

Abstract
Fungi are phylogenetically diverse organisms found in nearly every environment as key contributors to the processes of nutrient cycling and decomposition. To date, most fungal diversity has been documented from terrestrial habitats leaving aquatic habitats underexplored. In particular, comparatively little is known about fungi inhabiting freshwater lakes, particularly the benthic zone, which may serve as an untapped resource for fungal biodiversity. Advances in technology allowing for direct sequencing of DNA from environmental samples provide a new opportunity to investigate freshwater benthic fungi. In this study, we employed both culture-dependent and culture-independent methods to evaluate the diversity of fungi in one of the largest freshwater systems on Earth, the North American Laurentian Great Lakes. This study presents the first comprehensive survey of fungi from sediment from Lake Michigan and Lake Superior, resulting in 465 fungal taxa with only 7% of sequence overlap between these two methods. Additionally, culture-independent analyses of the ITS1 and ITS2 regions revealed 49% and 72%, respectively, of the OTUs did not match a described fungal taxonomic group below kingdom Fungi. The low level of sequence overlap between methods and high percentage of fungal taxa that can only be classified at the kingdom level suggests an immense amount of fungal diversity remains to be studied in these aquatic fungal communities.
Keywords: Ascomycota, aquatic fungi, Basidiomycota, freshwater, Great Lakes, next-gen sequencing, systematics
Introduction
Fungi are diverse organisms found in nearly every global environment as decomposers, pathogens, and symbionts. An estimated 1.5 million species of fungi inhabit the earth (Hawksworth, 1991), of which only 5% have been described to date (Mueller and Schmit, 2007). Much of the described fungal diversity has been documented from terrestrial environments leaving aquatic habitats seldom explored. Fungi are essential to ecosystems as key drivers of nutrient recycling; so understanding fungal diversity within aquatic ecosystems is paramount for understanding ecosystem processes (Shearer et al., 2007).
An estimated 3,000-4,150 species of fungi have been described from aquatic habitats (Shearer et al., 2007; Jones et al., 2014), compared to 98,000 described from terrestrial environments (Kirk et al., 2008). Fungi are found in diverse aquatic habitats including marine, brackish and freshwater environments, seas, wetlands, streams, and lakes. Similar to their roles in terrestrial environments, fungi in aquatic systems act as pathogens and decomposers of aquatic animals, insects, macrophytes, and algae (Hyde et al., 1998; Wong et al., 1998). Fungi in aquatic habitats can range from long-term residents that are adapted to complete their lifecycle in water (e.g. Chytridiomycota, aquatic hyphomycetes, and some yeasts), to transient fungal species that reside as propagules in water transported from a terrestrial source.
Freshwater environments account for nearly 10% of all biological diversity (Strayer and Dudgeon, 2010), but in regards to fungal diversity, they remain grossly understudied. The dynamics of fungi in lotic systems has been the focus of freshwater fungal research leaving freshwater lakes nearly unexplored (Wurzbacher et al., 2010). Freshwater lakes have unique habitats: littoral, pelagic, and benthic, each harboring unique communities of fungi (Wurzbacher et al., 2016). The littoral is subjected to high organic matter input leading to a diversity of fungi associated with plants and decomposition, the pelagic zone is home to more specialized groups of fungi that can survive as saprobes or parasites of plankton and algae, while the benthic zone is hypothesized to serve mainly as a reservoir for fungal spores (Wurzbacher et al., 2010). However, research in marine systems suggests fungal communities exist and are active in the benthic region (Edgcomb et al., 2011; Orsi et al., 2013). Although little overlap has been reported between freshwater and marine fungal species (Shearer et al., 2007), these results suggest that the benthic zone of freshwater lakes is likely to contain active fungal communities and serve as a new resource for discovering unique fungi.
One such region that holds great promise for describing fungal biodiversity is the North American Laurentian Great Lakes system, which holds approximately 18% of global surface freshwater (Fuller et al., 1995). To date, only 18 fungal species have been reported from Lake Michigan (Paterson, 1967; Kiziewicz and Nalepa, 2008). Paterson (1967) used bait “traps” placed at 26-31 m depths to capture Chytridiomycota while Kiziewicz and Nalepa (2008) collected water samples between 10-80 m to isolate Ascomycota, Chytridiomycota and Zygomycota. The goal of this research was to survey the benthic fungal community in Lake Michigan and Lake Superior by using culture-dependent and culture-independent techniques. Employing these methods in tandem can provide a more detailed snapshot of fungal communities. This study offers the first systematic inventory of fungi from sediments of two Great Lakes based on the use of both traditional culture-dependent techniques and current culture-independent environmental sequencing.
Methods
Sample Collection
Sediment samples were collected during the summer months of 2014, 2015, and 2016 from Lake Michigan (Fig. 1) and from Lake Superior (Fig. 2) in 2015 and 2016. One hundred and forty-five samples were taken from depths between 30-272 m from Lake Michigan, while 20 samples were taken from depths between 68-263 m from Lake Superior using a Wildco Ponar® dredge. The depth of the sediment samples ranged between 7-15 cm, but only the top 1 cm of the sediment surface layer was collected for analysis. The dredge was thoroughly washed and sanitized for at least three minutes in a 5% bleach solution between each sample collection. All equipment was swabbed with sterile Copan™ swabs before sample collection. Samples and swabs were stored on ice for up to four days before being sent overnight to the Illinois Natural History Survey. Samples were immediately processed as described below and subsequently stored at 7 °C (culture-dependent study) or -80 °C (culture-independent study). Maps of GPS coordinates were created using ArcMap™ and ArcGIS® software by Esri©.
Fig. 1.
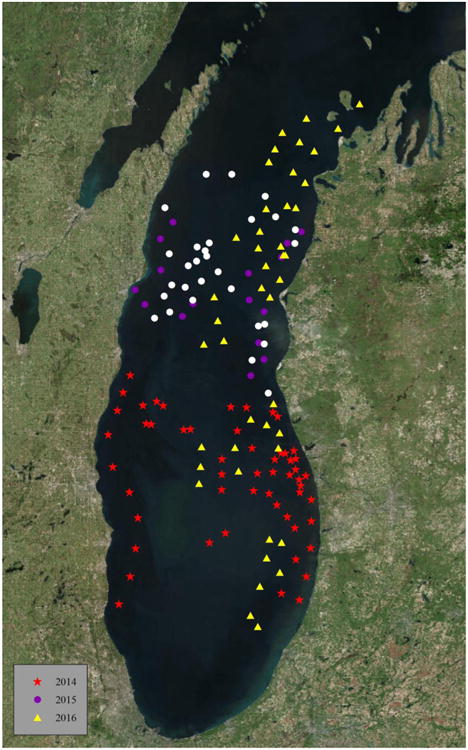
Location of sample sites from Lake Michigan. Sample locations were collected in 2014 (red stars), 2015 (purple dots) and 2016 (yellow triangles). All samples were analyzed using culture-dependent methods and a portion of samples from 2015 (white dots) was selected for culture-independent analysis.
