

Abstract
Twenty-five years ago, RET was identified as the primary driver of Multiple Endocrine Neoplasia Type 2 (MEN2) syndrome. MEN2 is characterized by several transformation events including pheochromocytoma, parathyroid adenoma and, especially penetrant, medullary thyroid carcinoma (MTC). Overall, MTC is a rare but aggressive type of thyroid cancer for which no effective treatment currently exists. Surgery, radiation, radioisotope treatment, and chemotherapeutics have all shown limited success and none of these approaches have proven durable in advanced disease. Non-mammalian models that incorporate the oncogenic RET isoforms associated with MEN2 and other RET-associated diseases have been useful in delineating mechanisms underlying disease progression. These models have also identified novel targeted therapies as single agents and as combinations. These studies highlight the importance of modeling disease in the context of the whole animal, accounting for the complex interplay between tumor and normal cells in controlling disease progression, as well as response to therapy. With convenient access to whole genome sequencing data from expanded thyroid cancer patient cohorts, non-mammalian models will become more complex, sophisticated and continue to complement future mammalian studies. In this review we explore the contributions of non-mammalian models to our understanding of thyroid cancer including MTC, with a focus on Danio rerio and Drosophila melanogaster—fish and fly—models.
Introduction
Twenty-five years after the seminal work linking oncogenic RET and Multiple Endocrine Neoplasia Type 2 (MEN2), the field has advanced well beyond what Donis-Keller et al (Donis-Keller et al. 1993) imagined. We now have a deeper understanding of the signals by which oncogenic RET drives tumorigenesis, we know that other genes such as BRAF can also be involved and, perhaps most importantly, we have multiple FDA-approved drugs that extend the lifespan of MEN2 patients. Nevertheless, many patient’s disease continues to advance due to emergent resistance or limited response to current therapeutics. Our growing knowledge regarding the mechanisms that drive MEN2-related transformation and the testing of a new generation of therapeutics holds hope that patients will have new and perhaps better therapeutic options. The goal of this review and other recent reviews (Vitale et al. 2017) is to consider how simple model systems have contributed to our understanding of MEN2 and RET-linked cancers including Medullary Thyroid Cancer (MTC), and how they can be used as we move forward in developing new therapeutic treatments.
MTC is defined as transformation of the thyroid’s parafollicular cells, also labeled ‘C cells’, and is the most consistently observed cancer in MEN2 patients. Nearly 100% of MEN2 patients develop MTC, accounting for approximately 20% of total MTC cases—and early surgical resection of the thyroid has improved survival (S. A. Wells Jr. et al. 2013; Samuel A. Wells Jr et al. 2015). Surgery is not an option for spontaneous cases, and developing targeted therapeutics has become a priority of the field. These efforts rely on model systems. Multiple mouse models have been developed to model thyroid cancers; in addition to xenograft approaches, transgenic models of PTC and MTC have been reported (Michiels et al. 1997; Sweetser et al. 1999; Acton et al. 2000; Smith‐Hicks et al. 2000; de Graaff et al. 2001; reviewed in Kirschner et al. 2016)). RET(C634R) or RET(M918T) mouse models were established to model MTC observed in MEN2A or MEN2B patient subtypes, respectively. These mouse models exhibited hyperplasia in a subset of animals. However penetrance of transformation was low, e.g., 13% for RET(M918T) after 11 months; some mouse transgenic lines required >20 months to present with MTC (Acton et al. 2000).
While these transgenic mouse models are breakthroughs in demonstrating the importance of RET-based transformation for MEN2 and MTC, the low penetrance and long time-to-onset has limited the use of these models for exploring networks or screening for therapeutics. In particular for therapeutics, cell lines following by xenografts are now often the preferred approach for identifying and validating candidate therapeutics. Information gained in non-mammalian models will need to be validated in these models.
Why study non-mammalian models of MEN2?
One approach to complex biological problems that has proven effective is the use of non-mammalian model organisms as tools. In development, neurobiology, and disease, C. elegans, Drosophila, and zebrafish have provided important mechanistic and therapeutic information. The purpose of these models is not to replace mammalian models but to complement them. For example, many of the molecular pathways that direct cancer including NOTCH, WNT, and HIPPO, as well as multiple members of the RAS signaling pathway were discovered (and named) in Drosophila. Regarding cancer, among the first examples of tumor suppressors were reported in Drosophila (Gateff and Schneiderman 1967, 1969; Gateff 1978).
Fish and flies provide powerful genetic tools for the study of disease including MEN2; these are further discussed below. Both systems are rapid and inexpensive, and whole animal experiments can be performed at larger scale than mice or rats. These advantages, bolstered by a large community that has developed a myriad of tools and data, allow diseases such as MEN2 to be studied with a view to the larger networks that drive the disease. Also as discussed below, both flies and fish have important limitations including the key point that details between fish, flies and humans can be different in ways that can affect therapeutic response. Perhaps non-mammalian models’ greatest advantage is that they can yield useful and sometimes surprising hypotheses that can guide experiments in mammalian models.
Zebrafish and models of thyroid cancer
The teleost Danio rerio has been used to explore development for more than three decades. Zebrafish are easy to raise in a laboratory setting and genetically tractable. Young embryos are mostly clear, allowing the details of embryonic development to be observed in situ. The ability to generate a large number of transgenic lines expressing targeted fluorescent markers allows for detailed study of the effects of genetic manipulations on the cell biology of specific tissues. By visualizing fluorescence-labelled thyroid cells during early development, zebrafish models permit the ability to monitor the effects of genetic changes or chemical treatment.
Systematic knockout efforts have generated a wealth of tools for exploring gene function (Driever et al. 1996; Haffter et al. 1996). Approximately 70% of human genes have a clear fish ortholog (Howe et al. 2013). Morpholino mediated transient gene knockdown as well as CRISPR mediated endogenous gene alteration provides the ability to perform a spectrum of genetic manipulations in the fish. With these advantages zebrafish has taken its place as a ‘canonical’ model system. More recently, the community has embraced using zebrafish to explore disease including therapeutics.
Fish thyroid cancer models
Teleost thyroids have striking similarities—and some differences—with our own thyroids. Similar to us they have both follicular cells and calcitonin-producing C cells. However, the two cell populations do not fuse but, rather, remain separate (Biddinger and Ray 1993; Alt et al. 2006). The C cells collect as ‘ultimobranchial bodies’ that are associated with the heart (Figure 1).
Figure 1.
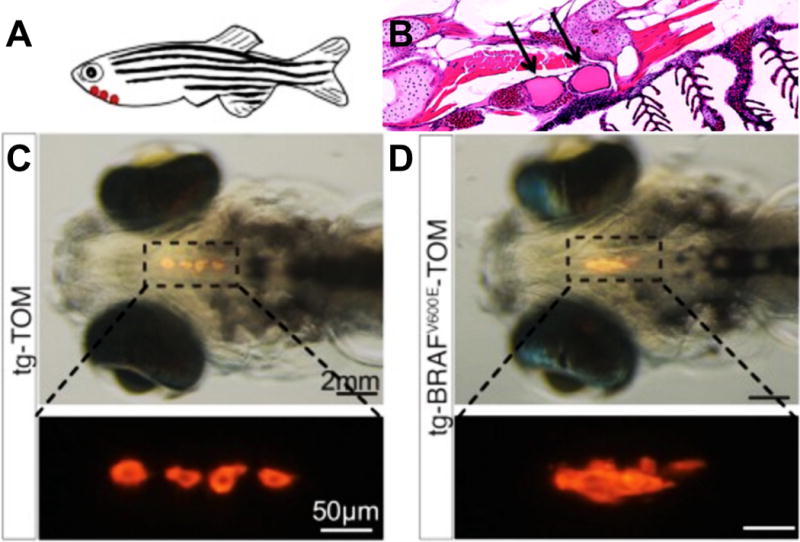
A, C, D: Targeting a fluorescent probe specifically to the thyroid (thyroglobulin-TdTomato) highlighted the zebrafish follicular cells. Thyroid structure was disrupted by co-targeting human BRAF(V600E). Adapted from Anelli et al, 2017. Panel B shows a sagittal section of adult thyroid follicles, which are associated with the ventral aorta and gils. From Bourque and Houvras, 2011.
Promising new zebrafish models of thyroid cancer have been developed. Modeling aspects of Papillary Thyroid Cancer (PTC), Houdras and colleagues demonstrated that targeted expression of the human BRAF oncogenic isoform BRAF(V600E) to thyrocytes led to multiple aspects of PTC (Anelli et al. 2017). Through targeted knockout, they demonstrated the functional importance of the epithelial-to-mesenchymal (EMT) regulator TWIST2 as a mediator of BRAF(V600E)-dependent transformation. They also demonstrated the ability of fish to respond to clinically relevant drugs, rescuing aspects of transformation with the BRAF pathway inhibitors dabrafenib and selumetinib.
