

Abstract
Copy number variants are deletions and duplications of a few thousand to million base pairs and are associated with extraordinarily high levels of autism spectrum disorder, schizophrenia, intellectual disability or attention-deficit/hyperactivity disorder. The unprecedented levels of robust and reproducible penetrance of copy number variants make them one of the most promising and reliable entry points to delve into the mechanistic bases of many mental disorders. However, the precise mechanistic bases of these associations still remain elusive in humans due to the many genes encoded in each copy number variant and the diverse associated phenotypic features. Genetically engineered mice have provided a technical means to ascertain precise genetic mechanisms of association between copy number variants and dimensional aspects of mental illnesses. Molecular, cellular and neuronal phenotypes can be detected as potential mechanistic substrates for various behavioral constructs of mental illnesses. However, mouse models come with many technical pitfalls. Genetic background is not well controlled in many mouse models leading to rather obvious interpretative issues. Dose alterations of many copy number variants and single genes within copy number variants result in some molecular, cellular and neuronal phenotypes without a behavioral phenotype or with a behavioral phenotype opposite to what is seen in humans. In this review, I discuss technical and interpretative pitfalls of mouse models of copy number variants and highlight well-controlled studies to suggest potential neuronal mechanisms of dimensional aspects of mental illnesses. Mouse models of copy number variants represent toeholds to achieve a better understanding of the mechanistic bases of dimensions of neuropsychiatric disorders and thus for development of mechanism-based therapeutic options in humans.
Keywords: copy number variants, autism spectrum disorder (ASD), schizophrenia, intellectual disability, attention deficit hyperactivity disorder(ADHD), Research Domain Criteria (RDoC)
Rare Genetic Variants
Copy number variants (CNVs), each occurring in less than 1% of a clinically diagnosed disorder, are associated with mental disorders with unprecedentedly high odds ratios. These include 1q21.1 deletion and duplication, 2p16.3 deletion, 3q29 deletion, 7p36.3 deletion and duplication, 7q11.23 duplication, 8q22.2 deletion, 9p24.3 deletion and duplication, 15q13.3 deletion, 16p11.2 deletion and duplication, 22q11.21 deletion and Xq28 duplication1–3. These CNVs are variably associated with all or some of mental disorders including autism spectrum disorder (ASD), schizophrenia, intellectual disability (ID) or attention-deficit/hyperactivity disorder (ADHD). Some of these CNVs are predominantly inherited (e.g., 1q21.1 duplication, 15q13.3 deletion, 16p11.2 duplication, 22q11.2 duplication) and others are de novo (e.g., 16p11.2 deletion, 17q12 deletion, 22q11.2 deletion)4.
While these genetic variants are solid entry points to research in precision medicine, a complete understanding of factors that cause incomplete penetrance and variable expressivity of a disorder is still lacking. Nonetheless, several promising hypotheses have been proposed. A CNV might become symptomatic when hit by a second CNV4,5. Moreover, given that common genetic variants en masse confer substantial risk for ASD and schizophrenia6–8, such factors in the genome might amplify or reduce the impact of a CNV. An experimental analysis of mouse models is consistent with the hypothesis that genetic backgrounds modify the impact of ASD-associated genes9–12. Environmental factors, as modifiers for the effects of CNVs, are likely to contribute to incomplete penetrance and variable expressivity11,13,14. Some environmental factors by themselves are also known to increase the probability of schizophrenia15 and ASD16.
DSM and RDoc
The clinical diagnosis of any mental disorder is based on observed clusters of elements considered to be essential for a given disorder. The Diagnostic and Statistical Manual was an attempt to standardize diagnosis based on itemized elements and non-parametric crossing of a diagnostic threshold. For examples, according to the DSM-5, one is diagnosed with schizophrenia, when two or more of “core” symptoms (i.e., delusions, hallucinations and disorganized speech, grossly disorganized or catatonic behavior and negative symptoms, among which at least one must be from delusions, hallucinations and disorganized speech) are present for a significant portion of time during a 1-month period.
There are a number of inconvenient facts about such categorical diagnoses by the DSM or International Classification of Diseases (ICD). First, treatment outcomes often ignore the boundary of so-defined disorders. Atypical neuroleptics are used to treat schizophrenia, bipolar disorder, and even anxiety disorders (e.g., sulpride). Second, the categorical division between mental disorders is inconsistent with genetics. Each CNV, in many cases, is associated with multiple mental disorders. Third, there is no mechanistic basis to group a certain set of symptoms for a given category. Certain drugs ameliorate only specific aspects of a disorder. For example, although typical neuroleptics soften psychosis and hallucinations, they are not effective for treatment of negative symptoms or cognitive impairments. Although atypical neuroleptics are effective for ameliorating both positive and negative symptoms of schizophrenia, they have little effect on cognitive impairments; they are effective for reducing aggression and repetitive behaviors of ASD, but do not improve defective social interaction and communication. Fourth, each disorder is highly heterogeneous. A combination of symptomatic elements and their severity widely vary individually. Fifth, even before one reaches a diagnostic criterion, individuals may exhibit many atypical behaviors and precursor symptoms, suggesting that the underlying biology precedes the onset of clinically defined disorders.