Fig. 2.
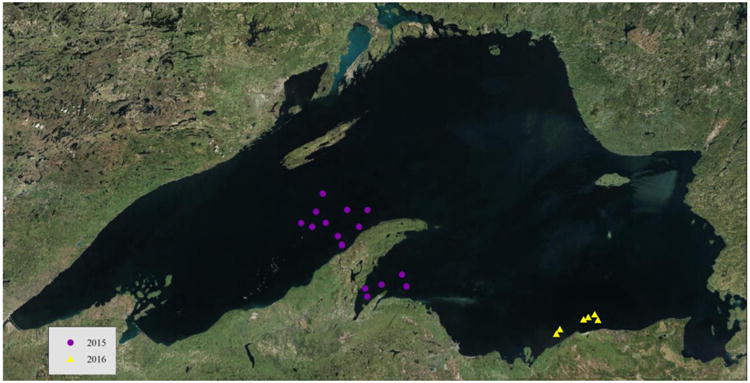
Location of sample sites from Lake Superior. Sample locations were collected in 2015 (purple dots) and 2016 (yellow triangles). All samples were analyzed using culture-dependent methods. All samples from 2015 were selected for culture-independent analysis.
Culture-dependent
Culturing
Approximately 1 g of sediment was used to generate serial dilutions (1/10, 1/100) using sterile distilled water. Next, 300 μL from each serial dilution was antiseptically spread onto the surface of a 90 mm Petri plate, wrapped with Parafilm®, and incubated at 14 °C to slow down the growth of fast-growing fungi. Sediment samples were plated on malt extract agar (Difco), tea agar (Mehrotra et al., 1982), and potato dextrose agar (Difco) supplemented with 97 % L-cysteine (Sigma) to encompass a variety of nutritional and redox levels. Negative control Copan™ swabs were streaked across malt extract agar. In addition, negative control plates were generated following the same lawn plating procedure described above, but using only sterile distilled water. Plates were monitored for up to one month for colony formation and fungal colonies which appeared morphologically different were transferred individually to separate 60 mm Petri plates containing potato dextrose agar until a pure culture was obtained. A sample of each isolate was prepared for DNA extraction, long-term storage, and subcultures. All media contained 50 mg/L chloramphenicol to prevent the growth of bacteria.
DNA extraction and sequencing
DNA was extracted by adding 200 μl of a 0.5 M NaOH solution to frozen mycelium tissue, which was ground for approximately 1 minute, incubated between 0-4 °C for approximately 20 minutes, and centrifuged at 16,800 rcf for 2 minutes. Five microliters of supernatant was added to 495 μL 100 mM Tris-HCL buffered with NaOH to pH 8.5-8.9 (Osmundson et al., 2013). PCR was performed using a Bio-Rad PTC 200 thermal cycler. A 25 μl reaction volume was used consisting of 12.5 μL GoTaq® Green Master Mix (Promega), 1 μl of each 10 μM primer ITS1F and ITS4 (Electronic Supplementary Material (ESM) Table S1), 3 μL Tris-HCL-DNA extraction solution and 7.5 μL nuclease free water. The following thermal cycle parameters were used: 94 °C for 2 minutes for initial denaturing, followed by 30 cycles of 94 °C for 30 seconds, 55 °C for 45 seconds, 72 °C for 1 minute with a final extension step at 72 °C for 10 minutes. Gel electrophoresis was carried out using a 1% TBE agarose gel with ethidium bromide to verify the presence of PCR product prior to purification. The resulting PCR product was purified using the Wizard® SV Gel and PCR Clean-up System (Promega). A BigDye® Terminator 3.1 cycle sequencing kit (Applied Biosystems Inc.) was used to sequence the entire ITS (internal transcribed spacer) region of nrDNA in one direction using the ITS5 (ESM Table S1) primer on an Applied Biosystems 3730XL high-throughput capillary sequencer.
Culture-independent
Sample Selection
A total of 48 sediment samples collected from Lake Michigan (Fig. 1) and Lake Superior (Fig. 2) in 2015 were selected for culture-independent environmental sequencing. Two negative controls were included for DNA extraction and sequencing.
DNA Extraction and Sequencing
Environmental DNA was extracted from approximately 0.5 g of sediment using MoBio PowerSoil DNA isolation kits following the Earth Microbiome protocol (Gilbert et al., 2010). The ITS1 and ITS2 regions of the nuclear ribosomal DNA were used to identify fungal taxa (Schoch et al., 2012). The high throughput Fluidigm Access Array (Brown et al., 2016) was used to amplify the ITS1 and ITS2 regions of the environmental samples using fungal specific primers (ESM Table S1). Fluidigm Access Array amplicons were sequenced using the Illumina Hi-Seq2500 platform using rapid 2 × 250nt paired-end reads. This platform provided an average of 33,955 paired reads per sample. All sequencing was performed at the W.M. Keck Center for Comparative and Functional Genomics at the University of Illinois.
