

Summary
Glycoproteins engaged in unproductive folding in the ER are marked for degradation by a signal generated by progressive demannosylation of substrate N-glycans that is decoded by ER lectins, but how the two lectins, OS9 and XTP3B, contribute to non-glycosylated protein triage is unknown. We generated cell lines with homozygous deletions of both lectins individually and in combination. We find that OS9 and XTP3B redundantly promote glycoprotein degradation and stabilize the SEL1L/HRD1 dislocon complex, that XTP3B profoundly inhibits the degradation of non-glycosylated proteins and that OS9 antagonizes this inhibition. The relative expression of OS9 and XTP3B and the distribution of glycan and non-glycan degrons within the same protein contribute to the fidelity and processivity of glycoprotein triage and therefore, determine the fates of newly synthesized proteins in the early secretory pathway.
eTOC blurb
The ERAD machinery must recognize misfolded proteins with vastly different structures, topologies, and folding rates. Van der Goot et al. show that the relative abundance of two lectins, OS9 and XTP3B, and the presence of glycan- and non-glycan degrons within a protein contribute to the fidelity and processivity of ER protein triage.
Introduction
Proteins destined for secretion or insertion into cellular membranes undergo conformational maturation in the endoplasmic reticulum (ER), where they are subject to covalent modifications including disulfide bond formation and N-linked glycosylation (McCaffrey and Braakman, 2016, Oka and Bulleid, 2013). Proteins that fail to correctly fold or assemble can be detrimental to cellular or organismal fitness and are destroyed by the ubiquitin-proteasome system (UPS) through a process called ER-associated degradation (ERAD) (McCracken and Brodsky, 1996). Understanding how this quality control surveillance process faithfully identifies and degrades folding-defective proteins, while facilitating efficient deployment of native proteins, is a central question in cell biology.
ERAD is mediated by a modular network centered around membrane-integrated E3 ubiquitin ligases that organize the complex machinery responsible for recognition of folding-defective proteins and their delivery to the cytosol for destruction by the ubiquitin-proteasome system (UPS) (Vembar and Brodsky, 2008, Xie and Ng, 2010, Olzmann et al., 2013). Although genetic and biochemical studies in yeast and mammals have identified many of the core components of the ERAD machinery involved in substrate recognition and degradation, how these components cooperate to distinguish terminally misfolded proteins from on-pathway folding species, given the immense diversity of these substrates in terms of structure, topology, and folding rates, is incompletely understood. It is clear, however, that asparagine-linked glycans play an important role in the maturation and quality control of the proteins to which they are attached (Helenius and Aebi, 2004, Xu and Ng, 2015).
The majority of proteins that enter the ER are co-translationally modified on asparagine residues by the covalent attachment of a three-branch, high-mannose glycan (Glc3-Man9-GlcNAc2) (Helenius and Aebi, 2004). Continuous removal of terminal α1,2-linked mannose residues from these glycans by slow-acting α-mannosidases has been proposed to serve as a “timer” that assesses how long a given protein has been engaged in unproductive folding attempts (Helenius and Aebi, 2004). The resulting trimmed glycan, bearing ≤7 mannose residues (Man5-7-GlcNAc2), exposes a terminal, C-branch, α1,6-linked mannose residue (Quan et al., 2008) that is recognized by ER-resident lectins that deliver the substrate to a membrane-integrated “dislocon” (Hebert et al., 2005, Xu and Ng, 2015). In yeast, the lectin Yos9 selectively binds to these trimmed glycans via a conserved mannose-6-phosphate receptor homology (MRH) domain (Szathmary et al., 2005, Kim et al., 2005, Bhamidipati et al., 2005) and directs them to the multispanning Hrd1 ubiquitin ligase via the adaptor protein Hrd3 (Denic et al., 2006). By sensing the simultaneous exposure of an unfolded domain and a terminal α1,6-linked mannose, Yos9 appears to function as a “gatekeeper” of this dislocation apparatus that ensures that proteins are committed to ERAD only after they have undergone unproductive folding attempts.
Metazoans have two orthologues of Yos9: OS9 and XTP3B (Erlectin; (Cruciat et al., 2006)). Both lectins physically interact with ERAD substrates and with the HRD1 E3 ubiquitin ligase complex via the adaptor protein SEL1L, the orthologues of yeast Hrd1 and Hrd3, respectively, (Christianson et al., 2008) and have thus been proposed to serve a similar gatekeeper function in mammalian cells (Bernasconi et al., 2008, Christianson et al., 2008, Hosokawa et al., 2008). The functional relationship between these two lectins, whether they have unique or redundant roles in ERAD, and the nature of their interactions with SEL1L is unclear. Several studies have reported that OS9 is essential, while XTP3B is dispensable for glycoprotein ERAD (Seaayfan et al., 2016, Christianson et al., 2008, Bernasconi et al., 2008, Mueller et al., 2008, Alcock and Swanton, 2009, Hosokawa et al., 2009, Tyler et al., 2012, Tang et al., 2014). One study reported that OS9 and XTP3B are functionally redundant (Bernasconi et al., 2010), while yet another concluded that XTP3B functionally opposes OS9, thereby protecting ERAD substrates from premature degradation (Fujimori et al., 2013). Thus, despite extensive study, the rules that govern the roles of OS9 and XTP3B in ERAD remain obscure and confusing, perhaps owing to technical issues like overexpression, which can drastically alter SEL1L/HRD1 complex stoichiometry (Hwang et al., 2017), or incomplete depletion by RNA silencing. It is also likely, however, that some of this ambiguity reflects real differences in the intrinsic signals by which different substrates are engaged by this complex protein quality control apparatus including the relative contributions of glycan and non-glycan degrons.
Some proteins synthesized in the secretory pathway are not naturally glycosylated but are nonetheless subject to HRD1-dependent ERAD (Okuda-Shimizu and Hendershot, 2007, Sato et al., 2012, Hoelen et al., 2015) indicating that both glycan-dependent and -independent modes of protein triage co-exist. Although Yos9 can bind both glycan and non-glycan polypeptide determinants on misfolded proteins in yeast (Buschhorn et al., 2004, Bhamidipati et al., 2005, Kim et al., 2005, Szathmary et al., 2005, Benitez et al., 2011, Jaenicke et al., 2011, Smith et al., 2014), a role for ER lectins in non-glycosylated protein ERAD in metazoans remains largely unaddressed. Moreover, while some glycosylated ERAD substrates are strongly stabilized by pharmacological inhibition of α−mannosidases, other substrates are only partially or negligibly stabilized by these drugs (Christianson et al., 2011), implying that ERAD of some glycoproteins may depend on hybrid signals containing both glycan- and peptide-based degrons.
Understanding the roles of ER lectins in ERAD is complicated further because the interaction between OS9 or XTP3B and SEL1L also exhibits both glycan-dependent (Satoh et al., 2010, Christianson et al., 2008) and glycan-independent features (Hosokawa et al., 2009, Fujimori et al., 2013). SEL1L has five predicted N-linked glycosylation sites that are highly conserved in both number and location. Whether any of these five glycans are subject to demannosylation and contribute to binding ER lectins has not been previously investigated.
In this study, we used CRISPR/Cas9 to knockout OS9 and XTP3B, individually and together, to investigate their roles in ERAD of a range of substrates with different topologies and glycosylation states. We find that these two lectins are essential to stabilize the SEL1L/HRD1 complex and are, individually, both sufficient and redundant to promote degradation of membrane or luminal glycoproteins. Non-glycosylated ERAD substrates, however, are degraded by a SEL1L-dependent process requiring neither lectin, suggesting that conformational determinants in folding- or assembly-defective proteins are decoded by factors in the ER other than OS9 and XTP3B. Remarkably, we find that non-glycoprotein degradation is strongly inhibited by XTP3B and that this inhibition is antagonized by OS9, suggesting a complex interplay between OS9 and XTP3B in ERAD substrate selection.
Results
Complex roles of OS9 and XTP3B in glycoprotein ERAD
To elucidate the role and functional relationship of OS9 and XTP3B in substrate recognition and delivery to the SEL1L/HRD1 complex, we used CRISPR/Cas9 genome editing to generate HEK293 cell lines in which the genes that code for these lectins were disrupted individually (OS9 KO, XTP3B KO) or together (DKO) (Fig. S1A and Tables S1–3). We also generated cell lines lacking SEL1L. Loss of expression of these proteins was confirmed by immunoblot analysis of whole-cell detergent lysates of two independent clonal cell lines generated with different guide RNA sequences (Fig. 1A and Fig. S1B). While acute loss of SEL1L has previously been reported to induce ER stress (Sun et al., 2014), Xbp1 mRNA splicing was not enhanced in the stable knockout cell lines and each cell line was able to activate an unfolded protein (UPR), suggesting that the UPR is neither impaired nor constitutively activated (Fig. S1C).
Figure 1. Complex roles of OS9 and XTP3B in glycoprotein ERAD.

(A) Immunoblot analysis of OS9 and XTP3B in cell lysates from wild type (WT) and two independent clonal knockout lines for each genotype. (B) Schematic representation of the topology and glycosylation status of the glycoproteins examined in this study. (C) Turnover of endogenous and exogenous ERAD substrates. Untransfected (CD147 and HLA-C) or transfected (A1ATNHK-HA) cells were treated with the protein synthesis inhibitors cycloheximide or emetine (PSI-chase) for the indicated times, and glycoprotein levels were determined by immunoblotting with the indicated antibodies. Mat=mature; CG=core-glycosylated; DeM=demannosylated. Quantification represents mean±SEM (n=3). (D and E) Effect of kifunensine on glycoprotein turnover. Untransfected (D) and A1ATNHK-HA transfected (E) cells were treated (~16 hrs) with kifunensine (KIF) prior to PSI-chase and immunoblot with the indicated antibodies. The vertical line indicates where a lane with protein marker was cut for simplicity and independent immunoblots are indicated by the dashed horizontal lines.. See also Fig. S1.