The Research Domain Criteria (RDoc) is an attempt to shift the paradigm17,18. This initiative is still a blue print for an alternative way of understanding mental disorders. It is based on a dimension composed of domain constructs, units of analysis and developmental trajectory. The domain constructs are observable behavioral and neurobiologic measures that form broad domains of function. For example, the social processes domain includes imitation, social dominance, attachment/separation fear and more. The cognitive domain includes attention, working memory, declarative memory and cognitive control. These domain constructs are independent of categorical classification of mental disorders and do not need to follow the preconceived grouping of symptoms to define mental disorders. Whether such constructs deviate sufficiently to call them ASD or schizophrenia—where to draw a cut-off beyond which to call something a disorder– is a matter of clinical convenience rather than mechanistic rationale. These constructs are to be examined at the levels of genes, molecules, cells, circuits, physiology, behavior, self-reports and paradigms. The NIH envisions that, through empirical testing and revision of constructs, valid dimensions will eventually form a basis for a new way of understanding and treating mental illnesses.
Mouse models of CNVs
Cell models developed from human iPSCs and 3D brain organoids provide a technical means to examine molecular and cellular phenotypes of CNVs, but many technical issues remain including their limited capacity to model only early development, inability to model neuronal functions in circuits in the brain or under environmental influences, high phenotypic variability and high spontaneous mutation rates in iPSCs. Researchers have capitalized on CNVs to develop mouse models with construct validity. As of October 2017, there are mouse models with the following CNVs with more being developed using CRISPR techniques: 16p11.2, 17p11.2 15q11–13, 15q13.3, 22q11.21 and 7q11.23. Behavioral, anatomic, neuronal, cellular and molecular correlates of these mouse models have been investigated.
Before discussing phenotypes of these mouse models, it is important to discuss one critical issue. Behavioral phenotypes are often viewed as variable and unreliable. While this might be the case compared to other phenotypes, this is also due to unreliable methods in which mice are generated and tested. Mouse models are maintained using various breeding methods (see Figure 1). One widely used method produces a non-congenic mouse line. A non-congenic mouse is F2 or later generations, resulting from crosses of mice with a mutation and a breeder mouse. In many cases, mutations are generated using ES cells of one inbred mouse (e.g., 129sv). A developed mouse is then crossed with a mouse line with high fecundity (e.g., C57BL/6J). The offspring then carry the genetic backgrounds of alleles of ES cells and alleles of a breeder strain. The chromosomal regions linked to the target genes are expected to contain more genomic materials of ES cells in mutant mice; the same region contains more alleles of a breeder mouse strain in wild-type littermates. Thus, mutant and wild-type littermates are expected to systematically differ in allelic composition in the vicinity of the targeted gene. In this sense, the “mixed” genetic background does not mean a randomly mixed genetic background, which would not pose interpretative issues, but rather represents a “biased” genetic background in the vicinity of the gene of interest between wild-type and mutant littermates. It has long been pointed out that these systematically “biased” genetic backgrounds pose an interpretation difficulty for phenotypes19–23.
Figure 1.
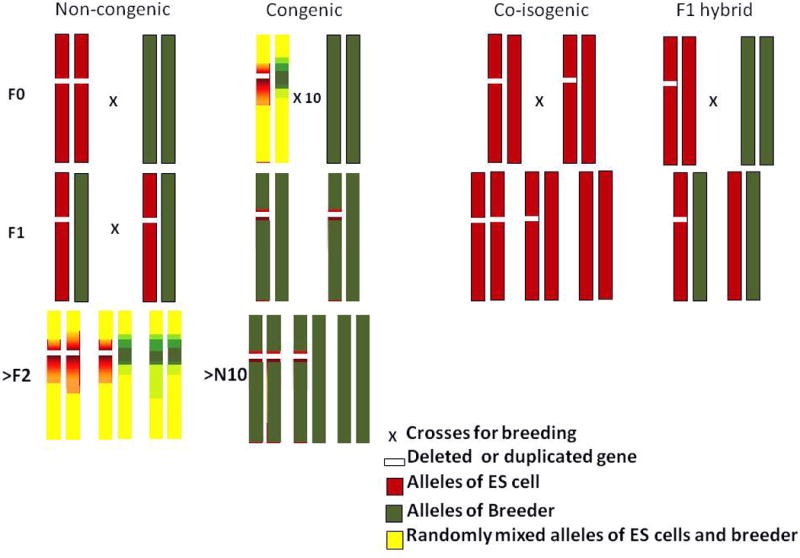
Genetic background of different breeding strategies.
The targeted gene (white band) is shown with background alleles originating from the embryonic stem (ES) cell donor strain (red), a breeder (green), and randomly mixed alleles of both parents (yellow) in non-congenic mice, congenic mice, co-isogenic mice and F1 hybrid mice. Non-congenic wild-type and mutant mice systematically differ in alleles flanking the targeted gene at the F2 generation due to recombination of alleles. By backcrossing such a mouse to the breeder for more than 10 generations (>10N), the genetic background of a congenic mouse is saturated with alleles of the breeder, thereby minimizing the systematic difference in the flanking regions between wild-type and mutant mice. A co-isogenic mouse is developed in ES cells of a mouse strain and bred with the same mouse line. The F1 hybrid is made by crossing a co-isogenic mutant mouse is crossed with another inbred mouse and the identical genetic background is present between wild-type and mutant mice at the F1 generation.