Sequence processing
The forward reads of ITS1 and ITS2 were analyzed using the QIIME bioinformatics pipeline default settings (QIIME v 1.8 (Caporaso, 2010)). We analyzed only forward reads for three reasons: 1) forward reads were higher quality as compared to reverse reads, 2) reads with large insertions would be removed from further analysis due to lack of paired-end overlap, and 3) maintaining as much of the high quality sequence data as possible would provide a more complete fungal inventory as compared to a subset of the sequence data. Default quality filtering (split_libraries_fastq.py) was performed on forward reads to eliminate reads with poor base-pair calling and length. Sequences were discarded at this step if sequences had more than three consecutive poor base calls, if there were any uncertain base calls (N's), and/or >1.5 maximum errors occurred in the sample barcode. Seventy-five percent of the total read length must be high quality base calls and the phred quality threshold was set to q=19. Open reference OTU (operational taxonomic unit) clustering was performed (pick_open_reference_otus.py) using a four step iterative approach based on 97% sequence identity for ITS regions. Sequences were clustered via the uclust method using default settings and prefiltered against the UNITE fungal reference database (Koljalg et al., 2013). The resulting OTU biom table was filtered to remove singletons (filter_otus_from_otu_table.py, n = 3) and control sample sequences (filter_samples_from_otu_table.py). The filtered biom table was subsequently used to filter the sequence FASTA file (filter_fasta.py) and the resulting FASTA file was compared against the NCBI nucleotide database using NCBI nBLAST (Altschul et al., 1990). The resulting XML file was processed using the NCBI BLAST parser tool (Ream and Kiss, 2013). The resulting representative OTU taxonomies from the UNITE fungal reference database and the NCBI nucleotide database were compared for congruency. Code for sequence processing is available at https://github.com/ebach/Great_Lake_Fungi. Raw and processed data files are publically available from the University of Illinois Databank at https://doi.org/10.13012/B2IDB-9320144_V2 (Miller, 2018).
Fungal Inventory
Culture-dependent
Species identifications were determined using the entire ITS gene region of nrDNA from those sequences with ≤ 2% nucleotide ambiguities. Sequences were searched using the NCBI nBLAST database and Latin binomials were assigned using top query coverage and maximum sequence identity.
Culture-independent
Species identifications were determined using partial ITS sequences (ITS1 or ITS2) compared to the UNITE fungal reference database and NCBI nBLAST database. When a consensus was not found between UNITE and NCBI, the consensus majority from the top ten nBLAST hits was assigned. Latin binomials were assigned based on ≥85% query coverage and ≥97% sequence identity as recommended by Nguyen et al. (2015). OTUs were classified at higher taxonomic level based on ≥85% query coverage, but <97% sequence identity. All OTUs with less than 85% query coverage were run through the RDP classifier (Wang et al., 2007) to ensure all sequences classified to the kingdom Fungi. OTUs with assigned taxonomy were binned into order and class using Index Fungorum (www.indexfungorum.org).
Method Comparison
A local blast search using ViroBlast (Deng et al., 2007) was conducted to further understand the overlap in sequence identities between culture-dependent and culture-independent methods. A local sequence database was created using 2,167 full length ITS sequences obtained from Great Lake fungal cultures generated over the three-year sampling period. The ITS1 and ITS2 sequences from the 2015 culture-independent analysis were compared against this local database. Matches were determined using the following criteria: ≥85% query coverage and ≥97% sequence identity.
The vegan package 2.4-3 (Oksanen et al., 2017) in R statistical program (R Core team 2013) was used to analyze fungal diversity among primer sets used in culture-independent sequencing. The ITS1 and ITS2 OTU datasets were rarified at 5,000 reads per sample to account for sequencing depth across samples. Alpha diversity was measured using the Chao1 index to estimate species richness within each sample (Gotelli and Colwell, 2011). A two-tailed t-test was performed to compare alpha diversity among primer sets.
To determine sampling efforts in the culture-dependent analysis, species delimitation was resolved by aligning culture-dependent sequences using PASTA (Mirarab, 2014), which estimates sequence alignments and trees simultaneously using MAFTT and RAxML (Stamatakis, 2014). The resulting tree was run in the Bayesian Poisson Tree Processes (bPTP) model with 500,000 iterations for species delimitation (Zhang, 2013). Results from the bPTP species delimitation were used to generate species accumulation curves using the vegan package in R-statistical platform.
Sampling efforts for culture-independent analysis were determined using fungal OTUs generated from the QIIME bioinformatics pipeline rarified at 5,000 reads per sample. The rarefied data set was used to determine species accumulation and diversity analysis using the vegan package in R-statistical platform. All analytical code is available at https://github.com/ebach/Great_Lake_Fungi.
Results
Fungal Inventory
Culture-dependent
Culture-dependent examination of sediment samples resulted in 2,167 sequenced fungal isolates over the course of 2014-2016 sampling years. Culturing resulted in 398 unique fungal species representing 185 genera, 40 orders, 13 classes and 3 phyla (ESM Table S2). The most common classes include Dothideomycetes, Eurotiomycetes, Leotiomycetes, and Sordariomycetes, which make up approximately 84% of all isolates identified to genus (Fig. 3). The classes Agaricostilbomycetes (<1%) and Exobasidiomycetes (<1%) were only recovered through culturing and not identified in our culture-independent data. Cladosporium cladosporoides, Paecilomyces inflatus, Penicillium chrysogenum, and Trichoderma spirale were obtained from negative control Copan™ swabs and were subsequently removed from further analyses. No fungal growth was observed on the negative control plates.
Fig. 3.
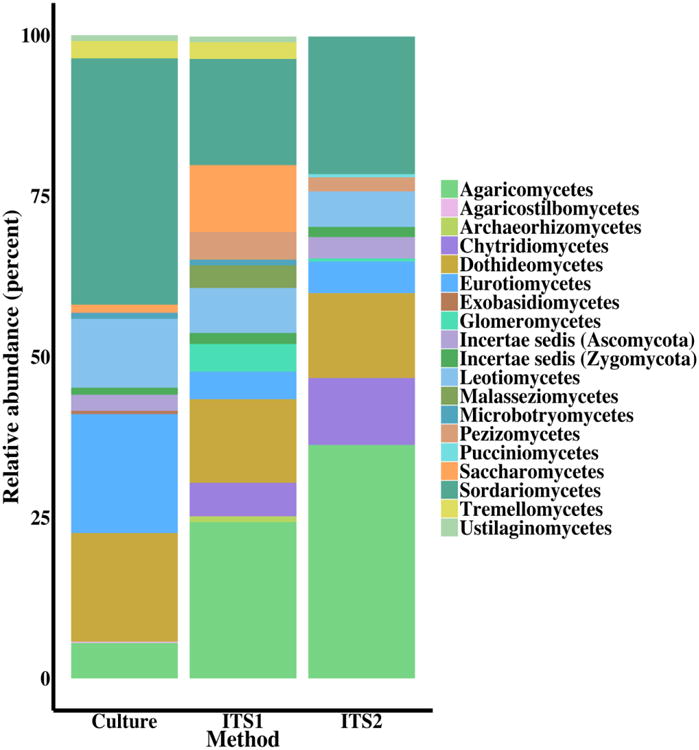
Summary of fungal classes by isolation method: culture-dependent and culture-independent by ITS region used for identification. Fungal isolates and OTUs unidentified at the class level were removed from this analysis including 11 (2.4%) from culture, 355 (75.5%) from ITS1, and 237 (56.6%) from ITS2.