Recently Yariv Houvras and colleagues have developed a model of RET-rearranged thyroid cancer in zebrafish (Y. Houvras, personal communication). Expression of the human CCDC6-RET fusion gene under control of a thyroid specific reporter led to significant neoplastic phenotypes within the first month of life.
Cancer cell grafting
In addition to developing genetic models to study cancer, zebrafish is also versatile as a model to graft human cancer cells. A number of studies have shown that a variety of cultured human cancer cells can be successfully grafted into fish (Barriuso, Nagaraju, and Hurlstone 2015). Some of the studies have further expanded the practical use of this approach and include: showing that transplanting human lung cancer cells into zebrafish allowed exploration of cancer cell invasiveness in-vivo (Moshal et al. 2011), orthotopic models of retinoblastoma cell invasion (Chen et al. 2015), and tumor induced vascularization studies (Zheng et al. 2016). Regarding endocrine cancers, grafting human MTC cells (MZ-CRC and TT) into the subepidermal cavity of zebrafish showed that the MTC cells could promote de novo angiogenesis and establish a vasculature surrounding the implanted tumor cells (Vitale et al. 2014). With a large number of human thyroid cancer cells available, from different subtypes, xenograft studies in zebrafish may prove useful for exploring the different mechanisms underlying these subtypes as well as their response to therapeutics (Cirello et al. 2017).
PDX
Recently zebrafish has also been successfully used as a host for implantation of patient derived tumor xenografted fragments (PDTX or PDX, (Cirello et al. 2017)). In principal, PDX models should more accurately represent the patient’s tumor genetic heterogeneity as implanted tumor lines can be used after only a few rounds of passaging. In PDX experiments with neuroendocrine tumors, e.g., pituitary adenomas, six of eight patient derived samples implanted successfully into host fish, a higher success rate than typical mice PDX models. This suggests that a fish strategy may prove useful when patient derived tumor samples are limited (Gaudenzi et al. 2017).
Therapeutics
The convenience of developing xenografted models of human cancers in zebrafish also makes them useful for drug discovery and exploring translational therapeutics. Injected human cancer cells have been used to study mechanisms of tumor cell dissemination and metastasis in zebrafish (reviewed in (Brown et al. 2017; Dang, Fogley, and Zon 2016); some of these models have been used for compound screens. Lead drug candidates as well as approved drugs have been identified in these models and, although their clinical relevance remains to be established, these efforts reinforce the potential for zebrafish to be used as a useful system for drug discovery. Given the powerful genetic and chemical biology screening tools available in zebrafish, a RET rearranged model of thyroid cancer should prove to be useful for further investigation into the mechanism of transformation and allow scientists to rapidly compare RET inhibitors or identify novel combinations for treatment.
Drosophila
Our laboratory has focused on the use of the fruit fly Drosophila as a tool to explore RET-dependent disease including MEN2 and MTC. Drosophila would seem an unlikely tool to explore MTC: while flies have analogs of many of our major organs, they do not have a thyroid. Nevertheless, flies have provided perhaps the most extensive exploration of RET-dependent disease of any non-mammalian model. Below, we consider the advantages and disadvantages of fly models, and review some of the insights fly models have provided.
Flies and cancer gene discovery
In 1967, Gateff and Schneiderman reported perhaps the first example of a tumor suppressor gene with the characterization of lethal giant larvae (lgl) (Gateff and Schneiderman 1967, 1969; Gateff 1978). Along with other pioneers such as Peter Bryant and colleagues they characterized a series of tumor suppressor genes that yielded tumors when homozygous mutant (reviewed in (Bilder 2004; Vaccari and Bilder 2009)). In subsequent years work by a large number of Drosophila laboratories has helped connect oncogenes and tumor suppressors to the details of tumor progression in situ. This work has been strongly informed by the exceptional body of work Drosophila has provided in understanding the signaling pathways that guide animal development. We recommend several excellent reviews that describe the contributions of flies to the cancer field (Sonoshita and Cagan 2017; Yadav, Srikrishna, and Gupta 2016; Herranz, Eichenlaub, and Cohen 2016; Vivek A. Rudrapatna, Cagan, and Das 2012). Here we focus on RET-based fly cancer models relevant to MEN2.
Conserved epithelia and disease genes
Flies have been used as models of hematopoietic and especially solid tumors, the latter of which represent the most common and often most difficult cancer type to treat. Fly tissues share extensive molecular and structural similarities with vertebrate tissues, and have served as models both for tissue assembly and for cell signaling pathways. Approximately 70% of human disease genes have a clear fly ortholog (Celniker and Rubin 2003; Reiter et al. 2001). The ability to manipulate sets of genes in small patches of cells—through technologies such as mitotic clones—has allowed fly researchers to explore the signaling connections between body, organs, tissues, and individual cells. It is in this context that model organisms such as flies can be useful for exploring the whole animal complexity of solid tumors. Fly MEN2 models provide a tool that—despite the lack of a thyroid—recapitulates several useful aspects of the aberrant signaling seen in MEN2-related tumors. Give important differences between man and fly, however, data in flies should be validated in human models.
Useful as cancer models
The development of Drosophila from an embryo to an adult requires approximately 10 days and an adult fly lives for two months. Flies are economical to raise, making the fly system ideal for analysis of mechanisms underlying cancer progression as well larger scale genetic and drug screens than in mice or rats. We routinely screen 1000 drug-dose points using 20,000 flies in 14 days to identify drugs or combinations for a particular cancer model. Complex fly cancer models with four or more mutations are readily developed; these complex models can provide insight into emergent cancer networks for breast, lung, colorectal, and thyroid cancers (e.g., (Uhlirova, Jasper, and Bohmann 2005; Read et al. 2005; Wu, Pastor-Pareja, and Xu 2010; Khoo et al. 2013; Hirabayashi, Baranski, and Cagan 2013; Bangi et al. 2016; Levine and Cagan 2016)). Fly models can also be used to screen for drugs and lead compounds (Vidal et al. 2005; Markstein et al. 2014; Willoughby et al. 2013; Edwards et al., 2011).
Perhaps the most attractive feature of using flies is access to their extensive genetic toolkit (Venken and Bellen 2014). The Drosophila community has a history of creating and sharing tools, and overall, they have developed a set of remarkable gene manipulation tools to allow (i) manipulation of any gene of interest, in (ii) nearly any tissue, at (iii) any stage of development.
Disadvantages
As with all model systems, Drosophila has significant limitations as a cancer model platform. The fly skeletal, lymphatic, and vasculature systems are different from that in vertebrates; consequently, key aspects of tumor progression including bone metastasis and intravasation/extravasation cannot be properly modeled in flies. Their immune system has similarities but also important differences with our own. And, of course, they lack a thyroid. Drosophila and human lineages diverged perhaps 300 million year ago and, as a result, perhaps the key disadvantage of using flies is that the details in signaling pathways and organs will have difficult-to-predict differences. Overall, we view flies as a ‘starting tool’ that can provide whole animal data quickly and inexpensively, but that represents only a first step in the discovery process.
The Drosophila RET(MEN2) signaling network: genetic screens
To model RETM918T—the RET isoform associated with MEN2B— Drosophila RETM955T was targeted to the developing eye, a well characterized epithelia in terms of cell-cell interactions and signal transduction (Read et al. 2005). RETM955T flies developed a ‘rough eye’ phenotype due to overproliferation, alterations in cell fate, and compensatory apoptosis. Targeting a broad cross section of the fly genome, multiple ‘genetic modifier screens’ identified 140 genes that mediate RETM955T-mediated transformation. This functional network included both positive and negative regulators of RET-mediated transformation. Multiple signaling pathways emerged including EGFR, RAS, SRC, JNK, NOTCH, etc., as well as cytoskeletal regulators and chromatin remodeling proteins, providing a broad view of the cellular networks that mediate RET-based transformation.
Interestingly, a collaboration that included medical geneticist Paul Goodfellow used the set of 140 Drosophila genetic modifiers to deconvolute a copy number variation analysis of MEN2 patient tumor samples (Read et al. 2005). Interestingly, two loci proved statistically significantly linked to increased risk of secondary pheochromocytomas when altered by copy number variation in MEN2 patients: the signaling kinase TNIK and the chromatin remodeling protein CHD3. This is consistent with the view that genomic alterations can affect MEN2 disease outcome. Overall, using Drosophila to identify candidate biomarkers and susceptibility loci represents an interesting potential use of Drosophila for cancer genomic studies, and has been used to deconvolute functional regulators in other diseases (e.g., (Pendse et al. 2013)). This set of genetic modifiers also offers an opportunity to better understand aspects of RET-mediated transformation; in succeeding years, we and others have systematically analyzed some of the mechanisms by which these genetic modifiers regulate RETMEN2-based transformation.