While as few as four generations of backcrossing would be expected to reduce genomic material from a strain of ES cells to approximately 12% of the entire genome (i.e.,), this estimate is based on regions unlinked to the mutated gene. A much higher percentage of genomic materials of ES cells would be expected to be retained in chromosomal regions linked to the mutated genes21. If phenotypes are qualitative (e.g., lack of an organ or appearance of pathology), unequal genetic background would not be much of an issue. However, in the majority of CNV studies, phenotypes are not qualitative ones but quantitatively deviate from the average of wild-type mice. If various inbred mice exhibit more or less similar quantitative behavioral traits, different genetic backgrounds would not be an issue, either. However, inbred mouse lines widely differ in various quantitative parameters relevant to mental disorders, including memory in the Morris water maze and the radial maze24–26, working memory26, prepulse inhibition26, social behaviors26,27, neonatal ultrasonic vocalization27–29 and seizures30.
The biased genetic background of non-congenic mice also complicates interpretation of anatomic and electrophysiological phenotypes. Inbred mouse lines exhibit considerable variation in anatomic structures: shapes and volumes of the corpus callosum, anterior commissure, hippocampal commissure, hippocampus and ventricle31–33, number of neurons24,33–36, number of neurotransmitter receptors25, shape and number of dendritic spines37 and the rate of various phases of adult neurogenesis24,38. Inbred mouse lines also differ in electrophysiological measures of synaptic plasticity39–41.
Molecules affected by a mutated gene are also confounded when genetic background is not controlled. Several elegant studies involving mutant mice revealed a robust biasing effect of genetic backgrounds on molecular differences between mutant and wild-type mice. Valor and Grant42 carefully examined differentially expressed genes between wild-type and mutant littermates of SAP102 and PSD-95. Most significantly differentiated genes were located in the vicinity of the deleted genes. The differential gene expression and alleles of ES cells were considerably reduced by backcrossing to C57BL/6J for ten generations. The same trend was observed in mice with mutations of Rab3A43, 17p11.2 deletion and duplication44, and mutation of Gtf2ird1, a gene encoded in 7q11.23 CNV45, and other mutations42.
These rigorous, carefully designed studies cast doubt on any observed phenotypic alterations–whether they are behavioral, anatomic, electrophysiologic, cellular or molecular–seen in non-congenic mutant mice, as solely reflecting the genuine effects of the targeted mutation. Any difference between non-congenic wild-type and mutant littermates could reflect allelic differences, the impact of the mutated gene, additive or epistatic effects of both, or some or all of these factors. It is possible that alleles in the flanking region of the gene of interest could be linked in humans as well, but humans are mostly outbred and the flanking region is expected to randomly differ between mutant carriers and non-carriers. This is not the case in non-congenic mice. There is a systematic bias in genetic background between mutant and wild-type littermates. Researchers often express the opinion that mutant mice with a “mixed” genetic background tend to exhibit more robust behavioral deficits and are thus ideal to investigate the impact of the mutated gene. However, the purpose of any analysis of a genetic mouse model should be to evaluate the impact of a targeted mutation on phenotypes; not to find the ‘robust’ phenotype regardless of its cause.
The strategies to circumvent this interpretative pitfall are congenic mouse, co-isogenic mouse and F1 hybrid mouse (see Fig. 1). The congenic strain uses a strategy in which a non-congenic mouse is backcrossed for ten generations or more to maximize the breeder alleles even in the vicinity of the targeted gene. Although allelic differences between wild-type and mutant mice are minimized and there is a considerably lesser degree of interpretative ambiguity, unequal alleles might still persist at the segments very proximal of the mutated gene in a congenic mouse line.
A co-isogenic mouse is developed in ES cells of one strain and bred in the same strain. ES cells derived from C57BL/6N are often used and bred with the same substrain to maintain the genetic background of wild-type and mutant mice as identical. An F1 hybrid is another strategy designed to keep the genetic background of mutant and wild-type littermates identical. The mutant mouse line with ES cell genetic background is crossed with another breeder mouse strain and their first generation offspring (F1) is used, so that both wild-type and mutant mice have one copy of a strain from which ES cells were derived and one copy of a strain used for breeding without recombination. An F1 hybrid makes it possible to critically evaluate the potential interaction between common alleles in the genetic background and the targeted mutation under a well-controlled experimental condition.
RDoc domain constructs in mouse models of CNVs
The RDoc domain construct coincidentally jibes well with what has long been used to assess behaviors in rodents. There are a number of aspects of neuropsychiatric disorders that are modeled in mice. These measures can be collected in mouse models along the developmental span under environmental influences. Thus, a good translational match between humans and experimental animals is possible with the RDoc as long as valid tasks are available to measure units of the same constructs in humans and experimental animals.
