

Abstract
Purpose of Review
Granulomatous-lymphocytic interstitial lung disease (GLILD) has classically been associated with common variable immune deficiency (CVID), but is increasingly being reported in other immunodeficiencies. We describe the second reported case of GLILD in a patient with 22q11.2 deletion syndrome (22q11.2DS) and review the recent literature surrounding GLILD.
Recent Findings
GLILD is characterized by granulomata and lymphoproliferation. Consensus statements and retrospective and case-control studies have better elucidated the clinicopathological and radiographic manifestations of GLILD, allowing for its differentiation from similar conditions like sarcoidosis. Gaps of knowledge remain, however, particularly regarding optimal management strategies. Combination therapies targeting T and B cell populations have recently shown favorable results.
Summary
GLILD is associated with poorer outcomes in CVID. Its recognition as a rare complication of 22q11.2DS and other immunodeficiencies therefore has important therapeutic and prognostic implications. Additional research is needed to better understand the natural history and pathogenesis of GLILD and to develop evidence-based practice guidelines.
Keywords: Granulomatous-lymphocytic interstitial lung disease, 22q11.2 deletion syndrome, Common variable immune deficiency, Primary immune deficiency, Sarcoidosis, Autoimmunity
Introduction
Immune dysregulation and autoimmunity are well-known complications of 22q11.2 deletion syndrome (22q11.2DS). The immunodeficiency is secondary to thymic dysplasia as a result of the chromosomal microdeletion, and it varies from the more common mild-to-moderate T cell lymphocytopenia to the rarer severe combined immunodeficiency phenotype. Additionally, 22q11.2DS often manifests with partial humoral deficiencies as well as autoimmune conditions, including, but not limited to, cytopenias, endocrinopathies, and arthritis [1]. Granulomatous-lymphocytic interstitial lung disease (GLILD) is a rare type of interstitial lung disease (ILD) with features of both granulomas and lymphoproliferation. GLILD is classically identified as a non-infectious complication of common variable immunodeficiency (CVID), a primary humoral immune deficiency defined by low immunoglobulin levels and poor specific antibody responses to vaccination [2••]. GLILD is now being increasingly recognized in other primary immunodeficiencies (PID) [3–9], partially secondary to improved care of patients with PID allowing for longer survival and the development of advanced complications. To our knowledge, GLILD has only been previously reported in a single patient with 22q11.2DS [10•]. We present here the second case of GLILD in a patient with 22q11.2DS followed by a review of the most recent literature on GLILD, highlighting the need to further clarify its natural history, pathogenesis, and optimal management strategies.
Case Report
An African-American male presented at 16 years of age to the rheumatology/immunology clinic for evaluation of granulomatous adenopathy and pulmonary nodules, with particular concern for sarcoidosis. Review of his history revealed a diagnosis of tetralogy of Fallot at birth with subsequent confirmation of 22q11.2DS by fluorescence in situ hybridization (FISH). The remainder of his childhood and adolescent years was complicated by speech delay, learning disabilities, and variable immune-mediated cytopenias, consistent with his diagnosis of 22q11.2DS [1]. He had no significant history of recurrent or opportunistic infections and available records of an immune system evaluation during his preadolescent years revealed decreased total CD3+ and CD3+CD4+ T cell numbers, decreased percentage of CD45RA+ naïve Tcells, normal T cell function, normal IgG, and normal specific antibody titers.
In regards to his cytopenias, he was noted to be persistently thrombocytopenic since infancy and intermittently anemic, neutropenic, and lymphopenic. Direct Coomb’s testing was variably positive, and this combined with a normocellular bone marrow led to the diagnosis of Evan’s syndrome. He was initially asymptomatic from the thrombocytopenia, but then developed recurrent episodes of epistaxis, some of which required medical and surgical intervention. This prompted sporadic combination treatment beginning at 11 years of age with intravenous immunoglobulin (IVIG), vincristine, and/or pulse corticosteroids to which he initially responded well. He ultimately required intermittent courses of rituximab for refractory thrombocytopenia at 13 and 15 years of age.
During a routine hematology visit prior to his referral, he reported decreased exercise tolerance secondary to exertional dyspnea and associated with peripheral cyanosis. An urgent cardiac evaluation yielded no cardiovascular cause for his symptoms, but a cardiac MRI did unexpectedly demonstrate bulky mediastinal lymphadenopathy (LAD), splenomegaly, and pulmonary nodules. Subsequent positron emission tomography computed tomography (PET-CT) revealed intensely FDG-avid supraclavicular, mediastinal, and retroperitoneal LAD; numerous bilateral scattered pulmonary nodules with a lower lobe predominance; and focal irregular areas of uptake in the spleen. Excisional biopsy of a mediastinal lymph node was not consistent with malignancy but instead revealed granulomatous inflammation, prompting concern for sarcoidosis after a thorough infectious disease workup for viral, bacterial, and fungal pathogens was negative.
At the time of his initial visit in rheumatology/immunology clinic, a thorough immune evaluation was performed and was remarkable for low naïve T cell numbers, decreased lymphoproliferative responses to mitogens and antigens, hypogammaglobulinemia, and suboptimal vaccine responses (Table 1). As a complete humoral immune system evaluation had not been performed for some time before his initial course of rituximab, it is unclear whether the humoral immune deficiency was secondary to his underlying 22q11.2DS or treatment with rituximab. Regardless, a diagnosis of combined immunodeficiency was made, and the patient was started on monthly IVIG replacement to maintain trough levels near or above 1000 mg/dL and monthly inhaled pentamidine for Pneumocystis pneumonia prophylaxis.
Table 1.