Culture-independent: ITS1 Primer Set
ITS1 sequencing produced 1,483,466 raw sequence reads (95% retention of post-quality control reads), rarified to 236,110 sequences. Sequences were assigned to 479 OTUs (Table 1) of which 73 (15.2%) were assigned a Latin binomial (ESM Table S2). Fifty (10.4%) OTUs were classified at higher taxonomic levels, and 347 OTUs (72.4%) did not match a described fungal taxonomic group below kingdom Fungi so were classified as fungal species. Nine (1.9%) OTUs with high query coverage were removed because they classified to Animalia or Rhizaria. The remaining 470 fungal OTUs comprise 47 genera, 27 orders, 15 classes, and 5 phyla. The most common classes include Agaricomycetes, Dothideomycetes, Saccharomycetes, and Sordariomycetes, which make up approximately 63% of the total identified genera (Fig. 3). The ITS1 primer set exclusively recovered Archaeorhizomycetes (<1%) and Malasseziomycetes (3.5%) that were not identified using the ITS2 primer set in culture-independent analysis or in culture-dependent analysis. Community analysis of presence/absence of culture-independent ITS1 OTUs demonstrated distinct fungal communities in Lake Michigan and Lake Superior (PERMANOVA; P=0.0001; Fig. 4A).
Table 1.
Overview of culture-dependent and culture-independent sequencing efforts. Culture-independent analysis is summarized by ITS region. Chao1 index was used to estimate species richness and standard error across samples.
| Method | Sequences | Fungal Species (or OTU) estimates | Chao1 estimates | Chao1 Standard Error | Shannon's Diversity (OTUs) | Evenness |
|---|---|---|---|---|---|---|
| Culture-dependent | 2,167 | 398-445 | 1-78 | 0-40 | ||
|
| ||||||
| Culture-independent | ||||||
|
| ||||||
| ITS1 | 1,483,466 | 470 | 5-92 | 0-35 | 0.01-2.43 | 0.004-0.705 |
| ITS2 | 896,295 | 419 | 4-65 | 0-30 | 0.17-2.17 | 0.08-0.98 |
Fig. 4.
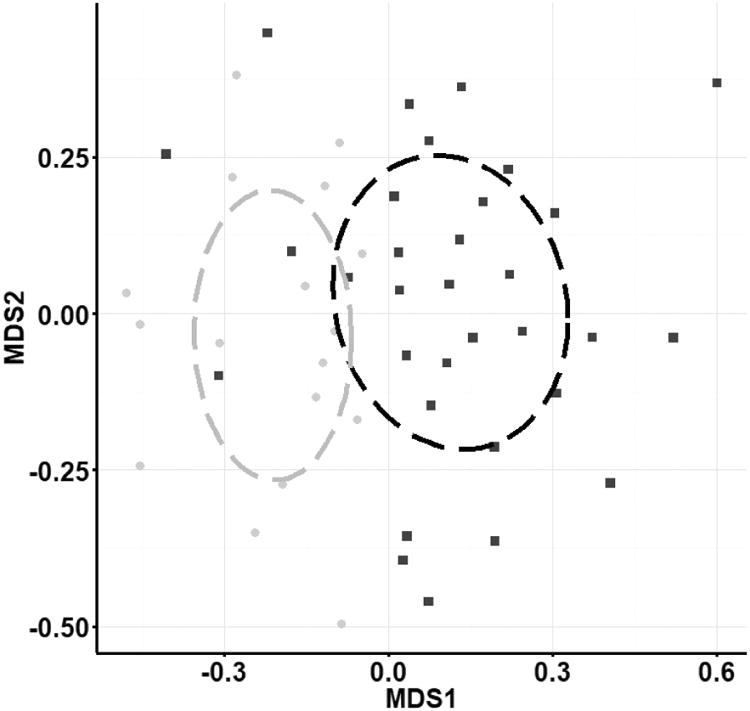
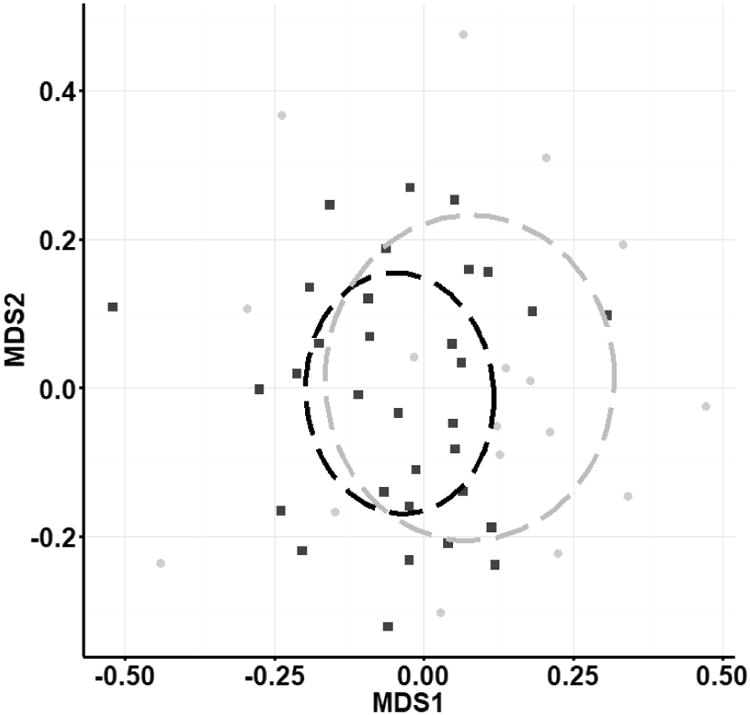
Non-metric multi-dimensional scaling ordination (Jaccard dissimilarity) of presence/absence of fungal OTUs sequenced from the a) ITS1 region and b) ITS2 region. Communities in Lake Michigan (black squares) and Lake Superior (gray squares) were different (PERMANOVA P<0.0006 for both). Stress scores were 0.29 (a) and 0.26 (b). Analysis of scaled abundance yielded similar results (Bray-Curtis similarity).