Because of the interpretative ambiguity of non-congenic mice, I am limiting our discussion to those studies in which genetic background is controlled using congenic mice, co-isogenic mice and F1 hybrids (see Figure 1). The studies listed in Table 1 are considered “primary” evidence that phenotypes can be, fairly unambiguously, ascribed to the mutated genes without the confounding effects of the biased genetic backgrounds. I exclude studies that 1) referred to previous studies that did not use F1 hybrid, co-isogenic or congenic mice (10 or more backcrossing), 2) stated that the mutant mouse was on a C57BL/6J or C57BL/6N background with a citation which used ES cells of 129sv inbred mouse lines and did not specify the number generations of backcrossing, 3) stated “a pure C57BL/6 genetic back ground” without defining what is meant by “pure” and in fact it included another genetic background. While those excluded studies are of interest, the phenotypes described in them cannot be unambiguously ascribed to the genes in question. Those phenotypes are likely to include the impact of the biased allelic distributions, particularly in the vicinity of the gene of interest, between wild-type and mutant mice. As the links between those background alleles and mental disorders are not established, such phenotypes cannot be pursued to examine the mechanistic basis of the genetic variants associated with mental disorders. In fact, many phenotypes reported in non-congenic mice are not replicated when genetic background is controlled. On the other hand, the studies discussed below provide a more solid basis to further delve into mechanistic bases of dimensional aspects of mental disorders.
Table 1.
Mouse models of CNVs and dimensional behavioral phenotypes
| CNV | Associated Diagnoses | Designation | ES or Zygote cell background | Additional Background | Age | PPI | WM | RL | SO | Anx | Pup Voc | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7q11.23 del | ID/DD/CM | Fkbp6-Gtf2i | G6 ES cell 129Sv | CD1 C57BL/6J N9 generation |
Not described | – | ↑ | ↓ | 116 | |||
| 15q11–13 dup | ID/DD/CM ASD SCZ |
patDp/+ Herc2-Mkrn3 |
129S7/SvEvBrd-Hprt<b-m2> | Not described | – | – | ↓ | ↓ | ↑ | ↑ | 60 | |
|
matDp/+ Herc2-Mkrn3 |
Not described | – | – | – | – | – | – | |||||
| 15q11–13 dup |
patDp/+ Herc2-Mkrn3 |
129S7/SvEvBrd-Hprt<b-m2> | C57BL/6J, congenic > 10 backcrosses |
9–13 weeks | – | – | ↑ | 61 | ||||
| 15q13.3 del | ID/DD/CM ASD SCZ |
Df(h15q13)/+ Fan1-Chrna7 |
C57BL/6NTac | 9–12 weeks | – | – | – | 62 | ||||
| 15q13.3 del |
15q13.3/7qC Fan-Chrna7 |
C57BL/6NTac | 4–7 months (17–30 weeks) | – | – | ↓ | ↓ | ↓ | 109 | |||
| 15q13.3 del |
Df(h15q13)/+ Fan1-Chrna7 |
C57BL/6NTac | 10–22 weeks | – | -↑ | -↓ | 63 | |||||
| 16p11.2 del | ID/DD/CM ASD SCZ |
Del/+ Sult1a1-Spn |
C57BL/6NTac | F1 hybrid with C3H/HeH | 12–20 weeks | ↓ | – | 132 | ||||
| 16p11.2 del |
Del/+ Sult1a1-Spn |
C57BL/6NTac | 12–20 weeks | – | ↓- | |||||||
| 16p11.2 dup | ID/DD/CM ASD SCZ BD |
Dup/+ Sult1a1-Spn |
C57BL/6NTac | F1 hybrid with C3H/HeH | 12–20 weeks | ↓ | – | |||||
| 16p11.2 dup |
Dup/+ Sult1a1-Spn |
C57BL/6NTac | 12–20 weeks | – | ↑- | |||||||
| 17p11.2 Del(SMS) | Df(11)17/+ Zfp179-Csn3 |
129S5/SvEvBrd mouse (129S5) | Congenic C57BL/6-TyrC-Brd 12 generations |
10 weeks | -↑ | ↓ | 44 | |||||
| 17p11.2 Dup (PTLS) | Dp(11)17/+ | 129S5/SvEvBrd mouse (129S5) | Congenic C57BL/6-TyrC-Brd 12 generations |
10 weeks | -↓ | ↑ | ||||||
| 17p11.2 Dup (PTLS) | Dp(11)17/+ | 129S5/SvEvBrd mouse (129S5) | Congenic C57BL/6-TyrC-Brd >10 generations |
8–12 weeks | ↑ | ↑ | ↓ | 137 | ||||
| 22q11.2 dup | ID/DD/CM ASD |
200kb Tg Gnb1l -Sept5 |
FVB | 2–4 months (9–17 weeks) and 5 weeks | ↓ | 133 | ||||||
| 22q11.2 dup | 190kb Tg Arvcf – Txnrd2 |
FVB | C57BL/6J, congenic 10 backcrosses |
1, 2 and 5 mo (4, 9 and 21 weeks) | – | ↓ | – | – | 58 | |||
| 22q11.2 del | ID/DD/CM ASD SCZ |
Df(16)A+/− Dgcr2-Hira |
129S7/SvEvBrd-Hprt<b-m2> | C57BL/6J, congenic 99.9% | 3–6 mo (13–26weeks) |
↓- | 110 | |||||
| 22q11.2 del |
Df(16)1/+ Dgcr2-Ufd1l |
129S7/SvEvBrd-Hrp<b-m2> | C57BL/6J, congenic >=10 backcrosses |
4–5 mo (17–21 weeks) |
↓ | 55 | ||||||
| 22q11.2 del |
Lgdel/+ Dgcr2-Hira |
129/Sv, C57BL/6J, SJL; 129S6/SvEvTac; FVB/N | C57BL/6; 129Sv; CD1; C57BL/6J > 25 backcrosses |
Not described | ↓ | 115 | ||||||
