

Ultralong room temperature phosphorescence is achieved from amorphous organic materials toward information encryption.
Abstract
Ultralong room temperature phosphorescence (URTP) emitted from pure amorphous organic molecules is very rare. Although a few crystalline organic molecules could realize URTP with long lifetimes (>100 ms), practical applications of these crystalline organic phosphors are still challenging because the formation and maintenance of high-quality crystals are very difficult and complicated. Herein, we present a rational design for minimizing the vibrational dissipation of pure amorphous organic molecules to achieve URTP. By using this strategy, a series of URTP films with long lifetimes and high phosphorescent quantum yields (up to 0.75 s and 11.23%, respectively) were obtained from amorphous organic phosphors without visible fluorescence and phosphorescence under ambient conditions. On the basis of the unique features of URTP films, a new green screen printing technology without using any ink was developed toward confidential information encryption and decryption. This work presents a breakthrough strategy in applying amorphous organic materials for URTP.
INTRODUCTION
Ultralong room temperature phosphorescence (URTP) has attracted considerable attention because its ultralong-lived excited state enables visual observation of long-lived emission from seconds to several hours (1, 2). Thus, URTP materials show highly potential applications in various fields such as biological imaging, organic light-emitting diodes (3, 4), information storage (5), photodynamic therapy (6), sensing devices (7), and security protection (8). For instance, the use of URTP materials for in vitro and in vivo imaging allows easy elimination of the interference from autofluorescence in cell organelles, as well as any background fluorescence (9, 10). However, developing highly efficient pure organic compounds with URTP is still a great challenge. In general, phosphors with URTP are based on metal-containing inorganic materials and organometallic complexes. Because (i) the generation of long-lived triplet excitons is not common in organic molecules without heavy atoms and (ii) triplet excitons in organic materials are highly localized and easily consumed by vibrational dissipation and oxygen (11–13), only a few pure organic molecules are able to emit efficient URTP (14–18).
Two vital factors should be considered carefully when designing novel pure organic molecules with URTP: (i) promoting the intersystem crossing (ISC) from the lowest excited singlet state (S1) to the triplet states (Tn) in organic molecules through efficient spin-orbit coupling and (ii) the suppression of nonradiative relaxation pathways from T1 to S0 as much as possible. On the basis of these molecular design rules, an effective approach to achieving URTP is through the formation of crystals from phosphor molecules. Bolton et al. (19) developed a cocrystal material with high–quantum yield (55%) URTP by enhancing the ISC through halogen bonding. Xie et al. (20) successfully designed a novel crystal with a URTP lifetime of up to 0.748 s by modulating the molecular packing in a single crystal. In addition, An et al. (21) explored a series of pure organic molecules through an H-aggregation method in crystals to achieve a phosphorescent lifetime of up to 1.35 s. In these cases, favorable molecular configuration and intermolecular interactions in organic crystals are necessary to achieve URTP under atmospheric conditions. Because manipulating molecular packing and interactions in crystal forms is quite restricted (17, 22–25), the development of pure amorphous organic materials for URTP is highly desired.
To achieve amorphous organic materials with URTP, an efficient approach is to induce the promotion of ISC and the suppression of molecular motions in these systems. Thus, embedding small molecular phosphors into rigid polymer matrices (17, 18, 22–25), steroidal molecules (22), and cavities of macrocycle hosts (26, 27) to obtain amorphous organic materials with URTP has been reported. Although some small host matrices could effectively minimize the quenching of triplet excitons, phase separation could easily occur. Phosphor deuteration could depress nonradiative transition, but the deuterium substitution process is often very difficult and complicated. On the other hand, film-forming polymer materials with persistent emission are attractive for optical device fabrication and processing (23). Therefore, enabling the restriction of nonradiative relaxation of amorphous organic compounds in polymer matrices to achieve URTP is an ideal solution.
Herein, a rational strategy has been developed to achieve pure amorphous organic materials with URTP. We designed a simple organic guest molecule, hexa-(4-carboxyl-phenoxy)-cyclotriphosphazene (donated as G), containing six extended benzoic acid arms (Fig. 1A). Although G cannot emit any visible fluorescence or phosphorescence under an ultraviolet (UV) lamp at room temperature, its six extended aromatic carbonyl units can provide enough n orbitals to trigger the ISC from S1 to Tn for a large amount of triplet-excitation generation (Fig. 1B). At the same time, –COOH groups of G are able to form hydrogen bonds (H-bonds) with a selected amorphous polymer matrix, that is, poly(vinyl alcohol) (PVA). To suppress nonradiative relaxation pathways of triplet excitations, we purposely introduced two types of intermolecular noncovalent H-bonds and one type of covalent cross-linked bond into the system (Fig. 1). The intermolecular H-bonds between guest molecules were introduced to promote ISC processes as well as to restrict the vibration of guest molecules. Other intermolecular H-bonds between PVA hosts and guests were incorporated to efficiently hamper their diffusional motions. The formation of cross-linked bonds between PVA hosts by UV irradiation could further compress their vibrational dissipation.
Fig. 1. Rational design strategy of URTP from amorphous organic materials.
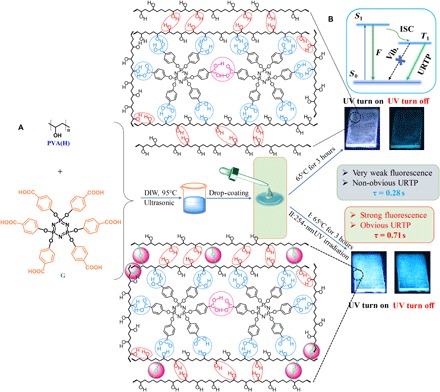
(A) Chemical structures of G and PVA and the fabrication of G-doped PVA films for URTP. (B) Schematic illustration of URTP processes in G-doped PVA films. DIW, deionized water.
Very weak URTP from G-doped PVA films was observed at room temperature, and the phosphorescent intensity (Ip), lifetime (τp), and quantum yield (Φp) were measured only up to 19.51 arbitrary units (a.u.), 0.28 s, and 2.85%, respectively. Significantly, outstanding URTP was achieved from G-doped PVA films after 254-nm light irradiation for 65 min, due to further suppression of vibrational dissipation by PVA cross-linking. The obtained values of Ip, τp, and Φp are as high as 315.47 a.u., 0.71 s, and 11.23%, which are about 16, 2.5, and 3.9 times higher than that of G-doped PVA films without cross-linking bonds (table S1), respectively. It should be noted that there was no observable fluorescence and phosphorescence emission for the original G and PVA matrix at room temperature. The URTP observation from an initial nonfluorescent and nonphosphorescent system might be ascribed to the idea that the formation of H-bonds and cross-linking bonds within G-doped PVA could effectively minimize the vibrational dissipation of the G molecule and the PVA polymer and promote the ISC process from S1 to Tn (Fig. 1B) (28, 29). Because persistent emission intensity of G-doped PVA films is highly dependent on the irradiation time, we successfully realized the confidential information encryption and decryption process by a green screen printing technology without using any inks. Because strong phosphorescence of G-doped PVA films could be quenched by water under ambient conditions and recovered by incubating at 65°C for several minutes, reversible on/off switching of the phosphorescence signal for multiple information encryption and decryption cycles was achieved.