We used translational shut-off to measure the turnover of HRD1-dependent ERAD clients in OS9 KO, XTP3B KO and DKO cells (Fig. 1B, C). CD147 is an endogenous, ubiquitously expressed, Type I integral membrane glycoprotein that forms a stoichiometric complex with the monocarboxylic acid transporters MCT1, MCT3, and MCT4 (Kirk et al., 2000). Due to inefficient assembly with its partners in the ER, excess ER-localized, core-glycosylated (CG) forms of CD147 are degraded by ERAD (Tyler et al., 2012) (Fig. 1C). CD147 (CG) was strongly stabilized in SEL1L KO cells, as expected for a substrate of HRD1-dependent ERAD. While deletion of OS9 or XTP3B individually had no detectable effect on CD147 stability, loss of both lectins (DKO) stabilized this substrate to the same extent observed in the SEL1L KO, suggesting that OS9 and XTP3B can functionally substitute for one another in degrading CD147. We identified HLA-C, another Type I integral membrane glycoprotein, as an endogenous ERAD substrate because of its increased association with endogenously TAP-tagged HRD1 (Hwang et al., 2017) upon pharmacological inhibition of the cytoplasmic AAA ATPase p97/VCP (Fig. S1D). Degradation of HLA-C was also strongly dependent on SEL1L and unaffected by loss of OS9. However, in contrast to CD147, knockout of both lectins only partially stabilized HLA-C, and XTP3B knockout individually led to a small but reproducible acceleration of HLA-C turnover (Fig. 1C). These effects were reproduced in independent clonal cell lines generated with different guide RNA sequences, rendering unlikely the possibility that they were caused by off-target mutations (Fig. S1E).
The partial effect of OS9/XTP3B DKO in stabilizing HLA-C, relative to the profound stabilization upon SEL1L KO, suggests that this substrate, unlike CD147 (which is stabilized to the same extent by DKO and SEL1L KO), may be targeted to the SEL1L/HRD1 complex by two routes, only one of which requires α1,6-mannose recognition. To test this, we studied the effect of kifunensine, a selective inhibitor of α-mannosidases (Elbein et al., 1990), on the turnover of both endogenous ERAD substrates. While kifunensine stabilized CD147 to the same extent as seen in DKO or SEL1L KO cells, it had an intermediate effect on HLA-C degradation, stabilizing it to roughly the same extent as observed in DKO cells (Fig. 1D). Thus, while both endogenous Type I substrates strongly depend on SEL1L, HLA-C degradation only partially depends on N-glycan trimming and recognition, suggesting that unassembled Type I membrane glycoproteins can be delivered to the SEL1L/HRD1 dislocon by at least two distinct routes that differ in their dependence on ER lectins and demannosylation.
To assess the involvement of OS9 and XTP3B in ERAD of luminal glycoproteins, we analyzed the turnover of the null Hong Kong variant of α-1-antitrypsin (A1ATNHK), a well-studied model ERAD substrate (Sifers et al., 1988). Translational shut-off confirmed that A1ATNHK is unstable in wild type cells, and its degradation is strongly dependent on SEL1L (Fig. 1C). Intriguingly, loss of ER lectins individually had opposite effects on A1ATNHK turnover, with OS9 KO stabilizing and XTP3B KO destabilizing the substrate to roughly the same extent. A1ATNHK was strongly stabilized in DKO cells (Fig. 1C) and to a lesser extent by kifunensine (Fig. 1E), suggesting that, like HLA-C, A1ATNHK degradation depends on a combination of glycan and non-glycan signals. XTP3B appears to have two different roles in decoding these signals: one that inhibits degradation in the presence of OS9 and one that promotes degradation in its absence.
Stability of the SEL1L/HRD1 complex depends on the presence of ER lectins but not their ability to bind glycans
The above findings establish that OS9 and XTP3B play partially redundant roles that are consistent with their proposed functions as adaptors that help to deliver terminally misfolded glycoproteins to the membrane ERAD machinery. It is also possible however that some of the phenotypes that we observe upon deletion of these lectins could reflect the loss of downstream machinery if they contributed to regulating the stability and/or activity of the SEL1L/HRD1 complex itself. HRD1 and SEL1L form a high molecular weight ERAD complex in the ER membrane (Hwang et al., 2017, Christianson et al., 2011, Mueller et al., 2008, Hosokawa et al., 2008, Christianson et al., 2008) and depend on each other for mutual stabilization (Sun et al., 2014, Iida et al., 2011). While disruption of either OS9 or XTP3B alone did not affect steady-state levels of SEL1L or HRD1, knockout of both lectins together resulted in a substantial decrease in SEL1L (75±7%; SEM; n=3) and HRD1 (47±9%; SEM; n=3) levels (Fig. 2A), accompanied by increases in EDEM1, EDEM3 (Fig. S2B) and OS9 (Fig. S2A). These changes in upstream and downstream SEL1L interactors are most likely the result of SEL1L destabilization in response to lectin depletion, as comparable changes were observed upon direct SEL1L targeting (Fig. S2A). Although SEL1L is very stable in WT HEK293 cells (t1/2 ≫ 6hr) we observed that SEL1L turnover was substantially accelerated (t1/2 ~4hr) in DKO cells (Fig. 2B). Thus, SEL1L stability is strongly dependent on ER lectins and that OS9 and XTP3B play redundant roles in maintaining the stability of the SEL1L/HRD1 ERAD complex.
Figure 2. Stability of the SEL1L/HRD1 complex depends on the presence of OS9 or XTP3B but not their MRH domains.

(A). Immunoblot analysis of cell lysates from wild type and the indicated knockout cell lines. Fold-change relative to wild type cells was calculated after normalization to GAPDH. (B) PSI-chase analysis of SEL1L. (C, D) Steady-state levels of SEL1L, HRD1, OS9, or XTP3B in cell lysates from WT cells, DKO cells, and DKO cells stably expressing the indicated OS9 constructs (C) or XTP3B constructs (D). Quantification in A and B represents mean±SEM (n=3). See also Fig. S2.
To investigate the mechanism of SEL1L destabilization we expressed wild-type or mutant OS9 and XTP3B in DKO cells. Expression of the two predominant isoforms of OS9, OS9.1/9.2 (Fig. 2C), or XTP3B (Fig. 2D) restored the steady-state levels of SEL1L and HRD1 to those observed in wild type cells, confirming that SEL1L deficiency in DKO cells is due solely to the loss of these lectins. Expression of OS9 variants that harbor mutations at Arg188 (R188A), a residue that directly contacts the exposed α1,6-mannose residues on substrate glycans, or the corresponding mutations in either or both MRH domains of XTP3B (Mikami et al., 2010, Satoh et al., 2010), also led to comparable rescue of SEL1L levels (Fig. 2C, D). Together, these data indicate that OS9 and XTP3B are functionally redundant in stabilizing the SEL1L/HRD1 ERAD complex and that this function does not require high affinity glycan binding.
Lectin-glycan interactions are required for ERAD of glycosylated substrates
To determine whether the effects of lectin knockout on glycoprotein ERAD are due to the dramatic reduction of SEL1L in DKO cells, we exploited the fact that expression of glycan binding-deficient variants of OS9 and XTP3B is sufficient to restore SEL1L levels in DKO cells. As expected, reintroduction of wild type OS9 (Fig. 3A) or XTP3B (Fig. 3B) restored the degradation of CD147 and HLA-C. Interestingly, we found that rescue by OS9.1 was less effective than OS9.2 in restoring CD147, but not HLA-C, degradation. This difference is not the result of a differential expression (data not shown) of the two splice variants, nor is it likely to be the result of integration site differences as similar findings were observed in independently isolated clonal cell lines (Fig. S3). Degradation of CD147 and HLA-C was also fully rescued by expression of an XTP3B MRH2 variant (R428A), indicating that this MRH domain is dispensable for both SEL1L stability and glycoprotein ERAD. By contrast, glycan binding-deficient MRH domain variants of OS9 (R188A) (Fig. 3A) or XTP3B MRH1 (R207A) (Fig. 3B) were completely deficient in rescuing the degradation of CD147 and HLA-C. These data show that, in contrast to their role in SEL1L stability, interaction of OS9 and XTP3B with glycans is essential for their function in ERAD of integral membrane glycoproteins.
Figure 3. Functional MRH-domain in OS9 and XTP3B is required for ERAD of glycosylated substrates.

(A and B) PSI-chase analysis of endogenous CD147 and HLA-C in WT, DKO, and DKO cells stably expressing the indicated OS9 (A) or XTP3B (B) variants. Quantification represents mean±SEM (n=3). Independent immunoblots are indicated by the dashed horizontal lines. See also Fig. S3.
XTP3B inhibits degradation of non-glycosylated substrates
Although most proteins synthesized in the secretory pathway are glycoproteins, some lack N-glycosylation sequons, but can nevertheless be degraded by SEL1L/HRD1-dependent ERAD when misfolded or unassembled (Okuda-Shimizu and Hendershot, 2007, Hoelen et al., 2015, Sato et al., 2012). Yos9 binds to both demannosylated ERAD substrates and to unfolded domains of peptides (Buschhorn et al., 2004, Bhamidipati et al., 2005, Kim et al., 2005, Szathmary et al., 2005, Benitez et al., 2011, Jaenicke et al., 2011), but its role in turnover of non-glycosylated proteins is unclear. To assess the role of ER lectins in the degradation of non-glycosylated proteins we studied the turnover of two non-glycosylated HRD1-dependent ERAD substrates (Fig. 4A).
Figure 4. Non-redundant roles of OS9 and XTP3B in ERAD of non-glycosylated substrates.