| 22q11.2 del |
Df(h22q11)/+ Dgcr2-Hira |
C57BL/6NTac | 6–26 weeks | ↓ | 56 | |||||||
| 22q11.2 del |
Df(h22q11)/+ Dgcr2-Hira |
C57BL/6NTac | 2–19 mo (9–81 weeks) |
↓-↑ | -↑ | 105 |
Age, age at which behavioral testing started, except for pup vocalization. SMS, Smith-Magenis Synrome; PTLS, Potocki-Lupski syndrome; ID, intellectual disability; DD, developmental delay; BD, bipolar disorder; CM, congenital malformations; ASD, autism spectrum disorder; SCZ, schizophrenia; PPI, prepulse inhibition; WM, working memory; RL, reversal learning; SO, social behaviors; Anx, Anxiety; Pup Voc, neonatal vocalization; Ref, reference; Del, deletion; dup, duplication; upward and downward arrows indicate increase and decrease. horizontal bar, non-significant phenotypes. Not included here are studies that used non-congenic mice or that stated mice were generated on a C57BL/6J mouse or “pure” C57BL/6 background, but cited articles did not create a mouse as such. Disorders associated with each CNV are based on1,2.
Sensorimotor gating in prepulse inhibition (PPI)
PPI scores are lower in individuals with schizophrenia, obsessive compulsive disorder (OCD), Tourette’s syndrome, bipolar disorder, ADHD, Huntington’s disease, Lewy body dementia and seizure disorder46. No consistent deficit has been noted in individuals with ASD. Children and adults with ASD without cognitive deficits have normal or even higher PPI47–52 except as reported in a study that used a specific parametric condition48. Children and adults with autism with cognitive deficits have normal PPI53,54. While they showed a deficit for a 120msec interval between a prepulse and pulse stimuli, in one set of samples, this was not found in a larger sample53. Thus, lower levels of PPI can be taken as a reliable dimensional aspect of psychiatric disorders except for ASD.
Several mouse models of CNVs exhibit defective PPI (see Table 1). Mouse models of 22q11.2 hemizygous deletion consistently show lower levels of PPI55,56 consistent with lower PPI scores in individuals with 22q11.2 hemizygosity57. Mice that over-express a segment of 22q11.2 are normal58, a finding in accordance with the observation that duplication of 22q11.2 does not increase the risk for schizophrenia and rather might be a protective factor for schizophrenia59. PPI is also normal in mouse models of 15q11–13 duplication60,61 and 15q13.3 deletion62,63. It remains unclear why these mouse models of 15q11–13 duplication and 15q13.3 deletion, which are associated with schizophrenia in humans, show normal PPI, but a lack of PPI deficits is consistent with a feature of ASD.
Working memory
While its precise construct structure is a matter of debate, working memory is measured via tasks that require the retention of information for a short period of time in a flexible manner. Working memory capacity typically expands from childhood to adulthood64–67. This developmental maturation process, however, starts to lag behind from adolescence (13–17 years) to adulthood (18 years and older) in individuals with ASD68–70. Working memory with high load and longer temporal span is more severely affected than other working memory tasks in ASD71–73.
Cognitive deficits of individuals with schizophrenia encompass diverse capacities, including attention, working memory, executive function, episodic memory, semantic memory, visual memory, verbal ability and learning, spatial memory and reasoning, face memory, emotion differentiation, verbal reasoning, list memory, processing speed and fluency74–78. However, various cognitive functions are not evenly impaired in patients with schizophrenia or 22q11 deletion74,79. Children and adolescents who later develop schizophrenia are particularly behind their peers in working memory, attention and processing speed80–87. Cognitive deficits start to appear a decade before–and worsen till—symptoms reach the diagnostic criteria of schizophrenia in individuals with 22q11.2 deletion88–93 or idiopathic schizophrenia80,84,85
Working memory, among various executive functions, is best correlated with IQ94–96 and is less developed, throughout development and in adulthood, in individuals with intellectual disability (ID)97–100. Weak working memory is also found in children with attention deficit hyperactive disorder (ADHD)101–104.