SRC
SRC is a well characterized oncogene that has been shown to promote progression of a broad palette of solid tumors (Ishizawar and Parsons 2004). SRC and its direct negative regulator CSK were identified as genetic modifiers of Drosophila RETM955T (Read et al. 2005). Using the fly eye assay as well as a wing epithelia assay, elevated SRC was found to promote migration, invasion, and metastasis-like cell behavior (Read, Bach, and Cagan 2004; Vidal, Larson, and Cagan 2006; Vidal et al. 2007). Most easily observed in the wing, the basal shift of transformed cells provides a useful in vivo assay for aspects of metastasis (Figure 2).
Figure 2.
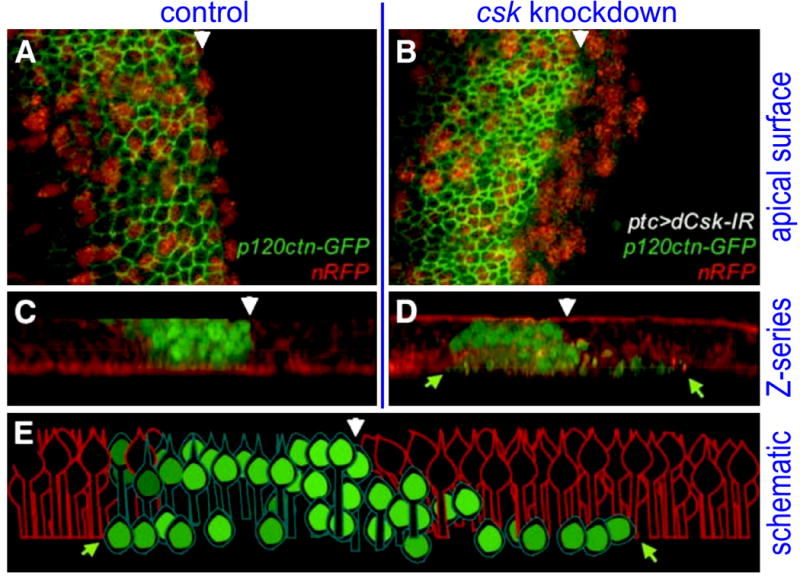
In contrast to control larval wing epithelia (panels A, C), activating SRC activity via CSK knockdown (B, D) led loss of apical junctions (P120-catenin, green), EMT, and basal migration of transformed cells. Red: nuclear-localized RFP. E: schematic of panel D. Adapted from Vidal et al, 2006.
Interestingly, activation of SRC—or other oncogenes such as RET—was found to have a ‘boundary effect’ at transformed/non-transformed boundaries: the SRC-transformed cells at tumor boundaries were the cells that shifted basally and migrated away from the original patched domain, suggesting that signals are passed between transformed and non-transformed cells. This phenomenon relied on pathways that included E-CADHERIN, RHO1, JNK, and matrix metalloproteases (Vidal, Larson, and Cagan 2006). This finding highlighted an important phenomenon relevant for cancer metastasis: that transformed cells adjacent to normal tissue, through a non-autonomous signaling mechanism, are potentially more invasive. This phenomenon gained support in fly/human studies of head and neck cancers (Heller et al. 2012); the role of interactions between normal thyroid cells and transformed C cells has yet to be explored.
SIN3A
SIN3A was identified as a potent tumor suppressor of fly RETM955T (Read et al. 2005). SIN3A’s role in regulating transcription has been studied comprehensively over the past decade but the mechanisms by which it regulates tumor progression is not fully understood. We found that SIN3A expression was downregulated in a broad cross-section of cancer types including RET-based tumors (T. K. Das, Sangodkar, et al. 2013). Again turning to fly models, our genetic, ChIP-seq, and molecular analysis in flies and human cancer cells showed that SIN3A is a suppressor of invasion downstream of RET as well as tumors driven by other RTKs.
SIN3A is a global transcriptional regulator that is increasingly linked to cancer (Bansal et al. 2016). In our fly cancer models SIN3A controlled a ‘gene cassette’ involved in invasion, making it an ideal point of deregulation to drive tumor progression. Our findings about SIN3A’s role regulating tumor progression has since been validated in a number of additional studies in humans and vertebrate models (Lewis et al. 2016; Takeda et al. 2015). Along with knockout of the SRC negative regulator CSK, knockout of SIN3A is one of the few examples of single gene loss that is sufficient to yield most aspects of transformation.
Caspases
The shift of RETMEN2, SRC activated, or SIN3A knockdown cells basally (Figure 2) also had another common feature: cells at the boundary contained high levels of cleaved, activated caspases (V. A. Rudrapatna, Bangi, and Cagan 2013). Most transformed cells were eventually—but not immediately—eliminated by apoptosis. Caspases are well characterized promoters of cell death and are considered important tumor suppressor pathways (Fuchs and Steller 2011).
The frequent presence of active caspases in invading/migrating cells suggested caspases could have yet another role in tumor progression beyond apoptosis. Blocking caspase activity in transformed cells enlarged the ‘primary’ wing tumor, but also prevented transformed cells from migrating, suggesting caspase activity could promote cell motility. When we activated caspases in epithelial cells and fine tuned their levels by co-expressing the caspase-inhibitor p35, otherwise wild-type cells showed migration/invasion into neighboring tissue (V. A. Rudrapatna, Bangi, and Cagan 2013). These cells activated MMPs and were dependent on the JNK-RHO pathway for migration (V. A. Rudrapatna, Bangi, and Cagan 2014). Similar observations have been made in human cells (e.g., (Liu et al. 2013)), supporting our findings and demonstrating how non-vertebrate model systems can shed novel cancer insights. Together this work highlights an unanticipated role for caspases: promoting cell migration in RET and SRC transformed cells. In one ‘goldilocks’ model, cancer networks activate must caspases at just the right level: too high levels promote apoptosis, while too low levels fail to fully promote migration.
NEK2
Chromosomal instability (CIN) has been proposed to promote tumor progression in various cancers including papillary thyroid cancer (PTC) and anaplastic thyroid cancer (ATC) (Mitsutake et al. 2005; Maric et al. 2011; Ragazzi et al. 2014). Centrosome amplification promotes CIN, which is postulated to be a source of genetic variation that fuels complex signaling and tumor growth (reviewed in (Negrini, Gorgoulis, and Halazonetis 2010)). In testing this hypothesis, we found that overexpression of the centrosomal component Nek2 in fly tissues promoted centrosome amplification but, in combination with activated point mutant RETM955T, it instead promoted metastasis. Nek2 upregulation also cooperated with other commonly activated tumor pathways including RAS and SRC to promote metastasis-like phenotypes(T. K. Das, Dana, et al. 2013). This data suggested that upregulation of components of the centrosome, including amplification of entire centrosomes, could in the presence of cancer drivers direct transformed cells to invade and metastasize, countering the ‘genetic-variation promoting’ model. Subsequent studies in vertebrate cells that selectively induced centrosome amplification have further supported this hypothesis (Cappello et al. 2014; Godinho et al. 2014).
RET(MEN2) signaling is distinct from RET fusions
25 year ago, Donis-Keller et al provided a window into the importance of RET-mediated transformation. The recent discovery of genomic RET fusions in thyroid, colorectal, and lung tumors has broadened the paradigm of RET-driven cancers. In most RET fusions the N-terminal portion is derived from another gene while the C-terminal domain includes the kinase domain of RET (Figure 3). The N-terminal domain usually consists of protein-protein (e.g, coiled-coiled) interaction domains with no known catalytic function; consequently, the RET kinase domain is considered the main signaling component of these proteins. Key examples include CCDC6-RET (associated primarily with PTC), NCOA4-RET (also PTC), and KIF5B-RET (lung cancer). RET fusions associated with PTC represent among the earliest described oncogenic fusions (Grieco et al. 1990; Santoro et al. 1994; Nikiforov and Nikiforova 2011).
Figure 3.
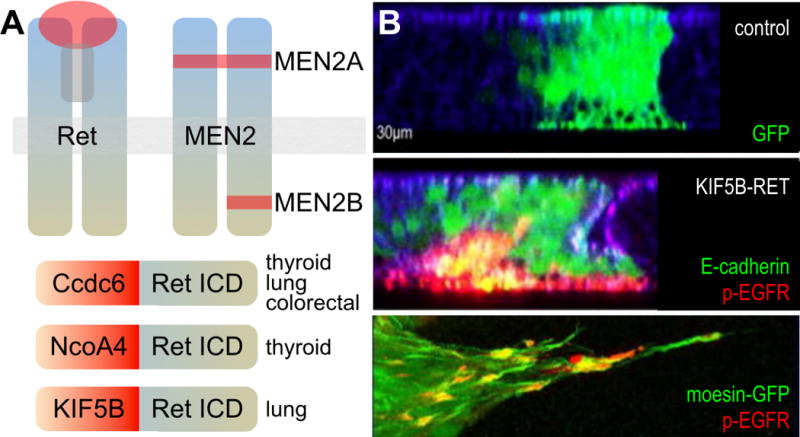
A. Oncogenic RET isoforms. ICD= intracellular domain. B. In contrast to controls, cells expressing KIF5B-RET (green) expressed phosphorylated EGFR as they underwent EMT and migrated basally. Bottom panel shows a closeup of invadopodia-like processes expressing phosphorylated EGFR (red) and phosphorylated SRC (not shown). Panel B adapted from Das and Cagan, 2017.