(A) Schematic representation of the topology and glycosylation status of the non-glycosylated proteins examined in this study. (B) PSI-chase analysis of the indicated transfected substrates ~72 hrs after transfection. Quantification represents mean±STDEV (n=2) for A1ATNHK-QQQ-HA and TTRD18G-HA or the mean±SEM (n=5) for TTRD18G/N98Q-HA. Independent immunoblots are indicated by the dashed horizontal lines. (C) Cells were transfected with either control (ctr) or HRD1 siRNA and subsequently with A1ATNHK-QQQ-HA and turnover analyzed by PSI-chase ~72 hrs later. Fold-changes in HRD1 levels were calculated relative to HRD1 levels in WT cells treated with ctr siRNA. Quantification represents the mean±STDEV (n=2).
Degradation of A1ATNHK-QQQ, an A1ATNHK variant engineered to lack all three glycosylation sequons (Cormier et al., 2009), was strongly SEL1L-dependent (Fig. 4B). In sharp contrast to glycosylated SEL1L/HRD1 clients, for which OS9 and XTP3B play important and redundant roles, we found that A1ATNHK-QQQ was robustly stabilized upon deletion of OS9 alone, and was slightly destabilized upon individual knockout of XTP3B. Remarkably, the strong stabilization of A1ATNHK-QQQ observed upon knockout of OS9 was completely suppressed by simultaneous deletion of XTP3B (DKO; Fig. 4B). A1ATNHK-QQQ degradation was robustly inhibited in DKO and wild type cells by silencing of HRD1, confirming that the restoration of A1ATNHK-QQQ degradation by co-deletion of XTP3B and OS9 is not due to diversion of this protein to a different E3 ligase complex (Fig. 4C). These data establish that OS9 and XTP3B are both dispensable for HRD1-dependent degradation of A1ATNHK-QQQ and, moreover, reveal a potent inhibitory function of XTP3B that can be neutralized by OS9.
TTRD18G, a folding-defective, disease-causing variant of the naturally non-glycosylated secretory protein transthyretin (TTR) is also a substrate for HRD1-dependent ERAD (Sekijima et al., 2005, Sato et al., 2012). We found that TTRD18G was strongly stabilized by knockout of SEL1L or OS9 (Fig. 4B) but unlike A1ATNHK-QQQ, this stabilization was unaffected by further loss of XTP3B , nor was TTRD18G stability affected by the individual deletion of XTP3B. Stabilization of TTRD18G following deletion of OS9 or SEL1L was accompanied by the appearance of a high molecular weight form of TTRD18G that is due to posttranslational N-glycosylation at a cryptic site (N98) that becomes exposed during prolonged non-productive residence in the ER (Sato et al., 2012). Therefore, even though TTR is normally non-glycosylated, TTRD18G is subject to degradation by both glycan-dependent and - independent ERAD machinery. To assess the relative contributions of glycan-dependent and - independent pathways to TTRD18G turnover, we mutated the cryptic glycosylation site to a glutamine (TTRD18G/N98Q). Although TTRD18G/N98Q was degraded more slowly than TTRD18G in wild type cells, it was stabilized to a similar extent in cells lacking SEL1L or OS9 and was destabilized upon deletion of XTP3B (Fig. 4B). Strikingly, as observed for A1ATNHK-QQQ, stabilization of TTRD18G/N98Q by OS9 knockout was suppressed by co-deletion of XTP3B, confirming that XTP3B can inhibit the degradation of non-glycosylated ERAD substrates. Thus, both glycan and non-glycan degrons can co-exist in the same molecule and OS9 and XTP3B play very different roles in decoding them.
These data establish that both OS9 and XTP3B are dispensable for SEL1L/HRD1-mediated ERAD of non-glycosylated proteins and reveal XTP3B to be a potent inhibitor of non-glycosylated protein degradation by a mechanism that is antagonized by OS9. OS9 does not require a functional MRH domain to neutralize XTP3B’s inhibitory function since degradation of TTRD18G/N98Q (Fig. S4A) and A1ATNHK-QQQ (Fig. S4B) in OS9 KO cells could be fully rescued with glycan-binding deficient OS9. To better understand this antagonism, we studied the effect of XTP3B overexpression on TTRD18G/N98Q and A1ATNHK-QQQ turnover in wild type cells that endogenously express both lectins. Transient transfection of wild type XTP3B resulted in increased steady-state levels of both TTRD18G/N98Q and A1ATNHK-QQQ and a substantial stabilization of both substrates (Fig. 5A, B), suggesting that turnover of non-glycosylated proteins depends on the relative levels of XTP3B and OS9. Interestingly, we found that A1ATNHK-QQQ was stable (t1/2~8hr) when expressed in K562 cells compared to its glycosylated counterpart A1ATNHK (t1/2~2.5hr; Fig. 5C), but that depleting endogenous XTP3B accelerated A1ATNHKQQQ degradation in K562 cells while depleting endogenous OS9 further stabilized A1ATNHK-QQQ (Fig. 5D, Fig. S4C). These effects were reproduced in independent cell lines generated with different guide RNA sequences, rendering unlikely the possibility that they were caused by off-target effects (Fig. S5D). Thus, the relative steady-state levels of XTP3B and OS9 influences the turnover of a non-glycosylated ERAD substrate, and may explain some of the cell type variability in the reported role of lectins in ERAD.
Figure 5. Inhibition of non-glycosylated protein ERAD by XTP3B MRH2.

(A, B) PSI-chase analysis 48 hrs after transfection with TTRD18G/N98Q-HA (A), A1ATNHK-QQQ-HA (B), XTP3B, XTP3BR207A/R428A or empty vector in wildtype cells. Quantification is based on one (A) or two (B) independent experiments for the empty vector and wild type XTP3B and one experiment for XTP3BR207A/R428A (B). Error bars represent STDEV. (C) Expression of A1ATNHK-GFP or A1ATNHK-QQQ-GFP in K562 cells was induced by growing cells in the presence of 0.1 μg/mL doxycycline (dox) for 16 hrs. Cells were subsequently treated with emetine for the indicated times. Protein turnover was determined by immunoblotting with an anti-GFP antibody. n=1. (D) K562 cells stably expressing dox-inducible A1ATNHK-QQQ-GFP and the indicated sgRNA were treated with 0.1 μg/mL doxycycline (dox) for 16 hours. Cells were subsequently treated with emetine and GFP median fluorescence intensity was measured by flow cytometry at the indicated times. Quantification is based on one experiment. (E) PSI-chase analysis of A1ATNHK-QQQ-HA in WT, DKO, and DKO cells stably expressing the indicated XTP3B constructs. Quantification represents the mean±STDEV (n=2). (F) Competition between OS9 and XTP3B for binding to SEL1L. Endogenous SEL1L was immunoprecipitated from 1% Triton X-100 lysates of wild type HEK293 cells transfected with OS9.2, XTP3B, or empty vector. SEL1L complexes were analyzed by immunoblotting with the indicated antibodies. Quantification of XTP3B and OS9 normalized to SEL1L is represented as mean±SEM (n=3). See also Fig. S4.
XTP3B inhibits non-glycosylated protein ERAD by competitive binding to SEL1L
The fact that relative abundance of OS9 and XTP3B affects substrate degradation, suggests that the lectins might be competing for interaction, either with the substrate itself or with a component of the ERAD machinery. While a requirement for a functional MRH domain for direct interaction with non-glycosylated substrates is unlikely, XTP3B could inhibit non-glycosylated protein ERAD via interaction with one or more glycosylated ERAD components. Indeed, we found that XTP3B (R207A/R428A) was less effective than wild type XTP3B at inhibiting turnover of TTRD18G/N98Q and A1ATNHK-QQQ (Fig. 5A, B). XTP3B (R207A) expression in DKO cells suppresses A1ATNHK-QQQ degradation to the same extent as wild type XTP3B, while expression of XTP3B (R428A) or XTP3B (R207A/R428A) only partially suppresses A1ATNHK-QQQ degradation (Fig. 5E), suggesting that MRH2 contributes to inhibition of non-glycosylated protein ERAD by XTP3B, possibly by direct interaction with trimmed glycans on a component of the ERAD machinery, most likely SEL1L. Strikingly, OS9.2 overexpression reduces the amount of endogenous XTP3B associated with SEL1L and, reciprocally, XTP3B overexpression reduces the amount of endogenous OS9.1 and OS9.2 associated with SEL1L (Fig. 5F and Fig. S4E). These data demonstrate that OS9 and XTP3B can compete for binding to SEL1L, suggest that these two lectins share common or overlapping interaction sites on SEL1L, and that the identity of the lectin occupying the binding site on SEL1L profoundly influences the turnover of nonglycosylated substrates.
SEL1L has five highly conserved, predicted N-linked glycans (Fig. 6A) and binding of OS9 and XTP3B to SEL1L is diminished by MRH domain mutations in these lectins (Christianson et al., 2008, Satoh et al., 2010). We determined the relative mannosylation state of four SEL1L glycopepetides by LC-MS/MS analysis of endogenous SEL1L immunoprecipitated from HEK293 cells. Glycopeptides corresponding to three out of four sites on SEL1L (N195, N272, N608) were composed almost exclusively of untrimmed or minimally trimmed high mannose (Man8-9) glycans while a single site (N431) was strongly enriched in demannosylated (Man5-7) glycans that have the potential to bind to MRH domains in OS9 or XTP3B (Fig. 6B). Interestingly, overexpressed SEL1L, whether full length (S-SEL1L) or truncated to lack the predicted transmembrane domain (S-SEL1L1-738KDEL), was demannosylated at N608 in addition to N431 (Fig. 6B and Fig. S5). SEL1L itself becomes an ERAD substrate when overexpressed (Iida et al., 2011) or following depletion of ER lectins (Fig. 2) raising the possibility that demannosylation of SEL1L glycans may contribute to its degradation when unassembled and that binding of OS9 or XTP3B could protect those glycans from demannosylation. In support of this idea, glycopeptides on endogenous SEL1L captured with S-tagged XTP3B, but not OS9 (Fig. 6C) were strongly enriched in N431-trimmed (Man5-7) glycans. This effect was not due to altered mannose trimming of bulk SEL1L in response to XTP3B overexpression as the fraction of trimmed glycans did not differ significantly between endogenous SEL1L immunoprecipitated from cells overexpressing an empty vector or XTP3B (Fig. S5B). Moreover, rescue of SEL1L KO cells with an S-tagged SEL1L mutant lacking the glycosylation sequon (N431Q), expressed at steady-state levels comparable to wildtype, reduced the amount of XTP3B but not OS9 that co-purified with SEL1L by S-affinity capture (Fig. 6D). Together, these data suggest that XTP3B binds preferentially to stable SEL1L molecules with N431-linked demannosylated glycans and that MRH2 contributes to this interaction.