RESULTS
The synthetic procedure of compound G is shown in fig. S1. G was obtained in 94% yield after three steps of reactions from p-hydroxybenzoic acid and a phosphonitrilic chloride trimer. Fourier transform infrared spectroscopy (FTIR), UV-visible (UV-Vis) spectroscopy, 1H/13C nuclear magnetic resonance (NMR) spectroscopy, and electrospray ionization mass spectrometry (ESI-MS) confirmed the desired molecule with high purity (fig. S2). A series of G-doped PVA films were fabricated using a simple drop-coating method from 100% hydrolyzed PVA (PVA-100) aqueous solution (10 g/liter) containing different concentrations of G (0.2, 0.5, 1, 3, 5, and 10 mg/ml in PVA-100 solution, denoted as PVA-100-0.2mg, PVA-100-0.5mg, PVA-100-1mg, PVA-100-3mg, PVA-100-5mg, and PVA-100-10mg, respectively). The detailed fabrication process (Fig. 1) of G-doped PVA films can be found in the “Preparation of G-doped PVA films” section. PVA was chosen as the polymer matrix in this work because it is a common polyhydroxy polymer that can easily form intermolecular and intramolecular H-bonds by itself or with various functionalized small organic dopants at room temperature. We did not observe any visible fluorescence or phosphorescence from a tetrahydrofuran (THF) solution or the solid state of G under ambient conditions. The resulting G-doped PVA thin films show fluorescence emission at around 460 nm and phosphorescence emission at 480 nm (fig. S3A and Fig. 2A). The phosphorescence spectra of G-doped PVA films (Fig. 2A) were obtained, and lifetimes of 0.04 to 0.28 s (Fig. 2B and table S1) were clearly realized at 480 nm emission to afford URTP. As such, a series of novel amorphous organic URTP materials were achieved successfully from phosphor G without any fluorescence or phosphorescence. We believe that this unique feature can be ascribed to strong intramolecular H-bonds of a rigid PVA matrix, as well as to strong intermolecular H-bonds between PVA chains and G, which effectively minimize the vibrational dissipation between PVA chains and the G molecule and enhance the ISC process from S1 to Tn as much as possible (Fig. 1B) (24, 30, 31).
Fig. 2. Photophysical properties of the PVA film doped with different concentrations of G.
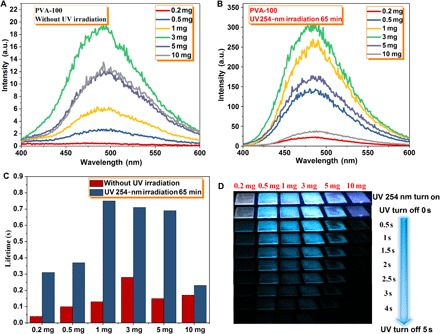
Phosphorescence spectra of the PVA film doped with different concentrations of G (λex = 280 nm) at room temperature. (A) Unirradiated. (B) 254-nm UV light irradiation for 65 min. (C) Phosphorescent lifetime of these G-doped PVA films at room temperature, monitored at 480 nm and λex = 280 nm. (D) Snapshots of these G-doped PVA films upon irradiation by a 254-nm UV lamp for 65 min, followed by recording URTP at different time intervals in the dark under ambient conditions.
To our surprise, the fluorescence (fig. S3B) and URTP properties of as-prepared G-doped PVA films were significantly enhanced upon UV irradiation of 254 nm for 65 min. After irradiating using a 254-nm UV lamp for 65 min, the values of Ip, τp, and Φp are as high as 22.83 a.u., 0.31 s, and 4.26% for PVA-100-0.2mg; 143.84 a.u., 0.37 s, and 6.44% for PVA-100-0.5mg; 249.69 a.u., 0.75 s, and 9.18% for PVA-100-1mg; 315.47 a.u., 0.71 s, and 11.23% for PVA-100-3mg; 175.09 a.u., 0.69 s, and 5.51% for PVA-100-5mg; and 35.39 a.u., 0.23 s, and 3.09% for PVA-100-10mg, respectively (fig. S4 and table S1), which are 2.6 to 58, 1.4 to 7.8, and 3 to 6 times (for Ip, τp, and Φp, respectively) higher than that of the corresponding unirradiated G-doped PVA films. This unique feature might be attributed to the formation of cross-linked bonds between PVA chains upon 254-nm UV irradiation, which further suppresses the vibrational dissipation of the PVA matrix (32, 33). For unirradiated or irradiated G-doped PVA films, the URTP properties are strongly dependent on the doping concentration of phosphor G. Figure 2 and table S1 show the effect of G with different doping concentrations. An optimum G concentration was determined to be 3 mg/ml in the PVA-100 aqueous solution (10 g/liter). When the concentration of G exceeded 3 mg/ml (that is, 5 and 10 mg/ml), the self-quenching effect resulted in low Ip and τp, and the PVA matrix cannot provide enough hydroxyl groups to minimize the vibrational motions of G through forming H-bonds. At very low concentrations, G molecules are not enough to facilitate necessary H-bonding between themselves for efficient spin-orbit coupling, thus resulting in minimal Ip and τp. Upon irradiation from a portable 254-nm UV lamp, all irradiated G-doped PVA films exhibit blue-green fluorescence in air at 298 K (Fig. 2D and movie S1). After the removal of the excitation source, these G-doped PVA films show persistent emission with a long duration of 3 to 5 s. The time-dependent luminescent spectra of G-doped URTP PVA films after turning off the UV light (delayed time: approximately 50 ms) at 298 K reveal that (fig. S5) the irradiated PVA-100-1mg film afforded a persistent emission time of 7.76 s (luminescent intensity at 1 a.u.), which is 2.3 times longer than that of the corresponding unirradiated film, further confirming that excellent amorphous organic URTP can be achieved through our developed strategy.
For unirradiated or irradiated G-doped PVA films, when the temperature dropped to 77 K (fig. S3D and table S1), both phosphorescent intensity and lifetime significantly increased because the cryogenic temperature can restrict the vibrational motions to decrease the nonradiative rate. In addition, an obvious blue shift of the maximum phosphorescence emission peak at 77 K was observed as compared to the phosphorescence at room temperature. Such blue shift emission should arise from the changes in the configuration of the transitions at cryogenic temperatures (20). Notably, the values of τp for the irradiated PVA-100-1mg film (0.75 s) and PVA-100-3mg film (0.71 s) at room temperature are even longer than that of the unirradiated PVA-100-1mg film (0.56 s) and PVA-100-3mg film (0.68 s) at 77 K, implying that the formation of abundant cross-linked bonds by UV irradiation is more efficient than that of the cryogenic temperature effect for the suppression of molecular motions between host and guest.
To better understand how UV irradiation influences the URTP properties of G-doped films, taking the PVA-100-3mg film as an example, its URTP properties under a 254-nm UV light at different irradiation times were studied in detail. It was notable that the URTP properties of the as-prepared PVA-100-3mg film were highly dependent on the irradiation time. Along with the increase of irradiation time from 0 to 65 min at 298 K, Ip, τp, Φp, and the persistent emission time of luminescent intensity at 1 a.u. gradually increased from 18.45 to 297.27 a.u., from 0.28 to 0.71 s, from 2.85 to 11.23%, and from 3.06 to 7.73 s, respectively (Fig. 3 and table S2). However, when the irradiation time exceeded 65 min, Ip, τp, and the persistent emission time of the PVA-100-3mg film gradually decreased. It was found that changes in Ip for the PVA-100-3mg film with a different irradiation time at 77 K were irregular, although the Ip values were higher than those at 298 K. The values of τp exhibit a similar trend to that at 298 K, except for the sample irradiated for 100 min. The τp value of the film irradiated for 65 min at 298 K was very close to that at 77 K, which were 0.71 and 0.88 s, respectively. These results further confirm that the formation of cross-linked bonds between the PVA matrix in this system plays a key role to achieve amorphous organic URTP materials by suppressing nonradiative relaxation pathways between hosts and guests. In addition, persistent emission for several seconds could be easily observed after the removal of the excitation source (Fig. 3D and movie S2). Thus, 65 min was taken as the optimum irradiation time for the PVA-100-3mg film.
