

Abstract
Baranovskiy et al. and Pellegrini argue that, based on structural data, the path for charge transfer through the [4Fe4S] domain of primase is not feasible. Our manuscript presents electrochemical data directly showing charge transport through DNA to the [4Fe4S] cluster of a primase p58C construct and a reversible switch in the DNA-bound signal with oxidation/reduction, which is inhibited by mutation of three tyrosine residues. Although the dispositions of tyrosines differ in different constructs, all are within range for microsecond electron transfer.
Our study (1) provides electrochemical and biochemical data for human DNA primase, supporting a proposal that [4Fe4S] cluster redox chemistry and DNA charge transport are elements in the binding and termination of the polymerase activity of the primase. We describe (i) electrochemical experiments on DNA-modified electrodes demonstrating that the p58C domain of DNA primase exhibits tighter binding with the [4Fe4S] cluster in the 3+ state compared with the 2+ state; (ii) mutagenesis experiments showing that when any of three tyrosine residues are mutated to phenylalanine, the ability to carry out the redox reaction on the cluster electrochemically is inhibited and also that charge transfer–deficient mutants exhibit reduced ability to initiate priming; and (iii) primer elongation and termination assays with native full-length primase showing that the introduction of a mismatch into the growing primer inhibits termination, which is consistent with the regulation of primase termination by mismatch-sensitive DNA-mediated charge transport. These data support our proposal that electron transfer between clusters in primase and polymerase α, mediated by the growing DNA/RNA duplex, contributes to handoff of the substrate between the two enzymes.
We emphasize that all electrochemistry and corresponding control experiments were performed on the truncated p58C construct, which contains the [4Fe4S] cluster, and all activity assays and corresponding control experiments were performed on the full-length p48/p58 enzyme. Neither Baranovskiy et al. (2) nor Pellegrini (3) directly dispute the electrochemical observations of charge transport through the DNA substrate to the iron-sulfur cluster of our p58 construct, nor do they dispute its inhibition by mutation of each of the three relevant tyrosine residues. Rather, they question the path for electron transfer through primase on the basis of differences in structures of p58C, the primase domain containing the [4Fe4S] cluster (4–6). The structure of human p58C (4) from which we identify conserved tyrosine residues participating in a charge transfer pathway contains residues 318 to 353 folded in a β-hairpin arrangement. Other structures of human (5) and yeast (6) p58C show these residues in an α-helical arrangement and show that conserved tyrosines (Y309, Y345, and Y347 for the human protein) in the charge transport pathway through the protein are spaced and oriented differently in different structures. However, both Comments do not appropriately consider the requirements for charge transport mediated by tyrosines in a protein. Charge transfer through protein, which is only weakly dependent on orientation, can occur over distances up to 15 Å on our proposed time scale and is most accurately estimated from the distances between tyrosine centroids (7). In fact, regardless of the differences in structures, the tyrosine centroids in all human and yeast p58C structures are within feasible range for microsecond electron transfer through the protein matrix.
In addition to differences in crystallization conditions, our human p58C construct differs in sequence from that of Baranovskiy et al.; our p58C construct corresponds to residues 272 to 464 from the human protein. Three additional residues on the N terminus of our construct—essentially positions 269, 270, and 271—are left from the His tag used to purify the protein. Tahirov uses a different truncation, including more residues on the N terminus but fewer on the C terminus (266 to 456). Our p58C construct was used for the electrochemistry and crystallography, but for all biochemical assays, we used the full human primase (no mutations).