In rodents, working memory is often evaluated as spontaneous and rewarded alternation with delay in a T-maze and win-shift responses in a radial maze in which the position of a baited arm is changed from trial to trial. Lower scores in tasks that include working memory have been observed in a segmental 22q11.2 duplication model in rewarded alternation with delay58 (see Table 1). A co-isogenic model of 22q11.2 hemizygous deletion exhibited increased scores of touchscreen delayed non-match to location, and lower scores in acquisition of T-maze non-match to sample; this mouse line did not differ from wild-type mice in working memory scores in a radial maze, spontaneous alternation or non-match-to -sample with delay in a T-maze105. However, in this study, mice were tested over a wide range of ages, from 2 months to 16 months. In particular, delayed non-match to location in a touch screen task and a T-maze task started from 8 and 7–16 months of age, respectively. The age of nineteen months in mice corresponds to approximately 56 years old in humans; 10–14 months of age in mice correspond to 38 to 47 years in humans106. As working memory deficits due to genetic variants appear in an age-dependent manner in mice58,107 and humans108 and baseline scores differ at different ages, testing in a much narrower range from 1 to 3 months of age might detect a phenotype. Working memory is normal in mouse models of paternal and maternal 15q11–13 duplication60,61, 15q13.3 deletion62,109, and 22q11.2 hemizygous deletion110 (see Table 1).
Flexibility and reversal learning
ASD is characterized by circumscribed interests and restricted and repetitive behaviors111. While not impaired in a simple discrimination task, individuals with an ASD diagnosis require more trials in reversal learning; this trend correlates with stereotyped, repetitive, or idiosyncratic speech and restricted, repetitive, and stereotyped patterns of behavior112.
The capacity for simple discrimination, extra-dimensional shift and reversal is impaired in individuals with schizophrenia and a reversal deficit is correlated with negative symptoms and disorganized thought, but not with positive symptoms113,114. Schizophrenic patients make more errors than controls in simple discrimination, initial compound discrimination and extra-dimensional shift and simple reversal, compound reversal, intradimensional reversal and extra-dimensional reversal.
A genetic mouse model of paternal 15q11–13 duplication exhibits resistance to reversal of learned behaviors in the Morris water maze and Barnes maze, although the original learning is intact; those with maternal duplication are normal in reversal of this task60 (see Table 1). A congenic mouse with 22q11.2 hemizygous deletion exhibits improved early-phase reversal learning, impaired late-phase and overall touch-screen reversal learning, and impaired discrimination performance115. A co-isogenic mouse model of 22q11.2 hemizygous deletion show a delay in acquisition of working memory in a standard working memory in a T-maze; however, they showed better performance in easy reversal learning and no phenotype in difficult reversal learning in a touch-screen task105. It should be noted that in this study, mice were tested in a wide age range from 2 to 8 months. Reversal learning was normal in a congenic 7q11.23 deletion mouse model116, a co-isogenic maternal 15q11–13 duplication model60, or a co-isogenic15q13.3 deletion model109 (see Table 1).
Social cognition
Defective reciprocal social interaction is one of the core symptomatic elements of ASD. Precise developmental charting is critical to correctly detect early signs of ASD, as the timing of appearance and maturity levels of various milestone features individually varies117. While incipient ASD infants start to show atypical development of social orienting and reciprocity (e.g., gaze to face and eye, social smiles, directed vocalizations and social engagement, imitation, response to name) at 12 to 18 months, they deviate from typically developing infants in social communication as early as 6 months118,119 and in memory-based face recognition from the age of 3 to 6 months120. Attention to social scenes and face is found altered in infants who are later diagnosed with ASD as early as 6 months in some cases121. More pronounced signs of communication delay and social difficulties become apparent by 3 years old. From 6 to 18 months of age, incipient ASD infants, compared to typically developing infants, exhibit atypical eye contact and reductions in imitation, response to name, social intent and social smiling122. During the period from 18 to 36 months of age, children with ASD display no pretend play, highly repetitive manipulation of a part of objects, a reduced joint attention and communication123. There is considerable improvement in theory of mind scores from 5 to 8 years of age in children with ASD124. Children with ASD eventually are able to track others’ mental states in false belief tasks, but ASD children require twice longer to reach the same level of theory of mind scores as typically developing children125. While adults with ASD can perform the theory of mind normally, they do not show automatic anticipatory eye gaze126, suggesting that ASD children do not acquire this capacity in the same way as typically developing children.
The development of social cognition, including perception of social cues such as face and voice, mentalizing and emotion regulation, consistently lags behind, throughout childhood to adulthood, in individuals with later diagnosis of psychosis and schizophrenia74,127–130. Defective social cognition is also seen in individuals with ADHD123, but different reasons might underlie social impairments seen in ASD and ADHD131.
Some of these features can be quantitatively measured in rodent tasks. In mice, naturalistic reciprocal affiliative social interaction and aggressive social interaction in a home-cage setting and “sociability” in a three-chamber task are routinely used. Given that adults with ASD are not quantitatively different from controls in theory of mind, the testing age is an important factor to consider. Altered social behaviors in humans with genetic variants are recapitulated in genetic mouse models of 15q11–13 paternal duplication60, 15q13.3 deletion109, 16p11.2 deletion and duplication132, 17p11.2 duplication44, and 22q11.2 duplication from GNB1L, TBX1, GP1BB and SEPT5133 (see Table 1).