Fig. 3. Irradiation time-dependent measurements of the photophysical properties of the PVA-100-3mg film.
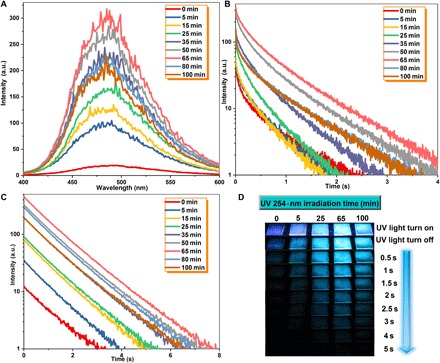
(A) Phosphorescence spectra of the PVA-100-3mg film upon light irradiation at different times (λex = 280 nm). (B) Phosphorescent decay profiles of the PVA-100-3mg film at different irradiation times at room temperature (monitored at 480 nm, λex = 280 nm). (C) Time-dependent luminescent spectra of the PVA-100-3mg film at different irradiation times followed by turning off the UV light (delayed time: approximately 50 ms) at room temperature. (D) Comparison of the emission observed at different time intervals before and after turning off the light excitation at room temperature.
FTIR spectra provide powerful and direct evidence for the formation of intermolecular H-bond interactions between PVA and fillers (32). As shown in fig. S6A, the νO–H value of pure PVA is 3253 cm−1, arising from intermolecular and intramolecular H-bond interactions. The νO–H value of PVA shows upward shifts upon the addition of different amounts of G, and about 27 cm−1 blue shift was observed for G-PVA-5mg. These observations reveal that the G molecule has a strong capability to form H-bonds with the PVA chain. To further probe into the nature of the obvious enhancement of URTP properties from G-doped PVA films under ambient conditions, 1H NMR experiments were conducted and analyzed. These as-prepared G-doped PVA films were scraped off, dissolved in dimethyl sulfoxide (DMSO)–d6 solvent, and passed through a filter before the NMR studies. Figure S7 shows 1H NMR spectra of the PVA-100 film, G, and unirradiated/irradiated G-doped PVA-100 films. The peak at 13.04 parts per million (ppm) ascribed to the –COOH proton of G completely disappeared in G-doped PVA films, even at high G-doped concentrations (PVA-100-5mg). In addition, three peaks that appeared at 4.25 to 4.69 ppm derived from isotactic, heterotactic, and syndiotactic structures of hydroxyl groups in the pure PVA-100 film became a broad peak at 4.48 ppm in G-doped PVA films (33), even at very low G-doped concentrations (PVA-100-1mg). These observations evidently prove the formation of strong intermolecular H-bonds between G and PVA-100 through carboxyl and hydroxyl groups (33). Aromatic protons of the G molecule in G-doped PVA films marked as B and C in Fig. 4 exhibit obvious shifts upon UV light irradiation. Along with the increase of G-doping concentration in PVA films, these protons show a trend to gradually recover to the original locations in pure G. A possible explanation is that, when increasing the G-doping concentration, the PVA matrix cannot provide enough hydroxyl groups to bind with the carboxyl groups of the G molecule through the formation of intermolecular H-bonds, thus resulting in the gradual recovery of aromatic proton positions.
Fig. 4. Schematic diagram for achieving URTP in G-doped PVA films and irradiation time-dependent 1H NMR spectra of the PVA-100-3mg film.
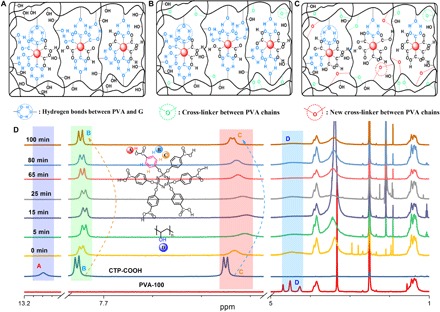
(A) Unirradiated G-doped PVA films with substantial H-bond formation from G-G, G-PVA, and PVA-PVA. (B) Irradiated G-doped PVA films by a 254-nm UV light (irradiation time ≤65 min) with cross-linked bond (C–O–C) formation between PVA chains. (C) Irradiated G-doped PVA films (irradiation time >65 min) with the rearrangement of H-bonds from G-G, G-PVA, and PVA-PVA. (D) 1H NMR spectra of G, PVA-100, and PVA-100-3mg films at different irradiation times in DMSO-d6. The inset shows the chemical structures of G (up) and PVA-100 (down).
On the basis of the obtained results, a rational mechanism for achieving amorphous organic URTP materials was proposed. First, the H-bonds formed between PVA-PVA, G-PVA, and G-G (Fig. 4A) were successfully proven by the disappearance of proton A, the shifting of protons B and C, and the broadening of proton D in NMR spectra of G-doped PVA films (fig. S7). The formation of massive amounts of H-bonds can effectively suppress the molecular motions from the PVA matrix and the G molecule as well as promote the ISC process of S1 to Tn. Thus, a series of G-doped PVA films as amorphous organic URTP materials with relatively weak phosphorescence were achieved, showing the longest τp of up to 0.28 s. Second, to further optimize the URTP materials, suppressing their vibrational dissipation is necessary. With the increase of irradiation time by the 254-nm UV light, protons B and C gradually shifted to upfield, from 7.84 ppm (0 min) to 7.82 ppm (65 min) for proton B, and from 7.81 ppm (0 min) to 7.80 ppm (65 min) for proton C (Fig. 4D). Meanwhile, three proton peaks at 4.25 to 4.69 ppm (D) derived from isotactic, heterotactic, and syndiotactic structures of hydroxyl groups in pure PVA-100 changed to a broad peak at 4.48 ppm, and the peak intensity gradually decreased with the increase of irradiation time. These results indicate the formation of cross-linked bonds (C–O–C) between PVA chains well (Fig. 4B). Thus, effective amorphous organic URTP materials were obtained through further minimizing molecular motions between PVA chains. The Ip, τp, and Φp values were enhanced 2.6 to 58, 1.4 to 7.8, and 3 to 6 times as compared with unirradiated G-doped PVA films, respectively. Additional evidence from FTIR, Raman, and x-ray photoelectron spectroscopy (XPS) further confirmed the formation of cross-linking between PVA chains in irradiated G-doped PVA films. In FTIR spectra, the peak of hydroxyl groups at 3000 to 3700 cm−1 gradually decreased upon increasing the irradiation time, and a new vibrational peak was observed at 1128 cm−1, due to the stretching vibration of C–O–C (fig. S6B) (34). A new Raman band that appeared at 1098 cm−1 was assigned to the C–O–C stretching vibration (fig. S6C) (35). The peak intensity of C–O–C at approximately 286.87 eV increased significantly upon the increase of irradiation time in the deconvoluted C1s XPS spectra of G-doped PVA films (fig. S8) (36).
