

SUMMARY
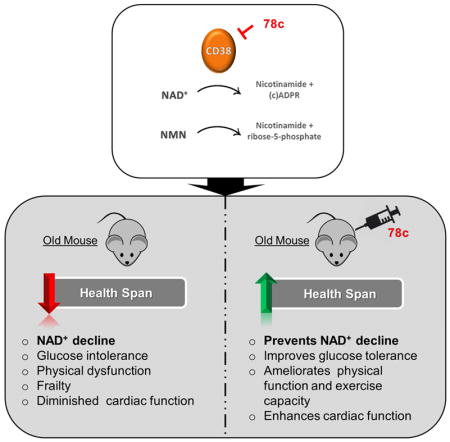
Aging is characterized by the development of metabolic dysfunction and frailty. Recent studies show that a reduction in nicotinamide adenine dinucleotide (NAD+) is a key factor for the development of age-associated metabolic decline. We recently demonstrated that the NADase CD38 has a central role in age-related NAD+ decline. Here we show that a highly potent and specific thiazoloquin(az)olin(on)e CD38 inhibitor, 78c, reverses age-related NAD+ decline and improves several physiological and metabolic parameters of aging, including glucose tolerance, muscle function, exercise capacity, and cardiac function in mouse models of natural and accelerated aging. The physiological effects of 78c depend on tissue NAD+ levels, and were reversed by inhibition of NAD+ synthesis. 78c increased NAD+ levels, resulting in activation of pro-longevity and health span-related factors including sirtuins, AMPK, and PARPs. Furthermore, in animals treated with 78c we observed inhibition of pathways that negatively affect health span, such as mTOR-S6K and ERK, and attenuation of telomere-associated DNA damage, a marker of cellular aging. Together, our results detail a novel pharmacological strategy for prevention and/or reversal of age-related NAD+ decline and subsequent metabolic dysfunction.
Keywords: CD38, NAD+, skeletal muscle, acetylation, SIRTUINS, exercise capacity, glucose, aging, and progeroid
In Brief
A reduction in nicotinamide adenine dinucleotide (NAD+) is associated with aging. XX et al show physiological and metabolic improvements of aging in old mice given with the small molecule 78c, which inhibits the NADase enzyme CD38. Mechanistically, mTORS6K/ERK and telomere-associated DNA damage pathways mitigate the NAD+ decline.
INTRODUCTION
NAD+ is a cofactor of key enzymes in glycolysis, the tricarboxylic acid cycle, and oxidative phosphorylation, participating in multiple redox reactions in cells. In addition, it serves as a substrate for several enzymes involved in cell signaling and DNA damage repair such as the sirtuins and poly (ADP-ribose) polymerases (PARPs) (Imai and Guarente, 2014; Verdin, 2015; Yoshino et al., 2017). The cellular NAD+ pool is controlled by a balance between the activity of NAD+-synthesizing and consuming enzymes (Aksoy et al., 2006; Bai et al., 2011; Bai et al., 2012; Barbosa et al., 2007; Grozio et al., 2013; Imai and Guarente, 2014; Mills et al., 2016; Nahimana et al.; 2009; Yang et al., 2007; Yoshino et al., 2011; Yoshino et al., 2017). Recent studies show that levels of NAD+ and its precursors decline during chronological aging and in progeroid syndromes, causing mitochondrial dysfunction and metabolic abnormalities (Braidy et al., 2011; Camacho-Pereira et al., 2016; Frederick et al., 2016; Gomes et al., 2013; Li et al., 2017; Massudi et al., 2012; Mills et al., 2016; Mouchiroud et al., 2013; Ramesey et al., 2008; Scheibye-Knudsen et al., 2014; Stein and Imai, 2014; Yoshino et al., 2011; Yoshino et al., 2017; Zhu et al., 2015). This decline in NAD+ levels is implicated in the development of age-related muscle dysfunction, glucose intolerance, and stem cell senescence (Cantó and Auwerx, 2012; Chini et al., 2016; Guarente, 2016; Imai and Guarente, 2014; Mills et al., 2016; Verdin, 2015; Schultz and Sinclair, 2016; Yoshino et al., 2017).
We recently reported that the NADase CD38 is one of the main enzymes responsible for age-related NAD+ decline in mammals, and that CD38 knockout (KO) mice are protected from this progressive deficit (Camacho-Pereira et al., 2016), indicating that CD38 may be an attractive target for the development of therapies to treat age-related metabolic dysfunction. Recently, thiazoloquin(az)olin(on)es, such as the small molecule 78c, have been identified as potent CD38 inhibitors (CD38i), (Becherer et al., 2015; Haffner et al., 2015). However, their mechanisms of action, specificity, and biological effects on age-related NAD+ decline and metabolic dysfunction have not been investigated. Here we demonstrate the potent anti-aging properties of the thiazoloquin(az)olin(on)e 78c. 78c reverses age-related NAD+ decline and ameliorates several metabolic, structural, and molecular features of aging in chronologically aged and progeroid mice. By kinetic analyses we show that 78c is highly potent and specific CD38i, with a reversible uncompetitive mechanism of action. We further demonstrate that 78c activates pro-longevity and health span-related factors, and inhibits those that negatively affect health span. In summary, these studies establish CD38 as a viable target for pharmacological NAD+-replacement therapy for age-related metabolic dysfunction.
RESULTS
78c is a potent, reversible, and uncompetitive inhibitor of CD38
We investigated the mechanism of inhibition of 78c and observed that 78c is a potent inhibitor of both human CD38 (recombinant, rhCD38) and murine CD38, with a Ki in the low nanomolar range (Fig. 1A–D). The inhibitory effect of 78c on CD38 NADase activity was reversible (Fig. S1A–B), and uncompetitive (Fig. 1B–C) decreasing both apparent Vmax and Km of CD38 for NAD+ (Fig. 1B). We confirmed this finding in assays using nicotinamide mononucleotide (NMN), a competitor of NAD+ and alternative substrate of CD38. Consistent with an uncompetitive inhibition model, NMN decreased the apparent Ki for 78c in a hyperbolic manner (Fig. S1C).
Figure 1. Characterization of 78c as a specific CD38i.

(A) Hydrolase activity of recombinant human CD38 (rhCD38) in the presence of 78c, using 1,N6-ethenoadenine dinucleotide (ε-NAD+) as substrate (n=3 experiments, IC50 17.7 nM). Inset shows the structure of 78c. R1=H; R2=Me; R3=trans-4- OCH2CH2OMe-cyclohexyl.
(B) Effect of different substrate concentration on 78c inhibition of rhCD38 hydrolase activity. Experimental data were fitted by steady state equations derived from the kinetic model in panel
(C) Kinetic model of CD38 inhibition by 78c. Model depicts 2 interconnected cycles, one for the hydrolytic activity of rhCD38 (left side), and the other for the cyclase activity of the enzyme as well as the inhibition by its products nicotinamide and ADP- ribose (right side). In the reaction scheme, E represents the enzyme CD38; k(1-7), the substrate binding constants; Ki, the inhibitor binding constant.
(D) NADase activity in tissue homogenates from 1-year-old WT and CD38 KO mice, assayed in the presence of 78c (n=3 experiments). IC50: liver (3.8 NM), skeletal muscle (4.4 nM), spleen (0.7 nM), brain (14.8 nM), ileum (0.4 nM), subcutaneous fat (5.2 nM). AFU= arbitrary fluorescence units.
(E) NAD+ levels in WT and CD38 KO MEFs treated for 24 hours with 0.2 μM 78c (n=4–6 experiments). NAD+ levels were calculated relative to control WT MEF.
(F) NAD+ levels in tissues of 1-year-old WT and CD38 catalytically inactive (CI) mice treated with 78c or vehicle (Control) for 8 days (n=4 mice per group). Spleen protein lysates of WT and CI mice were immunoblotted for CD38 and Tubulin (right panel).
(G) Activity of human recombinant PARP1 in the presence of 78c or the PARP inhibitor olaparib (n=4 experiments).
((H) NAD+ levels in A549 cells treated for 24 hours with 0.5 μM 78c and/or 5 μM olaparib (n=3–6 experiments).
(I) Activity of human recombinant SIRT1 in the presence of 100 nM 78c or 100 μM SIRT1 inhibitor suramin (n=3 experiments).
(J) Activity of human recombinant NAMPT in the presence of 100 nM 78c or 20 μM of NAMPT inhibitor FK866 (n=3 experiments).
All values are mean ± SEM. *P < 0.05. NS=not significant. IC50=half maximal inhibitory concentration. See also Figure S1.
To refine our understanding of the mechanism of inhibition, we next tested the effect of the CD38 reaction products on the inhibition of CD38 by 78c. CD38 degrades NAD+ not only via its hydrolase activity, that results in the generation of the products nicotinamide (NAM), and ADP-ribose (ADPR), but also via its ADP-ribosyl cyclase activity, which generates the calcium signaling molecule cyclic-ADP-ribose (cADPR) (Malavasi et al., 2008). We observed that NAM decreased the apparent V0 and increased the Ki for 78c (Fig. S1D). In contrast, neither ADPR nor cADPR had significant effects on these kinetic parameters (Fig. S1E–F).
Next, we compared the effects of 78c on the hydrolase and cyclase activities of CD38. As shown in Fig. S1G, 78c was 10-fold less potent against the cyclase activity than the hydrolase activity of rhCD38 (Ki 100 ± 28 nM vs. 9.7 ± 1.5 nM, respectively). We further examined the effect of 78c on the activity of the ADP-ribosyl cyclase purified from the sea slug Aplysia californica (Malavasi et al., 2008). This enzyme is a pure cyclase that generates cADPR and NAM from the substrate NAD+ and has 25% homology to human CD38 (Malavasi et al., 2008). The Aplysia enzyme was not inhibited by 78c at concentrations up to 50 nM (Fig. S1H).
To further establish 78c as a specific inhibitor of CD38, we tested the effect of 78c on the CD38 homologous enzyme CD157 (Malavasi et al., 2008). The role of CD157 (also known as BST-1) in NAD+ metabolism in mammalians is not completely understood. In contrast to CD38, CD157 is a slow turnover enzyme that accepts nicotinamide riboside (NR) as its preferential substrate (Preugschat et al., 2014). Thus, we tested the effect of 78c on the catalytic activity of CD157 using both NAD+ and NR as substrates and observed no effect of 78c in the CD157 catalytic activities (Fig. S1I). Collectively, these studies establish that 78c is a specific, reversible, and uncompetitive CD38i that preferentially inhibits CD38 NADase activity.
78c increases NAD+ levels through inhibition of CD38 NADase activity
Previous studies have shown that a single oral dose of 78c increases tissue NAD+ levels in the liver of young mice fed a high fat diet (Becherer et al., 2015; Haffner et al., 2015). However, it is not known if this effect is dependent on the presence or enzymatic activity of CD38. We used mouse embryonic fibroblasts (MEFs) derived from wild type (WT) and CD38 KO mice to test whether CD38 expression is essential for the effect of 78c on NAD+ levels. We observed that WT MEFs have lower basal levels of NAD+ compared to CD38 KO MEFs (Fig. 1E and Fig. S1J). 78c nearly doubled the amount of NAD+ in WT MEFs, but had no effect on cellular NAD+ levels in CD38 KO MEFs (Fig. 1E).
Next, we tested if the NAD+ -modulating effect of 78c is dependent on CD38 catalytic activity in vivo. We generated a novel CD38 “knock-in” mouse model containing a mutation at E230 of the CD38 gene, which results in expression of a catalytically inactive (CI) CD38 (Fig. 1F and Fig. S1K–N). Although the CD38-CI animals expressed levels of CD38 similar to the WT mice (see Western blot in Fig. 1F and Fig. S1N), they had no detectable CD38 NADase activity and were protected against age-related NAD+ decline (Fig. S1L–M). We further observed that in contrast to CD38-CI mice, WT mice treated with 78c showed a significant increase in NAD+ levels in several tissues (Fig. 1F). Thus, we conclude that 78c-induced tissue NAD+ boosting is dependent on the inhibition of the catalytic activity of CD38.
78c is a specific inhibitor of CD38 and does not directly affect the activity or expression of other enzymes involved in NAD+ metabolism
It has been previously proposed that increases in cellular NAD+ levels can be pharmacologically induced by inhibition of NAD+ degradation catalyzed by enzymes such as Poly (ADP-ribose) polymerase (PARPs) (Bai et al., 2011; Bai and Cantó, 2012; Imai and Guarente, 2014) or stimulation of enzymes involved in NAD+ synthesis such as NAMPT (Wang et al., 2014). Thus, to further characterize the mechanism by which 78c promotes increases in cellular NAD+ levels we investigated the effect of 78c in the activity and expression of key enzymes involved in NAD+ catabolism and anabolism.