Neonatal ultrasonic vocalization
Babies who are later diagnosed with ASD emit atypical preverbal vocalizations13,118,134,135. Neonatal vocalization, under conditions of maternal separation, has been examined using mouse models as a proxy of baby cries. Rodent pups emit vocal calls upon separation from dams and the dams use pup calls to locate and retrieve the pups. A number of genetic mouse models of ASD exhibit atypical neonatal vocalizations12,136. Mouse models of paternal 15q11–13 duplication60,15q13.3 deletion63,109 and 17p11.2 duplication137 show altered total numbers of neonatal calls (see Table 1). That atypical social communication functionally impacts the relationship of pups with mothers has recently been demonstrated in a mutant mouse that lacks one copy of Tbx1, a 22q11.2-encoded gene14,138.
Anxiety
Anxiety is considered a comorbid trait in ASD and schizophrenia, but the concept of comorbidity is a moot point in the dimensional evaluation of neuropsychiatric disorders, as the latter has no categorical classification of disorders, hence no such a thing as a core symptom or comorbid trait18. Various mouse models of CNVs exhibit heightened anxiety-related behaviors (see Table 1).
Factors affecting the magnitude and directions of behavioral phenotypes
Incomplete penetrance and variable expressivity are often found in mouse models even when genetic background is controlled139. Lack of phenotypes in some cases might be taken as evidence that a CNV represents a liability rather than a causative factor sufficient to result in phenotypes. However, many factors also need to be considered here for the apparently absent, weak or even opposite phenotypes in behavioral tasks.
Different species might not be equally sensitive to gene doses. While humans with 15q13.3 hemizygous deletion exhibit ASD at high rates, mice with hemizygous deletion of the homolog chromosome do not and it takes homozygous deletion to induce behavioral phenotypes relevant to ASD63.
The average score for any mouse behavioral task might not be a reliable way to detect CNV-linked phenotypes. Many CNVs have incomplete penetrance and variable expressivity in humans. While the penetrance of any disorder, including ASD, congenital malformation and developmental delay, is close to complete in 7q11.23 and 22q11.2 deletion cases, it is not for other CNVs140. Moreover, when individual diseases are separately considered, no CNV shows complete penetrance. It is thus not surprising that the overall average of a behavioral score, which represents a domain construct, does not differ between wild-type and mutant mice at the group basis; a subset of mice might exhibit phenotypes.
A host genetic background might powerfully modulate behavioral phenotypes. When the genetic background of 129SvJ is increased in Sept5 mutant mice, the baseline level of reciprocal social interaction increased and a phenotypic difference between wild-type and mutant littermates dissipated9,141,142. The apparent absence of a phenotype could be due to some modifying impact of alleles of 129SvJ on Sept5 mutation. This is different from the biased genetic background between wild-type and mutant mice, as congenic Sept5 mutant mice show statistically lower scores than wild-type littermates in reciprocal social interaction9,141,142. Similarly, when a congenic Fmr1 mutant mouse with C57BL/6J background is crossed with various inbred mouse lines, the impact of this gene mutation is erased or reversed on a F1 hybrid genetic background with A/J, DBA/2J, FVB/NJ, 129S1/SvImJ, or CD-1, attesting to the robust modifying effect of genetic background on phenotypes143. Shank3 mutation on co-isogenic C57BL/6N background manifests its phenotypic effects non-identically under different congenic genetic backgrounds of FVB/NTac and 129S6/SvEvTac144. Sittig and colleagues produced F1 crosses between male C57BL/6J mice heterozygous for null alleles of Cacna1c or Tcf7l2 and females of 30 inbred mouse lines145. Phenotypic expression differed widely under distinct F1 inbred hybrid backgrounds. This study is different from those that used non-congenic mutant mice. As an F1 generation was used, the genetic background is made homogenous between wild-type and mutant mice. Another elegant study by Herault and colleagues examined the impact of hemizygous deletion and duplication of mouse ortholog of human 16p11.2 CNV on behaviors under an F1 hybrid background. ASD-related social interaction deficits appeared under an F1 hybrid background of C57BL/6N and C3H/HeH, but not under co-isogenic C57BL/6N background132.
These mouse studies are conceptually provocative in that they suggest that the phenotypic appearance of a CNV is permitted under a specific genetic background9,11 or alternatively results from the collective impact of common alleles in the genetic background that can appear in the absent negative control of a gene(s) encoded in a CNV (Y. Herault, personal communication).
Environmental factors are another important modifier of behavioral phenotypes. In mice, when behavioral phenotypes relevant to ASD are not present, this could be due to housing conditions. The manner in which mutant and wild-type mice are housed exerts a profound impact on behavioral phenotypes. Yang and colleagues demonstrated that a mouse model of 16p11.2 deletion exhibited deficits in neonatal ultrasonic vocalization (USV) and adult reciprocal male-female social interaction when mutant mice were housed with wild-type mice, but not housed with the same genotype146. The composition of litter genotypes has also been shown to affect the phenotypic differences in olfactory responses to social odor in Nlgn3 mutant mice147.