Results of immunological workup
| Results (reference value) | |
|---|---|
| IgG (mg/dL) | 509 (600–1700) |
| IgM (mg/dL) | 242 (35–290) |
| IgA (mg/dL) | < 8 (40–400) |
| Diphtheria IgG titer (IU/mL) | 0.11 (≥ 0.15) |
| Tetanus IgG titer (IU/mL) | 0.48 (≥ 0.15) |
| Pneumococcal titers for PCV13 serotypes | 2/12 protective serotypes (> 70% protective) |
| Absolute lymphocyte count (cells/mcL) | 1000 (1500–5000) |
| Absolute CD3 count (cells/mcL) | 813 (915–3400) |
| Absolute CD4 count (cells/mcL) | 443 (510–2320) |
| Absolute CD8 count (cells/mcL) | 371 (180–1520) |
| Absolute CD19 count (cells/mcL) | 132 (105–920) |
| Absolute CD16/56 count (cells/mcL) | 251 (15–1080) |
| Absolute CD4+CD45+RA+ count (cells/mcL) | 13 (135–893) |
| Absolute CD8+CD45+RA+ count (cells/mcL) | 173 (83–653) |
| CD3% proliferation to PHA | 16% (> 58.5%) |
| CD3% proliferation to PWM | 5.0% (> 3.5%) |
| CD3% proliferation to CA | 0.0% (> 3.0%) |
| CD3% proliferation to TT | 2.3% (> 3.3%) |
In regard to his granulomatous inflammation, a review of the histopathology from his mediastinal lymph node biopsy was not convincing for sarcoidosis due to several atypical features not seen with sarcoidosis, including the ill-defined architecture of the granuloma, scattered areas of calcifications, and an area of necrosis. The patient’s symptoms spontaneously resolved and a high-resolution CT (HRCT) chest obtained 3 months after initial symptom onset revealed decrease in size and prominence of LAD, so additional workup was not pursued. Despite immunoglobulin replacement and control of infections, however, a routine PET-CT and CT chest obtained 1 year later revealed worsening and extensive pulmonary nodules and innumerable ground glass opacities in a basilar distribution; enlarged axillary LAD along with FDG-avid hilar and mediastinal LAD; and hypodense lesions in an enlarged spleen (Fig. 1a–d), suggesting a lymphoproliferative disease process. In spite of the radiographic deterioration, the patient remained asymptomatic, but spirometry and diffusion studies indicated moderate obstructive impairment with moderately reduced diffusion capacity for carbon monoxide (DLCO).
Fig. 1.
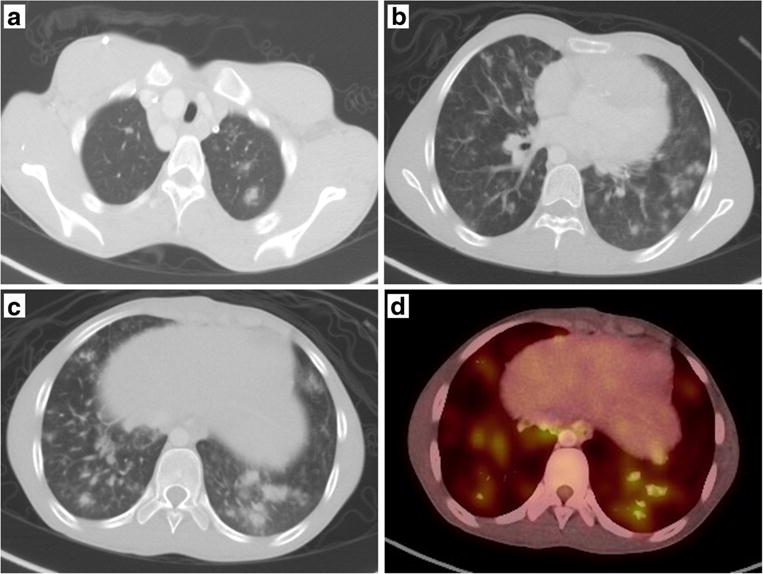
Radiographic features of GLILD. a–c The nonspecific nodular consolidations with surrounding ground glass opacity in the upper lobes (a), mid-lobes (b), and lower lobes (c) with an overall lower lobe predominance. The pulmonary nodules are FDG-avid as demonstrated in the PET scan image (d)
A pulmonary wedge resection via video-assisted thoracoscopic surgery (VATS) was subsequently performed for definitive diagnosis. Histopathological features consisted of lymphohistiocytic nodules containing poorly formed non-necrotizing granulomata, follicular bronchiolitis (FB), and lymphoid interstitial pneumonia (LIP) with diffuse alveolar damage (Fig. 2a–c). Immunophenotyping of lymphocytic regions revealed a T cell predominance (91%) with a 3:1 CD4:CD8 ratio and only 7% polytypic B cells. The combination of radiographic and histopathologic findings and exclusion of infectious, malignant, and rheumatologic causes led to the diagnosis of GLILD. Though asymptomatic, given the extent of his lymphoproliferation with associated cytopenias, the decision was made to treat with rituximab, which resulted in significant radiographic improvement.
Fig. 2.
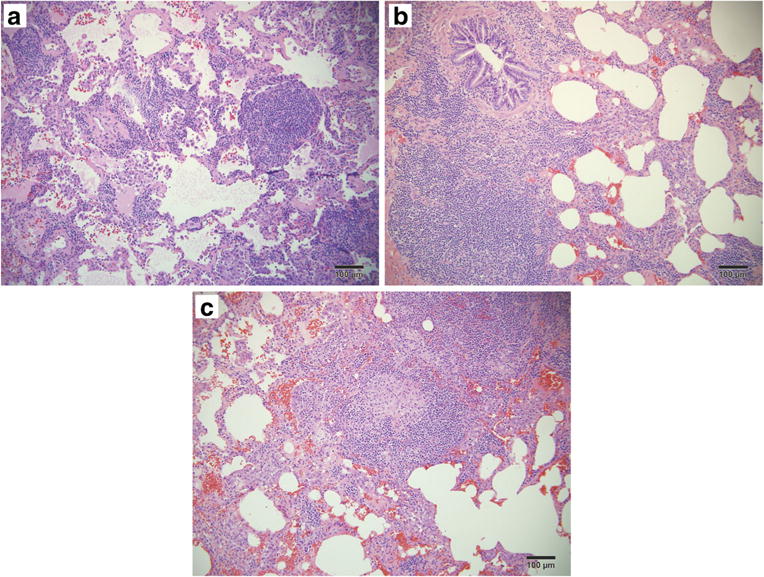
Histopathology from VATS pulmonary wedge resection. a–c Aggregates of lymphoid follicles and lymphocytic expansion of the interstitium (a), follicular bronchiolitis (b), and a loose non-necrotizing granuloma (c), features consistent with a diagnosis of GLILD
Since his initial diagnosis of GLILD, radiographic relapse of pulmonary nodularity and diffuse LAD with associated splenomegaly has occurred at regular intervals of approximately 8–9 months following each course of rituximab. Interestingly, repopulation of B cells detected peripherally has heralded each reoccurrence (Table 2), suggesting the need for both T cells and B cells for development of the granulomatous and lymphocytic features of GLILD. Thus, following his first reoccurrence, 6-mercaptopurine (6-MP) 0.5 mg/kg three times weekly was added to his therapeutic regimen to specifically target T cells while rituximab 375 mg/m2/dose was given weekly for 4 weeks at the times of relapse. Frequency and dose of 6-MP are often adjusted based on cytopenia and concerns for bone marrow suppression. As our patient continues to remain asymptomatic despite radiographic findings and given the morbidity and mortality associated with GLILD [2••], routine CTchest imaging is obtained every 3–6 months for monitoring.