PARP1 is the main PARP enzyme in mammalian tissues and utilize NAD+ during its catalysis (Bai et al., 2011; Bai and Cantó, 2012; Imai and Guarente, 2014). Thus, it is very important to clearly demonstrate that 78c has no direct inhibitory or stimulatory effects on this enzyme. This is of particular importance since several studies have proposed that PARP1 inhibition can be used to promote increases in cellular levels of NAD+ by decreasing its degradation (Bai et al., 2011; Bai and Cantó, 2012; Imai and Guarente, 2014). We first investigated if 78c had a direct effect on the enzymatic activity of PARP1 in vitro. While the PARP inhibitor olaparib completely inhibited the activity of this enzyme, 78c had no detectable effects on PARP1 activity (Fig. 1G). Next, we tested if inhibition of CD38 by 78c had an additive effect with PARP inhibition on cellular NAD+ levels. The PARP inhibitor olaparib increased cellular NAD+ levels by itself, and combination of olaparib with 78c produced an additive effect on the increase in cellular NAD+ (Fig. 1H), demonstrating that the 78c-mediated increase in NAD+ occurs independent of PARP activity. Additionally, we tested if pharmacological inhibition of PARP with olaparib in vivo would increase NAD+ levels in WT and CD38 KO mice. There was an increase in NAD+ levels in tissues of WT animals treated with olaparib (Fig. S1O), and the NAD+-boosting effects of olaparib were not only preserved in tissues of CD38 KO mice such as liver and spleen, but appeared to be even potentiated in skeletal muscle (Fig. S1O). These data together demonstrate that 78c does not directly influence the activity of PARP, and that its NAD+-boosting effects are mediated by a mechanism that is independent of PARP activity.
Next we determined the effect of 78c on the activity and expression of other enzymes involved in NAD+ metabolism (Fig. 1I–J and Fig. S1P). We tested the effect of 78c on the NAD+-consuming enzyme SIRT1 (as a representative member of the sirtuin family), and on NAMPT (the rate-limiting enzyme in the salvage pathway of NAD+ synthesis) (Fig 1I–J). Using recombinant SIRT1 and NAMPT, we saw that 78c does not directly inhibit or stimulate these enzymes (Fig. 1I–J). In contrast, SIRT1 activity was decreased by the known SIRT1 inhibitor suramin (Fig. 1I), and NAMPT activity was completely abolished by its specific inhibitor FK866 (Fig. 1J). Furthermore, animals treated with 78c showed no significant changes in the expression of genes involved in NAD+ catabolism and anabolism (Fig. S1P). Altogether, these data clearly demonstrate that 78c increases tissue NAD+ levels by a mechanism dependent on the inhibition of the NADase catalytic activity of CD38, and not via its effect on other NAD+ metabolizing enzymes. Thus, our studies establish 78c as a potent and specific CD38i.
Metabolic function is improved in aged mice treated with 78c
Having established that 78c is a specific CD38 inhibitor that increases NAD+ levels, we next explored the potential role of 78c as a pharmacological therapy for aging-related metabolic dysfunction. Aging is characterized by the development of several metabolic changes that may limit health span (Huffman et al. 2016; Imai and Guarente, 2014; Justice et al., 2016; Verdin, 2016). For example, aging leads to an increased incidence of glucose intolerance, insulin resistance, and diabetes, as well as a decrease in physical activity and exercise capacity, and impairment in skeletal muscle architecture and cardiac function (Huffman et al., 2016; Justice et al., 2016). Many of these parameters are proposed as important endpoints for studies aiming to characterize potential health span-promoting interventions (Huffman et al., 2016; Justice et al., 2016). Of note, many of these phenotypes are regulated by NAD+ availability and the activity of NAD+-dependent enzymes (Camacho-Pereira et al., 2016; Gomes et al., 2013; López-Otín et al., 2016; Mills et al., 2016; Yoshino et al., 2011; Yoshino et al., 2017; Zhang et al., 2016).
Thus, we tested the effect of 78c on metabolic parameters in mice at different ages. As expected, aged mice (2-year-old) had lower tissue NAD+ levels, and worse glucose tolerance compared to younger mice (Fig. 2A–C and Fig. S2A–D). 78c promoted a significant increase in NAD+ levels in tissues of aged mice, but had a negligible effect on NAD+ levels of young mice (3-month-old) (Fig. 2A and Fig. S2C–D). Furthermore, after treatment with 78c, we noted an improvement in glucose homeostasis parameters in 2-year-old mice without changes in body weight, but no effects in glucose tolerance in either 3-month-old or 1-year-old mice (Fig. 2B–F and Fig. S2A–B). In support of these observations, we detected lower insulin levels in aged mice treated with 78c vs vehicle treated controls (Fig. 2D). The effect of 78c on glucose tolerance in these 2-year-old mice appears to be mediated, at least in part, by an increase in insulin sensitivity, as demonstrated by the HOMA-IR index and insulin sensitivity test (Fig. 2E–F). We further observed that 78c induced a decrease in expression of hepatic glucose-6-phosphatase, an enzyme involved in gluconeogenesis and endogenous glucose production in diabetes (Fig. 2G). Consistent with the decrease in glucose-6-phosphatase, we found that aged mice treated with 78c had lower blood glucose levels in response to administration of pyruvate, a precursor in the synthesis of glucose in the liver, suggesting suppression of the gluconeogenesis pathway by 78c (Fig. 2H). With the exception of increased expression of Pgc1α in muscle, no other significant changes in expression of genes related to mitochondrial biogenesis or glucose metabolism were observed in tissues of 78c treated mice (Figure 2G and Fig. S2E–G).
Figure 2. 78c ameliorates metabolic dysfunction-associated features in chronologically aged mice.

Aged (2-year-old) mice were treated with vehicle (Control) or 78c for up to 14 weeks. (A) NAD+ levels in multiple tissues (n=4–13 mice per group) at the end of the treatment.
(B) Intraperitoneal glucose tolerance test (ipGTT) at baseline and after 7 weeks of treatment, and corresponding area under the curve (AUC) graph. Analysis by two-way repeated measures ANOVA with Bonferroni's post-tests shows significant interaction between the glucose curve for control and 78c-treated mice. Results in panels B and C show average of two independent experiments (n=18–24 mice per group).
(C) Weekly body weight measurements.
(D) Serum insulin measurement (ELISA) during ipGTT and corresponding AUC after 5 weeks of treatment (n=5 mice per group).
(E) Homeostatic model assessment index for insulin resistance (HOMA IR) (n=5 mice per group).
(F) Intraperitoneal insulin sensitivity test (ipIST) and corresponding AUC after 4 weeks of treatment (n=5 mice per group).
(G) Liver mRNA levels of glucose metabolism-related genes, determined by quantitative RT-PCR (n=6–19 mice per group) at the end of the treatment.
(H) Intraperitoneal pyruvate tolerance test (ipPTT) and corresponding AUC after 9 weeks of treatment (n=5 mice per group).
(I) ipGTT of 2-year-old mice after 4 weeks of treatment with vehicle (Control), 78c, FK866 (NAMPT inhibitor), or 78c+FK866 and corresponding AUC (n=5 mice per group). Statistical differences were determined using Two-way repeated measures ANOVA followed by multiple-comparison testing using Bonferroni’s post hoc analysis; *P < 0.05 Control compared with the 78c group. All values are mean ± SEM. *P < 0.05. NS=not significant. See also Figure S2.
We next determined whether the improvement in glucose tolerance produced by 78c was dependent on increases in tissue NAD+ levels. To test this hypothesis in vivo, we used FK866, an NAD+ biosynthesis inhibitor, to decrease tissue NAD+ levels. Steady state NAD+ levels are maintained by a dynamic equilibrium between synthesis and degradation (Aksoy et al., 2006; Bai et al., 2011; Bai et al., 2012; Barbosa et al., 2007; Chini et al., 2014; Grozio et al., 2013; Imai and Guarente, 2014; Nahimana et al., 2009; Yang et al., 2007; Yoshino et al., 2011; Yoshino et al., 2017). Interestingly, it appears that the turnover of NAD+ in cells is quite fast (Rechsteiner et al., 1978), and inhibition of its biosynthesis leads to a decrease in cellular NAD+ levels mediated by several of the NAD+ catabolic mechanisms including CD38, PARPs, sirtuins, and ADP-ribose transferases (ARTs). In fact, we and others have shown that NAD+ levels can be regulated by both biosynthetic and degradation inputs (Aksoy et al., 2006; Bai et al., 2011; Bai et al., 2012; Barbosa et al., 2007; Chini et al., 2014; Grozio et al., 2013; Imai and Guarente, 2014; Nahimana et al., 2009; Yang et al., 2007; Yoshino., 2011). As shown in Figure 2I, the beneficial effects of 78c on glucose metabolism were completely blocked by the co-administration of FK866.
To further characterize the impact of CD38 inhibition on metabolic parameters during aging, we tested the effect of 78c on the glucose homeostasis of P44+/+ mice, a progeroid model with overexpression of the p53 isoform p44 (Δ40p53) (Maier et al., 2004; Pehar et al., 2010). These mice develop glucose intolerance earlier in life than WT controls (Fig. S2H–K). Interestingly, NAD+ levels were lower in these animals than in WT controls, and CD38 NADase activity and CD38 expression were higher than age-matched controls (Fig. S2L–N). Treatment of the P44+/+ mice with 78c increased NAD+ levels in several tissues and improved glucose tolerance (Fig. S2O–P), without changes in food intake or weight of these mice (Fig. S2Q–R), and except for a small, but significant increase in oxygen consumption (VO2) and total energy expenditure (TEE), it did not cause other alterations in calorimetric parameters (Fig. S2R). Thus, we conclude that the CD38i 78c promotes an increase in tissue NAD+ levels and ameliorates age-related glucose intolerance in both natural aging and in a progeroid mouse model.
Age-related skeletal muscle dysfunction is ameliorated in mice treated with 78c
We next evaluated the effect of 78c on physical activity, exercise tolerance, and muscle architecture in mice of different ages. As shown in Figure 3, aging is associated with several changes in exercise capacity, physical activity, and muscle architecture. There was an age-dependent decrease in exercise capacity, spontaneous physical activity, and several changes in muscle architecture including: 1) changes in myofiber size; 2) an increase in centrally located nuclei; 3) infiltration of CD45+ inflammatory cells; and 4) an increase in abundance of necrotic myofibers (Fig. 3). Following chronic administration of 78c, both 1 and 2-year-old mice exhibited an increase in exercise capacity, as shown by a near-doubling of maximal distance ran and work performed (Fig. 3A). The time to exhaustion was also increased by nearly 30% in 2-year-old mice (Fig. 3A). There was an increase in maximal running speed that was statistically significant in 1-year-old mice but did not reach statistical significance in 2-year-old mice (Fig. 3A). In addition, both spontaneous physical activity and activity-related energy expenditure (AEE) were statistically significantly improved in 2-year-old mice treated with 78c (Fig. 3B). Interestingly, 78c treatment increased not only NAD+ levels and exercise tolerance, but also ATP and ATP/O2 coupling in skeletal muscle (Fig. 3A and Fig. S3A–D). In contrast, although 78c was able to promote an increase in NAD+ levels and exercise capacity in older mice, it had no significant effect on either NAD+ levels or exercise tolerance in young (3-month-old) mice (Fig. S2C–D and S3E–F).
Figure 3. 78c promotes muscle functional improvement and protection from damage and fibrosis in chronologically aged mice.

1-year-old mice and 2-year-old (aged) mice were treated with vehicle (Control) or 78c for up to 14 weeks.
(A) Physical performance assessment on a motorized treadmill. Measurements of distance, maximal running speed, running time, and work after 8 weeks of treatment (n=6–10 mice per group).
(B) Measurements of locomotor activity by Comprehensive Laboratory Animal Monitoring System (CLAMS) after 4 weeks of treatment: total activity, rearing, ambulation, and active energy expenditure (AEE) (n=6–9 mice per group).
(C) Representative images (upper panel) of laminin-immunostained skeletal muscle tissue sections from vehicle (Control) and 78c-treated 2-year-old mice. The white arrows point to areas with centrally located nuclei (DAPI nuclear staining /blue) within the myofibers marked by laminin (green). Graph shows number of centrally located nuclei (CLN) per unit area of muscle (n=6–9 mice per group).
(D) Representative images (upper panel) of skeletal muscle tissue sections of vehicle (Control) and 78c-treated 2-year-old mice immunostained for CD45. CD45-positive cells (red) are indicated by white arrows. Graph shows quantification of CD45-positive cells per unit area of muscle (n=6–10 mice per group).
(E) Representative images (upper panel) of skeletal muscle tissue sections of vehicle (Control) and 78c-treated 2-year-old mice immunostained for intracellular immunoglobulin G (IgG). IgG-positive fibers represent necrotic fibers (yellow arrows). Graph shows number of necrotic fibers per unit area of muscle (n=6–9 mice per group).
(F) Distribution of myofiber minimum Feret diameter in tibialis anterior (TA) muscle of vehicle (Control) and 78c-treated 1-year-old and 2-year-old mice (n = 6–9 mice per group; >300 fibers counted per mouse).
(G) Skeletal muscle mRNA levels of fibrosis and inflammation-related genes in aged mice, determined by quantitative RT-PCR (n = 5–6 mice per group, * P < 0.05, versus control mice).