The Comments also raise concerns over our interpretation of data obtained for a Y345F primase mutant, which results in reduced electron transfer efficiency. The argument is based primarily on the assignment of a hydrogen bond between Y345 and the triphosphate group at the 5′ end of the DNA/RNA substrate in a crystal structure (8). We find it puzzling that elimination of only one of more than 15 hydrogen bonds between p58C and the substrate is proposed as an explanation for the dramatic change in biochemical activity that we observe. In fact, binding assays in our laboratory comparing the full, wild-type primase enzyme and the Y345F primase variant using the RNA/DNA substrate similar to the one used in the crystal structure (8) show no detectable differences, as expected. Furthermore, the placement of the Y345 side chain in the structure, which lies at the center of their claim, may be more ambiguous than previously implied (8). Retrieving the coordinate files and electron density maps for the structures (5F0Q), we observe a very poor fit for this residue. High B factors for this residue relative to most other residues call into question whether the assigned hydrogen bond has high enough occupancy to substantiate the argument. Moreover, we have now carried out parallel biochemical experiments comparing primer synthesis of wild-type full primase and the charge transfer–deficient Y309F variant of full p48/p58 primase on single-stranded DNA (Fig. 1). Here, too, we observe significant inhibition of initiation in the mutant, just as we observed with the Y345F and Y345C primase variants. Thus, mutation of another tyrosine in the charge transfer pathway, one not interacting with any substrates, also inhibits initiation. Hence, inhibition of initiation is the result of inhibition of the redox switch.
Fig. 1. Wild-type (WT) p48/p58 versus p48/p58Y309F activity on ssDNA.
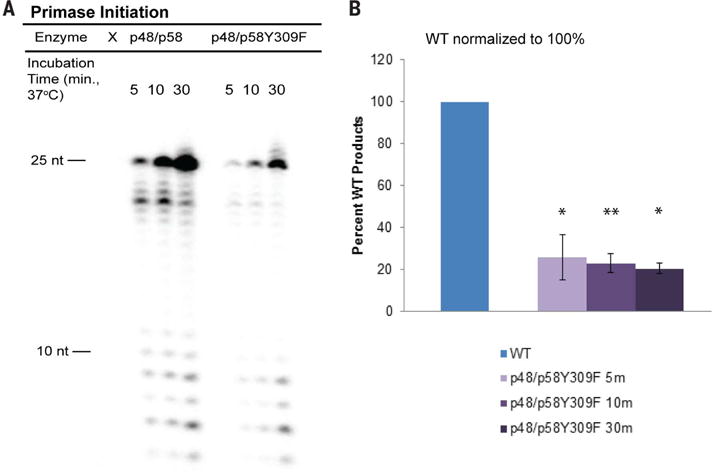
(A) Gel separation of products for primase initiation assay comparing WT and charge transfer–deficient primase. (B) Product quantifications for p48/p58Y309F, with WT primase (p48/p58) products normalized to one. Values shown are the mean of n = 3 trials; error bars represent standard deviation. *, 0.001 ≤ P < 0.005; **, P < 0.001; Student’s t test. All activity assays were performed under anaerobic conditions. Reactions contained 400 nM primase variant, 250 nM ssDNA, 188 μM uridine triphosphate (UTP), 112 μM cytidine triphosphate (CTP), 1 μM α−32P adenosine triphosphate (ATP) in 50 mM Tris, pH 8.0, 3 mM MgCl2.
Baranovskiy et al. criticize our choice of substrate for the electrochemical studies of p58C. Primase interacts with a range of DNA structures (4), enabling us to create a substrate that productively binds p58C and satisfies the technical criteria needed for DNA-mediated electrochemistry. Our substrates were designed for the primary objectives of the experiment: (i) to assess the ability of p58C to participate in DNA charge transport and (ii) to examine the differential effects of [4Fe4S] cluster oxidation state. The binding affinity of p58C for the substrate should therefore not be particularly strong, so that any changes in affinity between oxidized [4Fe4S]3+ and reduced [4Fe4S]2+ protein can be detected. A substrate similar to the construct containing a 3′ single-stranded DNA (ssDNA) overhang and 5′-triphosphate, which Baranovskiy et al. (9) observe p58C binds tightly, would obscure the observation of differences between the two redox states on the electrode. Moreover, as it contains a 5′-ssDNA overhang, our electrochemistry substrate is similar to the primed ends encountered by polymerase enzymes in cells.