Culture-independent: ITS2 Primer Set
ITS2 sequencing produced 896,295 raw sequence reads (95% retention of post-quality control reads) and 468 OTUs (Table 1), and 127 (27.1%) of these were assigned a Latin binomial (ESM Table S2). Sixty-two (13.2%) OTUs were classified at a higher taxonomic level and 230 OTUs (49.1%) did not match a described fungal taxonomic group below kingdom Fungi. Forty-nine (10.5%) OTUs with high query coverage were removed because their classification was outside the kingdom Fungi. The remaining 419 fungal OTUs comprise 53 genera, 26 orders, 10 classes, and 5 phyla. The most common classes include Agaricomycetes, Chytridiomycetes, Dothideomycetes, and Sordariomycetes making up approximately 83% of identified genera (Fig. 3). Representatives from the Pucciniomycetes (<1%) were only captured with the ITS2 primer set. Community analysis of ITS2 OTUs (presence/absence) also showed distinct fungal communities in Lake Michigan and Lake Superior (PERMANOVA; p=0.007; Fig. 4B), and analysis of scaled OTU abundance showed similar results.
Method Comparison
Despite having similar taxonomic identifications at the class level, a local query using the ViroBlast search tool resulted in 7% of culture-independent sequences matching fungal ITS sequences from our local database of 2,167 culture-dependent isolates. These 64 sequence matches contained Agaricomycetes (16%), Dothideomycetes (23%) and Sordariomycetes (45%). Comparing only the 48 sediment samples used in both culture-dependent and culture-independent analysis, 12 (1.3%) sequences obtained from culture-independent analysis match sequences that were obtained via culturing of these same samples.
Comparing the culture-independent primer sets, the ITS1 primer set identified 47 genera and 55 species, while the ITS2 primer set identified an additional 32 genera and 49 species (ESM Table S2). Estimated alpha diversity using the Chao1 index varied across samples approximating between 5-92 OTUs and 4-65 OTUs per sample for ITS1 and ITS2, respectively, with no significant difference in alpha diversity between primer sets (Table 1, two-tailed t-test, p=0.700). Similarly, culture-dependent alpha diversity measurements for the same 48 samples were estimated between 1-78 fungal species per sample site (Table 1). Measures of Shannon's diversity and evenness using relative abundance of sequences in OTUs ranged from 0.01-2.43 and 0.004-0.705, respectively for ITS1. Relative abundance of ITS2 OTUs yielded Shannon's diversity ranging from 0.17-2.17 and evenness ranging from 0.08-0.98 (Table 1).
The bPTP species delimitation model estimated 445 species with ≥ 97% Bayesian support from the culture-dependent study. This species delineation was subsequently used to generate a species accumulation curve, which shows a continuous increase in species accrual with each new sample (Fig. 5A). The culture-independent analysis estimated 470 species with increased species accumulation per sample (Fig. 5B).
Fig. 5.
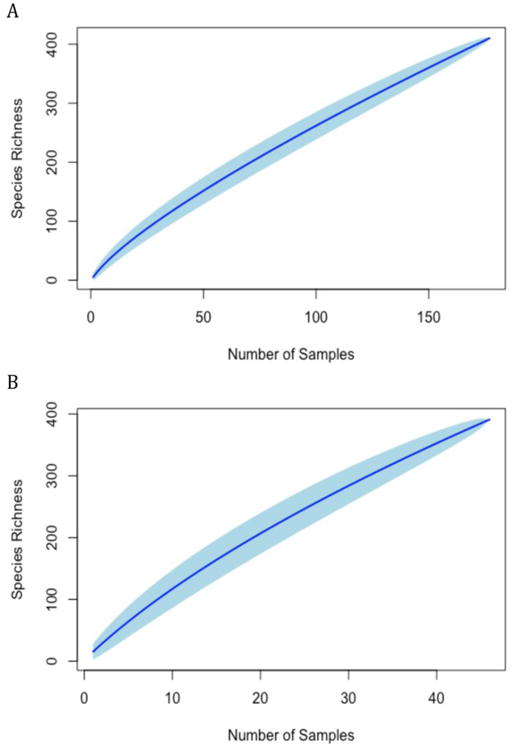
Species accumulation curves for culture-dependent (A) and culture-independent (B) datasets with 100 permutations. Confidence intervals at 95% are shown in light blue.
Discussion
This study, which reports the first inventory of fungi found in sediment from two Great Lakes, underscores the diversity of benthic fungal communities and the importance of including both culture-dependent and culture-independent identification approaches to assess the diversity of fungi occupying these aquatic habitats. By implementing both culture-dependent and culture-independent analyses, we detected 465 fungal taxa and only two, Aspergillus niger and Alternaria alternata, have been previously reported from water samples in Lake Michigan (Kiziewicz and Nalepa, 2008). This study has significantly increased our knowledge of fungal communities within the Great Lakes system contributing greatly to our understanding of fungal distribution and biodiversity in freshwater habitats.
Fungal Inventory
The fungal communities we observed are consistent with previous work from other freshwater habitats. Major phyla such as meiosporic and mitosporic Ascomycota, Basidiomycota, Chytridiomycota, and Zygomycota observed in this study are commonly found in freshwater habitats (Shearer et al., 2007; Wurzbacher et al., 2011). Previous studies of freshwater lakes have reported a high proportion of Chytridiomycota (Monchy et al., 2011, Wurzbacher et al. 2016), which was observed less frequently in this dataset. Chytridiomycota and other early diverging fungal lineages are not well identified using the ITS barcode (Schoch et al., 2012), and previous studies reporting high levels of Chytridiomycota used 18S and 28S rRNA regions (Monchy et al., 2011; Wurzbacher et al., 2016). It is also important to note our samples were taken from the benthic zone, which may be less hospitable to Chytridiomycota than the pelagic zone. In fact, a comprehensive summary of published 18S rRNA sequence data from aquatic fungal communities found that sediments did not appear to be dominated by any particular fungal group (Panzer et al., 2015). Taxa belonging to Glomeromycota were observed in our study and have been previously documented from the littoral region of freshwater lakes (Monchy et al., 2011) and shown to be associated with roots of aquatic macrophytes (Beck-Nielsen and Vindbak Madsen, 2001). Our study captured 24 previously documented freshwater species (ESM Table S2), but we believe that there may be additional freshwater species in the Great Lakes since 28% of the genera obtained in this study include freshwater species (Shearer and Raja, 2010). In addition, the high percentage of OTUs (i.e. 74% for ITS1 and 49% for ITS2) identified only at the kingdom level most likely represents undescribed, early diverging lineages such as Chytridiomycota, Cryptomycota, and Zoomycota that occur in this aquatic environment. However, the majority of our identified taxa (95%) were not previously reported freshwater taxa, suggesting some terrestrial fungi may transiently enter the benthic zone of aquatic systems.