Because hydroxyl groups in PVA chains are almost consumed by forming the cross-linked bonds (C–O–C) when the irradiation time exceeds 65 min, the H-bonds formed between PVA chains and the G molecule would be broken by further irradiation to release free hydroxyl groups, and subsequently, these free hydroxyl groups continue to form new cross-linked bonds (Fig. 4C). The fact that protons B and C in the PVA-100-3mg film nearly recovered to their original locations after 100 min of irradiation greatly verified this hypothesis (Fig. 4D). Although the formation of more cross-linked bonds can further suppress the vibrational dissipation of PVA chains, decreased H-bonds between G and PVA significantly increase the molecular motions of the G molecule and reduce the efficiency of ISC from S1 to Tn. Thus, the URTP properties of PVA films decreased when the irradiation time was 80 and 100 min.
Obvious changes of UV-Vis spectra in the 250- to 350-nm range for irradiated G-doped PVA films once again confirm the cross-linking in this system (fig. S9, A and C). In particular, the observed decrease in optical bandgap (Eg) upon increasing the irradiation time could be understood by the formation of stable charge transfer complexes (fig. S9, B and D). Like a defect in inorganic persistent emission materials, charge transfer complexes can effectively stabilize the triplet excitations to achieve persistent phosphorescence emission (1, 2, 21, 32, 35). The results of differential scanning calorimetry show that Tg of irradiated G-doped PVA films gradually increased with the increase in irradiation time (fig. S10A) (35). The temperature of 95% weight loss for all irradiated PVA films also gradually increased upon increasing the irradiation time (fig. S10B). These results are direct evidence of the interaction and cross-linking between the PVA chain and G; that is, the G-doped matrix becomes more rigid after UV irradiation. Powder x-ray diffraction (XRD) patterns of irradiated G-doped PVA films with different irradiation times are shown in fig. S10C. The diffraction intensity at 20.63° gradually decreased with the increase in irradiation time, whereas the intensity at 22.23° exhibited the opposite result. The variations in these XRD patterns reveal that UV irradiation changes the microstructure of G-doped PVA films with the formation of a cross-linking network (35). The changes in the microstructure of irradiated G-doped PVA films were also observed by scanning electron microscope (SEM) images (fig. S11). The surface is very smooth and uniform for unirradiated G-doped PVA films, even at high G-doping concentrations (PAV-100-3mg). A rough film surface and the formation of small-scale particles were found for irradiated films, and this observation became more and more obvious with the increase of irradiation time, which can be ascribed to the photocrosslinking reaction through the dehydration of hydroxyl groups in PVA chains (37). In short, all these results well confirm the formation of cross-linked bonds (C–O–C) in irradiated G-doped PVA films through the dehydration of hydroxyl groups in PVA chains.
To understand the conditions for the application, we tested the temperature-dependent and excitation wavelength–dependent phosphorescence spectra and decay profiles of G-doped PVA films. For unirradiated and irradiated PVA films, Ip and τp decreased almost linearly upon increasing the temperature (figs. S12 and S13 and table S3), and these G-doped PVA films still displayed good URTP properties even at high temperatures (363 K). As shown in fig. S13 (C and D), dramatic changes of Ip and τp can be easily observed in the temperature range of 293 to 363 K. To monitor thermal response reversibility of irradiated G-doped PVA films, the in situ phosphorescence spectra and decay profiles with a heating and cooling cycle in the temperature range of 293 to 363 K were recorded, and reversible Ip and τp were repeated in eight continuous cycles (fig. S12, C and D). These results clearly demonstrate that these films could act as efficient temperature sensors in the temperature range of 293 to 363 K. The excitation wavelength–dependent phosphorescence spectra and decay curves reveal that ultralong phosphorescence emission of unirradiated/irradiated G-doped PVA films at 480 nm could be efficiently excited within the range of 240 to 320 nm. A relatively high τp value (0.39 s) was still detected even if an excitation wavelength of 320 nm was used for irradiated G-doped PVA films (fig. S14 and table S4). These results clearly indicate that as-prepared G-doped PVA films could be used in various conditions.
To further validate our hypothesis, that is, the formation of H-bonds and cross-linked bonds plays a key role to achieve amorphous organic URTP, a negative control study was carried out. Eighty-seven percent and 80% hydrolyzed PVA (denoted as PVA-87 and PVA-80, respectively) were chosen as polymer matrices to fabricate G-doped PVA films. PVA-87 and PVA-80 have approximately 13 and 20% fewer H-bonding sites than PVA-100, which lead to some drawbacks as compared with the PVA-100 matrix. First, less opportunity to form interpolymeric H-bonding reduces the rigidity of the PVA matrix. Second, the decreased H-bonding sites also reduce the chances for the formation of H-bonds between PVA chains and the G molecule. Third, the chance for the formation of cross-linked bonds would be reduced because of fewer hydroxyl groups in PVA-87 and PVA-80 chains as compared with that of PVA-100. Thus, the suppression of rotational and vibrational motions of PVA-87 and PVA-80 chains should be less efficient than that of PVA-100 chains. As anticipated, G-doped PVA-87 and PVA-80 films presented a dramatic decrease in URTP properties at room temperature. The emission intensity at 480 nm decreases from 310.41 a.u. for the PVA-100-3mg film to 14.31 a.u. for the PVA-80-3mg film (Fig. 5A), the corresponding phosphorescence lifetime reduces from 0.71 to 0.24 s (Fig. 5B), and the phosphorescent quantum yield decreases from 11 to 1%. The time of persistent emission luminescence (emission intensity at 1 a.u.) after the removal of the excitation source (delayed time: approximately 50 ms) was 7.18 s for the PVA-100-3mg film, 2.21 s for the PVA-87-3mg film, and 1.34 s for the PVA-80-3mg film. In terms of the temperature-dependent and excitation wavelength–dependent phosphorescent intensity at 480 nm and lifetime (Fig. 5, C and D, and fig. S15, C and D), both PVA-87-3mg and PVA-80-3mg films exhibit dramatic reduction in Ip and τp due to a substantial decrease in the hydroxyl groups of PVA-87 and PVA-80 chains. In addition, a structurally similar polymer matrix without hydroxyl groups in the molecular chain, poly(vinyl acetate) (PVAc), was chosen as a host to prepare the G-doped PVAc films, and neither fluorescence nor phosphorescence could be observed. Some common polymer matrices without hydroxyl groups were also chosen as hosts, such as polystyrene, poly(methyl methacrylate), and poly(vinylpyrrolidone). Again, we did not observe any persistent emission from the corresponding G-doped polymer films. These results clearly demonstrate that the formation of intermolecular or intramolecular H-bonding and cross-linked bonds to minimize the vibrational dissipation of PVA chains and the G molecule is a very effective approach to achieving amorphous organic URTP materials. Notably, the phosphorescence spectra and decay curves at cryogenic temperatures show a similar trend to that at room temperature, whereas some enhancements of τp from room temperature to 77 K [0.28 for PVA-100-3mg (from 0.71 to 0.99 s), 0.39 for PVA-87-3mg (from 0.49 to 0.88 s), and 0.61 for PVA-80-3mg (from 0.24 to 0.85 s)] were observed, again confirming that the formation of enough H-bonds and cross-linked bonds to suppress nonradiative relaxation pathways between hosts and guests is very efficient.
Fig. 5. Photophysical properties of different G-doped PVA matrices upon irradiation by a 254-nm UV light for 65 min.