All values are mean ± SEM. Feret diameter linear regression curves were compared using a sum-of-squares F test, *P <0.05. Images show their corresponding scale bars. See also Figure S3.
To determine if the effects of 78c on exercise tolerance were also mediated by an increase in NAD+ levels, we treated 2-year-old mice with the NAD+ biosynthetic inhibitor FK866. We found that the 78c-mediated improvement in exercise capacity was completely prevented by co-administration of 78c and FK866 (Fig. S3A). Furthermore, increases in tissue NAD+ and ATP levels induced by 78c were also prevented by inhibition of NAMPT in vivo (Fig. S3B–C).
Accompanying these physiological improvements observed in aged mice, 78c treatment reversed several morphological features of aging in skeletal muscle (Fig. 3C–F). Strikingly, the number of centrally located nuclei, presence of inflammatory cell infiltrates, and number of necrotic myofibers decreased by more than 70% (Fig. 3C–E) in aged mice treated with 78c. Additionally, the tibialis anterior muscle of aged control mice exhibited increased numbers of hypotrophic fibers compared to 78c-treated aged mice, as expressed by the distribution of myofiber minimum feret diameters (Fig. 3F). Furthermore, expression of genes related to fibrosis was decreased in the skeletal muscle of aged mice treated with 78c (Fig. 3G). Consistent with the gene expression data, histological sections of muscle from aged control mice stained with the fibrosis marker wheat germ agglutinin (WGA) showed large fibrotic streaks, while samples from 78c–treated mice had nearly no signs of fibrosis (Fig. S3G). No significant changes were observed in muscle weight, fiber subtype distribution, or number of Pax7+ muscle satellite cells (Fig. S3H–J).
To further investigate the effect of 78c on muscle damage after exercise in 2-year-old mice, we performed a downhill running protocol (Fig. S3K-N). In accordance with our previous data, 78c also increased exercise capacity when mice were exercised to exhaustion in this protocol (Fig. S3K). Furthermore, animals on 78c had lower lactate levels after exercise (Fig. S3L). However, we did not observe a significant difference in markers of muscle tissue injury such as creatine kinase (CK) levels or in Evans Blue staining of two types of skeletal muscle from 78c and control animals that were subjected to this exercise protocol (Fig. S3M–N). These data indicates that 78c does not promote an obvious protection against acute muscle injury induced by exercise.
Age-related cardiac dysfunction is ameliorated in mice treated with 78c
Importantly, we also observed that aged (2-year-old) mice, but not young (3-month-old) mice, treated with 78c for 10 weeks had a significant improvement in several cardiac parameters as determined by echocardiography (Fig. S3O). Aged control mice showed a decline in ejection fraction (EF) and fractional shortening (FS), and an increase in left ventricle volume during systole (LVVs) and isovolumetric contraction time (IVCT) parameters related to cardiac straining when compared with young control mice (Fig. S3O). Treatment with 78c reversed these cardiac parameters and also restored cardiac muscle NAD+ to levels of young mice (Fig. S3O–P).
Our data together show that 78c can promote significant improvement in several physiological parameters including: glucose homeostasis, exercise capacity, muscle architecture and cardiac function in aged mice. Consistent with the health span data reported herein, we also observed that treatment with 78c increased overall survival in the Bub1bH/H animal model of accelerated aging (Baker et al., 2004) (Fig. S3Q).
CD38+ cells regulate NAD+ levels in CD38- cells by influencing the availability of NAD+ precursors
In view of the physiological and morphological improvements seen with 78c treatment, we next wanted to determine the mechanisms by which 78c increases NAD+ levels in cells and tissues. In particular, we explored if 78c directly inhibits CD38 in the main parenchymal cells of the respective tissues, or if the increases in NAD+ levels are mediated by inhibition of CD38-NADase activity in subpopulation(s) of cells present in these tissues, resulting in increased availability of NAD+ and its precursors to other cells. First, we found that CD38 was nearly undetectable in hepatocytes in vivo, in freshly isolated hepatocytes, and in the AML12 mouse hepatocyte cell line (Fig. 4A–B). Although immunofluorescent staining of mouse liver sections with an anti-CD38 antibody showed that hepatocytes did not stain for CD38, CD38+ cells were abundant in this tissue (Fig. 4A–B). The specificity of CD38 staining was confirmed in negative control experiments (Fig. S4A). CD38 staining in the liver was observed in both CD45+ cells (e.g. resident macrophages-Kupffer cells and other infiltrating/resident immune cells), and CD45− cells located in the sinusoidal region (e.g. endothelial cells, and stellate cells) (Fig. 4A). In particular, we found that freshly isolated mouse non-hepatocyte liver cells such as Kupffer cells are strongly positive for CD38 (Fig. 4B). These data is consistent with the data published by March et al. (2007) in rat livers.
Figure 4. CD38+ cells regulate NAD+ levels in CD38- cells by influencing the availability of NAD+ precursors.

(A) Immunofluorescent localization of CD38 (red) expression in mouse liver. Sections were co-stained for the pan-leukocyte marker CD45 (green). Hoechst-stained nuclei are shown in blue. Arrow heads (white) indicate lack of CD38 in hepatocytes. Arrows (yellow) show sinusoidal distribution of CD38. Right image depicts a lobular area with a centrally located immune cells cluster. CV= central vein. Scale bar represents 50 μm.
(B) Protein lysates of primary mouse hepatocytes, Kupffer cells and AML12 cells were immunoblotted with CD38 and Tubulin antibodies.
(C) NAD+, nicotinamide mononucleotide (NMN), and nicotinamide riboside (NR) levels in liver and skeletal muscle of aged mice treated with vehicle (Control) or 78c for 10 weeks. Metabolites were measured by high-performance liquid chromatography (HPLC)-mass spec (n=5–6 mice per group).
(D) Schematic drawing of the co-culture model. In co-culture experiments, CD38 null cells were plated in the lower chamber and CD38 positive cells were platted in the upper chamber. The lower and upper chambers were separated by a microporous membrane, as demonstrated by the drawing.
(E) AML12 cells were plated in the lower chamber of co-culture plates. Upper chamber had no cells (n=8). 0.5 μM 78c was added to the upper chamber, and both chambers were incubated together for 24 hours. Graph shows NAD+ levels in the AML12 cells.
(F) AML12 cells were plated in the lower chamber of co-culture plates. Upper chamber had Jurkat T cells. 0.5 μM 78c was added to the cells in the upper chamber 4 hours before addition of 100 μM NMN. 4 hours later, both chambers were incubated together for an additional 20 hours. Graphs shows NAD+ levels in the AML12 cells (n=3).
(G) AML12 cells were plated in the lower chamber of co-culture plates. 293T cells were plated in the upper chamber and then transfected with vector, CD38, or CD38.CI. 24 hours later, 100 μM NMN was added to the upper chamber. 4 hours later, both chambers were incubated together for an additional 20 hours. Graphs shows NAD+ levels in the AML12 cells (n=4–6).
(H) AML12 cells were plated in the lower chamber of co-culture plates. 293T cells were plated in the upper chamber and then transfected with vector or CD38 plasmid. 1 μM 78c was added to the upper chamber during transfection. 16 hours later, 100 μM Nicotinamide mononucleotide (NMN) was added to the upper chamber and 4 hours later both chambers were incubated for an additional 20 hours. Graphs shows NAD+ levels in the AML12 cells (n=8). (D). All values are mean ± SEM. * P < 0.05.
(I) AML12 cells were incubated in the presence or absence of 100 ng/ml rhCD38, 100 μM NMN, and 1 μM 78c for 18 hours. Graphs shows NAD+ levels in the AML12 cells (n=4).
(J) NAD+, NMN, and NR levels in liver of 2-year-old mice treated with vehicle (Control), 78c, NR, or 78c+NR. Mice received two doses of vehicle or 78c 22 hours and 6 hours prior to euthanasia. One dose of NR was given by gavage 6 hours before euthanasia. Metabolites were measured by high-performance liquid chromatography (HPLC)-mass spec (n=7–10 mice per group).
All values are mean ± SEM. *P < 0.05. NS=not significant. See also Figure S4.
Using flow cytometric analysis and immunostaining, we further explored the localization of CD38 expression in other tissues. We found that CD38 expression in skeletal and cardiac muscle is also localized mainly to non-parenchymal cells (Fig. S4B–F). In skeletal muscle the majority of CD38+ cells are endothelial (CD31+); however, CD38+ immune cells (CD45+) and putative fibro-adipogenic precursors (FAPs) and fibroblasts (CD31−/CD45−/Sca1+) are also abundant (Fig. S4C–E). Consistent with Boslett et al. (2018), the CD38+ cell population in the heart consisted of FAP and endothelial cells with few CD45+ cells represented (Fig. S4F). These data demonstrate that in all three tissues studied (i.e. liver, skeletal muscle, and heart) CD38 does not predominate on parenchymal cells, but rather endothelial, inflammatory, and FAP cells.
Due to the fact that we observed a clear skeletal muscle, cardiac, and liver phenotype in our studies, we hypothesized that inhibition of CD38 by 78c in the CD38+ cells in target tissues was sufficient to increase the availability of NAD+ and NAD+ precursors to CD38− parenchymal cells. Thus, we measured the levels of NAD+ and NAD+ precursors such as NMN and NR in tissues of mice that had been chronically treated with 78c (Figure 4C and Fig. S4G). Liver, skeletal muscle, and spleen of mice treated with 78c for 10 weeks showed a statistically significant increase in the levels of all three NAD+ metabolites, with the exception of NR levels in the liver, which had an increase that did not reach statistical significance (Fig. 4C and Fig. S4G). These results imply that CD38+ cells are likely capable of changing the availability of NAD+ and its precursors NMN and NR in the cellular microenvironment, thereby modulating the NAD+ levels in both CD38+ and nearby CD38− cells.
To test this hypothesis, we performed co-culture experiments with CD38+ cells in the top chamber and CD38− AML12 hepatocytes in the bottom chamber (Fig. 4D–H). Consistent with the lack of expression of CD38 in AML12 cells, treatment with 78c had no direct effect on NAD+ levels in these cells (Fig 4E). However, when AML12 cells were co-cultured with Jurkat T cells, which express endogenous CD38, 78c significantly increased cellular NAD+ levels on the AML12 cells (Fig. 4F). The increase induced by 78c was much higher when cells were also treated with the NAD+ precursor NMN suggesting that 78c, through inhibition of CD38, increases the availability of NMN to the CD38− AML cells (Fig. 4F).
To confirm if CD38+ cells modulate the availability of NAD+ precursors to CD38− cells, we added NMN to upper chambers containing 293T cells overexpressing: 1) control empty vector; 2) wild type CD38; or 3) CD38 CI. Interestingly, we observed that the addition of NMN to the upper chamber increased NAD+ levels only when CD38 was absent, or when a catalytically inactive CD38 (CI) was expressed in the upper chamber (Fig. 4G). When WT CD38 was present, NMN had no significant effect on NAD+ levels in AML12 cells located at the lower chamber (Fig. 4G). We also performed similar experiments in the presence and absence of 78c and measured the changes in NAD+ levels in the CD38− cells (AML12) in the bottom chamber (Fig. 4H). When CD38+ cells were present in the upper chamber, treatment with NMN did not promote an increase in NAD+ levels in the AML12 cells. However, treatment with 78c reversed the effect of the CD38+ cells on the NAD+-boosting effect of NMN in the AML12 cells (Fig. 4H).
To further determine that the effect of CD38 expressed in the top chamber cells was mediated by CD38 itself and not another factor, we added extracellular rhCD38 to the media of cultured AML12 cells and measured NAD+ levels following treatment with vehicle, NMN, or a combination of 78c and NMN (Fig. 4I). Addition of NMN significantly increased NAD+ levels in AML12 cells, and this effect was prevented by addition of rhCD38 to the culture media (Fig. 4I). Addition of 78c and NMN together to the media of AML12 cells treated with rhCD38 reversed this effect, leading to an increase in cellular NAD+ levels (Fig. 4I). The effect of rhCD38 and 78c was further explored at the cell signaling level. Treatment of AML12 cells with NMN in the presence of rhCD38 led to an increase in pan-protein acetylation and inhibition of the AMPK pathway (Fig. S4H). This effect was reversed by the co-administration of NMN with 78c (Fig. S4H).
Finally, we also performed experiments to demonstrate whether 78c could increase the availability of NAD+ precursors to tissues when animals were treated with the NAD+ precursor NR. Animals were treated with either vehicle, 78c, NR, or a combination of 78c and NR, and levels of NAD+, NMN, and NR were measured in tissues (Fig. 4J and Fig. S4I). We observed that the levels of all three metabolites were significantly increased by a combination of 78c and NR in both liver and spleen (Fig. 4J and Fig. S4I). These data combined demonstrate that 78c increases availability of NAD+ and its precursors to cells by inhibiting the activity of CD38 in the CD38+ cells.