Figure 6. XTP3B binds preferentially to SEL1L bearing a demannosylated glycan at Asn431.

(A) Schematic representation of the domain organization of SEL1L and a full-length and luminal S-tagged SEL1L construct. SS=Signal Sequence; S=S-peptide tag; TM=transmembrane domain. (B, C) Fraction of SEL1L tryptic N-glycopeptides bearing trimmed (Man5-7) glycans at the indicated sequons. Quantification of tryptic glycopeptides from endogenous SEL1L (B, C; n=2) immunoprecipitated from HEK293 cells or S-affinity purified (SAP) from cells expressing S--SEL1L (B; n=1), S-OS9.2 (C; n=2) or XTP3B-S (C; n=6) from transfected HEK293 cells. Error bars represent STDEV. Endogenous SEL1L IP data in B and C are the same. (D) S-SEL1L was affinity purified from 1% Triton X-100 lysates of SEL1L KO cells transfected with S-SEL1L, S-SEL1LN431Q, or empty vector. SEL1L complexes were analyzed by immunoblotting with the indicated antibodies. Quantification of XTP3B and OS9 was normalized to the amount of SEL1L precipitated and represented as mean±STDEV (n=3 for XTP3B; n=2 for OS9). Lanes in which transient S-SEL1L expression most closely reflected endogenous SEL1L levels were selected, lanes representing lower expression were removed. See also Fig. S5.
Discussion
Protein triage, the process by which terminally misfolded or unassembled proteins are committed for degradation while sparing native proteins and folding intermediates, must be able to accommodate the vast diversity in structures and folding kinetics of proteins that transit through the secretory pathway. Although progressive mannose trimming of substrate glycans and lectin recognition provides an elegant timing mechanism for ERAD, it is evident that additional, non-glycan signals also contribute to triage in the ER.
A model for ERAD triage
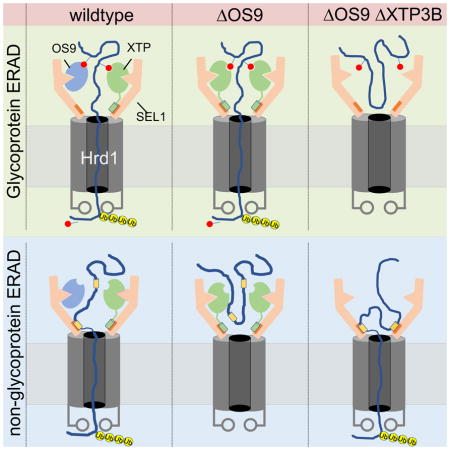
Our observation that OS9 and XTP3B play very different roles in decoding glycan- and non-glycan-dependent degrons in ERAD can be reconciled in a model (Fig. 7) that depicts the SEL1L/HRD1 dislocon based on the recently published cryoEM structure of yeast Hrd1/Hrd3 (Schoebel et al., 2017). This structure, onto which the conserved mammalian SEL1L sequence can be threaded with high confidence, reveals the dislocon complex to be a symmetrical dimer with two molecules of Hrd1 interacting through their transmembrane segments with two L-shaped molecules of Hrd3 that form a vestibule at the luminal side of the central Hrd1 pore through which proteins are presumably dislocated. This dimeric structure could permit the simultaneous binding of two lectins (OS9 and/or XTP3B) to each functional dislocon (Fig. 7A). Although the binding sites on SEL1L for OS9 and XTP3B are unmapped, our finding that OS9 and XTP3B can compete with one another for binding to SEL1L (Fig. 5F), supports the conclusion that these two lectins share common or overlapping interaction sites on SEL1L. The presence of two or more substrate binding sites on SEL1L could increase the avidity with which substrates containing multiple degrons bind to the dislocation complex, and promote processivity by helping to ensure that substrates, once engaged by SEL1L/HRD1, do not slip back into the lumen during dislocation. This dimeric structure implies that the MRH domains of OS9 and/or XTP3B can simultaneously engage two exposed α1,6-mannose degrons on the same glycosylated substrate. Our data suggest that exposed protein sequences that are buried within their native structures when correctly folded and assembled can also be engaged by SEL1L without the intervention of either lectin. This engagement could be mediated by chaperones like BiP or GRP94 which also bind to the HRD1-SEL1L complex (Christianson et al., 2008), or by direct interaction with SEL1L, perhaps at a site corresponding to the proposed substrate binding groove recently identified on the inner surface of Hrd3 (Schoebel et al., 2017). In either case we propose that XTP3B, but not OS9, occludes the direct or indirect access of substrates to SEL1L. Endogenous SEL1L/HRD1 elutes on Superose 6 size exclusion chromatography as a broad ~500 mDa peak that can be resolved into subcomplexes that differ in the relative amounts of associated OS9.1/9.2 and XTP3B (Hwang et al., 2017), suggesting that the distinctions between OS9 and XTP3B function observed here may be executed by distinct dislocon complexes composed of SEL1L/HRD1 dimers bound to different combinations of OS9.1, OS9.2 and XTP3B.
Figure 7. Role of ER lectins in ERAD.

(A) Schematic model of the organization of the SEL1L/HRD1 complex based on the Hrd1/Hrd3 cryoEM structure (Schoebel et al., 2017). OS9 and XTP3B are proposed to bind to a common site on SEL1L. MRH domains are depicted as semicircular indentations. Only one MRH domain is shown on XTP3B for simplicity. Orange box at base of SEL1L represents potential binding site for non-glycan degrons and XTP3B. (B) Three classes of proteins that differ in their dependence on glycan or non-glycan degrons are depicted in horizontal rows. Mutants used in this study are indicated in the vertical columns. “Wild type” (WT) situation is depicted as having a mixture of OS9 and XTP3B, but different combinations of the two lectins in wild type cells are likely. Efficient or inefficient ERAD is represent by six or one ubiquitin molecules attached to partially dislocated substrates, respectively. Reduced expression of SEL1L and HRD1 in DKO cells is indicated.
The substrates used in this study can be segregated into three classes based on the effects of genetic manipulation of OS9 and XTP3B expression (Fig. 7B). Class A substrates, represented by CD147, are glycoproteins that have a strict dependence on lectins and on mannosidase trimming for their turnover. These proteins are almost completely stabilized by inhibiting α-mannosidase activity with kifunensine, are maximally stabilized (compared to SEL1L knockout) in DKO cells, and can be degraded by either OS9 or XTP3B alone. CD147 degradation has an absolute requirement for at least one lectin and at least one functional MRH domain.
Class B proteins include A1ATNHK-QQQ and TTRD18G/N98Q, lack N-glycans and are degraded equally well in DKO and wild type cells, demonstrating that OS9 or XTP3B are completely dispensable for their degradation. Although OS9 has been reported to co-enrich with non-glycosylated substrates (Bernasconi et al., 2008, Hosokawa et al., 2009, Satoh et al., 2010), it is unclear if OS9 and XTP3B directly bind to non-glycan determinants on misfolded proteins, as has been suggested for Yos9 (Smith et al., 2014). Our data establish that such an interaction does not contribute functionally to the degradation of Class B substrates. Molecular chaperones such as BiP (Molinari and Helenius, 2000) or GRP94 could help maintain the solubility of Class B substrates in the ER lumen and prevent them from being secreted during repeated unproductive folding attempts, prior to engagement by SEL1L. Most surprisingly, our data reveal an unexpected role for XTP3B as a potent inhibitor of Class B protein ERAD.
Our data reveal the existence of a third class of ERAD substrates, Class C, that are targeted to ERAD by both glycan and non-glycan determinants. Class C proteins, which include HLA-C and A1ATNHK, are N-glycosylated, but are only partially stabilized in kifunensine-treated or DKO cells. TTRD18G represents a class B substrate that is converted to a Class C substrate by posttranslational glycosylation at a cryptic sequon. Degradation of Class C proteins is accelerated by knocking out XTP3B, suggesting that this lectin has dual activities in promoting and inhibiting the degradation of glycoproteins and non-glycosylated proteins, respectively. A previous study (Fujimori et al., 2013) reported acceleration of A1ATNHK clearance upon XTP3B knockdown in kifunensine-treated cells, leading the authors to conclude that XTP3B selectively inhibits the degradation of proteins bearing Glc1Man9GlcNAc2 glycans. Our designation of A1ATNHK as a Group C protein with hybrid degrons consisting of both glycan and non-glycan determinants is consistent with their findings but provides a more plausible explanation because only the non-glycan signal in A1ATNHK would be exposed in cells lacking mannosidase activity. These three classes account for the effects of knockout of XTP3B and OS9 on the ERAD substrates reported here and, most likely, for other folding- or assembly-defective substrates of HRD1-dependent ERAD. Additional ERAD substrate classes are also likely to exist, particularly for substrates of ubiquitin E3 ligases other than HRD1.