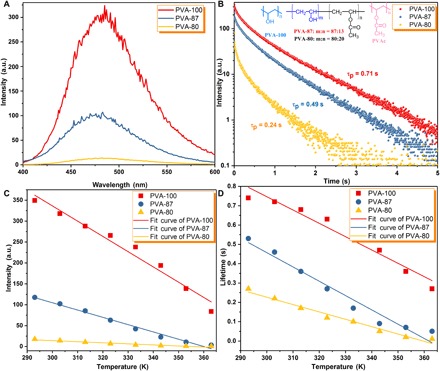
(A) Phosphorescence spectra of PVA-100-3mg, PVA-87-3mg, and PVA-80-3mg films upon irradiation by a 254-nm UV 1ight (λex = 280 nm) for 65 min. (B) Phosphorescent decay profiles of PVA-100-3mg, PVA-87-3mg, and PVA-80-3mg films (monitored at 480 nm, λex = 280 nm). The inset shows the chemical structures of PVA-100, PVA-87, PVA-80, and PVAc. (C) Plots of phosphorescence emission intensity at 480 nm versus temperature (293 to 363 K) for PVA-100-3mg, PVA-87-3mg, and PVA-80-3mg films (λex = 280 nm). (D) Plots of phosphorescent lifetime versus temperature (293 to 363 K) for PVA-100-3mg, PVA-87-3mg, and PVA-80-3mg films (monitored at 480 nm, λex = 280 nm).
To validate whether this rational design could be applied to various organic molecules, five potential phosphors (G1, G2, G3, G4, and G5) with a similar structure to G were successfully prepared (fig. S16). We could not observe any visible fluorescence and phosphorescence emission from G1 to G5 using a 254-nm UV light under ambient conditions. Surprisingly, obvious persistent blue-colored phosphorescence emission was observed by the naked eye when these compounds were doped into the PVA matrix to form G1-, G2-, G3-, G4-, and G5-doped PVA films. For G1, G4, and G5, persistent phosphorescence emission was achieved because of the formation of H-bonds between the carboxyl groups of G1, G4, and G5 and the hydroxyl groups of PVA chains to suppress their molecular motions. After irradiation for 65 min by a 254-nm UV lamp, phosphorescent intensity and lifetime were obviously enhanced because the vibrational dissipation between PVA chains was further compressed by cross-linked bonds. For G2 and G3, although strong H-bonds cannot be effectively formed between G2/G3 and PVA chains on account of less carboxyl groups in their molecular structures, persistent emission was still observed. This observation could be attributed to weak H-bonding formation between carbonyl groups in the G2/G3 and hydroxyl groups in the PVA matrix to fix their molecular motions. Better URTP properties were obtained after irradiation by a 254-nm UV lamp, especially for the G2-doped PVA film with a lifetime increase from 0.25 to 0.55 s. This phenomenon further proves that the cross-linking between PVA matrices by light irradiation plays a very important role to enhance URTP properties in amorphous organic materials.
DISCUSSION
The development of stimulus-responsive luminescent materials has attracted tremendous interest due to their potential applications in security protection such as information storage, encryption, and anti-counterfeiting. When using conventional luminescent materials, the data or information recorded directly by these materials is usually visible under either ambient conditions or UV light, which is harmful to their practical application for confidential information protection. Therefore, it remains a great challenge to develop alternative luminescent materials with good confidential encryption abilities for high-level information storage and security protection. Amorphous organic phosphorescent materials with ultralong lifetimes could be used as security inks to meet the requirements. Because persistent emission intensity of G-doped PVA films is highly dependent on the irradiation time, a novel green screen printing technology using G-doped PVA films without any inks was developed successfully. The detailed printing procedure is described as follows. First, compound G was dispersed into the PVA-100 aqueous solution (10 g/liter), and a homogeneous solution was obtained after ultrasonication for 30 min. Then, G-doped PVA films were fabricated through drop-coating the G-PVA solutions on clean glass substrates, followed by drying at 65°C for 3 hours. At this stage, the luminescence was weak and almost invisible. Second, a homemade screen plate with cartoon patterns (elephant, flower, and snowflake) or one leaf of fern was covered on G-doped PVA films. Third, to finish the screen printing, the covered PVA films were irradiated by a 254-nm UV lamp for 65 min. No patterns on the films were observed under sunlight. Clear patterns were observed upon exposure to the 254-nm UV lamp, and these patterns were still visible for several seconds after turning off the excitation source because of the existence of persistent emission. As compared with traditional screen printing technology that needs organic inks, the present method is green, convenient, and cheap. The unique feature with persistent emission makes these G-doped PVA films highly potential candidates for anti-counterfeiting and security applications.
Several complicated patterns could be easily printed on the basis of the green screen printing technology (Fig. 6, A and B), such as the logo of a lotus flower, the logo of Chongqing University of Technology, a bar code, and a QR code. These patterns were invisible under sunlight. However, clear patterns were observed under the 254-nm UV lamp (Fig. 6A and movie S3). To explore more advanced anti-counterfeiting technology, a common green emission material, aluminum–tris-quinolate (AlQ3), was introduced into the G-doped PVA films. Because the fluorescence emission of the G-doped PVA film could be completely covered by the strong green emission of AlQ3 under the 254-nm UV lamp, the lotus flower pattern prepared by green screen printing technology was invisible under the 254-nm UV lamp. A clear lotus flower pattern was visible when the 254-nm UV lamp was turned off, due to persistent emission of G-doped PVA films and the short lifetime of AlQ3 (τ < 10 ns) (Fig. 6C and movie S4). The lotus flower pattern gradually became invisible under ambient conditions, and the pattern completely disappeared after 60 min (Fig. 6D). Because water molecules in air could permeate into G-doped PVA films to break H-bonds between PVA and G at room temperature (fig. S17, A and B), about 11 cm-1 red shift for stretching vibration of the hydroxyl group (νO–H) and 1 cm-1 blue shift for the νC=O group were observed when the standing time of the lotus flower film was prolonged from 0 to 60 min. Reverse changes were found when the film was incubated at 65°C for different times, because water molecules inside the G-doped PVA film might move out to form new H-bonds between G and PVA. Corresponding changes from powder XRD patterns and SEM images under different conditions were also observed (fig. S17, C to G). These results indicate that water molecules could break the H-bond interaction between PVA-PVA and PVA-G, finally dissociating the matrix and phosphors. The dissolved phosphors cannot emit any visible fluorescence and phosphorescence (Fig. 6E). The lotus flower pattern could be gradually recovered by incubating at 65°C for several minutes. This reversible behavior was further confirmed by phosphorescence emission spectra under different conditions (fig. S18). On the basis of this behavior, reversible on/off switching of the phosphorescence signal for multiple information encryption and decryption cycles could be achieved.
Fig. 6. Luminescent G-doped PVA film patterns fabricated via green screen printing.
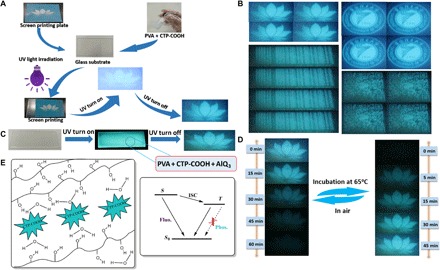
(A) Green screen printing without any inks using the lotus flower screen plate. First, G-doped PVA films were fabricated through drop-coating the G-PVA solution on a clean glass substrate, followed by drying at 65°C for 3 hours. Second, the screen plate of the lotus flower was covered on G-doped PVA films. Third, the covered PVA films were irradiated by a 254-nm UV lamp for 65 min to finish the screen printing progress. No patterns on the films were observed under sunlight. Clear patterns were observed upon exposure to the 254-nm UV lamp, and these patterns were still visible for several seconds after turning off the UV lamp. (B) Several complicated patterns by green screen printing technology after removing the excitation source. (C) More advanced anti-counterfeiting technology through doping AlQ3 into G-doped PVA films. (D) Reversible patterns of the lotus flower after removing the excitation source under different conditions. (E) Schematic illustration of the URTP process in the G-doped PVA films.