Baranovskiy et al. additionally suggest that a mismatch in the nascent primer inhibits initiation, but not truncation in the assay with a primed template substrate. On both well-matched and mismatched substrates, we observe a mixture of initiation and elongation products because primase binds to both ssDNA and primed DNA portions of the substrate (1). Primase initiation products (7 to 29 nucleotides) are synthesized on the ssDNA template used to generate a mismatched primer.
Baranovskiy et al. also criticize our assignment of the oxidized [4Fe4S]3+ species generated electrochemically. However, we show directly using cyclic voltammetry, as seen in figures 1 and 2 of (1), that we generate the oxidized [4Fe4S]3+ product. The cathodic peaks in figure 1 and figure 2 (1) furthermore show the reduction of an oxidized species on the DNA-modified electrode. Additionally, chemical oxidants with potentials similar to the potential applied to p58C can oxidize human p58C to the [4Fe4S]3+ state, as demonstrated by electron paramagnetic resonance spectroscopy (10), supporting our assignment of the [4Fe4S]3+ species. We were not able to perform binding affinity measurements with the electrochemically oxidized p58C because the oxidized protein is unstable over the long periods of time required for the measurement.
Finally, Baranovskiy et al. criticize our performing in vitro priming assays under anaerobic conditions that they argue do not correspond to the cellular environment. Anaerobic conditions were used to ensure that we had full control over the redox state of the [4Fe4S] cluster, which can be oxidized in the presence of air (11). In fact, controls performed under aerobic conditions gave similar results overall, although with greater scatter and lower precision.
Overall, the positioning of tyrosines 309, 345, and 347 in both structures of human p58C, irrespective of local conformation, suggests a feasible pathway for electron transfer through the [4Fe4S] protein. The electrochemical experiments with p58C variants, and the biochemical activity assays with corresponding full primase variants, illuminate the electron transfer chemistry performed by the primase [4Fe4S] cluster and the effect of this chemistry on DNA binding. Primer elongation assays with well-matched and mismatched template strands, moreover, demonstrate the regulatory role of DNA charge transport in termination. Thus, the concerns raised do not negate either our experimental observations of redox chemistry performed by the [4Fe4S] cluster or the proposal of a role for the redox chemistry, coupled to DNA charge transport, in regulating the activity of human DNA primase.
References
- 1.O’Brien E, et al. Science. 2017;355:eaag1789. [Google Scholar]
- 2.Baranovskiy AG, et al. Science. 2017;357:eaan2396. [Google Scholar]
- 3.Pellegrini L. Science. 2017;357:eaan2954. doi: 10.1126/science.aan2954. [DOI] [PubMed] [Google Scholar]
- 4.Vaithiyalingam S, Warren EM, Eichman BF, Chazin WJ. Proc Natl Acad Sci USA. 2010;107:13684–13689. doi: 10.1073/pnas.1002009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agarkar VB, Babayeva ND, Pavlov YI, Tahirov TH. Cell Cycle. 2011;10:926–931. doi: 10.4161/cc.10.6.15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sauguet L, Klinge S, Perera RL, Maman JD, Pellegrini L. PLOS ONE. 2010;5:e10083. doi: 10.1371/journal.pone.0010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winkler JR, Gray HB. J Am Chem Soc. 2014;136:2930–2939. doi: 10.1021/ja500215j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baranovskiy AG, et al. J Biol Chem. 2016;291:10006–10020. doi: 10.1074/jbc.M116.717405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baranovskiy AG, et al. J Biol Chem. 2016;291:4793–4802. doi: 10.1074/jbc.M115.704064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weiner BE, et al. J Biol Chem. 2007;282:33444–33451. doi: 10.1074/jbc.M705826200. [DOI] [PubMed] [Google Scholar]
- 11.Imlay JA. Mol Microbiol. 2006;59:1073–1082. doi: 10.1111/j.1365-2958.2006.05028.x. [DOI] [PubMed] [Google Scholar]