78c promotes changes in signaling pathways correlated with longevity and health span
One of the main goals of NAD+-replacement therapy in aging is to increase the availability of NAD+ to NAD+-dependent enzymes such as sirtuins (Cantó and Auwerx, 2012; Escande et al., 2013; Imai and Guarente, 2014; Li et. al., 2017; López-Otin et al., 2016; Schultz and Sinclair, 2016; Verdin, 2015; Yoshino et al., 2011; Yoshino et al., 2017; Zhang et al., 2016). Sirtuins are enzymes that remove specific covalent modification in lysine residues, such as acetylation, malonylation, and succinylation, in several proteins and have been implicated in the regulation of chromosomal integrity, DNA damage repair, metabolism, aging, health span, and longevity (Cantó and Auwerx, 2012; Escande et al., 2013; Imai and Guarente, 2014; Li et. al., 2017; López-Otin et al., 2016; Schultz and Sinclair, 2016; Verdin, 2015; Yoshino et al., 2011; Yoshini et al., 2017; Zhang et al., 2016). Therefore, first we tested the effect of 78c on sirtuin-dependent lysine modifications in cells. As predicted by the effect of CD38 on NAD+ levels, expression of CD38 in 293T cells increased acetylation of p65, a substrate of sirtuins 1, 2, and 6, and also pan-cellular acetylation (Fig. S5A–B). 78c reversed the effect of CD38 expression and promoted the deacetylation of lysines in p65 and other proteins (Fig. S5A–B). This effect was reversed by the non-specific sirtuin inhibitor nicotinamide (NAM) and semi-selective SIRT1 inhibitor EX-527 (Fig. S5A–B). These results suggest that 78c may increase the cellular availability of NAD+ to sirtuins via inhibition of CD38/NADase activity, thereby promoting NAD+-dependent lysine deacetylation of proteins. Importantly, this mechanism of action suggests that 78c may activate not only SIRT1, but also other NAD+-dependent health span and longevity-associated sirtuins. In fact, in the mitochondria, where SIRT3 but not SIRT1 is present, we observed that, akin to our published observations in CD38 KO mice (Camacho-Pereira et al., 2016), 78c promotes deacetylation of mitochondrial proteins (Fig. S5C). Next, we investigated whether 78c regulates lysine modifications in vivo. Compatible with our results in cells, treatment of aged mice with 78c promotes a decrease in pan-acetylation of lysines (Fig. 5A). Interestingly, we observed that 78c also promoted a decrease in pan-succinylation and pan-malonylation of lysine residues in muscle proteins of these mice (Fig. 5B–C). These data further support the concept that 78c-induced increase in tissue NAD+, leads to activation of multiple NAD+-dependent sirtuins such as the deacetylases (SIRT1, SIRT3, and SIRT6), and the demalonylases and desuccinylases, namely SIRT5 and 7.
Figure 5. 78c increases sirtuins and PARP activities, activates longevity signaling pathways, and decreases accumulation of DNA damage in vivo.

(A-C) 2-year-old (aged) mice were treated with vehicle (control) or 78c for 14 weeks. Protein lysates from skeletal muscle were immunoblotted with specific antibodies. Graph shows quantification of immunoblots.
(A) lysine acetylation (ack) and Tubulin (n=5 mice per group).
(B) succinyl lysine (suK) and GAPDH (n=6 mice per group).
(C) malonyl lysine (maK), Sirt5, and Tubulin (n=5–6 mice per group).
(D–J) Telomere Immuno-FISH analysis from liver (n=6–9 mice per group) and skeletal muscle (n=5 mice per group) of 2-year-old mice treated with vehicle (control) or 78c for 14 weeks.
(D) Mean number of telomere-associated DNA damage foci (TAF) per nuclei of hepatocytes.
(E) Percentage of TAF-positive cells (50–100 hepatocytes were counted).
(F) Mean number of γ-H2AX foci per nuclei of hepatocytes.
(G) Representative images of γH2A.X immuno-FISH in hepatocytes. Images are Huygens (SVI) deconvolved Z projections of stacks taken with a ×100 objective. White arrows indicate colocalization, and colocalizing foci are amplified in the right panel (amplified images are from single Z planes where colocalization was found). Scale bar: 10 μm.
(H) Mean number of TAF per nucleus of skeletal muscle cell.
(I) Percentage of TAF-positive cells (40–50 skeletal muscle cells were counted).
(J) Mean number of γ-H2AX foci per nucleus of skeletal muscle cell.
(K) Representative images ofγ-H2A.X immuno-FISH in myofibers. Images are Huygens (SVI) deconvolved Z projections of stacks taken with a ×100 objective. Yellow arrows indicate colocalization, and colocalizing foci are amplified in the right panel (amplified images are from single Z planes where colocalization was found). Scale bar: 10 μm.
All values are mean ± SEM. * P < 0.05. See also Figure S5.
Another NAD+-dependent class of enzymes implicated in aging and longevity are the PARPs. The roles of PARPs in aging and longevity, and cell injury are complex, It has been observed that PARP1 activity positively correlates with longevity in different species (Bakondi et al., 2011; Beneke et al., 2010; Noren-Hooten et al., 2012), In addition, data from a recent study demonstrate that PARP activity decreases during the aging process and that NAD+-boosting with NMN can restore PARP1 activity and promote health span (Li et al., 2017). Our own data confirms that NAD+-boosting with NMN promotes an increase in protein PARylation in vivo (Fig. S5D). Thus, we hypothesized that 78c-induced NAD+-boosting could also lead to increased availability of NAD+ to PARPs, promoting an increase in protein PARylation. To test this hypothesis, first we examined whether inhibition of CD38 by 78c stimulate PARP activity in cells. In 293T cells, with p53-mediated induction of PARylation, co-expression of CD38 caused a decrease in PARylation, and this effect was reversed by 78c (Fig. S5E). In contrast, the PARP inhibitor olaparib nearly completely blocked PARylation in these cells (Fig. S5E). Next we tested if in vivo treatment with 78c induced changes in protein PARylation, and we detected a robust increase in protein PARylation in the spleen of mice treated with 78c (Fig. S5F). Our combined data suggest that 78c-mediated inhibition of CD38 leads to an NAD+ boosting effect that promotes increase in activity of several NAD+-dependent enzymes including PARPs and sirtuins.
We further explored the effect of 78c on pro-longevity pathways, such as the AMPK pathway, and in pathways that negatively correlate with health span, such as the mTOR-p70S6K and ERK pathways (Huffman et al., 2016; Justice et al., 2016). 1-year-old 78c-treated mice exhibited significant activation of the pro-longevity AMPK pathway in spleen and skeletal muscle (Fig. S5F–G). Changes induced by 78c in the AMPK pathway were further confirmed in A549 cells and mouse bone marrow-derived macrophages, which express endogenous CD38 (Fig. S5H-I). On the other hand, 78c decreased the activation of the mTOR-p70S6K and ERK pathways in the spleen, and skeletal muscle of 1-year-old mice (Fig. S5F–G).
Our results combined indicate that 78c promotes an increase in tissue NAD+ levels which likely stimulates pan-activation of NAD+-dependent enzymes including several sirtuins and PARPs. Furthermore, 78c promotes activation of pro-longevity pathways, such as AMPK, and inhibition of pathways negatively correlated with longevity, such as p70S6K and ERK. These data also highlight the complex role of NAD+-boosting therapy and the multiple pathways that can be activated by increasing organismal NAD+ levels.
78c protects mice against accumulation of age-related DNA damage
Another important component of the aging process is the accumulation of DNA damage, particularly in telomeric DNA (Jurk et al., 2014). In contrast to the rest of the genome, telomere regions cannot be easily repaired due to inhibition of non-homologous recombination, resulting in a persistent DNA damage response which accumulates during aging. We explored the effect of 78c on the accumulation of telomere-associated foci (TAF), as determined by the co-localization of γH2A.X immunostaining with telomeres FISH (Jurk et al., 2014). Treatment of aged mice with 78c for 14 weeks significantly decreased TAF in both liver and skeletal muscle (Fig. 5D–K). This effect was observed both when we quantified the number of TAF per nuclei and the number of TAF-positive cells (Fig. 5D–K). Additionally, we noted a decreasing trend in total foci in animals treated with 78c (Fig. 5F, 5J) that did not reach statistical significance. Since TAF have been associated with cellular senescence in vivo, our data indicate that 78c may either prevent or reverse the accumulation of cellular senescence in tissues. However, we did not observe any significant changes in the expression of senescence markers such as p16, p53 or p21 in animals treated with 78c (RT-PCR, data not shown).
DISCUSSION
Our study reveals CD38 as a potential pharmacological target to reverse age-related NAD+ decline. By increasing tissue NAD+ levels, 78c ameliorates several metabolic, physiological, structural, and molecular features of aging. Recently, several reports have demonstrated that increasing NAD+ levels in tissues, through the use of NAD+ precursors such as NMN and NR, improves health span, mitigates age and metabolic disease-associated decline, and may increase longevity, thus highlighting the value and utility of NAD+-replacement therapy (Cantó and Auwerx, 2012; Li et al., 2017; Mills et al., 2016; Mouchiroud et al., 2013; Verdin, 2015; Yoshino et al., 2011; Zhang et al., 2016). We recently showed that CD38 is a key regulator of the pharmacokinetics and pharmacodynamics of NAD+ precursors (Camacho-Pereira et al., 2016). Thus, we believe that 78c may serve as a pharmacological tool to improve the effects of NAD+-replacement therapy with NAD+ precursors. Very interestingly, we demonstrate that treatment with 78c increases tissue availability of NAD+ precursors and that, although the effect of 78c in vivo requires the presence of functional CD38, the effect of CD38i can be transmitted even to non-CD38 positive cells in the tissue. This indicates a unique mechanism of action for this compound and for the role of CD38 in organismal NAD+ homeostasis. Also, because CD38 is mostly an ecto-enzyme, it appears that one of the effects of 78c is to inhibit the degradation of extracellular NAD+ and its precursors improving global organismal NAD+ homeostasis.
In conclusion, our study establishes CD38 as a viable pharmacological target for age-related metabolic decline by reversing the decrease in NAD+ associated with aging. Since NAD+ decline appears to have a causal role in age-related metabolic dysfunction and the development of age-related diseases, CD38 inhibitors appear to target a key cellular metabolic driver of the aging process. In fact, CD38i could potentially have a role as a novel treatment for age-related diseases, such as diabetes and insulin resistance. It is important to highlight that, in addition to small molecule CD38i such as the one described here, CD38 inhibition in humans has been recently targeted using biologicals such as CD38-specific blocking antibodies (Shallis et al., 2017). In fact, several of these antibodies are either being studied or have been approved by the FDA for the treatment of human cancers (Shallis et al., 2017). This demonstrates that some of the tools for inhibition or clearance of CD38 in humans are already available, and that the use of these agents could soon be translated for the treatment of age-related metabolic dysfunction.
LIMITATIONS OF STUDY
The specific mechanisms connecting the increases in NAD+ levels induced by 78c with physiological and metabolic functions were not fully delineated in our studies. Due to the fact that 78c induces increases in NAD+ levels in multiple tissues and activates many pro-health span pathways, determining which specific signaling pathway regulates each physiological functions is challenging. Also, in this study, the route of administration of 78c was intraperitoneally and the ideal therapeutic route would be orally. Finally, we did not study lifespan and longevity in natural aged animals. Validation of safety and efficacy of CD38i in human studies will be necessary for its potential future use in age-related NAD+ -decline
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Eduardo N. Chini (chini.eduardo@mayo.edu).
METHODS DETAILS
Reagents
Except when specified, all reagents and chemicals were purchased from Sigma-Aldrich.
Mouse Models
All protocols requiring animal manipulation were approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC), and studies were conducted in adherence to the NIH Guide for the Care and Use of Laboratory Animals. Mice were housed in standard cages at constant temperature and humidity, with 12 hour light-dark cycles, and were maintained on a normal chow diet (ND) (PicoLab 5053 Rodent Diet 20; Lab Diets) ad libitum, except when fasting was indicated. Male mice were used for these experiments.
The CD38 KO mice and P44+/+ progeroid mice have been described previously (Partida-Sánchez et al., 2001; Maier et al., 2004). The CD38 catalytically-inactive (CI) “knock-in” mice were generated at TransViragen (Chapel Hill, NC) using recombineering technology. Briefly, a bacterial artificial chromosome (BAC) transgene was generated in which amino acid E230 of the CD38 gene was converted to Q230 by point mutation. This mutation has been demonstrated to render CD38 catalytically inactive. Because the BAC also contains the CD157 gene, a translational STOP sequence was inserted into CD157 Exon 4 to block expression of CD157 from the BAC. The CD38 BAC transgene was injected into fertilized oocytes from homozygous CD38 KO mice (B6.129P2-Cd38tm1Lnd/J, The Jackson Laboratory) using a standard pronuclear microinjection protocol, and surviving eggs were implanted into the ampulla of pseudo-pregnant Swiss Webster (Taconic) females. Offspring were analyzed for the presence of the transgene by PCR using the assays listed in the table below. The transgenic founders were mated with homozygous CD38 KO mice for germline transmission assessment and transgene expression analysis.