Opposing roles of XTP3B’s MRH domains in triage
Whether either or both MRH domains of XTP3B possess glycan-binding activity has been controversial. One study (Yamaguchi et al., 2010) used binding of purified XTP3B-Fc fusion proteins containing deletions of either MRH1 or MRH2 to the surface of CHO cells deficient in enzymes that catalyze N-glycan synthesis, to conclude that MRH2 binds preferentially to fully trimmed (Man5GlcNAc2) glycans. Another study (Fujimori et al., 2013) analyzed the glycan specificity of purified, bacterially expressed, isolated MRH domains, concluding that MRH2 binds selectively to untrimmed glycans (Man9GlcNAc2). Neither study detected glycan-binding activity for MRH1. Our finding that XTP3BR428A but not XTP3BR207A can fully rescue glycoprotein ERAD in DKO cells is inconsistent with both of these published findings and strongly supports our conclusion that XTP3B’s MRH1 is essential for glycoprotein ERAD while MRH2 is dispensable. It is possible that MRH1 could functionally contribute to substrate recognition without individually exhibiting sufficient affinity to be detected by affinity capture, perhaps supporting an avidity based mechanism with multiple, distributed binding sites. Moreover, these discrepancies may also reflect the methodological limitations of using indirect analyses and recombinant fragments of overexpressed constructs. While our data do not demonstrate direct binding of the MRH domains to substrate glycans, the fact that both lectins can functionally replace each other suggests that OS9’s MRH domain and MRH1 on XTP3B have similar glycan-binding specificity and redundant roles in promoting degradation of proteins bearing trimmed glycans.
The observations that overexpressed XTP3B (R207A/R428A) (Figs. 5A and B) is less effective than wild type XTP3B at inhibiting turnover of non-glycosylated proteins, that rescue with XTP3B (R207A) in DKO cells inhibits degradation of a non-glycosylated protein to the same extent as wild type XTP3B, while expression of XTP3B (R428A) or XTP3B (R207A/R428A) only partially suppresses its degradation (Fig. 5E), suggest that this inhibitory effect is at least partially dependent on MRH2 and not MRH1. Because XTP3B pulldown enriches for SEL1L molecules with a trimmed glycan residue at Asn431, and because mutation of this residue reduces SEL1L’s interaction with XTP3B, it is tempting to speculate that XTP3B’s inhibitory effect depends on direct interaction between MRH2 and a population of SEL1L molecules containing trimmed glycans at Asn431. Further investigation, together with detailed structural analysis will be required to test this model.
Our data confirm a role for OS9 as an adaptor-lectin that recruits proteins with exposed α1,6-linked mannose to SEL1L/HRD1, corresponding closely to the function proposed for Yos9 in yeast ERAD. We find that, while XTP3B shares this conserved adaptor function, it plays a far more complex and nuanced role in substrate selection, likely reflecting the increased diversity and complexity of proteins that transit the secretory pathway in metazoans. We propose that the ability of XTP3B to inhibit ERAD of non-glycan containing proteins offers two potential benefits to facilitate quality control for this more diverse secretome. First, it is possible that in SEL1L complexes containing both lectins, XTP3B can reduce the affinity and dwell time for hydrophobic peptide determinants on Class B and C substrates by competing with substrates for a binding site on SEL1L. Second, it is possible that the inhibitory function of XTP3B could serve to slow the degradation of non-glycosylated (Class B) proteins which are not subject to a mannosidase “timer”, allowing them additional time to fold before being committed to degradation. A similar duality is found in yeast Yos9, which was shown to accelerate the degradation of N-glycosylated CPY* but to slow the degradation of the non-glycosylated variant, CPY*0000 (Benitez et al., 2011), suggesting that this ancient opposing functionality contained within a single protein evolved to become distributed between OS9 and XTP3B. Intriguingly, such inhibition of ERAD by XTP3B could also facilitate the triage of folding-defective mutants of natively non-glycosylated (Sato et al., 2012) or hypoglycosylated (Dersh et al., 2014) proteins like TTR or GRP94, respectively, by allowing them to become glycosylated at cryptic sequons that promote their degradation if damaged or unable to fold productively. Inhibiting Class C degradation by XTP3B could facilitate post-translational N-glycosylation (Shrimal et al., 2015) and consequently promote their degradation by an OS9 dependent process.
Our finding that inhibition of Class B/C ERAD by XTP3B can be antagonized by OS9 reveals negative cooperativity between these two lectins that can be most simply explained by competition for binding to SEL1L. The observation that A1ATNHK-QQQ degradation in wild type K562 cells is slower than its degradation in wild type HEK293 cells, but can be accelerated by reducing endogenous XTP3B expression, suggests that the relative expression of OS9 and XTP3B varies between cell types, perhaps providing insight into discrepant findings on the roles of ER lectins and may have biological significance with respect to the capacity of different cell types in degrading Class B/C proteins.
Finally, our data suggest that XTP3B could be a useful therapeutic target in conformational diseases like systemic amyloidoses and α1AT deficiency liver disease, where enhanced Class B/C ERAD could reduce formation of toxic aggregates. Alternatively, reducing the capacity to degrade Class B and C proteins could be effective in treating diseases like cystic fibrosis, where premature ERAD contributes to loss-of-function (Gelman and Kopito, 2003) or diabetes, where Hrd1-mediated ERAD of non-glycosylated preproinsulin contributes to autoantigen presentation (Hoelen et al., 2015).
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ron Kopito (kopito@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and transfection
HEK293 cells were cultured in DMEM containing 4.5 g/L glucose and L-glutamine (Mediatech), and supplemented with 10% FetalPlex animal serum complex (Gemini Bio-Products, West Sacramento, CA) at 37 °C and 5% CO2. A standard calcium-phosphate co-precipitation technique (Kingston et al., 2001) or X-tremeGENE HP DNA transfection reagent from Roche (06366244001) was used for transient plasmid transfections. Transient expression of ERAD substrates was monitored 48–72 hr post-transfections.
Cas9-BFP K562 human myeloma cells (a generous gift from Michael Bassik, Stanford University) stably expressing doxycycline-inducible A1ATNHK-GFP or A1ATNHK-QQQ -GFP were grown in RPMI 1640 medium (Corning) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 2 mM L-glutamine (Corning), 200 μg/mL Geneticin and 4 μg/mL Blasticidin. Cells were grown in a humidified incubator at 37°C and 5% CO2. A1ATNHK-GFP or A1ATNHK-QQQ-GFP expression was induced by growing cells in complete RPMI 1640 medium supplemented with 0.1 μg/mL doxycycline (dox; Sigma-Aldrich) for 16 hr.
Generation of knockout cell lines by CRISPR/Cas9 genome-editing
Clonal HEK293 knock-out cell lines were generated by co-transfection of a plasmid encoding both Cas9 and a guide RNA sequence targeting the gene of interest and a gene-specific homology-directed repair plasmid. 48–72 h post-transfection, stable cell lines were selected in medium containing 1–2 μg/mL Puromycin or 500 μg/mL Geneticin. Clonal cell lines were selected by limiting dilution after ~2 weeks in selection medium. Single-cell-derived clones were expanded under selective pressure and analyzed by immunoblotting. Cell lines generated in this study are listed in the Supplementary Methods (Table S3).
Generation of stable cell lines
To generate stable HEK293 cell lines expressing OS9.1, OS9.2, OS9.1R188A, OS9.2R188A, XTP3B, XTP3BR207A, XTP3BR428A, or XTP3BR207A/R428A, zeocin was added to cells ~48–72 h post-transfections at 400 μg/mL. Clonal cell lines were selected by limiting dilution after ~2 weeks in selection medium. Single-cell-derived clones were expanded under selective pressure and analyzed by immunoblotting. Cell lines generated in this study are listed in the Supplementary Methods (Table S4).
Lentiviral packaging and K562 viral transductions were performed essentially as previously described (Deans et al., 2016). Dox-inducible A1ATNHK-GFP or A1ATNHK-QQQ-GFP K562 cells were generated by infecting cells with a modified version of the pLVX-Tet-ON Advanced vector (Takara Bio) containing a pEF1α promoter. 72 hr after infection, cells were selected in complete RPMI 1640 medium supplemented with 400 μg/mL Geneticin for 10 days. Cells were subsequently infected with the pMCB497 plasmid (a generous gift from Michael Bassik, Stanford University) containing pTRE Tight-A1ATNHK-GFP or pTRE Tight-A1ATNHK-QQQ-GFP. Cells were passaged for 72 hr before selection in complete RPMI 1640 medium supplemented with 7.5 μg/mL Blasticidin for 10 days. For gene knockout studies, dox-inducible A1ATNHK-QQQ-GFP K562 cells were infected with individual sgRNAs in the pMCB320 vector (Morgens et al., 2017), passaged for 72 hr, and then selected in complete RPMI 1640 medium supplemented with 0.75 μg/mL Puromycin for 3 days.
METHOD DETAILS
Reagents
Puromycin was purchased from Enzo Life Sciences (BML-GR312-0050) and Geneticin (G418 sulfate), blasticidin and zeocin were purchased from Thermo Fisher Scientific (10131035, A1113903 and R250-01). Cycloheximide and emetine were purchased from Sigma-Aldrich (C7698 and E2375). Kifunensine was purchased from Tocris (3207). CB-5083 was generously provided by Cleave Biosciences (Burlingame, CA).
Plasmids
Expression constructs for A1ATNHK-HA, A1ATNHK-QQQ-HA, and TTRD18G-HA used in this study were derived from previously published expression plasmids (Christianson et al., 2008, Christianson et al., 2011) that were used as templates for PCR-based cloning into pcDNA3.1(+) (Thermo Fisher Scientific) using primers encoding an HA-tag (YPYDVPDYA). Point mutations were introduced by standard site-directed mutagenesis cloning techniques to generate constructs expressing TTRD18G/N98Q-HA. S-SEL1L, S-OS9.2, and XTP3B-S used in this study have been published previously (Christianson et al., 2008).