Patients with different RET point mutations or different RET fusions can show different levels and aspects of transformation. A key question is whether activating point mutant isoforms of RET and RET fusions promote tumorigenesis using similar mechanisms and,l whether they respond similarly to therapeutics. That is, would RET inhibition alone be sufficient as an optimal therapy against all oncogenic RET isoforms?
PTC fusions
Again using a dominant genetic modifier screening approach, fly models expressing the MEN2A and MEN2B RET isoforms in the fly eye showed similar though not quite identical functional networks (Read et al. 2005). Fusions, however, proved a different story: transformation of fly tissues expressing human CCDC6-RET vs. NCOA4-RET fusions relied on distinct genetic pathways for oncogenic signaling (Levinson and Cagan 2016). For example in a modifier screen of >200 tested kinases, mutations in four kinases modified CCD6-RET, nine modified NCOA4-RET, and only two kinases—fly orthologs of TEC and JNK—modified both CCDC6-RET and NCOA4-RET transformation. These functional networks were also significantly different from MEN2 forms of RET.
Similarly, in a survey of the efficacy of clinical and lead anti-cancer drugs CCDC6-RET and NCOA4-RET fly models responded to a different though partially overlapping set of drugs. For example, gefitinib and vandetanib more strongly suppressed CCDC6-RET induced lethality, while sorafenib and cabozantinib were more effective against NCOA4-RET induced lethality. Taken together these data illustrate the significant differences between CCDC6-RET and NCOA4-RET in both their signaling networks and drug response. The data also suggest that different RET fusions have different intracellular signaling networks that reflect more than simply activation of RET kinase activity.
KIF5B-RET fusion
A recent addition to the palette of RET fusions has been the identification of KIF5B-RET fusions in non-small cell lung cancer (NSCLC) patients (Figure 3) (Takeuchi et al. 2012; Ju et al. 2012). Preclinical vertebrate model studies have suggested KIF5B-RET can respond to RET-inhibitor therapy (Huang et al. 2016), yet in recent clinical trials many patients with KIF5B-RET NSCLC tumors responded poorly to RET-class drugs (Sarfaty et al. 2016; Gautschi et al. 2017). This suggests that therapies developed solely to target the fusion’s C-terminal RET kinase domain, without accounting for functions from the N-terminal partner domain, may show limited efficacy against KIF5B-RET tumors.
To explore these issues, we developed Drosophila and human lung cell line models to explore KIF5B-RET biology and also identify therapeutic options for KIF5B-RET tumors (Das and Cagan 2017). The full length KIF5B protein is a conventional kinesin heavy chain that regulates transport of a variety of cargoes to distinct cellular locations including axons and dendrites. In both fly and human cell lines, KIF5B-RET expression provoked both transformation and formation of unusually elongated filopodia-like processes and invadopodia (Figure 3). These unusual processes were not observed in RETM955T point mutants, or in the fusions CCDC6-RET and NCOA4-RET, indicating a distinct transformation network. Indeed, we demonstrated that the kinesin domain of KIF5B-RET, recruited and activated multiple RTKs—including EGFR, FGFR, and PDGFR—to RAB-containing vesicles, transporting these factors along with activated SRC into the aberrant cellular processes. The result was complex emergent signaling that required multi-targeting kinase inhibitors such as AD80 and sorafenib to more fully suppress the KIF5B-RET transformation network. The other RET-fusions, CCDC6-RET and NCOA4-RET, did not activate these pathways in a similar manner indicating the unique signaling capacity of KIF5B-RET oncogene (Das and Cagan 2017). These studies demonstrate that kinase fusion oncogenes can have unique and complex functions beyond their kinase domain activity. Therapeutic strategies for other kinase-fusion oncogenes may also benefit from understanding the signaling pathways activated by different domains of the fusion protein.
Drosophila as a tool to address current challenges of MEN2 cancer therapeutics
A key advantage of Drosophila is their willingness to thrive in small tubes or wells, allowing for moderately high-throughput whole animal drug screening. Combining this feature with powerful genetic tools has provided an opportunity to both identify lead compounds and also explore their whole animal mechanism of action (MOA).
Whole animal screening platform
A continuing challenge in cancer therapeutics is to identify drugs that selectively inhibit or kill cancer cells while sparing normal tissue. Whole animal screening platforms can help identify therapeutics that inhibit cancer cell growth (efficacy) without affecting animal viability (toxicity), i.e., optimized ‘therapeutic index’. Large scale whole animal screening in mammalian models are time consuming and cost prohibitive. The Drosophila RetM955T model offers a practical whole animal platform for testing candidate therapeutics.
Carlomagno, Santoro and colleagues demonstrated the efficacy of vandetanib against patient derived thyroid RETMEN2 cancer cells, a seminal step in developing therapies for RET-based cancers (Carlomagno et al. 2002). Fed to a Drosophila MEN2 model targeting RETM955T to the developing eye, vandetanib strongly suppressed the ‘rough eye’ phenotype (Figure 4) (Vidal et al. 2005). These flies were healthy and remained viable—other candidates showed poor efficacy or high toxicity—consistent with the view that vandetinib had an acceptable therapeutic index. This was the first indication that fly models of human thyroid neoplasias, similar to vertebrate models, could be used to assess lead therapeutic candidates.
Figure 4.
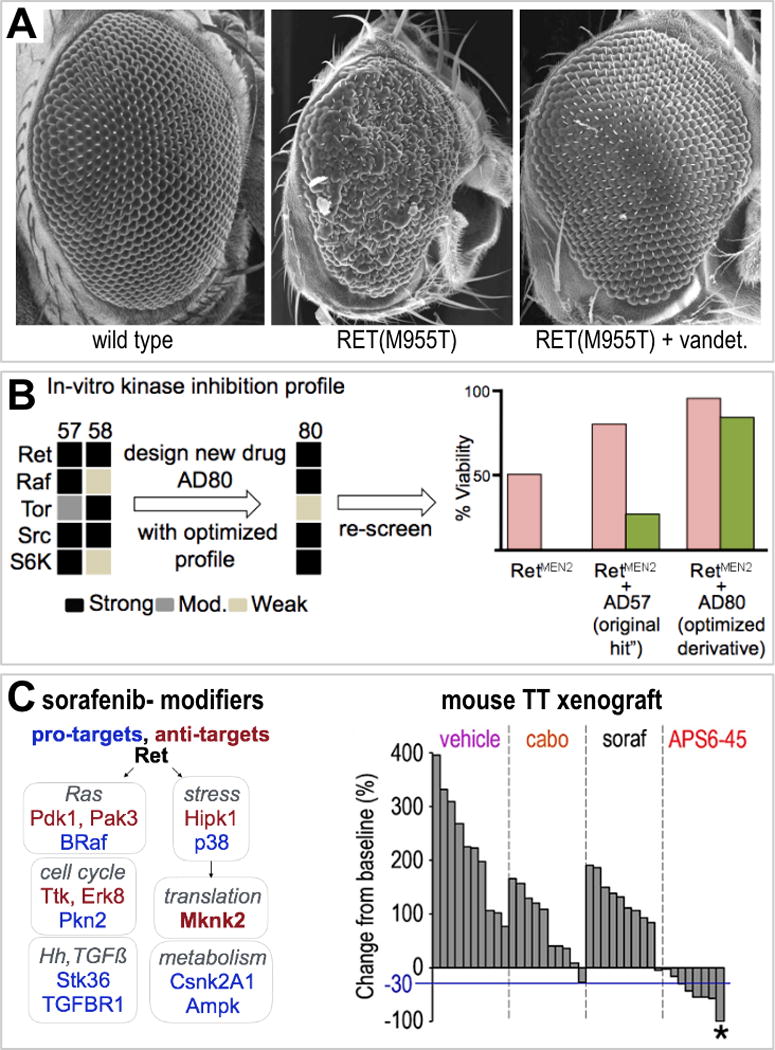
A. Vandetanib suppressed RET-mediated transformation of eye epithelia. B. AD80 acts through altered networks. C. Genetic identification of sorafenib’s pro/anti-target network led to the TCI lead APS6-45, which showed favorable mouse activity. Adapted from Read et al, 2005, Dar et al, 2012, Sonoshita et al, in press.
