectors. Gating between 0.3–6 ns was applied.
Histopathology, Biochemical and Genetic Studies
Diagnostic skin and muscle biopsies were performed according to standard protocols. Skeletal muscle biopsy was subject to routine diagnostic histopathological protocols including hematoxylin and eosin staining, as well as COX, SDH and sequential COX-SDH histochemistry. Whole mitochondrial genome sequencing was undertaken using muscle DNA. The presence of mitochondrial DNA rearrangements was investigated using an established, diagnostic real time PCR assay (He et al., 2002). Candidate screening of all exons and intronic regions of nuclear genes frequently associated with PEO and multiple mtDNA deletions (POLG1, GenBank: NM_002693; RRM2B, GenBank: NM_015713; SLC25A4, GenBank: NM_001151; TWNK, GenBank: NM_021830; POLG2, GenBank: NM_007215.3; TK2, GenBank: NM_004614.4; RNASEH1, GenBank: NM_002936) was performed, but no pathogenic variants were identified.
Whole Exome Sequencing, Analysis and Interpretation
Exome capture was attained using the Agilent Sure Select Human All Exon V5 (50Mb) capture kit, sequenced using the Illumina GAIIx platform in 75 base pair reads and aligned to the human reference genome (UCSC hg19). Called variants were restricted to exonic (coding) or splice-site variants with a MAF less than or equal to 0.01 (1%) from 378 in-house controls and external variant databases (ExAC, NHLBI ESP, 1000 Genomes). Autosomal dominant (heterozygous) and autosomal recessive (homozygous and compound heterozygous) inheritance were equally considered. Any rare variants in nuclear genes associated with mtDNA maintenance disorders were first examined. Next, variants were filtered using Gene Ontology (GO)-Terms to prioritise nuclear genes encoding proteins involved in mitochondrial function, DNA repair or replication. GO-Terms employed were the wildcard term ‘mitochondr*’, ‘DNA repair’, ‘replication’, ‘transcription’, ‘nucleotide’, ‘purine’, ‘pyrimidine’, ‘exonuclease’, ‘polymerase’, ‘topoisomerase’, ‘ligase’, ‘helicase’ and ‘nucleoside’. Copy number variants (CNVs) were also examined using the same GO-Terms. Identified heterozygous TOP3A (NM_004618.3) variants were confirmed by Sanger sequencing using forward and reverse primers flanked with universal M13-derived tags, according to standard protocols. Primers for Sanger sequencing are provided in Table S1.
Protein purification
The human Top3α sequence (or the Y362F or M100V variants), lacking the N-terminal 15 amino acids and with the additional of a C-terminal 6 × His tag, were cloned into the vector pBacPAK9 (Clontech) and used to prepare recombinant Autographa californica nuclear polyhedrosis virus stocks according to the manufacturer’s instructions. These viruses were used to infect Sf9 insect cells. Whole cell extracts were prepared by resuspending cell pellets in a lysis buffer of 20 mM Tris-HCl pH 8.0, 500 mM NaCl, 10 mM β-mercaptoethanol and 1 × proteinase inhibitors, and freeze-thawing in liquid nitrogen. The extract was cleared by centrifugation at 20,000 rpm for 30 min at 4°C using a Sorvall Surespin 630 rotor. The supernatant was loaded onto 2 mL of His-Select Nickel affinity gel (Sigma) equilibrated with buffer A (20 mM Tris-HCl pH 8.0, 400 mM NaCl, 10% glycerol, 10 mM β-mercaptoethanol and 1 × proteinase inhibitors) containing 10 mM imidazole, washed with buffer A (20 mM imidazole) and finally eluted with buffer A (250 mM imidazole). The eluate was further purified over a Superdex 200 16/600 (GE Healthcare Life Sciences) equilibrated with buffer B (20 mM HEPES pH 7.2, 10% glycerol, 1 mM dithiothreitol and 1 × proteinase inhibitors) containing 400 mM NaCl. The peak fractions were diluted 1:1 with buffer B (0 M NaCl) before being loaded onto HiTrap SP HP (GE Healthcare Life Sciences) equilibrated in buffer B (0.2 M NaCl). The column was eluted with a linear gradient (10 ml) of buffer B (0.2–1.2 M NaCl). The peak fractions were diluted 1:4 with buffer B (0 M NaCl) and finally loaded onto HiTrap Heparin HP (GE Healthcare Life Sciences) also equilibrated in buffer B (0.2 M NaCl). The column was eluted with a linear gradient (10 ml) of buffer B (0.2–1.2 M NaCl). Recombinant Top3α proteins eluted at approximately 700 mM NaCl.