CD38 E230Q PCR Assays
| Assay | BAC Region | Primer Sequences | Product Size | |
|---|---|---|---|---|
| BAC | WT | |||
| A | T7 end | CGGCCGCTAATACGACTCACTATAGGGAGA TTTATTTGAGACGGCTACGAAGGCTG |
362 bp | - |
| B | CD157 Insertion | AGTTCTAGAGCGGCCAGGGAGTTGCTTCCGAATGTATAGCC TCCACACATGCTCCACACTCGCTC |
553 bp | 385 bp |
| C | CD38 Insertion | CCAAGCTATCGAATTCGAAACTGAGATAAAGTGTGA ACAGTGCTGCCTAAAGACCTGCACACTTCCAAAGGTGCTGAA |
856 bp | 751 bp |
| D | Sp6 end | CCCTTTCAGGCACCCAGTTCTC CGCCTGGCCGTCGACATTTA |
428 bp | - |
Drug Treatments
CD38 inhibitor (78c)
78c was administered to C57BL/6 (3, 12, and 22 to 26 months old) and ICR (1-year-old) mice by intraperitoneal injection (i.p., 10 mg/kg/dose) twice daily over a period of 4 to 14 weeks. 3-month-old, 1-year-old and 2-year-old mice were treated with 78c for up to 14 weeks. Combination of 78c and FK866 was performed for 10 weeks. P44+/+ progeroid mice due to their accelerated aging were treated with 78c for 4 weeks. For the short treatment, mice received a 15 mg/kg/dose twice daily for 8 days. Control mice received vehicle (5% DMSO, 15% PEG400, 80% of 15% hydroxypropyl-γ-cyclodextrin (in citrate buffer pH 6.0)) injections. We also measured the concentration of 78c in multiple tissues and plasma. Samples and standards were extracted by protein precipitation with acetonitrile containing internal standards. The supernatant was diluted with 0.1% formic acid in water before injection into an HPLC-MS/MS system for separation and quantitation. The analytes were separated from matrix components using reverse phase chromatography on a 30x2.1 mm 5 μm Fortis Pace C18 using gradient elution at a flow rate of 0.8 mL/min. The tandem mass spectrometry analysis was carried out on SCIEX™ triple quadrupole mass spectrometer with an electrospray ionization interface, in positive ion mode. Data acquisition and evaluation were performed using Analyst® software (SCIEX™). The results of the measurements were: plasma (0.007 μg/mL); brain (0.000 μg/g); heart (0.003 μg/g); kidney (0.005 μg/g); liver (0.024 μg/g); pancreas (0.002 μg/g) and, spleen (0.0048 μg/g). We also observed that 78c could be detected in cellular extracts of cultured cells treated with different concentration of 78c.
PARP inhibitor
Olaparib (LC Laboratories) was given to, C57BL/6 and CD38 KO mice by i.p. injection (10 mg/kg/dose) once daily for 8 days. Control mice received vehicle injections (5% DMSO, 15% PEG400, and 80% of 15% hydroxypropyl-γ-cyclodextrin (in citrate buffer pH 6.0)).
NAMPT inhibitor
Aged C57BL/6 mice received FK866 (25 mg/kg/dose, i.p., once daily), 78c (10 mg/kg/dose, i.p., twice daily), or a combination of FK866 and 78c (same doses) for 10 weeks. Control mice received equivalent injections of vehicle for FK866 (1% Hydroxypropyl-β-cyclodextrin, 12% Propylene glycol) and vehicle for 78c (5% DMSO, 15% PEG400, 80% of 15% hydroxypropyl-γ-cyclodextrin (in citrate buffer pH 6.0)), a group was treated with 78c, and one group was treated with a combination of 78c (10 mg/kg/dose, i.p twice daily) and FK866 (25 mg/kg/dose, i.p once daily).
NAD+ precursors
For the treatment with nicotinamide mononucleotide (NMN), C57BL/6 mice received a single dose of NMN (500 mg/kg) or vehicle (PBS) by gavage. Mice were sacrificed after 2 hours, and tissues harvested. For the study with nicotinamide riboside (NR), aged C57BL/6 mice were pretreated with a single dose of 78c (10 mg/kg, i.p). Sixteen hours later, they received NR (100 mg/kg) by gavage and a second injection of 78c (10 mg/kg). Blood was collected just prior to administration of NR, and 30 min, 1 hour, 2 hours, and 6 hours later. Control mice received NR alone (200 mg/kg), 78c alone (10 mg/kg/dose, 2 doses), or vehicle (NR=PBS; 78c=5% DMSO, 15% PEG400, 80% hydroxypropyl-γ-cyclodextrin). The mice were sacrificed 6 hours after administration of 78c and NR, and tissues were collected.
Glucose and Insulin Assays
After 7 weeks of treatment, or otherwise specified, intraperitoneal glucose tolerance tests (ipGTT) were performed. Mice were fasted for 16 hours. Blood was collected from the tail vein before and 20, 30, 60, and 120 minutes after i.p. injection of 20% dextrose (1.5 g/kg body weight), and glucose levels were measured using an AlphaTrak II glucometer (Abbott Laboratories). For insulin sensitivity tests (IST) and the pyruvate tolerance test (PTT), following a 6-hour fast, mice were challenged with a single i.p. dose of 0.5 units/kg insulin or 1.5 g/kg of pyruvate, and blood glucose was measured at the intervals described above. The homeostatic model assessment of insulin resistance (HOMA-IR) was calculated using the equation [(G0 × I0)/405)], where G0 and I0 refer to 6 hour fasting plasma glucose and insulin (Berglund et al., 2008). Plasma insulin was determined using the Ultra-Sensitive Mouse Insulin ELISA kit (Crystal Chem) according to the manufacturer’s instructions.
Physical Function Analysis
Physical function was characterized by measuring running time, distance, maximal running speed, and work using a motorized treadmill (Columbus Instruments) after 8 weeks of treatment, or otherwise specified. The mice were acclimated to the treadmill for 3 consecutive days, 5 minutes per day, at a speed of 10 m/min and a grade of 5%. After a day of rest, animals ran on the treadmill at an initial speed of 5 m/min and 5% grade for 2 minutes, after which the speed was increased by 2 m/min every subsequent 2 minutes until the mice were exhausted. Exhaustion was defined as the inability of the mouse to remain on the treadmill despite an electrical shock stimulus and mechanical prodding. Another protocol was used to analyze creatine kinase and lactate levels before and after a downhill exhaustion treadmill run (1 hour, 2 hours and 96 hours post-exercise). Mice were acclimated to the treadmill as above, but at a downhill grade of −10° angle. After a day of rest, blood was collected from the ventral tail artery (“pre-exhaustion bleed”) and then mice ran using the downhill exhaustion protocol (warm-up 5 min at 3 m/min; downhill exhaustion run 5 min at 10 m/min, 5 min at 15 m/min, 5 min at 20 m/min, and up to 15 min at 25 m/min). For all treadmill experiments, running time was recorded, and running distance (a function of time and speed of the treadmill), work (the product of body weight [kg], gravity [9.81 m/s2], vertical speed [m/s × angle], and time [s] were calculated (LeBrasseur et al., 2009).
Metabolic and Behavioral Parameters
After 4 weeks of treatment, or otherwise specified, metabolic rates were measured at room temperature by indirect calorimetry of single animals in open-circuit “oxy-max” Comprehensive Laboratory Animal Monitoring System (CLAMS) chambers (Columbus Instruments) as described in Izumiya et al. (2008). On the day of the experiment, mice were weighed and acclimated in the chambers overnight. Over the next 48 hours, locomotor activity, oxygen consumption (VO2), and CO2 production (VCO2) were monitored in real time. Mice had food and water ad libitum during the first 24 hours, and were fasted during the subsequent 24 hours. Respiratory exchange ratio (RER = VCO2/VO2) and metabolic rate (MR = (3.815 + 1.232 × RER) × VO2) were calculated.
Echocardiography and image analysis
Echocardiography was performed as previously described (Weiss et al., 2006). Briefly, mice were minimally sedated with midazolam, 0.15 mg by subcutaneous injection. This produced a mouse that was conscious, mildly curious, but docile. The anterior chest was shaved and warmed gel was applied, to improve acoustic interface. The mouse was cradled in the investigator's left hand and images were obtained in real-time using a 15 MHz linear-array transducer, coupled to a Sonos 5500R ultrasonograph (Philips). Two-dimensional images were acquired at ~200 sec-1 in parasternal long- and short-axis planes, and stored off-line for subsequent analysis. Pulse-wave Doppler interrogation of mitral inflow was used to measure heart rate.
Images were analyzed in a blinded fashion as previously described and validated (Hill et al., 2000). Briefly, endo- and epicardial borders were traced electronically at end-diastole and end-systole using custom software developed for this purpose (Freeland Medical Systems, Indianapolis, IN). Left ventricular end-diastolic volume, end-systolic volume, stroke volume, ejection fraction, and mass were calculated using the bi-plane area-length method, previously validated.
Cell Culture, Plasmids, Transfection, and in vitro Drug Treatments
HEK293T and MEFs were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin (Life Technologies). A549 and Jurkat T cells were cultured in RPMI-1640 with 10% FBS and 1% penicillin/streptomycin. AML12 cells were grown in DMEM:F12 media with 10% FBS, 1% penicillin/streptomycin, 40 ng/mL dexamethasone, 0.005 mg/mL insulin, 0.005 mg/mL transferrin, and 5 μg/mL selenium. The human CD38 plasmid has been described before (Camacho-Pereira et al., 2016). The E226D inactivating mutation of this construct was generated by site-directed mutagenesis using the QuikChange Kit (Agilent Technologies). Flag-p65 and HA-p53 plasmids were from AddGene. Transfections were performed using Lipofectamine 2000 and 3000 (Life Technologies) according to the manufacturer’s instructions. HEK293T cells were transfected for 48 hours. Drug treatments were performed 20 hours after transfection began. HEK293T were treated with 0.5 μM 78c, 5 μM olaparib (LC Laboratories), 5 μM EX-527 (Tocris), or 5 μM nicotinamide for 24 hours. MEFs were treated with 0.2 μM 78c for 24 hours. A549 cells were treated for 24 hours with vehicle (DMSO), 78c (0.2–0.5 μM) or 5 μM olaparib. A549 and 293T cells were treated with drugs in media containing 0.5% FBS and MEFs were treated in media containing 10% FBS. To test the reversibility of the CD38i, A549 cells were treated with vehicle or 0.5 μM 78c for 16 hours. Then, cells were washed and incubated with (78c) or without (78c+release) 78c for another 8 hours. Control cells were left in vehicle for the whole treatment period. After treatment, cell lysates were prepared for measurements of CD38 activity. For the co-culture experiment, AML12 cells were plated in the lower chamber and HEK293T or Jurkat T cells were plated in the upper chamber. Transfections with CD38 plasmid were done as described above. 0.5–1 μM 78c was added to the upper chamber 4 hours before addition of 100 μM NMN. 4 hours later, both chambers were incubated together for an additional 20 hours. Then AML12 cells were collected for NAD+ measurements. In experiments where AML12 cells were incubated with recombinant CD38 in the cell culture media, the first step involved incubation of the recombinant protein (100ng/mL) with 1 μM 78c for 30 minutes at 37 °C in cell culture media containing 1% FBS. After 30 minutes, the recombinant protein-78c mixture was added to the cells and 100μM NMN was added. After 18 hours, AML12 cells were collected for NAD+ measurements or Western blot.
Isolation of Murine Embryonic Fibroblasts (MEFs) and Bone Marrow-Derived Macrophages
At day E13.5, C57BL/6 and CD38 KO embryos were harvested under sterile conditions according to a standard protocol. Briefly, embryonic internal organs (neural and hematopoietic tissue) were removed from each embryo by dissection. The residual tissue was lightly minced and then trypsinized at 37 °C for 15 min. MEF culture medium (see above) was added to cells. After confluence was reached, cells were frozen into cryovials. Mouse bone marrow-derived macrophages were isolated as described before (Matalonga et al., 2017). Male mice were used for these experiments.
Mitochondrial Isolation
HEK293T were transfected with vector, WT CD38, or catalytically inactive CD38 plasmid for 24 hours, followed by treatment with 0.5 μM 78c for 20 hours. Mitochondria were isolated using the Thermo Scientific Mitochondria Isolation Kit for Cultured Cells (#89874). Final pellets were lysed in NETN and immunoblotted with the indicated antibodies as described below in Western blot analysis.