OS9.1, OS9.2, and XTP3B with their endogenous signal sequences were PCR amplified from cDNA clones and cloned into pcDNA3.1(+) containing a zeocin selection marker. Point mutations to generate plasmids expressing OS9.1R188A, OS9.2R188A, XTP3BR207A, XTP3BR428A, and XTP3BR207A/R428A were generated with standard site-directed mutagenesis cloning techniques.
M757 vector (kindly provided by the Porteus lab) was used as a backbone to construct the homology-directed repair vectors used in this study. The vector was modified to contain a TAP tag for N-terminal tagging or a STOP cassette, sites for insertion of 5’ and 3’ homology arms, and a puromycin- or Geneticin-resistance gene under the control of the Ubiquitin C promoter. Selection cassettes were in between LoxP sequences to have the ability to excise the selection cassette after selection by Cre-Lox recombination. The amino acid sequence for the TAP tag is as follows: S-tag (KETAAAKFERQHMDS), PreScission protease cleavage site (LEVLFQGP), Strep-tag (WSHPQFEK), and linker and LoxP sequence (ACRITSYSIHYTKL). The STOP cassette contains 4 stop codons encoded in different reading frames. To generate gene-specific homology-directed repair vectors, ~500–900-bp 5’ and 3’ homologous arms were amplified by nested PCR using HEK293 genomic DNA as a template and were then sequentially cloned into the modified homology-directed repair vectors. Homology-directed repair vectors generated and used in this study are listed in Supplementary Table 1.
pX330-U6-Chimeric BB-CBh-hSpCas9 (Addgene plasmid #42230, gift from Feng Zhang) was used for expression of human codon-optimized SpCas9 and gene-specific guide RNA’s (Cong et al., 2013). We used an online CRISPR design tool (http://crispr.mit.edu) to select at least two independent guide RNA sequences targeting the first exon of the genes of interest. CRISPR constructs were generated with protocols published by the Zhang laboratory (http://www.genome-engineering.org/crispr) (see Supplementary Table 2).
All constructs were verified by sequencing and detailed sequence information will be made available upon request.
Translational shut-off assays
Cells were treated with cycloheximide or emetine, at a final concentration of 50 μg/mL or 10 μM, respectively, for the indicated times. For treatment with kifunensine, cells were incubated overnight (~16 hr) in the presence of kifunensine at a final concentration of 2.5 μg/mL after which emetine or cycloheximide treatment was started in the morning according to regular protocol. Cells were collected and washed in PBS after which they were lysed in lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 2.5 mM EDTA) containing 1% Triton X-100, 1% Decyl Maltose Neopentyl Glycol (DMNG, Anatrace NG322), or 1% SDS and protease inhibitors (Roche, 11697498001). Total protein concentration was determined for each sample with the Pierce BCA Protein Assay Kit (23225) according to manufacturer’s instructions. Equal amounts of total protein were analyzed by immunoblotting.
Affinity purification
HEK293 cells expressing endogenously TAP-tagged HRD1 (Hwang et al., 2017) were treated with the VCP/p97 inhibitor CB-5083 (Zhou et al., 2015) at a final concentration of 5 μM for 6 hr. HEK293 cells were harvested by scraping and washed in ice-cold PBS before lysis in buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 2.5 mM EDTA) containing 1% Decyl Maltose Neopentyl Glycol (DMNG, Anatrace NG322) and protease inhibitors (Roche, 11697498001) for 30 min at 4 °C. Tandem-affinity purification was performed as described previously (Hwang et al., 2017). HRD1 affinity-purified protein complexes were analyzed by immunoblotting.
SEL1L knockout cells were transiently transfected with S-SEL1L, S-SEL1LN431Q, or empty vector and were collected ~48 hr post-transfections by scraping. Cells were washed in ice-cold PBS before lysis in buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 2.5 mM EDTA) containing 1% Triton X-100 and protease inhibitors (Roche, 11697498001) for 10 min at 4 °C. Cell extracts with normalized protein amounts were incubated overnight with S-protein agarose. Affinity-purified complexes were washed three times with buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 2.5 mM EDTA) containing 0.5% Triton X-100 after which proteins were eluted from the beads by incubation at 65 °C for 10 min in 2X Laemmli buffer containing DTT at a final concentration of 100 mM. S-affinity purified SEL1L complexes were analyzed by immunoblotting.
Immunoprecipitation
Rabbit polyclonal anti-SEL1L (this study) was conjugated to Pierce Protein A/G Plus Agarose (20423) using the Pierce Crosslink IP Kit according to manufacturer’s instructions. To immunoprecipitate endogenous SEL1L, 1% Triton X-100 lysates from HEK293 cells with normalized protein amounts were incubated with anti-SEL1L conjugated beads and incubated ON. IPs were washed 3X in buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 2.5 mM EDTA) containing 0.5% Triton X-100. Proteins were eluted by heating proteins for 10 min at 65 °C in Laemmli buffer without reducing reagent. Samples were subsequent spun for 1 min at max. speed. Supernatant fractions were transferred to new tubes and dithiothreitol was added to a final concentration of 50 mM and samples were incubated for an additional 5 min at 65 °C.
SDS-PAGE and immunoblotting
Proteins were denatured in Laemmli buffer containing either 2-mercaptoethanol 2% (v/v) or 50 mM dithiothreitol by heating at 65 °C for 10 min. Samples were then separated by SDS-PAGE and transferred to nitrocellulose (Bio-Rad). Nitrocellulose membranes were blocked in 5% nonfat milk in PBS-T to reduce nonspecific antibody binding and incubated with primary antibodies diluted in PBS-T containing 0.1% Tween 20 and 4% BSA. Immunoreactivity was detected using fluorescent IRDye secondary antibodies and scanning by Odyssey imaging (Li-COR Biosciences). Band intensities were quantified by Image Studio™ Lite software (Li-COR Biosciences). For translational shut-off assays, bands were quantified by densitometry and protein levels at each time point were normalized to Tubulin or GAPDH. Percentage protein remaining was calculated relative to t = 0 hr for each cell line.
The following rabbit polyclonal antibodies were used for immunoblotting: anti-HLA-C (Abgent, AP17872c), anti-HRD1 (Proteintech, 13473-1-AP), anti-OS9 and anti-XTP3B (Christianson et al., 2011), anti-SEL1L (kindly provided by H. Ploegh and this study), anti-EDEM1 (Sigma-Aldrich, E8406), anti-ERManI (Proteintech, 15243), anti-GFP (D5.1) XP (Cell Signaling, 2956S) and anti-GAPDH (Millipore, AB516). The following mouse monoclonal antibodies were used: anti-CD147 clone A-12 (Santa Cruz Biotechnologies, sc-25273), anti-SEL1L (kindly provided by J. Christianson), anti-XTP3B (kindly provided by J. Christianson), anti-EDEM3 (Sigma-Aldrich, E0409), anti-HA clone 16B12 (BioLegend, 901501), anti-α-Tubulin clone DM1A (Sigma-Aldrich, T6199), and anti-GAPDH clone 6C5 (MilliPore, MAB374). The following secondary antibodies were used: IRDye800CW goat anti-mouse and rabbit IgG and IRDye680RD goat anti-mouse and rabbit IgG (Li-COR Biosciences, 925-32210, 925-32211, 925-68070, and 925-68071).
Small interfering RNA (siRNA) knockdown
Lipofectamine RNAiMAX from Thermo Fisher Scientific (13778030) was used for transient siRNA transfections according to manufacturer’s instructions. Silencer Select siRNAs were purchased from Thermo Fisher Scientific: SYVN1 (HRD1) s39020 and negative control no 1. Cells were analyzed ~48–72 h post-transfections.
Flow Cytometry
Cells were collected by centrifuging at 1,000 x g for 5 min, resuspended in PBS, and placed on ice. For each data point, at least 20,000 events were analyzed on a FACSCalibur (BD Biosciences) or an Aria II (BD Biosciences) equipped with a 488 laser. The median fluorescence intensity of the live cell population was quantified using FlowJo version 10.0.8 (Tree Star).
Xbp1 splicing assay
Cells were treated with Thapsigargin at a final concentration of 300 nM or DMSO for 2 hr. Cells were collected and washed in PBS after which RNA was extracted with an RNeasy Mini Kit from Qiagen (74104) according to the manufacturer’s instructions. 500 ng of total RNA was used for cDNA synthesis with Superscript™ IV Reverse Transcriptase from Thermo Fisher Scientific (18091050) according to manufacturer’s instructions. Spliced and unspliced Xbp1 was amplified by PCR with hXbp1-Fw and hXbp1-Rv and analyzed on a 2% agrose gel (TBE buffer).
LC-MS/MS analysis of affinity-captured SEL1L glycopeptides
HEK293 cells were plated in at least 4 – 15 cm tissue culture plates per sample and transfected the next day with plasmids encoding S-SEL1L(L753P), S-OS9.2, or XTP3B-S using calcium-phosphate precipitation (Kingston et al., 2001). A small amount of plasmid (0.75 μg/15 cm dish) was used in each transfection so that S-tagged proteins were expressed at near-endogenous levels. Cells were harvested and lysed in ice-cold lysis buffer (50 mM Tris-HCl, pH 7.3, 150 mM NaCl, 5 mM EDTA, plus protease inhibitors) containing 1% v/v Triton X-100. 10–20 mg of each lysate was pre-cleared by incubation with Sephadex G-100 beads for 1 h at 4°C and then incubated with S-protein agarose overnight at 4°C. To immunoprecipitate endogenous SEL1L, 1% Triton X-100 lysates from untransfected HEK293 cells were incubated with rabbit polyclonal anti-SEL1L (a kind gift from H. Ploegh, Whitehead Institute) conjugated to beads. SAPs and IPs were washed 4X in lysis buffer and twice with 50 mM NH4HCO3, pH 8.5, and then incubated in 50 μL 50 mM NH4HCO3, pH 8.5 containing 0.1% Rapigest for 24h at 37°C. Eluted proteins were incubated with 0.4 μg sequencing grade trypsin for 20 h at 37°C. Rapigest was precipitated by lowering the pH of the solution to ~2, centrifuging the samples for 10 min at 20,000 x g, and transferring detergent-free supernatants to glass tubes. Peptides were stored at −20°C prior to mass spectrometry analysis.