Electrophoretic mobility shift assay (EMSA)
Reactions (20 μl) contained 10 fmol ssDNA substrate (80 nt, labeled at the 5′ end with [γ-32P] ATP), 25 mM HEPES pH 7.5, 1 mM DTT, 100 μg/ml BSA, 10% glycerol, 70 mM NaCl and indicated amounts (0, 107.5, 215, 430 or 860 fmol) of the different Top3α variants. Reactions were incubated at 37°C for 15 min, then transferred to ice. Samples were separated on 8% PAGE-TBE gels at 150 V for 50 min in 0.5 × TBE running buffer at 4°C. Oligo sequence is provided in Table S1.
DNA relaxation assay
Top3α recombinant proteins were dialysed into a 1 × reaction buffer of 25 mM Tris-HCl pH 7.4, 5 mM MgCl2, 50 mM NaCl and 1 mM DTT, then BSA was added to 100 μg/ml. Reactions (30 μl) contained increasing amounts (1.75, 5 or 10 pmol) of Top3α in 1 × reaction buffer with 200 ng of freshly-prepared supercoiled pBluescript II SK(+) substrate. The reactions were incubated at 37°C for 45 min and then treated with 1 μL of proteinase K (20 mg/ml), 2 μL of 5% SDS and 2 μL of 50 mM EDTA at 37°C for 30 min before 6 μL of 6× Orange DNA loading Dye (Thermo Scientific) was added. Reactions were separated on 1% agarose TAE gels with or without 500 ng/ml ethidium bromide at 100 V for 4 hr. Gels run without ethidium bromide were subsequently stained with 500 ng/ml ethidium bromide in water for 30 min before being imaged under UV light.
Mapping of mtDNA rearrangements using deep sequencing
Total DNA from muscle (from the TOP3A patient and an unaffected control) was extracted and subjected to deep sequencing on a NextSeq 500 using the Nextera DNA library preparation kit (Illumina). A total of 57792541 and 58045634 reads were obtained for the TOP3A patient and wild-type samples, respectively. An initial alignment to chrM (rCRS assembly) and nuclear chromosomes was performed using bowtie2 (Langmead and Salzberg, 2012). Mitochondrial and unaligned reads were realigned to chrM using LAST (Kietbasa et al., 2011) to identify gapped alignments indicative of deleted or duplicated segments. The realignment gave a coverage depth of 2608× and 1059× for mtDNA from the TOP3A patient and control samples, respectively. Gapped alignments, indicative of potential breakpoints, were clustered using single-linkage hierarchical clustering, considering the maximum distance for the two breakpoints and using a distance threshold of 50 bp. Segments were classified as likely deletions or duplications based on read orientation and overlap with the two replication origins. Heteroplasmy was calculated as the fraction of gapped to wild-type reads at a given breakpoint, taking the average of the two breakpoints for each altered segment.