In addition, these films could serve as anti-counterfeit inks on account of their persistent emission properties (fig. S19). For example, when writing four letters “URTP” on a common printing paper, the letter “U” was written using the common rare-earth complex Eu(TTA)3 (dissolved in THF) with a short luminescent lifetime (<2 ms), and the other three letters “RTP” were written using our G-doped PVA aqueous solution with a very long luminescent lifetime (>200 ms). All these letters can be visible under a 254-nm UV light. Although the letter “U” cannot be observed because of the short lifetime of Eu(TTA)3 when turning off the UV light, the three letters “RTP” could still be visible under the same conditions.
In summary, we have presented a rational design to achieve long-lived phosphorescence emission using metal-free and heavy atom–free amorphous organic materials through the formation of strong intramolecular and intermolecular interactions to suppress nonradiative relaxation pathways. Our study has revealed that the cross-linked bonds between PVA chains formed under UV irradiation play a significant role to realize persistent phosphorescence emission (τp up to 0.75 s) in amorphous organic materials. Notably, the experimental data at cryogenic temperatures indicate that the suppression ability of cross-linked bonds to molecular motions is consistent with that at room temperature. Thus, a green, convenient, and low-cost screen printing technology with anti-counterfeiting potential has been developed on the basis of the unique features of the URTP films. The present design strategy might allow the development of next-generation amorphous organic materials with URTP capable of being applied in the fields of organic devices, bioimaging, information storage, and data security.
MATERIALS AND METHODS
Materials
PVA-100 [molecular weight (Mw) = 85,000 to 124,000 g/mol, 100% hydrolyzed; Sigma-Aldrich], PVA-87 (Mw = 13,000 to 23,000 g/mol, 87 to 89% hydrolyzed; Sigma-Aldrich), PVA-80 (Mw = 9000 to 10,000 g/mol, 80% hydrolyzed; Sigma-Aldrich), PVAc (average Mw = 100,000 g/mol; Sigma-Aldrich), and other chemicals were purchased commercially and used without further purifications unless otherwise specified.
Synthesis of hexa-(4-carbomethoxy-phenoxy)-cyclotriphosphazene (CTP-COOCH3)
Potassium carbonate (11.38 g, 0.082 mol) and methyl 4-hydroxybenzoate (6.54 g, 0.043 mol) in dry THF (120 ml) were added into a 250-ml round-bottom flask. The mixture solution was heated to 70°C under N2, and P3N3Cl6 (2.49 g, 0.007 mol) was added as soon as possible. The mixture was stirred at this temperature overnight. The solvent was removed by evaporation under a reduced pressure, and the obtained solid was redissolved in dichloromethane (70 ml). The organic phase was washed with deionized water and then dried over anhydrous MgSO4. Dichloromethane was removed by evaporation under a reduced pressure, the white powder was heated at 60°C in a vacuum oven overnight, and the desired product was obtained (6.1 g, yield 83.7%). 1H NMR (400 MHz, DMSO-d6, in ppm): δ, 7.77 to 7.79 (d, 12H, Ar-H), 7.03 to 7.06 (d, 12H, Ar-H), and 3.87 (s, 18H, –COOCH3). 13C NMR (CDCl3, 100 MHz, in ppm): δ, 165.98, 153.62, 131.35, 127.36, 120.57, and 52.30.
Synthesis of hexa-(4-carboxyl-phenoxy)-cyclotriphosphazene (CTP-COOH)
A quantity of sodium hydroxide (1.8 g, 45 mmol) was dissolved in deionized water (80 ml), and CTP-COOCH3 (5 g, 4.8 mmol) was dissolved in THF (100 ml). The two solutions were mixed and stirred at 70°C for 1.5 hours. After the reaction, THF was removed by a rotary evaporator, and the residual solution was poured into deionized water (300 ml), followed by using dilute hydrochloric acid to adjust pH to approximately 2 to 3. The product as a precipitate was obtained after stirring. The precipitate was collected by filtration, washed with deionized water, and dried at 60°C in a vacuum oven overnight. The desired ligand G was obtained as white powder (4.3 g, yield 94%). FTIR (KBr): v/cm−1, 3002.38, 2668.64, 2538.03, 1697.86, 1602.81, 1507.77, 1281.41, 1150.08, 947.66, 774.98, and 548.61. 1H NMR (400 MHz, DMSO-d6, in ppm): δ 13.05 (s, 6H,–COOH), 7.86 to 7.84 (d, 12H, Ar-H), and 7.00 to 7.03 (d, 12H, Ar-H). 13C NMR (300 MHz, DMSO-d6, in ppm): δ 166.69, 153.23, 131.82, and 128.82. ESI-MS: 980.09 (G + Na+).
Preparation of G-doped PVA films
PVA was dissolved in deionized water and heated to 95°C for 1.5 hours to afford the PVA aqueous solution (10 g/liter). Compound G with different concentrations (0.2, 0.5, 1, 3, 5, and 10 mg/ml) was added into the PVA solution. The mixed solutions were ultrasonicated for 0.5 hours, which were then drop-coated on precleaned quartz substrates and kept at 65°C for 3 hours to get the desired G-doped PVA films. Last, the resulting drop-coating films were irradiated under different periods of time (0, 5, 15, 25, 35, 50, 65, 80, and 100 min) using a 254-nm UV lamp (12 W).
Security protection procedure
Because the persistent emission intensity of G-doped PVA films is highly dependent on the irradiation time, the confidential information encryption and decryption could be easily realized. First, G-doped PVA films were prepared through drop-coating on precleaned glass substrates and dried at 65°C for 3 hours. Commercial screen plates with different patterns were covered on G-doped PVA films and then irradiated by a 254-nm UV lamp for 65 min to finish the screen printing (encryption process). No pattern on the films was observed under sunlight. However, clear patterns were observed upon exposure to the 254-nm UV lamp, and these patterns were still visible for several seconds after turning off the excitation source (decryption process). The URTP of G-doped PVA films gradually disappeared under ambient conditions because water molecules could break the PVA-PVA and PVA-G H-bonds. The URTP could be effectively recovered by incubating at 65°C for several minutes. On the basis of this behavior, reversible on/off switching of the phosphorescence signal for multiple information encryption and decryption cycles was achieved.