Rational polypharmacology
Vandetanib was the first drug to be approved for treatment of MTC in 2011; it has more recently been joined by cabozantinib (Razelle Kurzrock et al. 2011; S. A. Wells Jr. et al. 2012; R. Kurzrock et al. 2013). Both drugs are multi-targeting kinase inhibitors that target receptor tyrosine kinases including RET, VEGFR, KDR, and PDGFR as well as intracellular kinases including AXL, BTK, SRC, and MNK2. Many drugs owe a portion of their efficacy to ‘unintended polypharmacology’ (Amelio et al. 2017), and the importance of these additional targets to the success of vandetanib or cabozantinib is unknown. Although vandetanib and cabozantinib are effective at extending patient overall survival, patients can present with significant side effects and, in some cases, drug resistance can emerge.
The source of vandetanib and cabozantinib toxicity is not fully understood: toxicity likely derives from both unintended off-target activity and, perhaps, from on-target RET activity in normal tissues. One approach to address these issues is to develop leads that are more specific to oncogenic RET. Promising trials with ‘cleaner’ leads including LOXO-292 and BLU-667 are ongoing (Sabari, Siau, and Drilon 2017): reducing ‘off-target’ effects of drugs may lead to an improvement in overall therapeutic index, for example by reducing activity against potential liabilities such as KDR (Jain et al. 2010). An alternative approach is ‘tune’ multi-targeting (‘polypharmacological’) compounds to the oncogenic RET network while limiting whole body liabilities (Hopkins 2008; Knight, Lin, and Shokat 2010). Taking the latter approach, we collaborated with medicinal chemists Kevan Shokat and Arvin Dar to identify compounds that better inhibit the Ret signaling network in the context of the whole animal.
AD80
Screening for compounds that rescue RetM955T-mediated lethality led to identification of the DFG-out, Type II kinase inhibitor compound AD57 (Dar et al. 2012). We next used dominant modifier genetic screening to better understand the limitations and liabilities of AD57. Coupling in vitro kinase assays with genetic modifier screens, we identified RET, RAF, SRC, and mTOR as key AD57 targets. For example, removing a functional copy of the PI3K pathway regulator tor (RETM955T tor+/−) in the presence of AD57 increased whole animal toxicity and decreased efficacy. That is, a second generation compound should demonstrate reduced activity against the ‘anti-target’ mTOR.
Through chemical modeling, the Shokat laboratory predicted specific alterations to achieve a rebalancing of AD57’s targets. The resulting derivative compounds AD80 and AD81 showed reduced mTOR activity; they reduced PI3K activity by attacking the downstream kinase S6K (Dar et al. 2012). In human TT and MZ-CRC-1 cell lines, AD80 reduced viability approximately 150-fold more effectively than vandetanib; it also showed significantly improved efficacy in a TT-based mouse xenograft study when benchmarked against vandetanib. More recently, AD80 was shown to have exceptional efficacy in mammalian colorectal and lung cancer models containing RET fusions, models that proved resistant to more targeted RET drugs (Plenker et al. 2017). While these data are promising, importantly AD80 and its sister analogs have yet to be tested in a patient setting.
Sorafenib analog APS6-45
Our experience with developing AD80 and related compounds established a platform: identify an initial ‘hit’ through whole fly screening, then use genetics to help guide improvements. More recently we have extended this platform to include broader genetic screening and to pair whole fly screening and medicinal chemistry with computational chemistry (Sonoshita et al, in press). We focused on altering FDA approved compounds, to take advantage of their validated druggability.
Others’ work and our own screens identified the kinase inhibitor sorafenib as promising for treating RET-based MTC (Hong et al. 2008; Lam et al. 2010; Capdevila et al. 2012; de Castroneves et al. 2016). However, sorafenib’s liabilities—toxicity and emergent resistance—have limited its utility for MTC. Sorafenib shows a similar profile in a Drosophila RETM955T model: limited but significant efficacy but also significant toxicity. Using a dominant genetic modifier screen of the fly kinome to identify new activities (‘pro-targets’) and liabilities (‘anti-targets’) led to identification of an idealized sorafenib kinase profile (Sonoshita et al, in revision). Pairing computational docking studies and medicinal chemistry, new sorafenib analogs (‘tissue calibrated inhibitors’ or ‘TCIs’) were developed that showed exceptional activity and low toxicity in fly MTC models and in a TT cell-based mouse xenograft model of MTC (Figure 4).
This work further demonstrates that whole animal ‘calibrating’ of promising leads such as AD57 or sorafenib to RET-based disease networks may provide a useful approach to addressing drug liabilities including whole body toxicity, limited efficacy, and emergent resistance. It remains to be seen, however, whether the fly and mouse work will translate to patient response, specifically whether the druggable properties of sorafenib are retained while also transferring the therapeutic index improvements of the TCIs.
Perspective
In the past 25 years, the MEN2 field has been the beneficiary of a fruitful collaboration of clinicians, clinical scientists, and basic scientists. This blend of approaches has led to important advances in our understanding of MEN2 as well as useful first generation treatments. By identifying RET as a key driver of MEN2, Donis-Keller et al provided a blueprint for MEN2 studies, challenging the field to build on this knowledge to continually improve therapeutics. Modeling RET-activated pathways in MEN2 has also provided an exciting opportunity to model and understand other cancer types with oncogenic RET variants. This has provided a more complete picture of the full potential of tumorigenic RET signaling, including that of RET-fusions, allowing the field to explore both common and tailored therapeutic strategies across these different cancer paradigms. As with many other thriving fields, model organisms including mice, fish, and flies provide a useful discovery engine, helping provide new mechanistic understanding and new paths to therapeutics.
Acknowledgments
We thank members of the Cagan laboratory and especially Yariv Houvras for helpful discussions. This review was supported by National Institutes of Health grants R01-CA170495, R01-U54OD020353 and R01-CA109730, Department of Defense grant W81XWH-15-1-0111 and American Cancer Society grants 120886-PFM-11-137-01-DDC (T. D.) and 120616-RSGM-11-018-01-CDD (R. C.).
Footnotes
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
References
- Acton DS, Velthuyzen D, Lips CJ, Hoppener JW. Multiple Endocrine Neoplasia Type 2B Mutation in Human RET Oncogene Induces Medullary Thyroid Carcinoma in Transgenic Mice. Oncogene. 2000;19:3121–25. doi: 10.1038/sj.onc.1203648. [DOI] [PubMed] [Google Scholar]
- Alt Burkhard, Reibe Saskia, Feitosa Natalia M, Elsalini Osama A, Wendl Thomas, Rohr Klaus B. Analysis of Origin and Growth of the Thyroid Gland in Zebrafish. Developmental Dynamics: An Official Publication of the American Association of Anatomists. 2006;235(7):1872–83. doi: 10.1002/dvdy.20831. [DOI] [PubMed] [Google Scholar]
- Amelio Ivano, Lisitsa Andrey, Knight Richard A, Melino Gerry, Antonov Alexey V. Polypharmacology of Approved Anticancer Drugs. Current Drug Targets. 2017;18(5):534–43. doi: 10.2174/1389450117666160301095233. [DOI] [PubMed] [Google Scholar]
- Anelli Viviana, Villefranc Jacques A, Chhangawala Sagar, Martinez-McFaline Raul, Riva Eleonora, Nguyen Anvy, Verma Akanksha, et al. Oncogenic BRAF Disrupts Thyroid Morphogenesis and Function via Twist Expression. eLife. 2017 Mar;6 doi: 10.7554/eLife.20728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangi Erdem, Murgia Claudio, Teague Alexander GS, Sansom Owen J, Cagan Ross L. Functional Exploration of Colorectal Cancer Genomes Using Drosophila. Nature Communications. 2016 Nov;7:13615. doi: 10.1038/ncomms13615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal N, David G, Farias E, Waxman S. Emerging Roles of Epigenetic Regulator Sin3 in Cancer. Advances in Cancer Research. 2016 Mar;130:113–35. doi: 10.1016/bs.acr.2016.01.006. [DOI] [PubMed] [Google Scholar]