2D agarose gel electrophoresis
2D-AGE was carried out according to the method of Reyes et al. (2009). 8 μg total cellular DNA was restricted with 20 U of the indicated restriction enzymes in 400 μL reactions for 2 hr at 37°C. DNA was then ethanol precipitated, resuspended and loaded onto 0.4% agarose without ethidium bromide. First dimension gels were run at 27 V for 18 hr at RT, then DNA-containing lanes were excised, rotated 90° counterclockwise and 1% agarose containing 500 ng/ml ethidium bromide cast around the first-dimension gel slices. Second dimension gels were run at a constant 260 mA for 6 hr at 4°C and Southern blotted as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Nucleoid quantification and measurement
Images were deconvolved with the Huygens software prior to analysis. Analysis of nucleoid diameter was performed on 2D confocal and g-STED images by measuring the full width at half maximum (FWHM; Kukat et al., 2011) using the Adrian’s FWHM plugin from ImageJ. For each nucleoid, the diameter was calculated as the average of FWHM measurements of X and y axis. Analysis of nucleoids number per cell was performed on MAX-projections of confocal images by using the Analyze particles tool in ImageJ. Briefly, a region of interest (ROI) corresponding to a single cell was manually created and the resulting image was binarized prior to particle counting. Performance of the microscope and optimal depletion laser power were tested by using an 180ROROR multicolor nanoruler (GATTAquant, kindly provided by Dr. Christian Jünsgt). Images were then analyzed in ImageJ by calculating the distance between the Alexa594 spots, which position was generated on the local maxima. Representative pixel intensity profiles were generated in ImageJ. Image panels were assembled with Photoshop (Adobe). Unless otherwise stated, no other digital manipulation except brightness/contrast adjustment was applied.
Statistical analyses for confocal and g-STED
Statistical analyses as well as generation of frequency histogram profiles were performed in GraphPad Prism (San Diego, CA, USA). For nucleoids number and diameter analyses, significance of the differences among the RNAi conditions was assessed with the one-way ANOVA followed by the post hoc Tukey test.
mtDNA copy number measurements
Band intensities from storage phosphor images of Southern blots were quantified using Multigauge V3.0 (Fujifilm). Copy number is expressed as the ratio of signal from the mitochondrial probe normalized to the level of 28S rDNA, following subtraction of background signal.
Statistics
Error bars are expressed as SEM p values were calculated using the Student’s t test or one-way ANOVA, with the number of replicates provided in the figure legends. Significance thresholds used throughout the paper are * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001.
DATA AND SOFTWARE AVAILABILITY
The accession number for the sequencing data reported in this paper is ENA: PRJEB23559. The accession numbers for the identified TOP3A mutations reported in this paper are ClinVar: SCV000611605 and SCV000611606. Original imaging data have been deposited to Mendeley Data and are available at https://doi.org/10.17632/43wnnd4tjw.1.
Supplementary Material
Highlights.
Mitochondrial topoisomerase 3α separates mtDNA following replication
Mutations in TOP3A are a cause of human mitochondrial disease
mtDNA segregation proceeds via a hemicatenane formed at the origin of replication
Loss of Top3α impairs segregation of the mitochondrial nucleoid
Acknowledgments
This work was supported by the Swedish Research Council (M.F., C.M.G., E.L., and N.-G.L.), the Swedish Cancer Foundation (M.F., C.M.G., and E.L.), the European Research Council (Project ID: 683191 to M.F.); the IngaBritt and Arne Lundberg Foundation (M.F.), the Knut and Alice Wallenbergs Foundation (M.F., C.M.G., E.L., and N.-G.L.), the NIH (GM31819 and ESO13773 to J.D.G.), the Wellcome Centre for Mitochondrial Research (203105/Z/16/Z), the Medical Research Council (MRC) Centre for Translational Research in Neuromuscular Disease, the Mitochondrial Disease Patient Cohort (UK) (G0800674), the UK NIHR Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust, the MRC/EPSRC Molecular Pathology Node, and the UK NHS Highly Specialised Service for Rare Mitochondrial Disorders of Adults and Children (G.S.G., D.M.T., and R.W.T.). E.W.S. was funded by a Medical Research Council PhD studentship. P.F.C. is a Wellcome Trust Senior Fellow in Clinical Science (101876/Z/13/Z) and a UK NIHR Senior Investigator, who receives support from the Medical Research Council Mitochondrial Biology Unit (MC_UP_1501/2), the Medical Research Council (UK) Centre for Translational Muscle Disease research (G0601943), EU FP7 TIRCON, and the National Institute for Health Research (NIHR) Biomedical Research Centre based at Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge. We would like to acknowledge Dr. Christian Jünsgt and the CECAD imaging facility, and we thank Emma Watson, Gerald Pfeffer, and Alexia Chrysostomou for their help in the laboratory work-up of the patient. We would also like to thank Zofia Chrzanowska-Lightowlers and Robert N. Lightowlers for valuable discussion and reagents.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and one table and can be found with this article online at https://doi.org/10.1016/j.molcel.2017.11.033.