LC-MS/MS analyses were performed by online capillary reversed-phase separation of injected samples (0.3 mm x 15 cm C18 column) using an increasing gradient of acetonitrile in water (0% to 55% over 2h), with both solvents containing 0.1% formic acid. The chromatography system consisted of an Agilent 1100 capillary binary solvent delivery pump and solvent degasser, together with a HTC-Pal autosampler (Leap Technologies), which maintained queued samples in a thermoelectrically cooled sample tray. The effluent of the capillary column was fed directly into the electrospray source (modified with a low-flow metal needle kit) of a LTQ-Orbitrap XL hybrid mass spectrometer (Thermo Scientific). During a run, high-resolution MS1 scans were performed at nominal mass resolution setting of 60,000 (m/z 400), were acquired in positive-ion mode and saved as profile data. Tandem mass spectra (MS2) were performed in the LTQ linear ion-trap (top 5 nondynamically excluded ions from MS1 scan) during a run.
Glycopeptides were identified from accurate mass measurements, which were within 5 ppm of theoretical values, while the presence and amount of glycosylation was confirmed by neutral loss of hexose units in the MS2 spectra. In some cases additional confirmation of peptide sequence was obtained through MS3.
High-mannose glycoform relative amounts were determined in selected ion chromatograms for each glycopeptide. Percent abundance was calculated by dividing the normalized largest (NL) peak for each glycoform by the total of all glycoforms detected for the same glycopeptide.
QUANTIFICATION AND STATISTISTICAL ANALYSES
Throughout the manuscript, the data are represented as average ± SEM unless otherwise stated. N values represent the number of independent experiments performed. Intensities from bands detected by WB were quantified by Image Studio Lite software (Li-COR Biosciences). For translational shut-off assays, bands were quantified by densitometry and protein levels at each time point were normalized to Tubulin or GAPDH. Percentage protein remaining was calculated relative to t = 0 hr for each cell line.
Supplementary Material
Figure S1. Related to Figure 1. Characterization of knockout cell lines generated by CRISPR/Cas9-mediated genome editing.
Figure S2. Related to Figure 2. Destabilization of SEL1L/HRD1 complex upon loss of OS9 and XTP3B.
Figure S3. Related to Figure 3. OS9 isoform-specificity in targeting CD147 for ERAD.
Figure S4. Related to Figure 5. Inhibition of non-glycosylated protein ERAD by XTP3B is antagonized by OS9.
Figure S5. Related to Figure 6. Overexpressed SEL1L is demannosylated at Asn608.
Supplementary Tables 1-4
Highlights.
OS9 and XTP3B are redundant and essential for efficient glycoprotein ERAD
Glycoprotein ERAD is dependent on canonical lectin-glycan interaction
XTP3B inhibits ERAD of non-glycosylated proteins; inhibition is antagonized by OS9
The relative abundance of OS9 and XTP3B influences ERAD of non-glycoproteins
Acknowledgments
The authors thank the Porteus, Ploegh, Olzmann, and Bassik labs for reagents and technical advice, Olivia Liautaud for help with the experiments in K562 cells, and members of the Kopito lab for helpful discussions and critical comments on the manuscript. This work was supported by a grant from the National Institute of General Medical Sciences (R01GM074874) and a generous gift from the Becker Family Foundation to RRK. AG was supported by an EMBO postdoctoral fellowship (ALTF 721-2014) co-funded by the European Commission FP7 (Marie Curie Actions, EMBOCOFUND2012, GA-2012-600394). DEL was supported by postdoctoral fellowships from the NIH (F32GM113370) and the Alpha-1 foundation.
Footnotes
Author Contributions:
AG, MP, DL, performed the experiments. TS conducted the mass spectrometry. AG, MP, DL and RK analyzed and discussed the data and conceived of the experimental design. AG and RK wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ALCOCK F, SWANTON E. Mammalian OS-9 is upregulated in response to endoplasmic reticulum stress and facilitates ubiquitination of misfolded glycoproteins. J Mol Biol. 2009;385:1032–42. doi: 10.1016/j.jmb.2008.11.045. [DOI] [PubMed] [Google Scholar]
- BENITEZ EM, STOLZ A, WOLF DH. Yos9, a control protein for misfolded glycosylated and non-glycosylated proteins in ERAD. FEBS Lett. 2011;585:3015–9. doi: 10.1016/j.febslet.2011.08.021. [DOI] [PubMed] [Google Scholar]
- BERNASCONI R, GALLI C, CALANCA V, NAKAJIMA T, MOLINARI M. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J Cell Biol. 2010;188:223–35. doi: 10.1083/jcb.200910042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERNASCONI R, PERTEL T, LUBAN J, MOLINARI M. A dual task for the Xbp1-responsive OS-9 variants in the mammalian endoplasmic reticulum: inhibiting secretion of misfolded protein conformers and enhancing their disposal. J Biol Chem. 2008;283:16446–54. doi: 10.1074/jbc.M802272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BHAMIDIPATI A, DENIC V, QUAN EM, WEISSMAN JS. Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol Cell. 2005;19:741–51. doi: 10.1016/j.molcel.2005.07.027. [DOI] [PubMed] [Google Scholar]
- BUSCHHORN BA, KOSTOVA Z, MEDICHERLA B, WOLF DH. A genome-wide screen identifies Yos9p as essential for ER-associated degradation of glycoproteins. FEBS Lett. 2004;577:422–6. doi: 10.1016/j.febslet.2004.10.039. [DOI] [PubMed] [Google Scholar]
- CHRISTIANSON JC, OLZMANN JA, SHALER TA, SOWA ME, BENNETT EJ, RICHTER CM, TYLER RE, GREENBLATT EJ, HARPER JW, KOPITO RR. Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol. 2011;14:93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHRISTIANSON JC, SHALER TA, TYLER RE, KOPITO RR. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10:272–82. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONG L, RAN FA, COX D, LIN S, BARRETTO R, HABIB N, HSU PD, WU X, JIANG W, MARRAFFINI LA, ZHANG F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORMIER JH, TAMURA T, SUNRYD JC, HEBERT DN. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol Cell. 2009;34:627–33. doi: 10.1016/j.molcel.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRUCIAT CM, HASSLER C, NIEHRS C. The MRH protein Erlectin is a member of the endoplasmic reticulum synexpression group and functions in N-glycan recognition. J Biol Chem. 2006;281:12986–93. doi: 10.1074/jbc.M511872200. [DOI] [PubMed] [Google Scholar]
- DEANS RM, MORGENS DW, OKESLI A, PILLAY S, HORLBECK MA, KAMPMANN M, GILBERT LA, LI A, MATEO R, SMITH M, GLENN JS, CARETTE JE, KHOSLA C, BASSIK MC. Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat Chem Biol. 2016;12:361–6. doi: 10.1038/nchembio.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DENIC V, QUAN EM, WEISSMAN JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–59. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- DERSH D, JONES SM, ELETTO D, CHRISTIANSON JC, ARGON Y. OS-9 facilitates turnover of nonnative GRP94 marked by hyperglycosylation. Mol Biol Cell. 2014;25:2220–34. doi: 10.1091/mbc.E14-03-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELBEIN AD, TROPEA JE, MITCHELL M, KAUSHAL GP. Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J Biol Chem. 1990;265:15599–605. [PubMed] [Google Scholar]
- FUJIMORI T, KAMIYA Y, NAGATA K, KATO K, HOSOKAWA N. Endoplasmic reticulum lectin XTP3-B inhibits endoplasmic reticulum-associated degradation of a misfolded alpha1-antitrypsin variant. FEBS J. 2013;280:1563–75. doi: 10.1111/febs.12157. [DOI] [PubMed] [Google Scholar]
- GELMAN MS, KOPITO RR. Cystic fibrosis: premature degradation of mutant proteins as a molecular disease mechanism. Methods Mol Biol. 2003;232:27–37. doi: 10.1385/1-59259-394-1:27. [DOI] [PubMed] [Google Scholar]
- HEBERT DN, GARMAN SC, MOLINARI M. The glycan code of the endoplasmic reticulum: asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol. 2005;15:364–70. doi: 10.1016/j.tcb.2005.05.007. [DOI] [PubMed] [Google Scholar]