Table 2.
Lymphocyte percentages in correlation with each occurrence of GLILD
| Relapse #1 (T = 0) | CD3+ T cell % | CD19+ B cell % |
| T = −1 month | 86 | < 1 |
| T = 0 | 73 | 5 |
| Relapse #2 (T = 9 months) | CD3% | CD19% |
| T = 7 months | 80 | 1 |
| T = 9 months | 73 | 6 |
| Relapse #3 (T = 17 months) | CD3% | CD19% |
| T = 16 months | 83 | < 1 |
| T = 17 months | 72 | 3 |
Discussion and Literature Review
The overall clinical presentation of our patient is consistent with a diagnosis of 22q11.2DS with features including congenital cardiac anomaly, speech delay, learning disabilities, autoimmune cytopenias, and combined immunodeficiency. The diagnosis of GLILD was unexpected given that this is a complication that has mostly been associated with CVID and not 22q11.2DS. This case therefore demonstrates the challenges associated with diagnosis and management of this pulmonary complication and highlights the many questions that remain to be elucidated. While no diagnostic or treatment guidelines exist for GLILD, the British Lung Foundation and United Kingdom Primary Immunodeficiency Network (BLF/UK PIN) recently published consensus statements on the definition, diagnosis, and management of this clinicopathological entity based on the shared clinical experience of over 30 consultants, including immunologists, chest physicians, radiologists, and pathologists, who combined care for over 100 patients with GLILD [11••]. We will highlight some of these BLF/UK PIN consensus statements in our concise literature review on GLILD, with a focus on diagnosis and management as well comparing features of GLILD with those of sarcoidosis.
Definition of GLILD
Bates, et al. first coined the term granulomatous-lymphocytic interstitial lung disease in 2004 in reference to their subpopulation of CVID patients whose surgical lung biopsies consisted of both granulomatous and lymphoproliferative histologic patterns [2••] though reports of features of GLILD in association with immunodeficiency date back to 1973 [12•]. Additionally, certain clinical, laboratory, and radiographic features may suggest a diagnosis of GLILD even prior to histological analysis (as discussed in more detail below). To this effect, the BLF/UK PIN suggest the following consensus definition: “GLILD is a distinct clinico-radio-pathological ILD occurring in patients with CVID, associated with a lymphocytic infiltrate and/or granuloma in the lung, and in whom other conditions have been considered and where possible excluded” [11••]. While this consensus definition focuses on patients with CVID, we suggest its extrapolation to include other immunodeficiencies in which GLILD is becoming increasing recognized, such as in the case of our patient with 22q11.2DS.
Diagnosis of GLILD
While the natural history of GLILD is largely still unknown, increased morbidity and mortality in patients with CVID secondary to non-infectious complications including GLILD have been reported by several authors [2••, 13, 14]. In fact, median survival was reduced by 50% in the presence of GLILD even when compared to those with other types of ILD in a retrospective chart review of 69 CVID patients [2••]. This suggests the need for early recognition and accurate diagnosis for treatment initiation. The diagnostic clinical, radiographic, and histopathological features of GLILD are included within the BLF/UK PIN consensus definition and will be discussed here.
Clinical Features and Predictors of GLILD
Respiratory symptoms are often nonspecific and do not allow for delineation of underlying causes; though, dyspnea has been noted to correlate with a diagnosis of GLILD compared to other respiratory symptoms [2••]. Likewise, our patient’s presenting symptom was that of exertional dyspnea. Patients with GLILD, however, may often be asymptomatic [11••] as our patient has been over the last 2 years despite disease relapse radiographically. Similarly, pulmonary function testing may vary considerably from being normal to displaying a severe restrictive pattern with reduced diffusion capacity [2••, 11••, 15•]. Thus, a high index of suspicion for GLILD is necessary in the setting of other clinical and radiographic features.
A vast array of extra-pulmonary manifestations often accompany GLILD, including autoimmune hemolytic anemia (AIHA), idiopathic thrombocytopenia purpura (ITP), adenopathy, splenomegaly, hepatitis, inflammatory bowel disease, and polyarthritis [2••, 15•, 16, 17]. Some of these features, such as adenopathy and splenomegaly, histologically mimic the granulomatous lymphocytic processes occurring in the lungs, suggesting GLILD may be a pulmonary manifestation of a more multisystem granulomatous lymphocytic inflammatory disease [11••]. Mannina and colleagues compared 34 CVID patients with GLILD to 52 CVID patients without GLILD and found the presence of splenomegaly and polyarthritis to be independent predictors for the development of GLILD [16]. A similar study of 26 CVID patients also identified splenomegaly in addition to AIHA and ITP as potential risk factors for the development of GLILD [17]. Additionally, a recent analyses of CVID patients with autoimmune cytopenias in the United States Immunodeficiency Network (USIDNET) registry revealed that this manifestation was strongly associated with the presence of lymphoproliferation and granulomata [18]. While our patient’s comorbidities also included cytopenias and splenomegaly, it is unclear whether these features would be predictive for GLILD in non-CVID immunodeficient patients.