Western Blot Analysis
Tissue extracts or cells were homogenized and lysed in NETN buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) supplemented with 50 mM β-glycerophosphate, 5 mM NaF, a protease inhibitor cocktail (Roche), 5 mM nicotinamide, and 5 μM trichostatin A (TSA, Cell Signaling). For PARylation analysis only, 100 μM of tannic acid was added to the lysis buffer. After 30 min of incubation at 4°C, the samples were centrifuged at 12,000 rpm for 10 min at 4°C. The protein concentration of the supernatant was determined by BioRad protein assay. The lysates were separated by SDS-PAGE, and electrophoretically transferred to PVDF membranes (Immobilon®-P; Millipore). Membranes were immunoblotted with the indicated antibodies as shown in key resources table. Enhanced chemiluminescence detection was performed using SuperSignal West Pico or Femto Chemiluminescence Substrate (Thermo Scientific). Films were scanned and densitometry was performed using ImageJ.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| acetyl-lysine | Cell Signaling | Ab9441; RRID:AB_331805 |
| SirT1 (mouse) | Cell Signaling | Ab2028; RRID:AB_1196631 |
| SirT1 (human) | Cell Signaling | Ab2493; RRID:AB_2188359 |
| AMPK | Cell Signaling | Ab2532; RRID:AB_330331 |
| phospho-AMPK | Cell Signaling | Ab2535; RRID:AB_331250 |
| phospho-ACC | Cell Signaling | Ab3661; RRID:AB_330337 |
| p70 S6K | Cell Signaling | Ab2708; RRID:AB_390722 |
| phospho-p70 S6K | Cell Signaling | Ab9234; RRID:AB_2269803 |
| ERK | Cell Signaling | Ab4695; RRID:AB_390779 |
| phospho-ERK | Cell Signaling | Ab4370; RRID:AB_2315112 |
| γ-H2A.X | Cell Signaling | Ab9718; RRID:AB_2118009 |
| ACC | Abcam | Ab45172; RRID:AB_867475 |
| CD38 (human) | Abcam | Ab108403; RRID:AB_10890803 |
| acetyl-p65 | Abcam | Ab19870; RRID:AB_776753 |
| β-tubulin | Abcam | Ab15568; RRID:AB_2210952 |
| GAPDH | Ambion | AM 4300 |
| HA | Sigma | AbH3663; RRID:AB_262051 |
| Flag | Sigma | AbF1804; RRID:AB_262044 |
| CD38 (mouse) | Santa Cruz | Absc7049; RRID:AB_2275569 |
| AlexaFluor 647 rat anti- mouse CD38 | BD Pharmingen | Cat#562769 |
| FITC rat anti-mouse CD45 | BD Pharmingen | Cat#553080 |
| AlexaFluor 647 rat IgG2aκ isotype control | BD Pharmingen | Cat#557690 |
| FITC rat IgG2bκ isotypecontrol | BD Pharmingen | Cat#553988 |
| BA-D5 (Myosin Heavy | Developmental | AbBA-D5; RRID:AB_2235587 |
| Chain Type I Bovine) | Studies Hybridoma Bank, DSHB) | |
| BF-F3 (Myosin Heavy Chain Type IIB bovine) | DSHB | AbBF-F3; RRID:AB_2266724 |
| SC-71 (Myosin Heavy Chain Type IIA Bovine) | DSHB | AbSC-71; RRID:AB_2147165 |
| Pax7 | DSHB | AbPax7; RRID:AB_528428 |
| Laminin | Sigma Aldrich | Cat#L9393 |
| Alexa fluorescent conjugate 488 | Thermo Fisher | Cat#A-21202 |
| Alexa fluorescent conjugate 555 | Thermo Fisher | Cat#A-31570 |
| Alexa fluorescent conjugate 647 | Thermo Fisher | Cat#A-31571 |
| Cy-3-labelled telomere- specific (CCCTAA) peptide nucleic acid probe | Panagene | Cat#F1002-5 |
| CD45 VioGreen, mouse (clone: 30F11) | Miltenyi Biotec | 130-102-776 |
| Anti-succinyllysine | PTM Bio | PTM-401 |
| Anti-malonyllysine | PTM Bio | PTM-901 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Humulin R U-100 | Eli Lilly and Company | Cat#0002-8215 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | Cat#11668019 |
| 78c CD38 inhibitor | GSK | N/A |
| Olaparib | LC Laboratories | Cat#O-9201 |
| EX-527 | Tocris | Cat# 2780 |
| Nicotinamide | Sigma Aldrich | Cat# 10742 |
| cOmplete™ Protease Inhibitor Cocktail | Sigma Aldrich | Cat#CO-RO ROCHE |
| Nicotinamide 1,N6- ethenoadenine dinucleotide | Sigma Aldrich | Cat#41628 |
| β-Nicotinamide adenine dinucleotide hydrate | Sigma Aldrich | Cat#N0632 |
| Nicotinamide guanine dinucleotide sodium salt | Sigma Aldrich | Cat#N5131 |
| ADP-ribosylcyclase | Sigma Aldrich | Cat#A9106 |
| Recombinant human CD38 | R & D Systems | Cat#2404-AC-010 |
| β-Nicotinamide mononucleotide (NMN) | Sigma Aldrich | Cat#N3501 |
| NIAGEN®-nicotinamide riboside (NR) | Chromadex | N/A |
| FK866 | Sigma Aldrich | Cat#F8557 |
| Recombinant Human CD157 Protein | R & D Systems | Cat#Q10588 |
| Critical Commercial Assays | ||
| Ultra-Sensitive Mouse Insulin ELISA kit | Crystal Chem | Cat#90080 |
| SIRT1 activity assay kit | Enzo Lifesciences | Cat#BML-AK555-0001 |
| PARP1 activity assay kit | EMD Millipore | Cat#17-10149 |
| CycLex® NAMPT Colorimetric Assay Kit | MBL International | Cat#CY-1251 |
| RNeasy Plus Mini kit | Qiagen | Cat#74134 |
| QuantiTect Reverse Transcription kit | Qiagen | Cat#205310 |
| ATPlite Luminescence Assay | Perkin Elmer | Cat# 6016943 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat#69504 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | CRL-11268 |
| A549 | ATCC | CCL-185 |
| AML12 | ATCC | CRL-2254 |
| C57BL/6 CD38 KO MEFs | This paper | N/A |
| C57BL/6 MEFs | This paper | N/A |
| C57BL/6 bone marrow- derived macrophages | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: aged C57BL/6 | Aged Rodent Colony from National Institute on Aging | N/A |
| Mouse: C57BL/6 | Dr.Eduardo N. Chini, Mayo Clinic | N/A |
| Mouse: C57BL/6 CD38 catalytically-inactive (CI) | TransViragen | N/A |
| Mouse: C57BL/6 CD38KO | Dr.Eduardo N. Chini, Mayo Clinic (Partida- Sánchez et al., 2001) | N/A |
| Mouse: P44+/+ | Dr.Eduardo N. Chini, Mayo Clinic (Maier et al., 2004) | N/A |
| Mouse: Bub1bH/H | Dr. Jan van Deursen and Daren Baker, Mayo Clinic (Baker et al., 2004) | N/A |
| Oligonucleotides | ||
| Ccl2 | Applied Biosystems | Mm00441242_g2 |
| Cd38 | Applied Biosystems | Mm01220906_m1 |
| Col1a1 | Applied Biosystems | Mm00801666_g1 |
| Cox1 | Applied Biosystems | Mm04225243_g1 |
| Fgf21 | Applied Biosystems | Mm00840165_g1 |
| G6pc | Applied Biosystems | Mm00839363_m1 |
| Gck | Applied Biosystems | Mm00439129_m1 |
| Glut1 | Applied Biosystems | Mm00441480_m1 |
| Glut4 | Applied Biosystems | Mm00441480_m1 |
| Hk1 | Applied Biosystems | Mm00439344_m1 |
| Hk2 | Applied Biosystems | Mm00443385_m1 |
| Idh2 | Applied Biosystems | Mm00612429_m1 |
| Igf1 | Applied Biosystems | Mm00439560_m1 |
| Igf1r | Applied Biosystems | Mm00802831_m1 |
| Igfbp1 | Applied Biosystems | Mm00515154_m1 |
| Il6 | Applied Biosystems | Mm00446190_m1 |
| Ldha | Applied Biosystems | Mm01612132_g1 |
| Mdh2 | Applied Biosystems | Mm00725890_s1 |
| Mmp2 | Applied Biosystems | Mm00439498_m1 |
| Nadk | Applied Biosystems | Mm00446804_m1 |
| Nampt | Applied Biosystems | Mm00451938_m1 |
| Naprt1 | Applied Biosystems | Mm01205844_g1 |
| Nd4 | Applied Biosystems | Mm04225294_s1 |
| Ndufb5 | Applied Biosystems | Mm00452592_m1 |
| Nrf1 | Applied Biosystems | Mm01135606_m1 |
| Nrf2 | Applied Biosystems | Mm00477784_m1 |
| Pai1 | Applied Biosystems | Mm00435860_m1 |
| Parp1 | Applied Biosystems | Mm01321084_m1 |
| Parp2 | Applied Biosystems | Mm00456462_m1 |
| Pdha1 | Applied Biosystems | Mm00468675_m1 |
| Pdk1 | Applied Biosystems | Mm00554300_m1 |
| Pkm | Applied Biosystems | Mm00834102_gH |
| Pgc1a | Applied Biosystems | Mm00447180_m1 |
| Ppara | Applied Biosystems | Mm00440939_m1 |
| Sdha | Applied Biosystems | Mm01352366_m1 |
| Sirt1 | Applied Biosystems | Mm00490758_m1 |
| Sirt3 | Applied Biosystems | Mm00452131_m1 |
| Sirt6 | Applied Biosystems | Mm01149042_m1 |
| Tgfb1 | Applied Biosystems | Mm01178820_m1 |
| Tnfa | Applied Biosystems | Mm00443258_m1 |
| Gapdh | Applied Biosystems | 4352932E |
| Hprt | Applied Biosystems | Mm01545399_m1 |
| Tbp | Applied Biosystems | Mm00446971_m1 |
| Recombinant DNA | ||
| Plasmid: Flag-vector | Addgene | Modification of pIRES2-EGFP Cat#6029-1 |
| Plasmid: Flag-p65 | Addgene | #20012 |
| Plasmid: Flag-CD38 | Camacho-Pereira et al., 2016 | Human CD38 clone into modified pIRES2-EGFP |
| Plasmid: HA-p53 | This paper | Cloned into a modified pCMV/myc/HA vector from pcDNA3 Flag p53 (Addgene) |
| Plasmid: Flag-CD38-CI | This paper | E226D mutation of Flag-CD38 construct |
| Software and Algorithms | ||
| GraphPad Prism 6 | GraphPad Software | https://www.graphpad.com/scientificsoftware/prism |
| Image J Softonic | Softonic | https://imagej.net/ |
Lactate and Creatinine Kinase Levels
Plasma lactate concentration was determined using the lactate colorimetric assay kit (Abcam). The OD was measured at 450 nm and the standard curve plot (nmol/well vs. OD 450 nm) was then generated. Finally, the lactate concentrations were determined as follows: C = La/Sv (nmol/μL or mM), where La is the lactic acid amount (nmol) and Sv is the sample volume (μL) in the well. For creatinine kinase measurements, serum was added to creatinine kinase reagent (StanBio) according to the manufacturer’s protocol. The resulting mixture was assayed for CK activity using an Epoch 2 microplate reader (BioTek Instruments).
NAD+ Levels
To determine intracellular NAD+ levels, 3 × 106 cells or approximately 20 mg of tissue was homogenized in 10% trichloroacetic acid (TCA). Samples were centrifuged at 12,000 rpm for 2 min at 4oC. The supernatants were collected, and the pellets were resuspended in 0.2 N NaOH for protein determination. TCA was removed with organic solvent (3 volumes 1,1,2-trichloro-1,2,2-trifluroethane: 1 volume trioctylamine) in a ratio of 2 volumes of organic solvent to 1 volume of sample. After phase separation, the top aqueous layer containing NAD+ was recovered and the pH was corrected by addition of 1M Tris pH 8.0. Detection of NAD+ was performed as described before using a cycling assay (Aksoy et al., 2006). We have previously demonstrated that the NAD+ cycling assay is as sensitive and specific as the UPLC-mass spectroscopy assay (Camacho-Pereira et al., 2016).
NR, NAD+, and NMN Measurements by HPLC-mass spectroscopy
Skeletal muscle from aged C57BL/6 mice treated with vehicle (control), 78c, NR, or 78c+NR was extracted with organic solvent and neutralized with 1M Tris pH 8.0. For measurements, the HPLC was at a flow rate of 0.25 mL/min with 99% buffer A from 0–3 min, a linear gradient to 99% buffer A/1% buffer B (100% methanol) from 3–20 min, 80% buffer A/20% buffer B from 20–21 min, a linear gradient to 30% buffer A/70% buffer B from 21–28 min at 0.35 mL/min, 99% buffer A/1% buffer B from 28–31 min, and a linear gradient to 99% buffer A from 31–37 min at 0.25 mL/min. The protocol was adjusted based on previous studies (Yoshino and Imai, 2013). Concentrations were quantitated based on the peak area compared to a standard curve and normalized to protein content in the tissue sample.