Method Comparison
Culture-dependent and culture-independent methods captured different portions of fungal diversity in the Great Lakes, which is not surprising given each method's biases. Culture-dependent methods are biased toward the selection of faster-growing fungal species and those that grow on the selected medium. In contrast, culture-independent methods are biased toward fungal species that provide sufficient amounts of amplifiable DNA and exhibit optimal primer match. Despite these biases, the integration of both techniques has become a valuable tool in understanding fungal community composition.
Both methods recovered high proportions of taxa assigned to Dothideomycetes and Sordariomycetes (Ascomycota). However, culture-independent sequencing obtained a greater proportion of Agaricomycetes (Basidiomycota). This is expected since serial dilution plating is best used to obtain fungal spores from soil (Warcup, 1955). Historically, this method is poor at recovering Basidiomycota (Warcup, 1959), which can explain why Basidiomycota were recovered less frequently using culture-dependent techniques. Although culture-dependent and culture-independent methods captured a similar proportion of higher taxonomies (Dothideomycetes and Sordariomycetes), sequence level comparison only resulted in a 7% overlap indicating that each method is capturing a unique subset of these higher taxonomic groups. Similar results have been shown in marine sediment, reporting unique fungal assemblages associated with traditional culturing and targeted environmental sequencing methods where only seven out of 24 genera were recovered using both methods (Zhang et al., 2014).
The use of ITS1 and ITS2 primer sets in culture-independent analysis identified unique taxa increasing overall fungal diversity obtained from the Great Lakes. Implementation of multiple primer sets has been reported to identify different taxa and increase overall fungal diversity in culture-independent analyses in marine systems (Singh et al., 2011). The ITS1 region tends to be more variable across taxonomic groups compared to the ITS2 region (Nilsson et al., 2008), which may explain differences in the number of OTU clusters between primer sets. Blaalid et al. (2013) reported a higher number of OTU clusters using the ITS1 gene region compared to ITS2. Similar to our study, Blaalid et al. (2013) also found similar taxonomic composition at higher classification levels, but differences in lower-level genus identifications between primer sets. This difference could be attributed to PCR biases from ITS1 and ITS2 primer sets which may preferentially amplify Ascomycota or Basidiomycota (Bellemain et al., 2010). In this system, implementing multiple primer sets is a necessary step to understanding fungal communities. Although each primer set in this study identified unique taxa, estimated species richness was comparable between the primer sets. However, we did observe high variation in estimated species richness across samples. This variation suggests potential spatial heterogeneity where fungal communities may appear as ‘hotspots’ in sediment of the Great Lakes.
A significant portion of next generation sequencing resulted in OTUs identified to only the kingdom level, attributed to low query coverage or no matches in UNITE or NCBI databases. As of April 2017, NCBI contained molecular data for approximately 44,000 of the 120,000 described fungal species (www.ncbi.nlm.nih.gov/taxonomy), indicating that these OTUs could represent described species that are underrepresented in molecular databases, such as those early diverging lineages that oftentimes lack an ITS sequence. However, Hibbett et al. (2009) claims this alone does not encompass all unidentified OTUs and that these OTUs are more likely novel fungal taxa. The lack of strong taxonomic resolution for a significant portion of the sequencing data is currently a disadvantage of environmental DNA sequencing.
Species accumulation curves for both culture-dependent and culture-independent analysis (Fig. 5A,B) showed a continued increase in species count with each new sample indicating that additional sampling is needed to achieve a more complete inventory of fungal communities in the Great Lakes.
Conclusion
This study has presented the first comprehensive survey of fungi in sediments from two Great Lakes as determined by culture-dependent methods in combination with culture-independent methods. We show that the majority of identified taxa have not been reported from aquatic systems, which may implicate transient terrestrial fungi as a possible source of benthic zone fungal diversity. However, culture-independent analyses recovered a large portion of OTUs identified only to the kingdom Fungi, which may represent previously described, cryptic aquatic taxa or novel fungal taxa, especially in the Chytridiomycota, Cryptomycota, and Zoomycota, remaining to be discovered. The low level of sequence overlap between methods and high percentage of fungal taxa that can only be classified at the kingdom level suggests an immense amount of fungal diversity remains to be studied in these aquatic fungal communities. Additional study is warranted in order to determine how environmental conditions are influencing fungal communities in freshwater benthos and whether or not these communities are metabolically active in these lake sediments.