Low IgA levels, high IgM levels, low switched memory B cells, increased CD21low B cells, decreased CD3+ and CD8+ T cells, and skewing of T cells towards a memory phenotype have all individually been correlated with a diagnosis of GLILD in CVID patients [2••, 13, 15•, 16, 17]. Small sample sizes and inconsistencies among these studies, however, likely led to disagreement among consultants of the BLF/UK PIN consensus statements regarding further immunological testing beyond that necessary to characterize a patient’s immune deficiency [11••]. On the other hand, this group did agree that genetic mutation analysis should be pursued following a diagnosis of GLILD. Flow cytometry analysis did reveal skewing of CD4+ T cells towards a memory phenotype in our patient, but more importantly highlighted significantly decreased naïve T cells consistent with the thymic dysplasia of 22q11.2DS. B cell phenotyping was not performed as this was likely to be of limited diagnostic or therapeutic utility, given prior treatment with multiple courses of rituximab would make interpretation of results difficult.
Radiographic Features of GLILD
HRCT of the chest was unanimously considered essential in the diagnostic workup in CVID patients with signs and symptoms concerning for GLILD in the BLF/UK PIN consensus statements [11••]. Given that individuals may be asymptomatic with normal lung function testing, it could also be argued that HRCT of the chest should be routinely and intermittently obtained for screening purposes in CVID patients and possibly other PID. Evaluation of 30 CT scans from CVID patients with GLILD revealed three predominant pulmonary features: nodules in a centrilobular or random distribution, ground glass opacifications often with a lower lobe predominance, and bronchiectasis [19]. Fine to course reticulation with fibrotic features was also often observed in a cohort of GLILD patients with restrictive lung disease and/or impaired diffusion capacity [20]. Additionally, Torigian and colleagues demonstrated that the widespread nodules seen in GLILD also have a lower lobe predilection and noted the additional feature of interlobular septal widening of the mid-to-lower lung fields [21].
Unfortunately, the spontaneous waxing and waning of these radiographic features [21] presents a challenge in and often delays the diagnosis of GLILD. This was demonstrated in our patient who had spontaneous radiographic improvement of pulmonary nodularity and LAD over a 3-month time period, which also correlated with symptom resolution. Thus, his lung biopsy for definitive diagnosis did not occur until about 12 months after initial symptom onset when repeat CT chest demonstrated increased disease burden. Moreover, while the radiographic patterns noted above are consistently identified in patients with GLILD, there is still considerable overlap with disease entities that should be considered in the differential diagnosis, including infection, ILD from other causes, sarcoidosis, and lymphoma. This considerable overlap underscores the supposition that no radiographic findings are considered sufficient for diagnosis of GLILD without histopathology from tissue biopsy [11••].
Histopathological Features of GLILD
Lung biopsy via VATS is considered the test of choice for definitive diagnosis in the workup of GLILD based on the BLF/UK PIN consensus statements [11••]. Though granulomatous lymphocytic inflammation may occur in other organs, the lung is still considered the preferred biopsy site, likely due to the broader differential diagnosis of pulmonary disease and limited tissue sampling from other sites. Indeed, trans-bronchial biopsy of a mediastinal lymph node in our patient revealed only the granulomatous features of the disease, there-by preventing accurate diagnosis until a pulmonary wedge resection was performed.
Recently, Rao and colleagues thoroughly detailed the histopathological and immunohistochemical features of VATS-guided or open lung biopsies obtained from 16 CVID patients with GLILD [22•]. Nodular peribronchiolar and diffuse interstitial lymphocytic inflammation was demonstrated in all cases with expansion of the interstitium occurring in severe cases. These features are consistent with the FB and LIP patterns that had been ascribed to GLILD a decade prior [2••]. In addition to lymphocytic inflammation, non-necrotizing granulomata were also present in 15/16 cases examined [22•]. These were randomly distributed throughout the lung parenchyma in a basilar distribution, varied from being well-formed to poorly formed and consisted of epithelioid histiocytes with occasional multinucleated giant cells. Additional features found in the vast majority of cases (87.5 and 75%, respectively) included organizing pneumonia and interstitial fibrosis. Figure 2 demonstrates some of these same histological patterns in the lung biopsy of our patient.
Immunohistochemical staining identified CD3+CD4+ T cells as the predominant lymphocyte present within the peribronchial and interstitial inflammation with B cells representing only a minor component [22•]. In contrast, CD20+ and PAX5+ B cells surrounded by a small ring of CD4+ T cells characterized the nodular components of GLILD. Interestingly, nuclear FoxP3 expression, identifying regulatory T cells, was absent in the lungs of all patients, a feature that likely contributes to the pathogenesis of GLILD, which has yet to be elucidated. The histological presence of both T cells and B cells in the lungs of patients with GLILD has important implications in the management of this pathologic entity as will be discussed later.
GLILD Versus Sarcoidosis
As mentioned previously, the differential diagnosis of GLILD is quite broad and includes, but is not limited to, infection, organizing pneumonia, lymphoid interstitial pneumonia, sarcoidosis, and lymphoma [11••]. Consideration and evaluation for some or all of these diagnoses in patients in whom GLILD is being considered is essential for proper diagnosis and subsequent treatment. As multisystemic involvement of noncaseating granulomas is a prominent feature in both GLILD and sarcoidosis and given the prevalence of sarcoidosis worldwide [23], misdiagnosis of GLILD as sarcoidosis is not uncommon [24•]. GLILD, however, differs considerably from sarcoidosis epidemiologically, clinically, radiographically, histologically, and in treatment response [25, 26•].