Enzymatic Activities
CD38 hydrolase activity with nicotinamide 1,N6-ethenoadenine dinucleotide (ε-NAD+) as substrate, and CD38 cyclase activity with nicotinamide guanine dinucleotide (NGD), a β-NAD analog that converts to the fluorescent product cyclic GDP ribose (cGDPR), were performed as described by (Aksoy et al., 2006; Graeff et al., 1994). ADP-ribosyl cyclase activity of purified cyclase from Aplysia ovotestis extracts was measured using 0.1 μg/mL of Aplysia cyclase and NGD as substrate. Unless otherwise indicated in the figures, ε-NAD+ concentration was 50 μM and NGD concentration was 200 μM. When performed with human recombinant enzymes, CD38 was purchased from R & D Systems. When performed in tissues, samples were prepared as described above for Western blot and assayed in the absence or presence of 78c at concentrations of 1, 10, 25, and 50 nM. Nicotinamide measurements were performed with a coupled enzyme assay as described by (Smith et al, 2009). When NMN was used as a substrate, the concentration was 1 mM. When NR was used as a substrate, the concentration was 0.5 mM.
For testing reversibility of the CD38 inhibition, human recombinant CD38 was pre-incubated for 30 min in different conditions: control media, in the presence of 100 μM ε-NAD+, 25 nM 78c, and both ε-NAD+ and 78c. Afterward, the enzyme was diluted 100-fold with control media and the activity assayed in 50 μM ε-NAD+ in the absence and presence of 78c.
Hydrolysis of NR by CD157 was measured by monitoring the decrease in absorbance at 262 nm, as described by Preugschat et al., 2014, using 30 μM NR or NAD+ as substrate. Commercially available kits were used for measurement of SIRT1 activity (Enzo Lifesciences), PARP1 activity (EMD Millipore), and NAMPT activity (MBL International).
Pharmacokinetic Modeling and Fitting of 78c
Steady-state equations of the reaction cycle scheme for CD38 were derived from the kinetic scheme presented in Fig. 1c by using the King-Altman algorithm as described in (Segel et al., 1975). Because CD38 has minimal cyclase activity with ε-NAD+ as a substrate, and 78c has very little effect on such activity, for the sake of simplicity only the cycle on the left of the scheme (hydrolytic activity of CD38) was used to derive the equations. The kinetic scheme accounts for the uncompetitive inhibition of CD38 by 78c, the kinetic properties of CD38, and the effect of either product, nicotinamide (NAM) or ADP-ribose (ADPr), on the kinetics of inhibition. For ε-NAD+-78c interaction:
Where
And
For the NAM-78c interaction (at [ADPr] = 0):
Where Vmax app = k1k2k3[eNAD]/(k2k3 + k–1k3 + k1k3 [eNAD] + k–1k2 [NAM] + k1k–2 [eNAD] [NAM]
And ki app = ki * (1+(k−1+k2)/k1 [eNAD] + (k–2 [NAM]/k3)*(1+k–1/k1[eNAD]))
For the interaction ADPr-78c (at [NAM] = 0):
Where
And
Quantification of mRNA and Mitochondrial DNA
RNA was isolated from mouse liver and skeletal muscle using the RNeasy Plus Mini kit (Qiagen). cDNA was synthesized using QuantiTect Reverse Transcription kit (Qiagen). Quantitative real-time PCR was performed using commercially available TaqMan gene expression probes (Applied Biosystems, see table below), according to the manufacturer’s instructions, on a BioRad CFX384 thermal cycler. The relative mRNA abundance of target genes was calculated by the 2(−ddCq) method (Livak and Schmittgen, 2001). The expression changes were calculated relative to control.
Total DNA was extracted from 10–20 mg of gastrocnemius muscle and liver using the DNeasy Blood & Tissue Kit (Qiagen). PCR was performed as described above using Taqman probes for Cox1 (mitochondrial complex IV), Nd4 (mitochondrial complex I), and Gapdh. Mitochondrial copy number was calculated as the ratio of Cox1 or Nd4 to Gapdh.
TaqMan Gene Expression Assays
| Gene Symbol | Probe ID |
|---|---|
| Ccl2 | Mm00441242_g2 |
| Cd38 | Mm01220906_m1 |
| Col1a1 | Mm00801666_g1 |
| Cox1 | Mm04225243_g1 |
| Fgf21 | Mm00840165_g1 |
| G6pc | Mm00839363_m1 |
| Gck | Mm00439129_m1 |
| Glut1 | Mm00441480_m1 |
| Glut4 | Mm00441480_m1 |
| Hk1 | Mm00439344_m1 |
| Hk2 | Mm00443385_m1 |
| Idh2 | Mm00612429_m1 |
| Igf1 | Mm00439560_m1 |
| Igf1r | Mm00802831_m1 |
| Igfbp1 | Mm00515154_m1 |
| Il6 | Mm00446190_m1 |
| Ldha | Mm01612132_g1 |
| Mdh2 | Mm00725890_s1 |
| Mmp2 | Mm00439498_m1 |
| Nadk | Mm00446804_m1 |
| Nampt | Mm00451938_m1 |
| Naprt1 | Mm01205844_g1 |
| Nd4 | Mm04225294_s1 |
| Ndufb5 | Mm00452592_m1 |
| Nrf1 | Mm01135606_m1 |
| Nrf2 | Mm00477784_m1 |
| Pai1 | Mm00435860_m1 |
| Parp1 | Mm01321084_m1 |
| Parp2 | Mm00456462_m1 |
| Pdha1 | Mm00468675_m1 |
| Pdk1 | Mm00554300_m1 |
| Pkm | Mm00834102_gH |
| Pgc1a | Mm00447180_m1 |
| Ppara | Mm00440939_m1 |
| Sdha | Mm01352366_m1 |
| Sirt1 | Mm00490758_m1 |
| Sirt3 | Mm00452131_m1 |
| Sirt6 | Mm01149042_m1 |
| Tgfb1 | Mm01178820_m1 |
| Tnfa | Mm00443258_m1 |
| Gapdh | 4352932E |
| Hprt | Mm01545399_m1 |
| Tbp | Mm00446971_m1 |
Liver and Skeletal Muscle Immunohistochemistry
Liver
OCT-embedded liver tissue was cryosectioned at 10 μm thickness, fixed in MeOH:acetone (1:1) for 5 min at −20°C. Sections were blocked with 10% normal donkey serum in PBS-T for 1 hour at RT, and then incubated overnight at 4°C with fluorescently tagged primary antibodies in 1% donkey serum. The following antibodies from BD Pharmingen were used: AlexaFluor 647 rat anti-mouse CD38 (562769), FITC rat anti-mouse CD45 (553080), AlexaFluor 647 rat IgG2aκ isotype control (557690), and FITC rat IgG2bκ isotype control (553988). Nuclei were stained with Hoechst 33342. Images were obtained with a LSM 780 confocal microscope, and image capture was performed with ZEN 2.1 black software (Zeiss) using standardized exposure settings.
Muscle
Frozen gastrocnemius muscle and tibialis anterior (TA) muscle tissue sections (flash frozen; 8 μm thickness) were post-fixed in 4% paraformaldehyde for 10 min at room temperature (RT) prior to immunostaining. Tissue sections were permeabilized with 0.2% Triton-X100 in PBS followed by blocking with 3% BSA in PBS. Primary antibody incubations occurred at RT for 90 min followed by incubation with secondary antibody at RT for 30 min in 3% BSA in PBS. The following antibodies were used in these experiments: BA-D5 (Developmental Studies Hybridoma Bank, DSHB), BF-F3 (DSHB), SC-71 (DSHB), Pax7 (DSHB), Laminin (Sigma 4HB-2). Secondary antibodies were all Alexa fluorescent conjugates (488, 555, or 647) from Invitrogen. Images were acquired on a Nikon Ts2 inverted fluorescence microscope or a Zeiss LSM 700 confocal microscope and processed using Nikon analysis software or ImageJ/Fiji.
Skeletal Muscle and Heart Flow Cytometry Analysis
Hindlimb muscles and heart from 5 month old FVB mice were digested using 0.2% w/v collagenase II (Gibco) at 37°C for 75 min. The cells were prepared from the digested tissues by filtering through 100 μm, 70 μm, and 40 μm cell strainer consecutively, followed by washing with PBS. The cells were incubated in staining buffer (PBS, 0.5% w/v BSA, and 2mM EDTA) with FcR blocking reagent (Miltenyi Biotec, 1:10) at 4°C for 10 min. The cells were then incubated with anti-CD38-Alexa Fluor 647 (BD Biosciences, 562769, 1:50), anti-CD31-FITC (Miltenyi Biotec, 130-102-519, 1:10), anti-CD45-VioGreen (Miltenyi Biotec, 130-110-803, 1:25) and Anti-Sca1-PE-Cy7 antibodies in 4°C for 10 min. Samples were mixed with propidium iodide solution (Miltenyi Biotec, 1:100) for dead cell staining. Flow cytometry was performed using MACSQuant analyzer 10. Data were analyzed with Flowjo v10 software. Unstained sample was used for gating.
Evans Blue Dye Injection and Histologic Evaluation
Mice were given intraperitoneal injections of 1% Evans blue dye (EBD) at 1% of body mass (0.25mL/25g) in PBS, pH 7.5 sterilized by passage through a Millex®-GP 0.22 μm filter (Millipore, Bedford, MA, USA). Mice were injected with EBD one hour post-exercise. At day 4, mice were euthanized and tissues were harvested. Tibialis anterior (TA) and gastrocnemius (GA) muscles of the hind limbs were removed, sliced transversely at the maximum diameter, embedded in Tissue-Tek OCT mounting media (Miles Laboratories), snap frozen in liquid nitrogen, and stored at −80°C. Frozen sections (10 μm) were cut at −25°C on a Leica (CM1850UV) cryostat, dipped in cold acetone (−20°C) for 1 min and then air-dried at RT. The sections were then dipped into xylene and mounted with Histomount (National Diagnostics) and a glass coverslip. All slides were stored in reduced light conditions to minimize photo bleaching. Images of Evans blue/Hoechst 33342 sections were obtained with a LSM 780 confocal microscope (Carl Zeiss Microscopy, LLC) and a Plan-Apochromat 20x/0.8 M27 objective using excitation wavelengths of 561/405 nm and emissions of 573–700/415-470 nm, respectively. Image capture was performed with ZEN 2.1 black software (Zeiss) using standardized exposure settings. Per animal, 10 images of different areas were used to quantify the percentage of area where extravasation of EBD was detected. Using ImageJ software (NIH), each image was split to its RBG stacks format. All images had their thresholds adjusted to the same parameters.
Telomere Immuno-FISH
Immuno-FISH was performed as described in (Hewitt et al., 2012). Briefly, following immunostaining against γ-H2A.X, tissue sections were washed in PBS and fixed in 4% formaldehyde aqueous buffered solution for 20 min. Tissue sections were then dehydrated with 70%, 90%, 100% ethanol solutions. Samples were denatured for 10 min at 80°C in hybridization buffer [70% dei onized formamide, 25 mM MgCl2, 1 M Tris pH 7.2, 5% blocking reagent (Roche, Welwyn, UK)] containing 4 ng/μL Cy- 3-labelled telomere-specific (CCCTAA) peptide nucleic acid probe (Panagene, F1002-5), followed by hybridization for 2 hours at RT. The slides were washed for 10 min with 70% formamide in 2xSSC, followed by a wash in 2xSSC, and a final wash in PBS for 10 min. Nuclei were visualized with DAPI and sections were mounted and imaged in a Leica DM5500B fluorescence microscope. In depth Z stacking was used (a minimum of 40 optical slices with 100× objective) followed by Huygens (SVI) deconvolution. Telomere-associated Foci (TAF) - showing co-localization between γ-H2A.X and telomere probe were quantified manually. Approximately 50 to 100 hepatocytes and 40–50 muscle fibers were quantified per animal.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were expressed as the mean± SEM. Statistical analyses were performed using unpaired, two-tailed Student's t test for two populations’ means at a single time point. For repeated measurements (ipGTT, ipIST), two-way ANOVA tests for repeated measurement followed by Bonferroni post-tests were performed. Survival was analyzed according to the Kaplan-Meier method, and differences in their distribution were evaluated by the log-rank (Mantel-Cox) test and the Gehan-Breslow-Wilcoxon test. Myofiber Feret diameter linear regression curves were compared using a sum-of-squares F test. P values<0.05 were regarded as significant (*P<0.05). Analyses were performed using GraphPad Prism 6.
Supplementary Material
HIGHLIGHTS.
Highly potent and specific CD38 inhibitor, 78c, prevents age-related NAD+ decline
Treatment of old mice with 78c improved physiological and metabolic parameters.
Inhibition of CD38 promotes an increase in NAD+ and its precursors in tissue.
78c is a novel NAD+ -boosting therapy to prevent age-related NAD+ decline.