Supplementary Material
Acknowledgments
Research reported in this publication was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health (grant number R01GM107490). Maps were created using ArcGIS® software by Esri©. ArcGIS® and ArcMap™ are the intellectual property of Esri© and are used herein under license. All rights reserved. For more information about Esri® software, please visit www.esri.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Beck-Nielsen D, Vindbak Madsen T. Occurrence of vesicular-arbuscular mycorrhiza in aquatic macrophytes from lakes and streams. Aquat Bot. 2001;71:141–148. http://dx.doi:10.1016/S0304-3770(01)00180-2. [Google Scholar]
- Bellemain E, Carlsen T, Brochmann C, Coissac E, Taberlet P, Kauserud H. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol. 2010;10:189. doi: 10.1186/1471-2180-10-189. http://dx.doi:Artn189\rDoi10.1186/1471-2180-10-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaalid R, Kumar S, Nilsson RH, Abarenkov K, Kirk PM, Kauserud H. ITS1 versus ITS2 as DNA metabarcodes for fungi. Mol Ecol Resour. 2013;13:218–224. doi: 10.1111/1755-0998.12065. http://dx.doi:10.1111/1755-0998.12065. [DOI] [PubMed] [Google Scholar]
- Brown SP, Ferrer A, Dalling JW, Heath KD. Don't put all your eggs in one basket: a cost-effective and powerful method to optimize primer choice for rRNA environmental community analyses using the Fluidigm Access Array. Mol Ecol Resour. 2016;16:946–956. doi: 10.1111/1755-0998.12507. http://dx.doi:10.1111/1755-0998.12507. [DOI] [PubMed] [Google Scholar]
- Caporaso JG. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. http://dx.doi:10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Nickle DC, Learn GH, Maust B, Mullins JI. ViroBLAST: A stand-alone BLAST web server for flexible queries of multiple databases and user's datasets. Bioinformatics. 2007;23:2334–2336. doi: 10.1093/bioinformatics/btm331. http://dx.doi:10.1093/bioinformatics/btm331. [DOI] [PubMed] [Google Scholar]
- Edgcomb VP, Beaudoin D, Gast R, Biddle JF, Teske A. Marine subsurface eukaryotes: the fungal majority. Environ Microbiol. 2011;13:172–183. doi: 10.1111/j.1462-2920.2010.02318.x. http://dx.doi:10.1111/j.1462-2920.2010.02318.x. [DOI] [PubMed] [Google Scholar]
- Fuller K, Shear H. The Great Lakes: An Environmental Atlas and Resource Book. Government of Canada and the United State Environmental Protection Agency. 1995:51. [Google Scholar]
- Gardes M, Bruns TD. ITS primers with enhanced specificity for basidiomycetes -application to the identification of mycorrhizae and rusts. Mol Ecol. 1993;2:113–118. doi: 10.1111/j.1365-294x.1993.tb00005.x. [DOI] [PubMed] [Google Scholar]
- Gilbert JA, Meyer F, Jansson J, Gordon J, Pace N, Tiedje J, Ley R, Fierer N, Field D, Kyrpides N, Glöckner FO, Klenk HP, Wommack KE, Glass E, Docherty K, Gallery R, Stevens R, Knight R. The Earth Microbiome Project: Meeting report of the “1 EMP meeting on sample selection and acquisition” at Argonne National Laboratory October 6, 2010. Stand Genomic Sci. 2010;3:249–53. doi: 10.4056/aigs.1443528. http://dx.doi:10.4056/aigs.1443528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotelli NJ, Colwell RK. Estimating Species Richness. In: Magurran AE, McGill BJ, editors. Biological Diversity: Frontiers in Measurement and Assessment. Oxford University Press; 2011. pp. 39–54. http://dx.doi:10.2307/3547060. [Google Scholar]
- Hawksworth DL. The fungal dimension of biodiversity: magnitude, significance, and conservation. Mycol Res. 1991;95:641–655. http://dx.doi:10.1016/S0953-7562(09)80810-1. [Google Scholar]
- Hibbett DS, Ohman A, Kirk PM. Fungal ecology catches fire. New Phytol. 2009;184:279–282. doi: 10.1111/j.1469-8137.2009.03042.x. http://dx.doi:10.1111/j.1469-8137.2009.03035.x. [DOI] [PubMed] [Google Scholar]
- Hyde KD, Jones EBG, Leaño E, Pointing SB, Poonyth AD, Vrijmoed LLP. Role of fungi in marine ecosystems. Biodivers Conserv. 1998;7:1147–1161. http://dx.doi:10.1023/A:1008823515157. [Google Scholar]
- Ihrmark K, Bödeker ITM, Cruz-Martinez K, Friberg H, Kubartova A, Schenck J, Strid Y, Stenlid J, Brandström-Durling M, Clemmensen KE, Lindahl BD. New primers to amplify the fungal ITS2 region – evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol Ecol. 2012;82:666–677. doi: 10.1111/j.1574-6941.2012.01437.x. https://doi.org/10.1111/j.1574-6941.2012.01437.x. [DOI] [PubMed] [Google Scholar]
- Jones EBG, Hyde KD, Pang LK. Introduction Freshwater Fungi and Fungal-like Organisms. Walter de Gruyter; Berlin: 2014. [Google Scholar]
- Kirk PM, Cannon PF, Minter DW, Stalpers JA. Dictionary of the fungi. 10th. CABI Publishing; Wallingford, UK: 2008. [Google Scholar]
- Kiziewicz B, Nalepa TF. Some fungi and water molds in waters of Lake Michigan with emphasis on those associated with the benthic amphipod Diporeia spp. J Great Lakes Res. 2008;34:774–780. http://dx.doi:10.3394/0380-1330-34.4.774. [Google Scholar]
- Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AFS, Bahram M, Bates ST, Bruns TD, Bengtsson-Palme J, Callaghan TM, Douglas B, Drenkhan T, Eberhardt U, Dueñas M, Grebenc T, Griffith GW, Hartmann M, Kirk P, Kohout P, Larsson E, Lindahl BD, Lücking R, Martín MP, Matheny PB, Nguyen NH, Niskanen T, Oja J, Peay KG, Peintner U, Peterson M, Põldmaa K, Saag L, Saar I, Schüβler A, Scott JA, Senés C, Smith ME, Suija AVE, Taylor DLEE, Telleria MT, Weiss M, Larsson KH. Towards a unified paradigm for sequence-based identification of Fungi. Mol Ecol. 2013;22:5271–5277. doi: 10.1111/mec.12481. http://dx.doi:10.1111/mec.12481. [DOI] [PubMed] [Google Scholar]
- Mehrota BS, Bisht NS, Harsh NSK. Utilization of waste tea leaves for the growth and maintenance of cultures of wood decaying fungi. Natl Acad Sci Lett. 1982;5:87–88. [Google Scholar]
- Miller AN. Next-gen sequencing and metadata analyses of Great Lakes fungal data. University of Illinois at Urbana-Champaign; 2018. https://doi.org/10.13012/B2IDB-9320144_V2. [Google Scholar]
- Mirarab S, Nguyen N, Warnow T. PASTA: Ultra-Large Multiple Sequence Alignment. In: Sharan R, editor. Research in Computational Molecular Biology RECOMB 2014 Lecture Notes in Computer Science. Vol. 8394. Springer; 2014. pp. 177–191. [Google Scholar]
- Monchy S, Sanciu G, Jobard M, Rasconi S, Gerphagnon M, Chabé M, Cian A, Meloni D, Niquil N, Christaki U, Viscogliosi E, Sime-Ngando T. Exploring and quantifying fungal diversity in freshwater lake ecosystems using rDNA cloning/sequencing and SSU tag pyrosequencing. Environ Microbiol. 2011;13:1433–1453. doi: 10.1111/j.1462-2920.2011.02444.x. http://dx.doi:10.1111/j.1462-2920.2011.02444.x. [DOI] [PubMed] [Google Scholar]
- Mueller GM, Schmit JP. Fungal biodiversity: What do we know? What can we predict? Biodivers Conserv. 2007;16:1–5. http://dx.doi:10.1007/s10531-006-9117-7. [Google Scholar]
- Nguyen NH, Smith D, Peay K, Kennedy P. Parsing ecological signal from noise in next generation amplicon sequencing. New Phytol. 2015;205:1389–1393. doi: 10.1111/nph.12923. http://dx.doi:10.1111/nph.12923. [DOI] [PubMed] [Google Scholar]
- Nilsson RH, Kristiansson E, Ryberg M, Hallenberg N, Larsson KH. Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol Bioinform Online. 2008;4:193–201. doi: 10.4137/ebo.s653. http://dx.doi:10.4137/EBO.S653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H. Vegan: community ecology package. R package version 2.4-3. 2017 https://CRAN.R-project.org/package=vegan.