- Barriuso Jorge, Nagaraju Raghavendar, Hurlstone Adam. Zebrafish: A New Companion for Translational Research in Oncology. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2015;21(5):969–75. doi: 10.1158/1078-0432.CCR-14-2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddinger PW, Ray M. Distribution of C Cells in the Normal and Diseased Thyroid Gland. Pathology Annual. 1993;28(Pt 1):205–29. [PubMed] [Google Scholar]
- Bilder David. Epithelial Polarity and Proliferation Control: Links from the Drosophila Neoplastic Tumor Suppressors. Genes & Development. 2004;18(16):1909–25. doi: 10.1101/gad.1211604. [DOI] [PubMed] [Google Scholar]
- Bourque C, Houvras Y. Hooked on zebrafish: insights into development and cancer of endocrine tissues. Endocr Relat Cancer. 2011 Aug 30;18(5):R149–64. doi: 10.1530/ERC-11-0099. [DOI] [PubMed] [Google Scholar]
- Brown Hannah K, Schiavone Kristina, Tazzyman Simon, Heymann Dominique, Chico Timothy Ja. Zebrafish Xenograft Models of Cancer and Metastasis for Drug Discovery. Expert Opinion on Drug Discovery. 2017;12(4):379–89. doi: 10.1080/17460441.2017.1297416. [DOI] [PubMed] [Google Scholar]
- Capdevila Jaume, Iglesias Lara, Halperin Irene, Segura Angel, Martínez-Trufero Javier, Ángeles Vaz Maria, Corral Jesús, et al. Sorafenib in Metastatic Thyroid Cancer. Endocrine-Related Cancer. 2012;19(2):209–16. doi: 10.1530/ERC-11-0351. [DOI] [PubMed] [Google Scholar]
- Cappello P, Blaser H, Gorrini C, Lin DCC, Elia AJ, Wakeham A, Haider S, et al. Role of Nek2 on Centrosome Duplication and Aneuploidy in Breast Cancer Cells. Oncogene. 2014;33(18):2375–84. doi: 10.1038/onc.2013.183. [DOI] [PubMed] [Google Scholar]
- Carlomagno F, Vitagliano D, Guida T, Ciardiello F, Tortora G, Vecchio G, Ryan AJ, Fontanini G, Fusco A, Santoro M. ZD6474, an Orally Available Inhibitor of KDR Tyrosine Kinase Activity, Efficiently Blocks Oncogenic RET Kinases. Cancer Research. 2002;62:7284–90. [PubMed] [Google Scholar]
- de Castroneves Luciana Audi, Negrão Marcelo Vailati, de Freitas Ricardo Miguel Costa, Papadia Carla, Lima José Viana, Jr, Fukushima Julia T, Simão Eduardo Furquim, et al. Sorafenib for the Treatment of Progressive Metastatic Medullary Thyroid Cancer: Efficacy and Safety Analysis. Thyroid: Official Journal of the American Thyroid Association. 2016;26(3):414–19. doi: 10.1089/thy.2015.0334. [DOI] [PubMed] [Google Scholar]
- Celniker Susan E, Rubin Gerald M. The Drosophila Melanogaster Genome. Annual Review of Genomics and Human Genetics. 2003;4:89–117. doi: 10.1146/annurev.genom.4.070802.110323. [DOI] [PubMed] [Google Scholar]
- Chen Xiaoyun, Wang Jian, Cao Ziquan, Hosaka Kayoko, Jensen Lasse, Yang Huasheng, Sun Yuping, Zhuang Rujie, Liu Yizhi, Cao Yihai. Invasiveness and Metastasis of Retinoblastoma in an Orthotopic Zebrafish Tumor Model. Scientific Reports. 2015 Jul;5:10351. doi: 10.1038/srep10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirello Valentina, Gaudenzi Germano, Grassi Elisa S, Colombo Carla, Vicentini Leonardo, Ferrero Stefano, Persani Luca, Vitale Giovanni, Fugazzola Laura. Tumor and Normal Thyroid Spheroids: From Tissues to Zebrafish. Minerva Endocrinologica. 2017 Jan; doi: 10.23736/S0391-1977.17.02610-4. [DOI] [PubMed] [Google Scholar]
- Dang Michelle, Fogley Rachel, Zon Leonard I. Identifying Novel Cancer Therapies Using Chemical Genetics and Zebrafish. Advances in Experimental Medicine and Biology. 2016;916:103–24. doi: 10.1007/978-3-319-30654-4_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AC, Das TK, Shokat KM, Cagan RL. Chemical Genetic Discovery of Targets and Anti-Targets for Cancer Polypharmacology. Nature. 2012;486:80–84. doi: 10.1038/nature11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Tirtha Kamal, Cagan Ross Leigh. KIF5B-RET Oncoprotein Signals through a Multi-Kinase Signaling Hub. Cell Reports. 2017;20(10):2368–83. doi: 10.1016/j.celrep.2017.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das TK, Dana D, Paroly SS, Perumal SK, Singh S, Jhun H, Pendse J, Cagan RL, Talele TT, Kumar S. Centrosomal Kinase Nek2 Cooperates with Oncogenic Pathways to Promote Metastasis. Oncogenesis. 2013 Sep;2:e69. doi: 10.1038/oncsis.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das TK, Sangodkar J, Negre N, Narla G, Cagan RL. Sin3a Acts through a Multi-Gene Module to Regulate Invasion in Drosophila and Human Tumors. Oncogene. 2013;32:3184–97. doi: 10.1038/onc.2012.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, Howe JR, Moley JF, Goodfellow P, Wells SA., Jr Mutations in the RET Proto-Oncogene Are Associated with MEN 2A and FMTC. Human Molecular Genetics. 1993;2:851–56. doi: 10.1093/hmg/2.7.851. [DOI] [PubMed] [Google Scholar]
- Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, Stemple DL, Stainier DY, et al. A Genetic Screen for Mutations Affecting Embryogenesis in Zebrafish. Development. 1996 Dec;123:37–46. doi: 10.1242/dev.123.1.37. [DOI] [PubMed] [Google Scholar]
- Edwards A, Gladstone M, Yoon P, Raben D, Frederick B, Su TT. Combinatorial Effect of Maytansinol and Radiation in Drosophila and Human Cancer Cells. Dis Model Mech. 2011;4(4):496–503. doi: 10.1242/dmm.006486. Epub 2011/04/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs Yaron, Steller Hermann. Programmed Cell Death in Animal Development and Disease. Cell. 2011;147(4):742–58. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gateff E. Malignant Neoplasms of Genetic Origin in Drosophila Melanogaster. Science. 1978;200(4349):1448–59. doi: 10.1126/science.96525. [DOI] [PubMed] [Google Scholar]
- Gateff E, Schneiderman HA. Developmental Studies of a New Mutant of Drosophila Melanogaster-Lethal Malignant Brain Tumor (1 (2) GL4) In American Zoologist. 1967;7:760. +. Soc Integrative Comparative Biology. [Google Scholar]
- Gateff E, Schneiderman HA. Neoplasms in Mutant and Cultured Wild-Tupe Tissues of Drosophila. National Cancer Institute Monograph. 1969 Jul;31:365–97. [PubMed] [Google Scholar]
- Gaudenzi Germano, Albertelli Manuela, Dicitore Alessandra, Würth Roberto, Gatto Federico, Barbieri Federica, Cotelli Franco, et al. Patient-Derived Xenograft in Zebrafish Embryos: A New Platform for Translational Research in Neuroendocrine Tumors. Endocrine. 2017;57(2):214–19. doi: 10.1007/s12020-016-1048-9. [DOI] [PubMed] [Google Scholar]
- Gautschi Oliver, Milia Julie, Filleron Thomas, Wolf Juergen, Carbone David P, Owen Dwight, Camidge Ross, et al. Targeting RET in Patients With RET-Rearranged Lung Cancers: Results From the Global, Multicenter RET Registry. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. 2017;35(13):1403–10. doi: 10.1200/JCO.2016.70.9352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho Susana A, Picone Remigio, Burute Mithila, Dagher Regina, Su Ying, Leung Cheuk T, Polyak Kornelia, Brugge Joan S, Théry Manuel, Pellman David. Oncogene-like Induction of Cellular Invasion from Centrosome Amplification. Nature. 2014;510(7503):167–71. doi: 10.1038/nature13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graaff E, de Srinivas S, Kilkenny C, D’Agati V, Mankoo BS, Costantini F, Pachnis V. Differential Activities of the RET Tyrosine Kinase Receptor Isoforms during Mammalian Embryogenesis. Genes & Development. 2001;15(18):2433–44. doi: 10.1101/gad.205001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G, Fusco A, Vecchio G. PTC Is a Novel Rearranged Form of the Ret Proto-Oncogene and Is Frequently Detected in Vivo in Human Thyroid Papillary Carcinomas. Cell. 1990;60:557–63. doi: 10.1016/0092-8674(90)90659-3. [DOI] [PubMed] [Google Scholar]
- Haffter P, Granato M, Brand M, Mullins MC, Hammerschmidt M, Kane DA, Odenthal J, et al. The Identification of Genes with Unique and Essential Functions in the Development of the Zebrafish, Danio Rerio. Development. 1996 Dec;123:1–36. doi: 10.1242/dev.123.1.1. [DOI] [PubMed] [Google Scholar]