To highlight similarities and differences between GLILD and sarcoidosis, Bouvry and colleagues retrospectively matched 20 cases with GLILD in a 3:1 ratio with 60 controls with sarcoidosis [26•]. Though CVID is primarily a disease of Caucasians and the prevalence of sarcoidosis is greatest in the African-American population [23], no ethnic differences were noted in this case-control study. Clinically, the presence of recurrent infections, associated autoimmune diseases such as cytopenias, crackles on lung auscultation, and hepatosplenomegaly were statistically more prominent in subjects with GLILD. Extra-pulmonary manifestations of granulomatous disease manifest more frequently in patients with GLILD [26•], affecting the lymph nodes, spleen, and liver most often [2••, 15•, 16, 17]. In contrast, cutaneous granulomas and uveitis represent the more common extra-pulmonary manifestations of sarcoidosis [27]. Notably, pulmonary function testing, peripheral lymphocyte counts, and serum angiotensin converting enzyme levels did not differ between the two groups [26•]. Immunoglobulin levels may be used to distinguish the two disease entities as hypogammaglobinemia is a cardinal feature of patients with GLILD associated with CVID while polyclonal hypergammaglobulinemia is frequently seen in sarcoidosis.
Radiographically, pulmonary nodules in a random distribution with a lower lobe predominance and surrounded by a ground glass opacity to create a “halo” sign predominate in GLILD [26•], as demonstrated in our patient in Fig. 1. In contrast, micronodules in a perilymphatic distribution with an upper lobe predominance are characteristic features of sarcoidosis. Additionally, bronchiectasis is a feature more frequently identified in patients with GLILD. Histologically, LIP and FB are characteristic of GLILD but rarely seen in sarcoidosis and should, therefore, increase the index of suspicion for GLILD when discovered in combination with non-caseating granulomas, which are often indistinguishable between the two disease entities. Finally, overall survival in those with GLILD is significantly reduced compared to those with sarcoidosis [26•], likely due to a combination of factors including the natural history of the disease, existing comorbidities, and treatment response. While GLILD results in increased morbidity and mortality and is often resistant to corticosteroid therapy [2••], up to a half of patients with pulmonary sarcoidosis experience spontaneous remission while the vast majority of the remainder respond favorably to corticosteroids [28, 29]. Therefore, accurate diagnosis of granulomatous pulmonary disease has important therapeutic and prognostic implications.
Management of GLILD
The underlying pathogenesis of GLILD is incompletely understood, rendering it difficult to select effective therapeutic agents to target disease processes. Proposed mechanisms of pathogenesis over decades include autoreactive T cells or chronic antigenic stimulation of T cells [30, 31] in combination with low switched memory B cells [32], abnormal cytokine profiles [33], and a dysregulated response to human herpes virus-8 [34]. The histological identification of both T cells and B cells in the lungs of patients with GLILD [15•, 22•] and the radiographic relapse noted in our patient following repopulation of B cells, suggest that both lymphocyte populations act in concert in the formation of a dysregulated cytokine environment that supports lymphoproliferation. The lack of regulatory T cells identified histologically [22•] may further contribute to the unchecked proliferation of these abnormal lymphocytes. Enhanced understanding of the mechanisms underlying the development of GLILD represents a key area of study needed to improve the care of patients with this pulmonary complication.
Optimization of immunoglobulin replacement and first-line treatment with corticosteroids are agreed-upon therapeutic strategies among the BLF/UK PIN consensus statements [11••]. While immunoglobulin replacement may be effective for some patients [35], progression of disease is frequently seen, as in our patient. Moreover, effectiveness of corticosteroids is variable [2••, 15•, 36], often necessitating treatment with additional therapeutic agents. Though no guidelines exist on which drugs should be selected following treatment failure with corticosteroids, the BLF/UK PIN consensus statement consultants agree with azathioprine, rituximab, and mycophenolate as reasonable second-line agents [11••]. No prospective randomized clinical trials assessing therapies in GLILD have been performed. Chase and colleagues performed a retrospective chart review of 7 patients with steroid-resistant GLILD who were subsequently treated with a combination of rituximab and azathioprine (or 6-MP in those unable to tolerate azathioprine) for 18 months to target both histologically-identified B and T cell populations, respectively. Significant reductions in radiographic features and improvements in lung function were noted following therapy [15•]. Despite the treatment of our patient with rituximab and 6-MP, he continues to demonstrate a relapsing and remitting course, which may be a feature of the natural history of the disease. Individual case reports and small case series with the use of additional agents, such as cyclosporine, anti-TNF inhibitors, and abatacept, have yielded favorable results [37–40], suggesting that treatment should be individualized in the context of comorbidities and underlying genetic mutations if known.
In addition to a lack of standardized therapies, little agreement exists on the length of therapy or even in which patients to initiate therapy. Though he was asymptomatic, we opted to treat our patient given his young age, extent of lymphoproliferation identified on PET-CT, impaired DLCO, and the increased morbidity and mortality associated with GLILD. The BLF/UK PIN consensus statements note agreement on treatment of patients with deteriorating lung function regardless of the presence or absence of symptoms but no agreement on treatment in patients with abnormal but stable lung function [11••]. This suggests a wide array of treatment practices and highlights the need for development of evidence-based guidelines. While gas transfer was the preferred test for monitoring treatment response and disease progression in the BLF/UK PIN consensus statements [11••], a recent case report suggests the use of PET-CT to combine functional information regarding metabolically active disease with anatomical imaging to more accurately access disease burden [41•].
Conclusion
While historically identified as a non-infectious complication of CVID, GLILD is now increasingly being reported in other PID, including Kabuki syndrome [3], LRBA mutations [4], X-linked inhibitor of apoptosis [5], CTLA4 mutations [6], Good’s syndrome [ 7], RAG mutations [8 ], hypogammaglobulinemia of unclear etiology in infancy [9], and 22q11.2DS [10•]. Moreover, GLILD may even be the first manifestation of an underlying PID [42•]. To our knowledge, this is only the second reported case of GLILD in a 22q11.2DS patient. Though the natural history of GLILD is unknown, the associated comorbidities and poor outcomes highlight the need for early recognition and treatment. Thus, clinicians should maintain a high index of suspicion for GLILD as a rare phenotype of not only CVID but all PID as this has important therapeutic and prognostic implications.