Supplementary Material
Acknowledgments
Funding: This work was financially supported by the Natural Science Foundation of China (no. 21404017), the special program of Chongqing Science and Technology Commission (nos. cstc2016shmszx80052 and cstc2017zdcy-zdyfX0007), and the Science and Technology Research Program of Chongqing Municipal Education Commission (no. KJ1709223). We also appreciate the support from the Singapore Academic Research Fund (nos. RG19/16, RG121/16, and RG11/17) and the Singapore Agency for Science, Technology and Research Advanced Manufacturing and Engineering Individual Research Grant (no. A1783c0007). C.Y. acknowledges the scholarship support from the China Scholarship Council (no. 201508505177). Author contributions: Y.S., C.Y., and Y.Z. conceived the project. Y.S., S.Z.F.P., and C.Y. designed and performed most of the experiments. Y.S., Y.L., X.Z., C.Y., and Y.Z. analyzed the data. D.J., W.K.O., W.Q.L., and G.L. performed the XPS, FTIR, thermogravimetric analysis, and XRD experiments. Y.S. and C.Y. carried out the syntheses. Y.S. and C.Y. prepared the manuscript. S.Z.F.P., D.J., W.Q.L., and Y.Z. revised the manuscript. All the authors discussed the results and commented on the manuscript. C.Y. and Y.Z. supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The data sets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaas9732/DC1
Supplementary Materials and Methods
fig. S1. Synthetic procedure of compound G.
fig. S2. Characterization of compound G.
fig. S3. Fluorescence and phosphorescence spectra of G-doped PVA films.
fig. S4. Phosphorescence decay profiles of G-doped PVA films.
fig. S5. Photophysical properties of G-doped PVA films.
fig. S6. FTIR and Raman spectra of G-doped PVA films.
fig. S7. 1H NMR spectra of compounds.
fig. S8. XPS spectra of the PVA-100-3mg film.
fig. S9. UV-Vis spectra of the PVA-100-3mg film.
fig. S10. Thermal properties and XRD patterns of the PVA-100-3mg film.
fig. S11. SEM images of G-doped PVA films.
fig. S12. Photophysical properties of the PVA-100-3mg film.
fig. S13. Temperature-dependent photophysical properties of the PVA-100-3mg film.
fig. S14. Excitation wavelength–dependent photophysical properties of the PVA-100-3mg film.
fig. S15. Photophysical properties of G-doped different PVA matrix films.
fig. S16. Photophysical properties and molecular structures.
fig. S17. FTIR spectra, XRD patterns, and SEM images of the PVA-100-3mg film under different conditions.
fig. S18. Phosphorescence emission spectra of patterns for the lotus flower under different conditions.
fig. S19. Green screen printing procedures without using any inks.
table S1. Photophysical data of PVA films by doping different concentrations of G.
table S2. Photophysical data of the PVA-100-3mg film at different irradiation times under a 254-nm UV lamp.
table S3. Photophysical data of the PVA-100-3mg film at different temperatures.
table S4. Photophysical data of the PVA-100-3mg film at different excitation wavelengths under ambient conditions.
movie S1. Appearance of the emitting G-doped PVA films when UV light is on and off.
movie S2. Appearance of the emitting PVA-100-3mg film when UV light is on and off.
movie S3. Imaging of the logo of the lotus flower.
movie S4. Imaging of the logo of the lotus flower containing AlQ3.
Reference (38)
REFERENCES AND NOTES
- 1.Pan Z., Lu Y.-Y., Liu F., Sunlight-activated long-persistent luminescence in the near-infrared from Cr3+-doped zinc gallogermanates. Nat. Mater. 11, 58–63 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Van den Eeckhout K., Smet P. F., Poelman D., Persistent luminescence in Eu2+-doped compounds: A review. Materials 3, 2536–2566 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uoyama H., Goushi K., Shizu K., Nomura H., Adachi C., Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 492, 234–238 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Zhang Q., Li B., Huang S., Nomura H., Tanaka H., Adachi C., Efficient blue organic light-emitting diodes employing thermally activated delayed fluorescence. Nat. Photonics 8, 326–332 (2014). [Google Scholar]
- 5.Kishimura A., Yamashita T., Yamaguchi K., Aida T., Rewritable phosphorescent paper by the control of competing kinetic and thermodynamic self-assembling events. Nat. Mater. 4, 546–549 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Collins H. A., Khurana M., Moriyama E. H., Mariampillai A., Dahlstedt E., Balaz M., Kuimova M. K., Drobizhev M., Yang V. X. D., Phillips D., Rebane A., Wilson B. C., Anderson H. L., Blood-vessel closure using photosensitizers engineered for two-photon excitation. Nat. Photonics 2, 420–424 (2008). [Google Scholar]
- 7.Kamtekar K. T., Monkman A. P., Bryce M. R., Recent advances in white organic light-emitting materials and devices (WOLEDs). Adv. Mater. 22, 572–582 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Yang Z., Mao Z., Zhang X., Ou D., Mu Y., Zhang Y., Zhao C., Liu S., Chi Z., Xu J., Wu Y.-C., Lu P.-Y., Lien A., Bryce M. R., Intermolecular electronic coupling of organic units for efficient persistent room-temperature phosphorescence. Angew. Chem. Int. Ed. Engl. 55, 2181–2185 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai S., Shi H., Li J., Gu L., Ni Y., Cheng Z., Wang S., Xiong W.-., Li L., An Z., Huang W., Visible-light-excited ultralong organic phosphorescence by manipulating intermolecular interactions. Adv. Mater. 29, 1701244 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Fateminia S. M. A., Mao Z., Xu S., Yang Z., Chi Z., Liu B., Organic nanocrystals with bright red persistent room-temperature phosphorescence for biological applications. Angew. Chem. Int. Ed. Engl. 56, 12160–12164 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Zhao W., He Z., Lam J. W. Y., Peng Q., Ma H. L., Shuai Z., Bai G., Hao J., Tang B. Z., Rational molecular design for achieving persistent and efficient pure organic room-temperature phosphorescence. Chem 1, 592–602 (2016). [Google Scholar]
- 12.Gong Y., Chen G., Peng Q., Yuan W. Z., Xie Y., Li S., Zhang Y., Tang B. Z., Achieving persistent room temperature phosphorescence and remarkable mechanochromism from pure organic luminogens. Adv. Mater. 27, 6195–6201 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Yang J., Ren Z., Xie Z., Liu Y., Wang C., Xie Y., Peng Q., Xu B., Tian W., Zhang F., Chi Z., Li Q., Li Z., AIEgen with fluorescence–phosphorescence dual mechanoluminescence at room temperature. Angew. Chem. Int. Ed. Engl. 56, 880–884 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Chen X., Xu C., Wang T., Zhou C., Du J., Wang Z., Xu H., Xie T., Bi G., Jiang J., Zhang X., Demas J. N., Trindle C. O., Luo Y., Zhang G. Q., Versatile room-temperature-phosphorescent materials prepared from N-substituted naphthalimides: Emission enhancement and chemical conjugation. Angew. Chem. Int. Ed. Engl. 55, 9872–9876 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Shoji Y., Ikabata Y., Wang Q., Nemoto D., Sakamoto A., Tanaka N., Seino J., Nakai H., Fukushima T., Unveiling a new aspect of simple arylboronic esters: Long-lived room-temperature phosphorescence from heavy-atom-free molecules. J. Am. Chem. Soc. 139, 2728–2733 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Gong Y., Zhao L., Peng Q., Fan D., Yuan W. Z., Zhang Y., Tang B. Z., Crystallization-induced dual emission from metal- and heavy atom-free aromatic acids and esters. Chem. Sci. 6, 4438–4444 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H., Yao X., Ma X., Tian H., Amorphous, efficient, room-temperature phosphorescent metal-free polymers and their applications as encryption ink. Adv. Opt. Mater. 4, 1397–1401 (2016). [Google Scholar]