- Heller E, Hurchla MA, Xiang J, Su X, Chen S, Schneider J, Joeng KS, et al. Hedgehog Signaling Inhibition Blocks Growth of Resistant Tumors through Effects on Tumor Microenvironment. Cancer Research. 2012;72:897–907. doi: 10.1158/0008-5472.CAN-11-2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz Héctor, Eichenlaub Teresa, Cohen Stephen M. Cancer in Drosophila: Imaginal Discs as a Model for Epithelial Tumor Formation. Current Topics in Developmental Biology. 2016 Feb;116:181–99. doi: 10.1016/bs.ctdb.2015.11.037. [DOI] [PubMed] [Google Scholar]
- Hirabayashi S, Baranski TJ, Cagan RL. Transformed Drosophila Cells Evade Diet-Mediated Insulin Resistance through Wingless Signaling. Cell. 2013;154:664–75. doi: 10.1016/j.cell.2013.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D, Ye L, Gagel R, Chintala L, El Naggar AK, Wright J, Kurzrock R. Medullary Thyroid Cancer: Targeting the RET Kinase Pathway with Sorafenib/tipifarnib. Molecular Cancer Therapeutics. 2008;7:1001–6. doi: 10.1158/1535-7163.MCT-07-2422. [DOI] [PubMed] [Google Scholar]
- Hopkins AL. Network Pharmacology: The next Paradigm in Drug Discovery. Nature Chemical Biology. 2008;4:682–90. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- Howe Kerstin, Clark Matthew D, Torroja Carlos F, Torrance James, Berthelot Camille, Muffato Matthieu, Collins John E, et al. The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature. 2013;496(7446):498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Qingling, Schneeberger Valentina E, Luetteke Noreen, Jin Chengliu, Afzal Roha, Budzevich Mikalai M, Makanji Rikesh J, et al. Preclinical Modeling of KIF5B-RET Fusion Lung Adenocarcinoma. Molecular Cancer Therapeutics. 2016 Aug; doi: 10.1158/1535-7163.MCT-16-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawar Rumey, Parsons Sarah J. C-Src and Cooperating Partners in Human Cancer. Cancer Cell. 2004;6(3):209–14. doi: 10.1016/j.ccr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Jain L, Sissung TM, Danesi R, Kohn EC, Dahut WL, Kummar S, Venzon D, et al. Hypertension and Hand-Foot Skin Reactions Related to VEGFR2 Genotype and Improved Clinical Outcome Following Bevacizumab and Sorafenib. Journal of Experimental & Clinical Cancer Research: CR. 2010;29:95. doi: 10.1186/1756-9966-29-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK, Kim YT, Kim JI, Kang JH, Seo JS. A Transforming KIF5B and RET Gene Fusion in Lung Adenocarcinoma Revealed from Whole-Genome and Transcriptome Sequencing. Genome Research. 2012;22:436–45. doi: 10.1101/gr.133645.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo P, Allan K, Willoughby L, Brumby AM, Richardson HE. In Drosophila, RhoGEF2 Cooperates with Activated Ras in Tumorigenesis through a Pathway Involving Rho1-Rok-Myosin-II and JNK Signalling. Disease Models & Mechanisms. 2013;6:661–78. doi: 10.1242/dmm.010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner Lawrence S, Qamri Zahida, Kari Suresh, Ashtekar Amruta. Mouse Models of Thyroid Cancer: A 2015 Update. Molecular and Cellular Endocrinology. 2016 Feb;421:18–27. doi: 10.1016/j.mce.2015.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Zachary A, Lin Henry, Shokat Kevan M. Targeting the Cancer Kinome through Polypharmacology. Nature Reviews Cancer. 2010;10(2):130–37. doi: 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzrock R, Atkins J, Wheler J, Fu S, Naing A, Busaidy N, Hong D, Sherman S. Tumor Marker and Measurement Fluctuations May Not Reflect Treatment Efficacy in Patients with Medullary Thyroid Carcinoma on Long-Term RET Inhibitor Therapy. Annals of Oncology: Official Journal of the European Society for Medical Oncology/ESMO. 2013;24(9):2256–61. doi: 10.1093/annonc/mdt177. [DOI] [PubMed] [Google Scholar]
- Kurzrock Razelle, Sherman Steven I, Ball Douglas W, Forastiere Arlene A, Cohen Roger B, Mehra Ranee, Pfister David G, et al. Activity of XL184 (Cabozantinib), an Oral Tyrosine Kinase Inhibitor, in Patients with Medullary Thyroid Cancer. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. 2011;29(19):2660–66. doi: 10.1200/JCO.2010.32.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam ET, Ringel MD, Kloos RT, Prior TW, Knopp MV, Liang J, Sammet S, et al. Phase II Clinical Trial of Sorafenib in Metastatic Medullary Thyroid Cancer. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. 2010;28:2323–30. doi: 10.1200/JCO.2009.25.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine Benjamin D, Cagan Ross L. Drosophila Lung Cancer Models Identify Trametinib plus Statin as Candidate Therapeutic. Cell Reports. 2016;14(6):1477–87. doi: 10.1016/j.celrep.2015.12.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson Sarah, Cagan Ross L. Drosophila Cancer Models Identify Functional Differences between Ret Fusions. Cell Reports. 2016;16(11):3052–61. doi: 10.1016/j.celrep.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis Monica J, Liu Jianzhong, Libby Emily Falk, Lee Minnkyong, Crawford Nigel PS, Hurst Douglas R. SIN3A and SIN3B Differentially Regulate Breast Cancer Metastasis. Oncotarget. 2016;7(48):78713–25. doi: 10.18632/oncotarget.12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Yan-Rong, Sun Baocun, Zhao Xiu-Lan, Gu Qiang, Liu Zhi-Yong, Dong Xue-Yi, Che Na, Mo Jing. Basal Caspase-3 Activity Promotes Migration, Invasion, and Vasculogenic Mimicry Formation of Melanoma Cells. Melanoma Research. 2013;23(4):243–53. doi: 10.1097/CMR.0b013e3283625498. [DOI] [PubMed] [Google Scholar]
- Maric Irena, Viaggi Silvia, Caria Paola, Frau Daniela V, Degan Paolo, Vanni Roberta. Centrosomal and Mitotic Abnormalities in Cell Lines Derived from Papillary Thyroid Cancer Harboring Specific Gene Alterations. Molecular Cytogenetics. 2011 Nov;4:26. doi: 10.1186/1755-8166-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markstein Michele, Dettorre Samantha, Cho Julio, Neumüller Ralph A, Craig-Müller Sören, Perrimon Norbert. Systematic Screen of Chemotherapeutics in Drosophila Stem Cell Tumors. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(12):4530–35. doi: 10.1073/pnas.1401160111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels FM, Chappuis S, Caillou B, Pasini A, Talbot M, Monier R, Lenoir GM, Feunteun J, Billaud M. Development of Medullary Thyroid Carcinoma in Transgenic Mice Expressing the RET Protooncogene Altered by a Multiple Endocrine Neoplasia Type 2A Mutation. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(7):3330–35. doi: 10.1073/pnas.94.7.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsutake Norisato, Knauf Jeffrey A, Mitsutake Shin, Mesa Cleo, Jr, Zhang Lei, Fagin James A. Conditional BRAFV600E Expression Induces DNA Synthesis, Apoptosis, Dedifferentiation, and Chromosomal Instability in Thyroid PCCL3 Cells. Cancer Research. 2005;65(6):2465–73. doi: 10.1158/0008-5472.CAN-04-3314. [DOI] [PubMed] [Google Scholar]
- Moshal Karni S, Ferri-Lagneau Karine F, Haider Jamil, Pardhanani Pooja, Leung Tinchung. Discriminating Different Cancer Cells Using a Zebrafish in Vivo Assay. Cancers. 2011;3(4):4102–13. doi: 10.3390/cancers3044102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini Simona, Gorgoulis Vassilis G, Halazonetis Thanos D. Genomic Instability–an Evolving Hallmark of Cancer. Nature Reviews Molecular Cell Biology. 2010;11(3):220–28. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- Nikiforov Yuri E, Nikiforova Marina N. Molecular Genetics and Diagnosis of Thyroid Cancer. Nature Reviews Endocrinology. 2011;7(10):569–80. doi: 10.1038/nrendo.2011.142. [DOI] [PubMed] [Google Scholar]