Strides have been made in recent years to characterize the clinical, radiographic, and histological features of GLILD, which allow for its differentiation from other disease entities, such as sarcoidosis. Nonetheless, important gaps of knowledge remain. Robust and consistent clinical predictors of disease development and effective treatment strategies have yet to be prospectively identified. Additionally, it is unclear whether those identified in CVID patients will extrapolate to patients with other immunodeficiencies. Elucidation of the pathogenesis of GLILD will likely not only uncover predictive factors but will also help guide optimal therapeutic regimens while clarity of the natural history of the disease will further refine treatment and monitoring protocols. Though several drugs, including combination chemotherapy with rituximab and azathioprine, have yielded favorable results, whether this alters the morbidity and mortality associated with the disease is uncertain.
Though the BLF/UK PIN consensus statements on the definition, diagnosis, and management of GLILD in CVID patients are not evidence-based, they do represent the first step towards the development of essential practice guidelines. The need for additional research is highlighted by the practice variabilities reported. Though the rarity of this complication in the immunodeficient population, which itself represents a small constituent of the overall population, provides an impedance to further study, active involvement of multidisciplinary subspecialists caring for immunodeficiency patients in patient registries, such as the USIDNET, provides one way to overcome this hurdle. Thus, concerted efforts to gather and report data, including those that have resulted in unsatisfactory outcomes, will further aid in the understanding of this challenging disease for both clinicians and patients.
Abbreviations
- 22q11.2DS
22q11.2 deletion syndrome
- GLILD
Granulomatous-lymphocytic interstitial lung disease
- ILD
Interstitial lung disease
- CVID
Common variable immune deficiency
- PID
Primary immune deficiency
- FISH
Fluorescence in situ hybridization
- IVIG
Intravenous immunoglobulin
- LAD
Lymphadenopathy
- PET-CT
Positron emission tomography computed tomography
- HRCT
High-resolution CT
- VATS
Video-assisted thoracoscopic surgery
- DLCO
Diffusion capacity for carbon monoxide
- FB
Follicular bronchiolitis
- LIP
Lymphoid interstitial pneumonia
- 6-MP
6-Mercaptopurine
- BLF/UK PIN
British Lung Foundation and United Kingdom Primary Immunodeficiency Network
- AIHA
Autoimmune hemolytic anemia
- ITP
Idiopathic thrombocytopenia purpura
- USIDNET
United States Immunodeficiency Network
Footnotes
This article is part of the Topical Collection on Autoimmunity
Compliance with Ethical Standards
Conflict of Interest: The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent: This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, et al. 22q11.2 deletion syndrome. Nature reviews Disease primers. 2015;1:15071. doi: 10.1038/nrdp.2015.71. https://doi.org/10.1038/nrdp.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2••.Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol. 2004;114(2):415–21. doi: 10.1016/j.jaci.2004.05.057. https://doi.org/10.1016/j.jaci.2004.05.057. First coined the term GLILD in patients with CVID and reported that a diagnosis of GLILD is associated with increased morbidity and early mortality. [DOI] [PubMed] [Google Scholar]
- 3.De Dios JA, Javaid AA, Ballesteros E, Metersky ML. An 18-year-old woman with Kabuki syndrome, immunoglobulin deficiency and granulomatous lymphocytic interstitial lung disease. Conn Med. 2012;76(1):15–8. [PubMed] [Google Scholar]
- 4.Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90(6):986–1001. doi: 10.1016/j.ajhg.2012.04.015. https://doi.org/10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steele CL, Dore M, Ammann S, Loughrey M, Montero A, Burns SO, et al. X-linked inhibitor of apoptosis complicated by granulomatous lymphocytic interstitial lung disease (GLILD) and granulomatous hepatitis. J Clin Immunol. 2016;36(7):733–8. doi: 10.1007/s10875-016-0320-3. https://doi.org/10.1007/s10875-016-0320-3. [DOI] [PubMed] [Google Scholar]
- 6.Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (New York, NY) 2014;345(6204):1623–7. doi: 10.1126/science.1255904. https://doi.org/10.1126/science.1255904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jensen ML, Bendstrup E, Hilberg O. Granulomatous-lymphocytic interstitial lung disease and recurrent sinopulmonary infections in a patient with Good’s syndrome. BMJ case reports. 2015;2015 doi: 10.1136/bcr-2014-205635. https://doi.org/10.1136/bcr-2014-205635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchbinder D, Baker R, Lee YN, Ravell J, Zhang Y, McElwee J, et al. Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol. 2015;35(2):119–24. doi: 10.1007/s10875-014-0121-5. https://doi.org/10.1007/s10875-014-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adeleye A, Kelly M, Wright NA, Yu W, Anselmo MA. Granulomatous lymphocytic interstitial lung disease in infancy. Can Respir J. 2014;21(1):20–2. doi: 10.1155/2014/904675. https://doi.org/10.1155/2014/904675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10•.Mather MW, Hayhurst H, Bacon CM, Cole TS, Pan-Hammarstrom Q, Misbah S, et al. Mutation of TNFRSF13B in a child with 22q11 deletion syndrome associated with granulomatous lymphoproliferation. J Allergy Clin Immunol. 2015;135(2):559–61. doi: 10.1016/j.jaci.2014.07.025. https://doi.org/10.1016/j.jaci.2014.07.025. The first and only previously published report of GLILD in a patient with 22q11.2DS. [DOI] [PubMed] [Google Scholar]