- 18.Zhang T., Chen H., Ma X., Tian H., Amorphous 2-bromocarbazole copolymers with efficient room-temperature phosphorescent emission and applications as encryption ink. Ind. Eng. Chem. Res. 56, 3123–3128 (2017). [Google Scholar]
- 19.Bolton O., Lee K., Kim H.-J., Lin K. Y., Kim J., Activating efficient phosphorescence from purely organic materials by crystal design. Nat. Chem. 3, 205–210 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Xie Y., Ge Y., Peng Q., Li C., Li Q., Li Z., How the molecular packing affects the room temperature phosphorescence in pure organic compounds: Ingenious molecular design, detailed crystal analysis, and rational theoretical calculations. Adv. Mater. 29, 1606829 (2017). [DOI] [PubMed] [Google Scholar]
- 21.An Z., Zheng C., Tao Y., Chen R., Shi H., Chen T., Wang Z., Li H., Deng R., Liu X., Huang W., Stabilizing triplet excited states for ultralong organic phosphorescence. Nat. Mater. 14, 685–690 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Hirata S., Totani K., Zhang J., Yamashita T., Kaji H., Marder S. R., Watanabe T., Adachi C., Efficient persistent room temperature phosphorescence in organic amorphous materials under ambient conditions. Adv. Funct. Mater. 23, 3386–3397 (2013). [Google Scholar]
- 23.Lee D., Bolton O., Kim B. C., Youk J. H., Takayama S., Kim J., Room temperature phosphorescence of metal-free organic materials in amorphous polymer matrices. J. Am. Chem. Soc. 135, 6325–6329 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Kwon M. S., Lee D., Seo S., Jung J., Kim J., Tailoring intermolecular interactions for efficient room-temperature phosphorescence from purely organic materials in amorphous polymer matrices. Angew. Chem. Int. Ed. Engl. 53, 11177–11181 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Kwon M. S., Yu Y., Coburn C., Phillips A. W., Chung K., Shanker A., Jung J., Kim G., Pipe K., Forrest S. R., Youk J. H., Gierschner J., Kim J., Suppressing molecular motions for enhanced room-temperature phosphorescence of metal-free organic materials. Nat. Commun. 6, 8947 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma X., Cao J., Wang Q., Tian H., Photocontrolled reversible room temperature phosphorescence (RTP) encoding β-cyclodextrin pseudorotaxane. Chem. Commun. 47, 3559–3561 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Chen H., Ma X., Wu S., Tian H., A rapidly self-healing supramolecular polymer hydrogel with photostimulated room-temperature phosphorescence responsiveness. Angew. Chem. Int. Ed. Engl. 53, 14149–14152 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Sun X., Zhang B., Li X., Trindle C. O., Zhang G., External heavy-atom effect via orbital interactions revealed by single-crystal x-ray diffraction. J. Phys. Chem. A 120, 5791–5797 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Lucenti E., Forni A., Botta C., Carlucci L., Giannini C., Marinotto D., Previtali A., Righetto S., Cariati E., H-Aggregates granting crystallization-induced emissive behavior and ultralong phosphorescence from a pure organic molecule. J. Phys. Chem. Lett. 8, 1894–1898 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Jiang K., Zhang L., Lu J., Xu C., Cai C., Lin H., Triple-mode emission of carbon dots: Applications for advanced anti-counterfeiting. Angew. Chem. Int. Ed. Engl. 55, 7231–7235 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Jiang K., Wang Y., Cai C., Lin H., Activating room temperature long afterglow of carbon dots via covalent fixation. Chem. Mater. 29, 4866–4873 (2017). [Google Scholar]
- 32.Wang Y., Li T., Ma P., Bai H., Xie Y., Chen M., Dong W., Simultaneous enhancements of UV-shielding properties and photostability of poly(vinyl alcohol) via incorporation of sepia eumelanin. ACS Sustain. Chem. Eng. 4, 2252–2258 (2016). [Google Scholar]
- 33.Mori Y., Honda T., Lu R., Hayakawa N., Miyakoshi T., Ultraviolet degradation of poly(vinyl alcohol) used in restoration of historical and cultural properties. Polym. Degrad. Stab. 114, 30–36 (2015). [Google Scholar]
- 34.Belmonte G. K., Charles G., Strumia M. C., Weibel D. E., Permanent hydrophilic modification of polypropylene and poly(vinyl alcohol) films by vacuum ultraviolet radiation. Appl. Surf. Sci. 382, 93–100 (2016). [Google Scholar]
- 35.Sheela T., Bhajantri R. F., Ravindrachary V., Rathod S. G., Pujari P. K., Poojary B., Somashekar R., Effect of UV irradiation on optical, mechanical and microstructural properties of PVA/NaAlg blends. Radiat. Phys. Chem. 103, 45–52 (2014). [Google Scholar]
- 36.He J., Chen J. P., Cu(II)-Imprinted poly(vinyl alcohol)/poly(acrylic acid) membrane for greater enhancement in sequestration of copper ion in the presence of competitive heavy metal ions: Material development, process demonstration, and study of mechanisms. Ind. Eng. Chem. Res. 53, 20223–20233 (2014). [Google Scholar]
- 37.Peng M., Xiao G., Tang X., Zhou Y., Hydrogen-bonding assembly of rigid-rod poly(p-sulfophenylene terephthalamide) and flexible-chain poly(vinyl alcohol) for transparent, strong, and tough molecular composites. Macromolecules 47, 8411–8419 (2014). [Google Scholar]
- 38.Ravindrachary V., Ismayil, Nayak S. P., Dutta D., Pujari P. K., Free volume related fluorescent behavior in electron beam irradiated chalcone doped PVA. Polym. Degrad. Stab. 96, 1676–1686 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaas9732/DC1
Supplementary Materials and Methods
fig. S1. Synthetic procedure of compound G.
fig. S2. Characterization of compound G.
fig. S3. Fluorescence and phosphorescence spectra of G-doped PVA films.
fig. S4. Phosphorescence decay profiles of G-doped PVA films.
fig. S5. Photophysical properties of G-doped PVA films.
fig. S6. FTIR and Raman spectra of G-doped PVA films.
fig. S7. 1H NMR spectra of compounds.
fig. S8. XPS spectra of the PVA-100-3mg film.
fig. S9. UV-Vis spectra of the PVA-100-3mg film.
fig. S10. Thermal properties and XRD patterns of the PVA-100-3mg film.
fig. S11. SEM images of G-doped PVA films.
fig. S12. Photophysical properties of the PVA-100-3mg film.
fig. S13. Temperature-dependent photophysical properties of the PVA-100-3mg film.
fig. S14. Excitation wavelength–dependent photophysical properties of the PVA-100-3mg film.
fig. S15. Photophysical properties of G-doped different PVA matrix films.
fig. S16. Photophysical properties and molecular structures.
fig. S17. FTIR spectra, XRD patterns, and SEM images of the PVA-100-3mg film under different conditions.
fig. S18. Phosphorescence emission spectra of patterns for the lotus flower under different conditions.
fig. S19. Green screen printing procedures without using any inks.
table S1. Photophysical data of PVA films by doping different concentrations of G.
table S2. Photophysical data of the PVA-100-3mg film at different irradiation times under a 254-nm UV lamp.
table S3. Photophysical data of the PVA-100-3mg film at different temperatures.
table S4. Photophysical data of the PVA-100-3mg film at different excitation wavelengths under ambient conditions.
movie S1. Appearance of the emitting G-doped PVA films when UV light is on and off.
movie S2. Appearance of the emitting PVA-100-3mg film when UV light is on and off.
movie S3. Imaging of the logo of the lotus flower.
movie S4. Imaging of the logo of the lotus flower containing AlQ3.
Reference (38)