AUTHOR CONTRIBUTIONS
Conceptualization, T.J.N. and C.M.G.; Methodology, E.W.S., S.B., P.F.C., and E.L.; Formal Analysis, T.J.N., C.A.N., E.M., E.W.S., S.B., and E.L.; Investigation, T.J.N., C.A.N., E.M., E.W.S., G.S.G., D.M.T., E.H., and M.F.; Resources, G.S.G., D.M.T., P.F.C., and R.W.T.; Writing – Original Draft, T.J.N. and C.M.G.; Writing – Review & Editing, C.A.N., E.W.S., G.S.G., R.W.T., and J.D.G.; Visualization, T.J.N., C.A.N., E.M., E.W.S., R.W.T., and J.D.G.; Supervision, G.S.G., P.F.C., N.-G.L., E.L., M.F., R.W.T., J.D.G., and C.M.G.; Funding Acquisition, N.-G.L., M.F., R.W.T., J.D.G., and C.M.G.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Akman G, Desai R, Bailey LJ, Yasukawa T, Dalla Rosa I, Durigon R, Holmes JB, Moss CF, Mennuni M, Houlden H, et al. Pathological ribonuclease H1 causes R-loop depletion and aberrant DNA segregation in mitochondria. Proc Natl Acad Sci USA. 2016;113:E4276–E4285. doi: 10.1073/pnas.1600537113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albring M, Griffith J, Attardi G. Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc Natl Acad Sci USA. 1977;74:1348–1352. doi: 10.1073/pnas.74.4.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissière A, Campos Y, Rivera H, de la Aleja JG, Carroccia R, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain. 2008;131:338–351. doi: 10.1093/brain/awm298. [DOI] [PubMed] [Google Scholar]
- Aravind L, Leipe DD, Koonin EV. Toprim—a conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res. 1998;26:4205–4213. doi: 10.1093/nar/26.18.4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44(D1):D1251–D1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Plank JL, Dombrowski CC, Kowalczykowski SC. Decatenation of DNA by the S. cerevisiae Sgs1-Top3-Rmi1 and RPA complex: a mechanism for disentangling chromosomes. Mol Cell. 2012;47:886–896. doi: 10.1016/j.molcel.2012.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douarre C, Sourbier C, Dalla Rosa I, Brata Das B, Redon CE, Zhang H, Neckers L, Pommier Y. Mitochondrial topoisomerase I is critical for mitochondrial integrity and cellular energy metabolism. PLoS ONE. 2012;7:e41094. doi: 10.1371/journal.pone.0041094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elachouri G, Vidoni S, Zanna C, Pattyn A, Boukhaddaoui H, Gaget K, Yu-Wai-Man P, Gasparre G, Sarzi E, Delettre C, et al. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. 2011;21:12–20. doi: 10.1101/gr.108696.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505:335–343. doi: 10.1038/nature12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffart S, Cooper HM, Tyynismaa H, Wanrooij S, Suomalainen A, Spelbrink JN. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum Mol Genet. 2009;18:328–340. doi: 10.1093/hmg/ddn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulaouic H, Roulon T, Flamand O, Grondard L, Lavelle F, Riou JF. Purification and characterization of human DNA topoisomerase IIIalpha. Nucleic Acids Res. 1999;27:2443–2450. doi: 10.1093/nar/27.12.2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson CM, Falkenberg M, Larsson NG. Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem. 2016;85:133–160. doi: 10.1146/annurev-biochem-060815-014402. [DOI] [PubMed] [Google Scholar]
- Hanai R, Caron PR, Wang JC. Human TOP3: a singlecopy gene encoding DNA topoisomerase III. Proc Natl Acad Sci USA. 1996;93:3653–3657. doi: 10.1073/pnas.93.8.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon FG, DiGate RJ, Kowalczykowski SC. RecQ helicase and topoisomerase III comprise a novel DNA strand passage function: a conserved mechanism for control of DNA recombination. Mol Cell. 1999;3:611–620. doi: 10.1016/s1097-2765(00)80354-8. [DOI] [PubMed] [Google Scholar]