Acknowledgments
This work was supported in part by grants from the Ted Nash Long Life Foundation, the Glenn Foundation for Medical Research via the Paul F. Glenn Laboratories for the Biology of Aging at the Mayo Clinic, a grant from Calico Laboratories, the Mayo Foundation, National Institutes of Health (NIH) grants from the National Institute of Aging (NIA, grant AG-26094), the Pancreatic Cancer SPORE project from NIH/NCI to E.N.C (grant: CA102701-08), the Mayo Clinic Center for Cell Signaling in Gastroenterology (NIDDK P30DK084567), and National Institute of Arthritis, Musculoskeletal, and Skin Diseases (NIAMS, grant AR-66696). JFP and DJ are supported by BBSRC grant BB/K017314/1, MRC Arthritis UK funded center CIMA, and NUIA. We would like to thank GSK and Calico for providing the CD38i 78c.
Footnotes
AUTHOR CONTRIBUTIONS
E.N.C. and D.B. generated the hypothesis of the manuscript. E.N.C. generated the concept of the manuscript. E.N.C., M.G.T. and C.C.S.C. were involved in design and conduction of all experiments. M.G.T., C.C.S.C., K.S.K., G.M.W., and M.R. performed cell experiments and Western-blots. M.G.T., K.S.K., and G.M.W. performed PCRs. M.G.T., C.C.S.C., K.S.K., A.C., and G.C.O. performed enzymatic studies and nucleotide measurement experiments. M.G.T., K.S.K., G.C.O., and K.Z.H. performed animal studies. R.H. and J.D.M. performed echocardiographic measurements and analysis. Cardiac studies were performed by A.S., D.S.C., J.J.B., J.Z., A.S., D.S.C. and J.D.D. performed skeletal muscle immunohistochemistry experiments. D.J. and J.F.P. performed telomere immuno-FISH experiments. D.B. performed pharmacokinetic characterization of 78c and development of CI transgenic mouse. All authors contributed to writing the manuscript.
Declaration of interests: Dr. Chini holds a patent on the use of CD38 inhibitors for metabolic diseases.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun. 2006;345:1386–1392. doi: 10.1016/j.bbrc.2006.05.042. [DOI] [PubMed] [Google Scholar]
- Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P, Cantó C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012;16:290–295. doi: 10.1016/j.cmet.2012.06.016. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2014;36:744–49. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- Bakondi E, Catalgol B, Bak I, Jung T, Bozaykut P, Bayramicli M, Ozer NK, Grune T. Age-related loss of stress-induced nuclear proteasome activation is due to low PARP-1 activity. Free Radic Biol Med. 2011;50:86–92. doi: 10.1016/j.freeradbiomed.2010.10.700. [DOI] [PubMed] [Google Scholar]
- Barbosa MTP, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, Chini EN. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007;21:3629–3639. doi: 10.1096/fj.07-8290com. [DOI] [PubMed] [Google Scholar]
- Becherer JD, Boros EE, Carpenter TY, Cowan DJ, Deaton DN, Haffner CD, Jeune MR, Kaldor IW, Poole JC, Preugschat F, et al. Discovery of 4-Amino-8-quinoline Carboxamides as Novel, Submicromolar Inhibitors of NAD-Hydrolyzing Enzyme CD38. J Med Chem. 2015;58(17):7021–56. doi: 10.1021/acs.jmedchem.5b00992. [DOI] [PubMed] [Google Scholar]
- Beneke S, Scherr AL, Ponath V, Popp O, Bürkle A. Enzyme characteristics of recombinant poly(ADP-ribose) polymerases-1 of rat and human origin mirror the correlation between cellular poly(ADP-ribosyl)ation capacity and species-specific life span. Mech Ageing Dev. 2010;131:366–369. doi: 10.1016/j.mad.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Berglund ED, Li CY, Poffenberger G, Ayala JE, Fueger PT, Willis SE, Jewell MM, Powers AC, Wasserman DH. Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes. 2008;57(7):1790–9. doi: 10.2337/db07-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age-related changes in NAD+ metabolism oxidative stress and SIRT1 activity in wistar rats. PLOS ONE. 2011;6:e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Boslett J, Hemann C, Christofi FL, Zweier JL. Characterization of CD38 in the major cell types of the heart: endothelial cells highly express CD38 with activation by hypoxia-reoxygenation triggering NAD(P)H depletion. Am J Physiol Cell Physiol. 2018;314(3):C297–c309. doi: 10.1152/ajpcell.00139.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho-Pereira J, Tarragó MG, Chini CC, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, Chini EN. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016;23(6):1127–39. doi: 10.1016/j.cmet.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Auwerx J. Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacol Rev. 2012;64(1):166–87. doi: 10.1124/pr.110.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini CC, Guerrico AM, Nin V, Camacho-Pereira J, Escande C, Barbosa MT, Chini EN. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res. 2014;20(1):120–130. doi: 10.1158/1078-0432.CCR-13-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini CC, Tarragó MG, Chini EN. NAD and the aging process: Role in life, death and everything in between. Mol Cell Endocrinol. 2016;455:62–74. doi: 10.1016/j.mce.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, O'Neil L, White TA, Sinclair DA, Chini EN. Flavonoid Apigenin Is an Inhibitor of the NAD+ase CD38: Implications for Cellular NAD+ Metabolism, Protein Acetylation, and Treatment of Metabolic Syndrome. Diabetes. 2013;62(4):1084–93. doi: 10.2337/db12-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick DW, Loro E, Liu L, Davila A, Jr, Chellappa K, Silverman IM, Quinn WJ, 3rd, Gosai SJ, Tichy ED, Davis JG, et al. Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab. 2016;24(2):269–82. doi: 10.1016/j.cmet.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155(7):1624–38. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeff RM, Mehta K, Lee HC. GDP-ribosyl cyclase activity as a measure of CD38 induction by retinoic acid in HL-60 cells. Biochem Biophys Res Commun. 1994;205:722–727. doi: 10.1006/bbrc.1994.2725. [DOI] [PubMed] [Google Scholar]
- Grozio A, Sociali G, Sturla L, Caffa I, Soncini D, Salis A, Raffaelli N, De Flora A, Nencioni A, Bruzzone S. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J Biol Chem. 2013;288(36):25938–49. doi: 10.1074/jbc.M113.470435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. The resurgence of NAD+ Science. 2016;352(6292):1396–7. doi: 10.1126/science.aag1718. [DOI] [PubMed] [Google Scholar]
- Haffner CD, Becherer JD, Boros EE, Cadilla R, Carpenter T, Cowan D, Deaton DN, Guo Y, Harrington W, Henke BR, et al. Discovery, Synthesis, and Biological Evaluation of Thiazoloquin(az)olin(on)es as Potent CD38 Inhibitors. J Med Chem. 2015;58(8):3548–71. doi: 10.1021/jm502009h. [DOI] [PubMed] [Google Scholar]
- Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. 2012;3:708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE, Weiss RM. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2854–2862. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- Huffman DM, Justice JN, Stout MB, Kirkland JL, Barzilai N, Austad SN. Evaluating Health Span in Preclinical Models of Aging and Disease: Guidelines, Challenges, and Opportunities for Geroscience. J Gerontol A Biol Sci Med Sci. 2016;71(11):1395–1406. doi: 10.1093/gerona/glw106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumiya Y, Hopkins T, Morris C, Sato K, Zeng L, Viereck J, Hamilton JA, Ouchi N, LeBrasseur NK, Walsh K. Fast/Glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab. 2008;7(2):159–72. doi: 10.1016/j.cmet.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice J, Miller JD, Newman JC, Hashmi SK, Halter J, Austad SN, Barzilai N, Kirkland JL. Frameworks for Proof-of-Concept Clinical Trials of Interventions That Target Fundamental Aging Processes. J Gerontol A Biol Sci Med Sci. 2016;71(11):1415–1423. doi: 10.1093/gerona/glw126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, Saretzki G, Fox C, Lawless C, Anderson R, et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat Commun. 2014;2:4172. doi: 10.1038/ncomms5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM, Brown TA. Myostatin inhibition enhances the effects of exercise on performance and metabolic outcomes in aged mice. J Gerontol A Biol Sci Med Sci. 2009;64:940–948. doi: 10.1093/gerona/glp068. [DOI] [PubMed] [Google Scholar]
- Li J, Bonkowski MS, Moniot S, Zhang D, Hubbard BP, Ling AJY, Rajman LA, Qin B, Lou Z, et al. A conserved NAD+ binding pocket that regulates protein-protein interactions during aging. Science. 2017;355:1312–1317. doi: 10.1126/science.aad8242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2-ΔΔCt method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- López-Otín C, Galluzzi L, Freije JM, Madeo F, Kroemer G. Metabolic Control of Longevity. Cell. 2016;166(4):802–21. doi: 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M, Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18(3):306–19. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88(3):841–86. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- March S, Graupera M, Rosa Sarrias M, Lozano F, Pizcueta P, Bosch J, Engel P. Identification and functional characterization of the hepatic stellate cell CD38 cell surface molecule. Am J Pathol. 2007;170(1):176–87. doi: 10.2353/ajpath.2007.051212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLOS ONE. 2012;7:e42357. doi: 10.1371/journal.pone.0042357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matalonga J, Glaria E, Bresque M, Escande C, Carbo JM, Kiefer K, Vicente R, Leon TE, Beceiro S, Pascual-Garcia, et al. The Nuclear Receptor LXR Limits Bacterial Infection of Host Macrophages through a Mechanism that Impacts Cellular NAD Metabolism. Cell Rep. 2017;18(5):1241–1255. doi: 10.1016/j.celrep.2017.01.007. [DOI] [PubMed] [Google Scholar]
- Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, Apte RS, Uchida K, et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016;24(6):795–806. doi: 10.1016/j.cmet.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. 2013;154(2):430–41. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahimana A, Attinger A, Aubry D, Greaney P, Ireson C, Thougaard AV, Tiornelund J, Dawson KW, Dupuis M, Duchosal MA. The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic malignancies. Blood. 2009;113:3276–3286. doi: 10.1182/blood-2008-08-173369. [DOI] [PubMed] [Google Scholar]
- Noren Hooten N, Fitzpatrick M, Kompaniez K, Jacob K, Moore B, Nagle J, Barnes J, Lohani A, Evans M. Coordination of DNA repair by NEIL1 and PARP-1: a possible link to aging. Aging (Albany NY) 2012;4:674–685. doi: 10.18632/aging.100492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partida-Sánchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, Kusser K, Goodrich S, Howard M, Harmsen A, et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med. 2001;7(11):1209–16. doi: 10.1038/nm1101-1209. [DOI] [PubMed] [Google Scholar]
- Pehar M, O'Riordan KJ, Burns-Cusato M, Andrzejewski ME, del Alcazar CG, Burger C, Scrable H, Puglielli L. Altered longevity-assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell. 2010;9(2):174–90. doi: 10.1111/j.1474-9726.2010.00547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preugschat F, Carter LH, Boros EE, Porter DJ, Stewart EL, Shewchuk LM. A pre-steady state and steady state kinetic analysis of the N-ribosyl hydrolase activity of hCD157. Arch Biochem Biophys. 2014;564:156–163. doi: 10.1016/j.abb.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Ramsey KM, Mills KF, Satoh A, Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell. 2008;7:78–88. doi: 10.1111/j.1474-9726.2007.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechsteiner M, Hillyard D, Olivera BM. Turnover of nicotinamide adenine dinucleotide in cultures of human cells. J Cell Physiol. 1978;88(2):207–17. doi: 10.1002/jcp.1040880210. [DOI] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, Dunn CA, Singh N, Veith S, Hasan-Olive MM, et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014;20(5):840–55. doi: 10.1016/j.cmet.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz MB, Sinclair DA. Why NAD(+) Declines during Aging: It's Destroyed. Cell Metab. 2016;23(6):965–6. doi: 10.1016/j.cmet.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segel I John Wiley and Son Inc. Steady-state kinetics of multireactant enzymes. Wiley-Interscience. Chapter 9: Enzyme kinetics, behavior and analysis of rapid equilibrium and steady-state enzyme systems 1975 [Google Scholar]
- Shallis RM, Terry CM, Lim SH. The multi-faceted potential of CD38 antibody targeting in multiple myeloma. Cancer Immunol Immunother. 2017;66(6):697–703. doi: 10.1007/s00262-017-1990-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BC, Hallows WC, Denu JM. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal Biochem. 2009;394(1):101–9. doi: 10.1016/j.ab.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, Imai S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014;33:1321–1340. doi: 10.1002/embj.201386917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350(6265):1208–13. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, Mirzaei H, Pieper AA, Ready JM, McKnight SL. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell. 2014;158(6):1324–1334. doi: 10.1016/j.cell.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation. 2006;114(19):2065–9. doi: 10.1161/CIRCULATIONAHA.106.634139. [DOI] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130(6):1095–107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Baur JA, Imai SI. NAD+ Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2017 doi: 10.1016/j.cmet.2017.11.002. S1550–4131(17)30670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Imai S. Accurate measurement of nicotinamide adenine dinucleotide (NAD+) with high-performance liquid chromatography. Methods Mol Biol. 2013;1077:203–215. doi: 10.1007/978-1-62703-637-5_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D'Amico D, Ropelle ER, Lutolf MP, et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352(6292):1436–43. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci. 2015;112:2876–2881. doi: 10.1073/pnas.1417921112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.