- Orsi W, Biddle JF, Edgcomb V. Deep sequencing of subseafloor eukaryotic rRNA reveals active fungi across marine subsurface provinces. PLoS One. 2013;8:e56335. doi: 10.1371/journal.pone.0056335. http://dx.doi:10.1371/journal.pone.0056335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmundson TW, Eyre CA, Hayden KM, Dhillon J, Garbelotto MM. Back to basics: An evaluation of NaOH and alternative rapid DNA extraction protocols for DNA barcoding, genotyping, and disease diagnostics from fungal and oomycete samples. Mol Ecol Resour. 2013;13:66–74. doi: 10.1111/1755-0998.12031. http://dx.doi:10.1111/1755-0998.12031. [DOI] [PubMed] [Google Scholar]
- Panzer K, Yilmaz P, Weiβ M, Reich L, Richter M, Wiese J, Schmaljohann R, Labes A, Imhoff JF, Glöckner FO, Reich M. Identification of habitat-specific biomes of aquatic fungal communities using a comprehensive nearly full-length 18S rRNA dataset enriched with contextual data. PLoS One. 2015;10:1–20. doi: 10.1371/journal.pone.0134377. http://dx.doi:10.1371/journal.pone.0134377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson R. Benthic and planktonic phycomycetes from northern Michigan. Mycologia. 1967;59:405–416. [Google Scholar]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2013. http://www.R-project.org. [Google Scholar]
- Ream D, Kiss AJ. NCBI / GenBank BLAST Output XML Parser Tool 2013 [Google Scholar]
- Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci. 2012;109:6241–6246. doi: 10.1073/pnas.1117018109. http://dx.doi:10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer CA, Descals E, Kohlmeyer B, Kohlmeyer J, Marvanová L, Padgett D, Porter D, Raja HA, Schmit JP, Thorton HA, Voglymayr H. Fungal biodiversity in aquatic habitats. Biodivers Conserv. 2007;16:49–67. http://dx.doi:10.1007/s10531-006-9120-z. [Google Scholar]
- Shearer CA, Raja HA. Freshwater Ascomycetes Database. [Accessed March 2017];2010 http://fungi.life.illinois.edu/
- Singh P, Raghukumar C, Verma P, Shouche Y. Fungal community analysis in the deep-sea sediments of the central Indian basin by culture-independent approach. Microb Ecol. 2011;61:507–517. doi: 10.1007/s00248-010-9765-8. http://dx.doi:10.1007/s00248-010-9765-8. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strayer DL, Dudgeon D. Freshwater biodiversity conservation: recent progress and future challenges. J North Am Benthol Soc. 2010;29:344–358. http://dx.doi:10.1899/08-171.1. [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. http://dx.doi:10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warcup JH. On the origin of colonies of fungi developing on soil dilution plates. Trans Br Mycol Soc. 1955;38:298–301. http://dx.doi:10.1016/S0007-1536(55)80075-7. [Google Scholar]
- Warcup JH. Studies on Basidiomycetes in soil. Trans Br Mycol Soc. 1959;42:45–52. http://dx.doi:10.1016/S0007-1536(59)80065-6. [Google Scholar]
- White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninshy JJ, White TJ, editors. PCR Protocols: A guide to methods and applications. Academic Press; 1990. pp. 315–322. [Google Scholar]
- Wong MKM, Goh TK, Hodgkiss IJ, Hyde KD, Ranghoo VM, Tsui CKM, Ho WH, Wong WSW, Yuen TK. Role of fungi in freshwater ecosystems. Biodivers Conserv. 1998;7:1187–1206. http://dx.doi:10.1023/A:1008883716975. [Google Scholar]
- Wurzbacher C, Bärlocher F, Grossart H. Fungi in lake ecosystems. Aquat Microb Ecol. 2010;59:125–149. http://dx.doi:10.3354/ame01385. [Google Scholar]
- Wurzbacher C, Kerr J, Grossart HP. Aquatic Fungi. Grillo O, Venora G, editors. The Dynamic Processes of Biodiversity: Case Studies of Evolution and Spatial Distribution, InTech, Rijeka, Croatia. 2011:227–258. http://dx.doi:10.5772/23029.
- Wurzbacher C, Warthmann N, Bourne E, Attermeyer K, Allgaier M, Powell JR, Detering H, Mbedi S, Grossart HP, Monaghan M. High habitat-specificity in fungal communities in oligo-mesotrophic, temperate Lake Stechlin (North-East Germany) MycoKeys. 2016;16:17–44. http://dx.doi:10.3897/mycokeys.16.9646. [Google Scholar]
- Zhang J, Kapli P, Pavlidis P, Stamatakis A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics. 2013;29(22):2869–2876. doi: 10.1093/bioinformatics/btt499. http://dx.doi:10.1093/bioinformatics/btt499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XY, Tang GL, Xu XY, Nong XH, Qi SH. Insights into deep-sea sediment fungal communities from the East Indian ocean using targeted environmental sequencing combined with traditional cultivation. PLoS One. 2014;9(10):e109118. doi: 10.1371/journal.pone.0109118. http://dx.doi:10.1371/journal.pone.0109118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.