- 11••.Hurst JR, Verma N, Lowe D, Baxendale HE, Jolles S, Kelleher P, et al. British Lung Foundation/United Kingdom Primary Immunodeficiency Network consensus statement on the definition, diagnosis, and management of granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2017;5(4):938–45. doi: 10.1016/j.jaip.2017.01.021. https://doi.org/10.1016/j.jaip.2017.01.021. These consensus statements represent the largest collection of shared clinical experience in the multi-disciplinary care of patients with GLILD and are the first step towards development of practice guidelines. [DOI] [PubMed] [Google Scholar]
- 12•.Liebow AA, Carrington CB. Diffuse pulmonary lymphoreticular infiltrations associated with dysproteinemia. Med Clin North Am. 1973;57(3):809–43. doi: 10.1016/s0025-7125(16)32278-7. One of the first reports of the lymphoproliferative features seen in GLILD in a patient with dysproteinemia. [DOI] [PubMed] [Google Scholar]
- 13.Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–86. doi: 10.1182/blood-2007-11-124545. https://doi.org/10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 14.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119(7):1650–7. doi: 10.1182/blood-2011-09-377945. https://doi.org/10. 1182/blood-2011-09-377945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15•.Chase NM, Verbsky JW, Hintermeyer MK, Waukau JK, Tomita-Mitchell A, Casper JT, et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID) J Clin Immunol. 2013;33(1):30–9. doi: 10.1007/s10875-012-9755-3. https://doi.org/10. 1007/s10875-012-9755-3. This retrospective chart review of 7 patients with GLILD that were unresponsive to conventional therapy with corticosteroids demonstrated the utility of combination chemotherapy treatment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mannina A, Chung JH, Swigris JJ, Solomon JJ, Huie TJ, Yunt ZX, et al. Clinical predictors of a diagnosis of common variable immunodeficiency-related granulomatous-lymphocytic interstitial lung disease. Ann Am Thorac Soc. 2016;13(7):1042–9. doi: 10.1513/AnnalsATS.201511-728OC. https://doi.org/10.1513/AnnalsATS.201511-728OC. [DOI] [PubMed] [Google Scholar]
- 17.Hartono S, Motosue MS, Khan S, Rodriguez V, Iyer VN, Divekar R, et al. Predictors of granulomatous lymphocytic interstitial lung disease in common variable immunodeficiency. Ann Allergy, Asthma Immunol: Off Publ Am Coll Allergy, Asthma, Immunol. 2017;118(5):614–20. doi: 10.1016/j.anai.2017.01.004. https://doi.org/10.1016/j.anai.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Feuille EJ, Anooshiravani N, Sullivan KE, Fuleihan RL, Cunningham-Rundles C. Autoimmune cytopenias and associated conditions in CVID: a report from the USIDNET registry. J Clin Immunol. 2017;38(1):28–34. doi: 10.1007/s10875-017-0456-9. https://doi.org/10.1007/s10875-017-0456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka N, Kim JS, Bates CA, Brown KK, Cool CD, Newell JD, et al. Lung diseases in patients with common variable immunodeficiency: chest radiographic, and computed tomographic findings. J Comput Assist Tomogr. 2006;30(5):828–38. doi: 10.1097/01.rct.0000228163.08968.26. https://doi.org/10. 1097/01.rct.0000228163.08968.26. [DOI] [PubMed] [Google Scholar]
- 20.Park JE, Beal I, Dilworth JP, Tormey V, Haddock J. The HRCT appearances of granulomatous pulmonary disease in common variable immune deficiency. Eur J Radiol. 2005;54(3):359–64. doi: 10.1016/j.ejrad.2004.09.005. https://doi.org/10.1016/j.ejrad.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 21.Torigian DA, LaRosa DF, Levinson AI, Litzky LA, Miller WT., Jr Granulomatous-lymphocytic interstitial lung disease associated with common variable immunodeficiency: CT findings. J Thorac Imaging. 2008;23(3):162–169. doi: 10.1097/RTI.0b013e318166d32f. doi: https://doi.org/10.1097/RTI.0b013e318166d32f. [DOI] [PubMed] [Google Scholar]
- 22•.Rao N, Mackinnon AC, Routes JM. Granulomatous and lymphocytic interstitial lung disease: a spectrum of pulmonary histopathologic lesions in common variable immunodeficiency—histologic and immunohistochemical analyses of 16 cases. Hum Pathol. 2015;46(9):1306–14. doi: 10.1016/j.humpath.2015.05.011. https://doi.org/10.1016/j.humpath.2015.05.011. Characterized the spectrum of histopathologic and immunohistochemical features of GLILD through a retrospective chart review of 16 cases and highlighted the absence of FOXP3+ T regulatory cells in lung biopsies of patients with GLILD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carmona EM, Kalra S, Ryu JH. Pulmonary sarcoidosis: diagnosis and treatment. Mayo Clin Proc. 2016;91(7):946–54. doi: 10.1016/j.mayocp.2016.03.004. https://doi.org/10.1016/j.mayocp.2016.03.004. [DOI] [PubMed] [Google Scholar]
- •24.Shanks AM, Alluri R, Herriot R, Dempsey O. Misdiagnosis of common variable immune deficiency. BMJ case reports. 2014 doi: 10.1136/bcr-2013-202806. 2014(apr01 1):bcr2013202806. https://doi.org/10.1136/bcr-2013-202806. This case report emphasized the clinical and histologic overlap of GLILD and sarcoidosis, often leading to misdiagnosis. [DOI] [PMC free article] [PubMed]
- 25.Verbsky JW, Routes JM. Sarcoidosis and common variable immunodeficiency: similarities and differences. Semin Respir Crit Care Med. 2014;35(3):330–5. doi: 10.1055/s-0034-1376862. https://doi.org/10.1055/s-0034-1376862. [DOI] [PubMed] [Google Scholar]
- 26•.Bouvry D, Mouthon L, Brillet PY, Kambouchner M, Ducroix JP, Cottin V, et al. Granulomatosis-associated common variable immunodeficiency disorder: a case-control study versus sarcoidosis. Eur Respir J. 2013;41(1):115–22. doi: 10.1183/09031936.00189011. doi: https://doi.org/10.1183/09031936.00189011. The clinical, radiographic, and histologic features as well as the clinical course of GLILD and sarcoidosis are compared and contrasted in this case-control study. [DOI] [PubMed] [Google Scholar]