- HELENIUS A, AEBI M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–49. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- HOELEN H, ZALDUMBIDE A, VAN LEEUWEN WF, TORFS EC, ENGELSE MA, HASSAN C, LEBBINK RJ, DE KONING EJ, RESSSING ME, DE RU AH, VAN VEELEN PA, HOEBEN RC, ROEP BO, WIERTZ EJ. Proteasomal Degradation of Proinsulin Requires Derlin-2, HRD1 and p97. PLoS One. 2015;10:e0128206. doi: 10.1371/journal.pone.0128206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOSOKAWA N, KAMIYA Y, KAMIYA D, KATO K, NAGATA K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J Biol Chem. 2009;284:17061–8. doi: 10.1074/jbc.M809725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOSOKAWA N, WADA I, NAGASAWA K, MORIYAMA T, OKAWA K, NAGATA K. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J Biol Chem. 2008;283:20914–24. doi: 10.1074/jbc.M709336200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HWANG J, WALCZAK CP, SHALER TA, OLZMANN JA, ZHANG L, ELIAS JE, KOPITO RR. Characterization of protein complexes of the endoplasmic reticulum-associated degradation E3 ubiquitin ligase Hrd1. J Biol Chem. 2017;292:9104–9116. doi: 10.1074/jbc.M117.785055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IIDA Y, FUJIMORI T, OKAWA K, NAGATA K, WADA I, HOSOKAWA N. SEL1L protein critically determines the stability of the HRD1-SEL1L endoplasmic reticulum-associated degradation (ERAD) complex to optimize the degradation kinetics of ERAD substrates. J Biol Chem. 2011;286:16929–39. doi: 10.1074/jbc.M110.215871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAENICKE LA, BRENDEBACH H, SELBACH M, HIRSCH C. Yos9p assists in the degradation of certain nonglycosylated proteins from the endoplasmic reticulum. Mol Biol Cell. 2011;22:2937–45. doi: 10.1091/mbc.E10-10-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM W, SPEAR ED, NG DT. Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol Cell. 2005;19:753–64. doi: 10.1016/j.molcel.2005.08.010. [DOI] [PubMed] [Google Scholar]
- KINGSTON RE, CHEN CA, OKAYAMA H. Calcium phosphate transfection. Curr Protoc Immunol. 2001;Chapter 10(Unit 10):13. doi: 10.1002/0471142735.im1013s31. [DOI] [PubMed] [Google Scholar]
- KIRK P, WILSON MC, HEDDLE C, BROWN MH, BARCLAY AN, HALESTRAP AP. CD147 is tightly associated with lactate transporters MCT1 and MCT4 and facilitates their cell surface expression. EMBO J. 2000;19:3896–904. doi: 10.1093/emboj/19.15.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCAFFREY K, BRAAKMAN I. Protein quality control at the endoplasmic reticulum. Essays Biochem. 2016;60:227–235. doi: 10.1042/EBC20160003. [DOI] [PubMed] [Google Scholar]
- MCCRACKEN AA, BRODSKY JL. Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J Cell Biol. 1996;132:291–8. doi: 10.1083/jcb.132.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIKAMI K, YAMAGUCHI D, TATENO H, HU D, QIN SY, KAWASAKI N, YAMADA M, MATSUMOTO N, HIRABAYASHI J, ITO Y, YAMAMOTO K. The sugar-binding ability of human OS-9 and its involvement in ER-associated degradation. Glycobiology. 2010;20:310–21. doi: 10.1093/glycob/cwp175. [DOI] [PubMed] [Google Scholar]
- MOLINARI M, HELENIUS A. Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science. 2000;288:331–3. doi: 10.1126/science.288.5464.331. [DOI] [PubMed] [Google Scholar]
- MORGENS DW, WAINBERG M, BOYLE EA, URSU O, ARAYA CL, TSUI CK, HANEY MS, HESS GT, HAN K, JENG EE, LI A, SNYDER MP, GREENLEAF WJ, KUNDAJE A, BASSIK MC. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat Commun. 2017;8:15178. doi: 10.1038/ncomms15178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUELLER B, KLEMM EJ, SPOONER E, CLAESSEN JH, PLOEGH HL. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc Natl Acad Sci U S A. 2008;105:12325–30. doi: 10.1073/pnas.0805371105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OKA OB, BULLEID NJ. Forming disulfides in the endoplasmic reticulum. Biochim Biophys Acta. 2013;1833:2425–9. doi: 10.1016/j.bbamcr.2013.02.007. [DOI] [PubMed] [Google Scholar]
- OKUDA-SHIMIZU Y, HENDERSHOT LM. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol Cell. 2007;28:544–54. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLZMANN JA, KOPITO RR, CHRISTIANSON JC. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol. 2013:5. doi: 10.1101/cshperspect.a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUAN EM, KAMIYA Y, KAMIYA D, DENIC V, WEIBEZAHN J, KATO K, WEISSMAN JS. Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol Cell. 2008;32:870–7. doi: 10.1016/j.molcel.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATO T, SAKO Y, SHO M, MOMOHARA M, SUICO MA, SHUTO T, NISHITOH H, OKIYONEDA T, KOKAME K, KANEKO M, TAURA M, MIYATA M, CHOSA K, KOGA T, MORINO-KOGA S, WADA I, KAI H. STT3B-dependent posttranslational N-glycosylation as a surveillance system for secretory protein. Mol Cell. 2012;47:99–110. doi: 10.1016/j.molcel.2012.04.015. [DOI] [PubMed] [Google Scholar]
- SATOH T, CHEN Y, HU D, HANASHIMA S, YAMAMOTO K, YAMAGUCHI Y. Structural basis for oligosaccharide recognition of misfolded glycoproteins by OS-9 in ER-associated degradation. Mol Cell. 2010;40:905–16. doi: 10.1016/j.molcel.2010.11.017. [DOI] [PubMed] [Google Scholar]
- SCHOEBEL S, MI W, STEIN A, OVCHINNIKOV S, PAVLOVICZ R, DIMAIO F, BAKER D, CHAMBERS MG, SU H, LI D, RAPOPORT TA, LIAO M. Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature. 2017;548:352–355. doi: 10.1038/nature23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEAAYFAN E, DEFONTAINE N, DEMARETZ S, ZAAROUR N, LAGHMANI K. OS9 Protein Interacts with Na-K-2Cl Co-transporter (NKCC2) and Targets Its Immature Form for the Endoplasmic Reticulum-associated Degradation Pathway. J Biol Chem. 2016;291:4487–502. doi: 10.1074/jbc.M115.702514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEKIJIMA Y, WISEMAN RL, MATTESON J, HAMMARSTROM P, MILLER SR, SAWKAR AR, BALCH WE, KELLY JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- SHRIMAL S, CHEREPANOVA NA, GILMORE R. Cotranslational and posttranslocational N-glycosylation of proteins in the endoplasmic reticulum. Semin Cell Dev Biol. 2015;41:71–8. doi: 10.1016/j.semcdb.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIFERS RN, BRASHEARS-MACATEE S, KIDD VJ, MUENSCH H, WOO SL. A frameshift mutation results in a truncated alpha 1-antitrypsin that is retained within the rough endoplasmic reticulum. J Biol Chem. 1988;263:7330–5. [PubMed] [Google Scholar]
- SMITH MH, RODRIGUEZ EH, WEISSMAN JS. Misfolded proteins induce aggregation of the lectin Yos9. J Biol Chem. 2014;289:25670–7. doi: 10.1074/jbc.M114.583344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUN S, SHI G, HAN X, FRANCISCO AB, JI Y, MENDONCA N, LIU X, LOCASALE JW, SIMPSON KW, DUHAMEL GE, KERSTEN S, YATES JR, 3RD, LONG Q, QI L. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci U S A. 2014;111:E582–91. doi: 10.1073/pnas.1318114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZATHMARY R, BIELMANN R, NITA-LAZAR M, BURDA P, JAKOB CA. Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol Cell. 2005;19:765–75. doi: 10.1016/j.molcel.2005.08.015. [DOI] [PubMed] [Google Scholar]
- TANG HY, HUANG CH, ZHUANG YH, CHRISTIANSON JC, CHEN X. EDEM2 and OS-9 are required for ER-associated degradation of non-glycosylated sonic hedgehog. PLoS One. 2014;9:e92164. doi: 10.1371/journal.pone.0092164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TYLER RE, PEARCE MM, SHALER TA, OLZMANN JA, GREENBLATT EJ, KOPITO RR. Unassembled CD147 is an endogenous endoplasmic reticulum-associated degradation substrate. Mol Biol Cell. 2012;23:4668–78. doi: 10.1091/mbc.E12-06-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VEMBAR SS, BRODSKY JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–57. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIE W, NG DT. ERAD substrate recognition in budding yeast. Semin Cell Dev Biol. 2010;21:533–9. doi: 10.1016/j.semcdb.2010.02.007. [DOI] [PubMed] [Google Scholar]
- XU C, NG DT. Glycosylation-directed quality control of protein folding. Nat Rev Mol Cell Biol. 2015;16:742–52. doi: 10.1038/nrm4073. [DOI] [PubMed] [Google Scholar]
- YAMAGUCHI D, HU D, MATSUMOTO N, YAMAMOTO K. Human XTP3-B binds to alpha1-antitrypsin variant null(Hong Kong) via the C-terminal MRH domain in a glycan-dependent manner. Glycobiology. 2010;20:348–55. doi: 10.1093/glycob/cwp182. [DOI] [PubMed] [Google Scholar]
- ZHOU HJ, WANG J, YAO B, WONG S, DJAKOVIC S, KUMAR B, RICE J, VALLE E, SORIANO F, MENON MK, MADRIAGA A, KISS VON SOLY S, KUMAR A, PARLATI F, YAKES FM, SHAWVER L, LE MOIGNE R, ANDERSON DJ, ROLFE M, WUSTROW D. Discovery of a First-in-Class, Potent, Selective, and Orally Bioavailable Inhibitor of the p97 AAA ATPase (CB-5083) J Med Chem. 2015;58:9480–97. doi: 10.1021/acs.jmedchem.5b01346. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Related to Figure 1. Characterization of knockout cell lines generated by CRISPR/Cas9-mediated genome editing.
Figure S2. Related to Figure 2. Destabilization of SEL1L/HRD1 complex upon loss of OS9 and XTP3B.
Figure S3. Related to Figure 3. OS9 isoform-specificity in targeting CD147 for ERAD.
Figure S4. Related to Figure 5. Inhibition of non-glycosylated protein ERAD by XTP3B is antagonized by OS9.
Figure S5. Related to Figure 6. Overexpressed SEL1L is demannosylated at Asn608.
Supplementary Tables 1-4