- Pendse J, Ramachandran PV, Na J, Narisu N, Fink JL, Cagan RL, Collins FS, Baranski TJ. A Drosophila Functional Evaluation of Candidates from Human Genome-Wide Association Studies of Type 2 Diabetes and Related Metabolic Traits Identifies Tissue-Specific Roles for dHHEX. BMC Genomics. 2013;14:136. doi: 10.1186/1471-2164-14-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenker Dennis, Riedel Maximilian, Brägelmann Johannes, Dammert Marcel A, Chauhan Rakhee, Knowles Phillip P, Lorenz Carina, et al. Drugging the Catalytically Inactive State of RET Kinase in RET-Rearranged Tumors. Science Translational Medicine. 2017;9(394) doi: 10.1126/scitranslmed.aah6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragazzi Moira, Ciarrocchi Alessia, Sancisi Valentina, Gandolfi Greta, Bisagni Alessandra, Piana Simonetta. Update on Anaplastic Thyroid Carcinoma: Morphological, Molecular, and Genetic Features of the Most Aggressive Thyroid Cancer. International Journal of Endocrinology. 2014 Aug;2014:790834. doi: 10.1155/2014/790834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read RD, Bach EA, Cagan RL. Drosophila C-Terminal Src Kinase Negatively Regulates Organ Growth and Cell Proliferation through Inhibition of the Src, Jun N-Terminal Kinase, and STAT Pathways. Molecular and Cellular Biology. 2004;24:6676–89. doi: 10.1128/MCB.24.15.6676-6689.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read RD, Goodfellow PJ, Mardis ER, Novak N, Armstrong JR, Cagan RL. A Drosophila Model of Multiple Endocrine Neoplasia Type 2. Genetics. 2005;171:1057–81. doi: 10.1534/genetics.104.038018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. A Systematic Analysis of Human Disease-Associated Gene Sequences in Drosophila Melanogaster. Genome Research. 2001;11(6):1114–25. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudrapatna VA, Bangi E, Cagan RL. Caspase Signalling in the Absence of Apoptosis Drives Jnk-Dependent Invasion. EMBO Reports. 2013;14:172–77. doi: 10.1038/embor.2012.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudrapatna VA, Bangi E, Cagan RL. A Jnk-Rho-Actin Remodeling Positive Feedback Network Directs Src-Driven Invasion. Oncogene. 2014 doi: 10.1038/onc.2013.232. [DOI] [PubMed] [Google Scholar]
- Rudrapatna Vivek A, Cagan Ross L, Das Tirtha K. Drosophila Cancer Models. Developmental Dynamics: An Official Publication of the American Association of Anatomists. 2012;241(1):107–18. doi: 10.1002/dvdy.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari Joshua K, Siau Evan D, Drilon Alexander. Targeting RET-Rearranged Lung Cancers with Multikinase Inhibitors. Oncoscience. 2017;4(3-4):23–24. doi: 10.18632/oncoscience.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro M, Dathan NA, Berlingieri MT, Bongarzone I, Paulin C, Grieco M, Pierotti MA, Vecchio G, Fusco A. Molecular Characterization of RET/PTC3; a Novel Rearranged Version of the RETproto-Oncogene in a Human Thyroid Papillary Carcinoma. Oncogene. 1994;9(2):509–16. [PubMed] [Google Scholar]
- Sarfaty Michal, Moore Assaf, Neiman Victoria, Dudnik Elizabeth, Ilouze Maya, Gottfried Maya, Katznelson Rivka, et al. RET Fusion Lung Carcinoma: Response to Therapy and Clinical Features in a Case Series of 14 Patients. Clinical Lung Cancer. 2016 Oct; doi: 10.1016/j.cllc.2016.09.003. [DOI] [PubMed] [Google Scholar]
- Smith-Hicks Constance L, Sizer Kurt C, Powers James F, Tischler Arthur S, Costantini Frank. The EMBO Journal. 4. Vol. 19. EMBO Press; 2000. C‐cell Hyperplasia, Pheochromocytoma and Sympathoadrenal Malformation in a Mouse Model of Multiple Endocrine Neoplasia Type 2B; pp. 612–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoshita M, Cagan RL. Modeling Human Cancers in Drosophila. Current Topics in Developmental Biology. 2017;121:287–309. doi: 10.1016/bs.ctdb.2016.07.008. [DOI] [PubMed] [Google Scholar]
- Sweetser DA, Froelick GJ, Matsumoto AM, Kafer KE, Marck B, Palmiter RD, Kapur RP. Ganglioneuromas and Renal Anomalies Are Induced by Activated RET(MEN2B) in Transgenic Mice. Oncogene. 1999;18(4):877–86. doi: 10.1038/sj.onc.1202376. [DOI] [PubMed] [Google Scholar]
- Takeda Haruna, Wei Zhubo, Koso Hideto, Rust Alistair G, Chin Christopher, Yew Kuan, Mann Michael B, Ward Jerrold M, Adams David J, Copeland Neal G, Jenkins Nancy A. Transposon Mutagenesis Identifies Genes and Evolutionary Forces Driving Gastrointestinal Tract Tumor Progression. Nature Genetics. 2015;47(2):142–50. doi: 10.1038/ng.3175. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, et al. RET, ROS1 and ALK Fusions in Lung Cancer. Nature Medicine. 2012;18:378–81. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- Uhlirova M, Jasper H, Bohmann D. Non-Cell-Autonomous Induction of Tissue Overgrowth by JNK/Ras Cooperation in a Drosophila Tumor Model. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13123–28. doi: 10.1073/pnas.0504170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccari Thomas, Bilder David. At the Crossroads of Polarity, Proliferation and Apoptosis: The Use of Drosophila to Unravel the Multifaceted Role of Endocytosis in Tumor Suppression. Molecular Oncology. 2009;3(4):354–65. doi: 10.1016/j.molonc.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken Koen JT, Bellen Hugo J. Chemical Mutagens, Transposons, and Transgenes to Interrogate Gene Function in Drosophila Melanogaster. Methods. 2014;68(1):15–28. doi: 10.1016/j.ymeth.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Larson DE, Cagan RL. Csk-Deficient Boundary Cells Are Eliminated from Normal Drosophila Epithelia by Exclusion, Migration, and Apoptosis. Developmental Cell. 2006;10:33–44. doi: 10.1016/j.devcel.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Vidal M, Warner S, Read R, Cagan RL. Differing Src Signaling Levels Have Distinct Outcomes in Drosophila. Cancer Research. 2007;67:10278–85. doi: 10.1158/0008-5472.CAN-07-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Wells S, Ryan A, Cagan R. ZD6474 Suppresses Oncogenic RET Isoforms in a Drosophila Model for Type 2 Multiple Endocrine Neoplasia Syndromes and Papillary Thyroid Carcinoma. Cancer Research. 2005;65:3538–41. doi: 10.1158/0008-5472.CAN-04-4561. [DOI] [PubMed] [Google Scholar]
- Vitale Giovanni, Gaudenzi Germano, Circelli Luisa, Manzoni Marco F, Bassi Andrea, Fioritti Niccolò, Faggiano Antongiulio, Colao Annamaria, NIKE Group Animal Models of Medullary Thyroid Cancer: State of the Art and View to the Future. Endocrine-Related Cancer. 2017;24(1):R1–12. doi: 10.1530/ERC-16-0399. [DOI] [PubMed] [Google Scholar]
- Vitale Giovanni, Gaudenzi Germano, Dicitore Alessandra, Cotelli Franco, Ferone Diego, Persani Luca. Zebrafish as an Innovative Model for Neuroendocrine Tumors. Endocrine-Related Cancer. 2014;21(1):R67–83. doi: 10.1530/ERC-13-0388. [DOI] [PubMed] [Google Scholar]
- Wells SA, Jr, Pacini F, Robinson BG, Santoro M. Multiple Endocrine Neoplasia Type 2 and Familial Medullary Thyroid Carcinoma: An Update. The Journal of Clinical Endocrinology and Metabolism. 2013;98:3149–64. doi: 10.1210/jc.2013-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SA, Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, Baudin E, et al. Vandetanib in Patients with Locally Advanced or Metastatic Medullary Thyroid Cancer: A Randomized, Double-Blind Phase III Trial. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology. 2012;30:134–41. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells Samuel A, Jr, Asa Sylvia L, Dralle Henning, Elisei Rossella, Evans Douglas B, Gagel Robert F, Lee Nancy, et al. Revised American Thyroid Association Guidelines for the Management of Medullary Thyroid Carcinoma. Thyroid: Official Journal of the American Thyroid Association. 2015;25(6):567–610. doi: 10.1089/thy.2014.0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby LF, Schlosser T, Manning SA, Parisot JP, Street IP, Richardson HE, Humbert PO, Brumby AM. An in Vivo Large-Scale Chemical Screening Platform Using Drosophila for Anti-Cancer Drug Discovery. Disease Models & Mechanisms. 2013;6:521–29. doi: 10.1242/dmm.009985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Pastor-Pareja JC, Xu T. Interaction between Ras(V12) and Scribbled Clones Induces Tumour Growth and Invasion. Nature. 2010;463:545–48. doi: 10.1038/nature08702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarish Kumar Yadav, Srikrishna Saripella, Gupta Subash Chandra. Cancer Drug Development Using Drosophila as an in Vivo Tool: From Bedside to Bench and Back. Trends in Pharmacological Sciences. 2016;37(9):789–806. doi: 10.1016/j.tips.2016.05.010. [DOI] [PubMed] [Google Scholar]
- Zheng Ming-Wu, Zhang Chun-Hui, Chen Kai, Huang Mei, Li Ya-Ping, Lin Wan-Ting, Zhang Rong-Jie, et al. Preclinical Evaluation of a Novel Orally Available SRC/Raf/VEGFR2 Inhibitor, SKLB646, in the Treatment of Triple-Negative Breast Cancer. Molecular Cancer Therapeutics. 2016;15(3):366–78. doi: 10.1158/1535-7163.MCT-15-0501. [DOI] [PubMed] [Google Scholar]