- 27.Baughman RP, Teirstein AS, Judson MA, Rossman MD, Yeager H, Jr, Bresnitz EA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1885–9. doi: 10.1164/ajrccm.164.10.2104046. https://doi.org/10.1164/ajrccm.164.10.2104046. [DOI] [PubMed] [Google Scholar]
- 28.Gibson GJ, Prescott RJ, Muers MF, Middleton WG, Mitchell DN, Connolly CK, et al. British Thoracic Society Sarcoidosis study: effects of long term corticosteroid treatment. Thorax. 1996;51(3):238–47. doi: 10.1136/thx.51.3.238. https://doi.org/10.1136/thx.51.3.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gottlieb JE, Israel HL, Steiner RM, Triolo J, Patrick H. Outcome in sarcoidosis. The relationship of relapse to corticosteroid therapy. Chest. 1997;111(3):623–31. doi: 10.1378/chest.111.3.623. [DOI] [PubMed] [Google Scholar]
- 30.Levinson AI, Hopewell PC, Stites DP, Spitler LE, Fudenberg HH. Coexistent lymphoid interstitial pneumonia, pernicious anemia, and agammaglobulinemia. Arch Intern Med. 1976;136(2):213–6. https://doi.org/10.1001/archinte.1976.03630020067014. [PubMed] [Google Scholar]
- 31.Kohler PF, Cook RD, Brown WR, Manguso RL. Common variable hypogammaglobulinemia with T-cell nodular lymphoid interstitial pneumonitis and B-cell nodular lymphoid hyperplasia: different lymphocyte populations with a similar response to prednisone therapy. J Allergy Clin Immunol. 1982;70(4):299–305. doi: 10.1016/0091-6749(82)90066-5. https://doi.org/10.1016/0091-6749(82)90066-5. [DOI] [PubMed] [Google Scholar]
- 32.Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, Vlkova M, Hernandez M, Detkova D, Bos PR, Poerksen G, von Bernuth H, Baumann U, Goldacker S, Gutenberger S, Schlesier M, Bergeron-van der Cruyssen F, le Garff M, Debre P, Jacobs R, Jones J, Bateman E, Litzman J, van Hagen PM, Plebani A, Schmidt RE, Thon V, Quinti I, Espanol T, Webster AD, Chapel H, Vihinen M, Oksenhendler E, Peter HH, Warnatz K. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111(1):77–85. doi: 10.1182/blood-2007-06-091744. doi: https://doi.org/10.1182/blood-2007-06-091744,1. [DOI] [PubMed] [Google Scholar]
- 33.Mullighan CG, Fanning GC, Chapel HM, Welsh KI. TNF and lymphotoxin-alpha polymorphisms associated with common variable immunodeficiency: role in the pathogenesis of granulomatous disease. J Immunol. 1997;159(12):6236–41. [PubMed] [Google Scholar]
- 34.Wheat WH, Cool CD, Morimoto Y, Rai PR, Kirkpatrick CH, Lindenbaum BA, et al. Possible role of human herpesvirus 8 in the lymphoproliferative disorders in common variable immunodeficiency. J Exp Med. 2005;202(4):479–84. doi: 10.1084/jem.20050381. https://doi.org/10.1084/jem.20050381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hasegawa M, Sakai F, Okabayashi A, Sato A, Yokohori N, Katsura H, et al. Intravenous immunoglobulin monotherapy for granulomatous lymphocytic interstitial lung disease in common variable immunodeficiency. Intern Med (Tokyo, Japan) 2017;56(21):2899–902. doi: 10.2169/internalmedicine.7757-16. https://doi.org/10.2169/internalmedicine.7757-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boursiquot JN, Gerard L, Malphettes M, Fieschi C, Galicier L, Boutboul D, et al. Granulomatous disease in CVID: retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J Clin Immunol. 2013;33(1):84–95. doi: 10.1007/s10875-012-9778-9. https://doi.org/10.1007/s10875-012-9778-9. [DOI] [PubMed] [Google Scholar]
- 37.Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (New York, NY) 2015;349(6246):436–40. doi: 10.1126/science.aaa1663. https://doi.org/10.1126/science.aaa1663. [DOI] [PubMed] [Google Scholar]
- 38.Franxman TJ, Howe LE, Baker JR., Jr Infliximab for treatment of granulomatous disease in patients with common variable immunodeficiency. J Clin Immunol. 2014;34(7):820–7. doi: 10.1007/s10875-014-0079-3. https://doi.org/10.1007/s10875-014-0079-3. [DOI] [PubMed] [Google Scholar]
- 39.Hatab AZ, Ballas ZK. Caseating granulomatous disease in common variable immunodeficiency treated with infliximab. J Allergy Clin Immunol. 2005;116(5):1161–2. doi: 10.1016/j.jaci.2005.08.041. https://doi.org/10.1016/j.jaci.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 40.Davies CW, Juniper MC, Gray W, Gleeson FV, Chapel HM, Davies RJ. Lymphoid interstitial pneumonitis associated with common variable hypogammaglobulinaemia treated with cyclosporin a. Thorax. 2000;55(1):88–90. doi: 10.1136/thorax.55.1.88. https://doi.org/10.1136/thorax.55.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41•.Jolles S, Carne E, Brouns M, El-Shanawany T, Williams P, Marshall C, et al. FDG PET-CT imaging of therapeutic response in granulomatous lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID) Clin Exp Immunol. 2017;187(1):138–45. doi: 10.1111/cei.12856. https://doi.org/10.1111/cei.12856. This case report suggests that FDG PET-CT imaging may prove to be a useful imaging modality that combines both functional and anatomical information in the long-term monitoring of patients with GLILD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42•.Tashtoush B, Memarpour R, Ramirez J, Bejarano P, Mehta J. Granulomatous-lymphocytic interstitial lung disease as the first manifestation of common variable immunodeficiency. Clin Respir J. 2016;12(1):337–43. doi: 10.1111/crj.12511. https://doi.org/10.1111/crj.12511. Highlights the need to maintain a high index of suspicion for GLILD as this may be the first manifestation of an underlying immune deficiency. [DOI] [PubMed] [Google Scholar]