- He L, Chinnery PF, Durham SE, Blakely EL, Wardell TM, Borthwick GM, Taylor RW, Turnbull DM. Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acids Res. 2002;30:e68. doi: 10.1093/nar/gnf067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson B, Vinograd J. Catenated circular DNA molecules in HeLa cell mitochondria. Nature. 1967;216:647–652. doi: 10.1038/216647a0. [DOI] [PubMed] [Google Scholar]
- Hudson G, Amati-Bonneau P, Blakely EL, Stewart JD, He L, Schaefer AM, Griffiths PG, Ahlqvist K, Suomalainen A, Reynier P, et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008;131:329–337. doi: 10.1093/brain/awm272. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Ban-Ishihara R, Maeda M, Matsunaga Y, Ichimura A, Kyogoku S, Aoki H, Katada S, Nakada K, Nomura M, et al. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol Cell Biol. 2015;35:211–223. doi: 10.1128/MCB.01054-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kie1basa SM, Wan R, Sato K, Horton P, Frith MC. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011;21:487–493. doi: 10.1101/gr.113985.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukat C, Wurm CA, Spåhr H, Falkenberg M, Larsson NG, Jakobs S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci USA. 2011;108:13534–13539. doi: 10.1073/pnas.1109263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukat C, Davies KM, Wurm CA, Spåhr H, Bonekamp NA, Kühl I, Joos F, Polosa PL, Park CB, Posse V, et al. Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc Natl Acad Sci USA. 2015;112:11288–11293. doi: 10.1073/pnas.1512131112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SC, Uchiyama LF, Nunnari J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science. 2016;353:aaf5549. doi: 10.1126/science.aaf5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low RL, Orton S, Friedman DB. A truncated form of DNA topoisomerase IIbeta associates with the mtDNA genome in mammalian mitochondria. Eur J Biochem. 2003;270:4173–4186. doi: 10.1046/j.1432-1033.2003.03814.x. [DOI] [PubMed] [Google Scholar]
- Pallotti F, Lenaz G. Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol. 2007;80:3–44. doi: 10.1016/S0091-679X(06)80001-4. [DOI] [PubMed] [Google Scholar]
- Panyutin IG, Hsieh P. The kinetics of spontaneous DNA branch migration. Proc Natl Acad Sci USA. 1994;91:2021–2025. doi: 10.1073/pnas.91.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plank JL, Chu SH, Pohlhaus JR, Wilson-Sali T, Hsieh TS. Drosophila melanogaster topoisomerase IIIalpha preferentially relaxes a positively or negatively supercoiled bubble substrate and is essential during development. J Biol Chem. 2005;280:3564–3573. doi: 10.1074/jbc.M411337200. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Sun Y, Huang SN, Nitiss JL. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol. 2016;17:703–721. doi: 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Yasukawa T, Cluett TJ, Holt IJ. Analysis of mitochondrial DNA by two-dimensional agarose gel electrophoresis. Methods Mol Biol. 2009;554:15–35. doi: 10.1007/978-1-59745-521-3_2. [DOI] [PubMed] [Google Scholar]
- Reyes A, Melchionda L, Nasca A, Carrara F, Lamantea E, Zanolini A, Lamperti C, Fang M, Zhang J, Ronchi D, et al. RNASEH1 mutations impair mtDNA replication and cause adult-onset mitochondrial encephalomyopathy. Am J Hum Genet. 2015;97:186–193. doi: 10.1016/j.ajhg.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzier C, Bannwarth S, Chaussenot A, Chevrollier A, Verschueren A, Bonello-Palot N, Fragaki K, Cano A, Pouget J, Pellissier JF, et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain. 2012;135:23–34. doi: 10.1093/brain/awr323. [DOI] [PubMed] [Google Scholar]
- Sarbajna S, West SC. Holliday junction processing enzymes as guardians of genome stability. Trends Biochem Sci. 2014;39:409–419. doi: 10.1016/j.tibs.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Satoh M, Kuroiwa T. Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell. Exp Cell Res. 1991;196:137–140. doi: 10.1016/0014-4827(91)90467-9. [DOI] [PubMed] [Google Scholar]
- Shi Y, Dierckx A, Wanrooij PH, Wanrooij S, Larsson NG, Wilhelmsson LM, Falkenberg M, Gustafsson CM. Mammalian transcription factor A is a core component of the mitochondrial transcription machinery. Proc Natl Acad USA. 2012;109:16510–16515. doi: 10.1073/pnas.1119738109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart L, Redinbo MR, Qiu X, Hol WG, Champoux JJ. A model for the mechanism of human topoisomerase I. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- Suski C, Marians KJ. Resolution of converging replication forks by RecQ and topoisomerase III. Mol Cell. 2008;30:779–789. doi: 10.1016/j.molcel.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thresher R, Griffith J. Electron microscopic visualization of DNA and DNA-protein complexes as adjunct to biochemical studies. Methods Enzymol. 1992;211:481–490. doi: 10.1016/0076-6879(92)11026-f. [DOI] [PubMed] [Google Scholar]
- Tsai HZ, Lin RK, Hsieh TS. Drosophila mitochondrial topoisomerase III alpha affects the aging process via maintenance of mitochondrial function and genome integrity. J Biomed Sci. 2016;23:38. doi: 10.1186/s12929-016-0255-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscomi C, Zeviani M. MtDNA-maintenance defects: syndromes and genes. J Inherit Metab Dis. 2017;40:587–599. doi: 10.1007/s10545-017-0027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lyu YL, Wang JC. Dual localization of human DNA topoisomerase IIIalpha to mitochondria and nucleus. Proc Natl Acad Sci USA. 2002;99:12114–12119. doi: 10.1073/pnas.192449499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Feng L, Hsieh TS. Drosophila topo IIIalpha is required for the maintenance of mitochondrial genome and male germ-line stem cells. Proc Natl Acad Sci USA. 2010;107:6228–6233. doi: 10.1073/pnas.1001855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Bachrati CZ, Ou J, Hickson ID, Brown GW. Human topoisomerase IIIalpha is a single-stranded DNA decatenase that is stimulated by BLM and RMI1. J Biol Chem. 2010;285:21426–21436. doi: 10.1074/jbc.M110.123216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeviani M, Servidei S, Gellera C, Bertini E, DiMauro S, DiDonato S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature. 1989;339:309–311. doi: 10.1038/339309a0. [DOI] [PubMed] [Google Scholar]
- Zhang H, Barceló JM, Lee B, Kohlhagen G, Zimonjic DB, Popescu NC, Pommier Y. Human mitochondrial topoisomerase I. Proc Natl Acad Sci USA. 2001;98:10608–10613. doi: 10.1073/pnas.191321998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhang YW, Yasukawa T, Dalla Rosa I, Khiati S, Pommier Y. Increased negative supercoiling of mtDNA in TOP1mt knockout mice and presence of topoisomerases II031 and IIβ in vertebrate mitochondria. Nucleic Acids Res. 2014;42:7259–7267. doi: 10.1093/nar/gku384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Rothstein R. Holliday junctions accumulate in replication mutants via a RecA homolog-independent mechanism. Cell. 1997;90:87–96. doi: 10.1016/s0092-8674(00)80316-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.