Testing age should also be considered. Depending on phenotypes, the gene might affect testing results at specific ages more preferably. ASD patients overcome defective mentalizing by adulthood126. Working memory shows developmental maturation from childhood to adulthood in humans108 and during the corresponding period from 1 to 2 months of age in mice58,107 and copy number alterations of 22q11.2-encoded genes impact the maximal maturation at 2 months, but not at 1 month of age107 in a similar manner as seen in individuals with alleles of a 22q11.2 gene108.
Behavioral phenotypes of single gene mutant models
It remains mostly unclear how individual genes encoded in each CNV affect dimensional aspects of mental disorders. On one hand, one can assume that all CNV-encoded genes contribute to dimensions of neuropsychiatric disorders. On the other hand, one can also assume that not all CNV-encoded genes are responsible for dimensional behavioral deviations. These alternative views cannot be critically tested using mouse models of the entire region of a CNV.
Due to the pioneering work involving mutant mouse models of 22q11.2-encoded segments and single genes11,148–150, much more is understood about the way individual 22q11.2 CNV-encoded genes affect dimensions of mental illnesses than other CNVs. However, mutant mice used in many studies were non-congenic lines and phenotypes cannot be unambiguously ascribed to the impacts of targeted genes. Thus, here, I discuss primary evidence in which the genetic background of a mutant mouse is either co-isogenic, an F1 hybrid or congenic with 10 or more generations of backcrossing. To date, such mutant mice with many 22q11.2 genes encoded in the commonly deleted 1.5 Mb region are available and have been tested in the behavioral constructs listed (Fig. 2). Attempts to provide phenotypic profiles of all mouse genes by the International Mouse Phenotyping Consortium (http://www.mousephenotype.org/) will accelerate similar analyses of other single genes of this and other CNVs. Critically, mice developed by the IMPC are co-isogenic mice with C57BL/6N background151, thereby avoiding the interpretive issues of non-congenic mice. In fact, several previously published data of non-congenic mice are not replicated in the co-isogenic mutant mice of the IMPC152.
Figure 2.
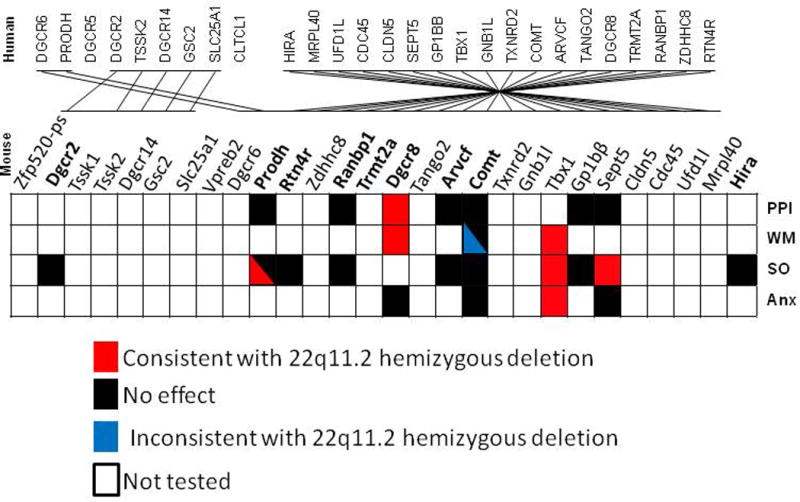
Impact of deletions of individual 22q11.2 gene on various behavioral constructs relevant to mental disorders. Bold gene names indicate that congenic or co-isogenic mice have been developed. When mice are tested for the selected phenotypes, they are left blank. Deletions of genes cause phenotypes consistent with (red) and opposite to (blue) what is seen in humans with 22q11.2 hemizygous deletions. Deletions cause no detectible effect on a phenotype (black). Phenotypes have not been examined (blank). Non-congenic mice are not included in analysis. Non-congenic mice that are backcrossed for less than 10 generations or carry alleles of both C57BL/6N and C57BL/6J are not included, as these C57BL/6 substrains differ in many behavioral measures due to allelic differences189–192. Abbreviations: PPI, prepulse inhibition; WM, working memory; SO, affiliative social interaction, sociability, neonatal and adult vocalization and adult aggression; Anx, anxiety-like behaviors. Dgcr2152; Prodh152,153; Rtn4r152; Ranbp1152; Dgcr8154,155; Arvcf152; Comt152,157–160;Tbx1138,156; Gp1bb152; Sept59,141,152; Hira152.
Several genes have been shown to contribute to dimensional constructs of individuals with 22q11.2 hemizygosity (Fig. 2). Deficiency of Prod alters adult vocalization in females152, but has no detectable effect on PPI152,153 or aggression152. Dgcr8 heterozygous mice are impaired in prepulse inhibition154,155 and working memory155, but not thigmotaxis, an anxiety-related behavior in an open field155; social interaction was not tested. Tbx1 heterozygosity results in defective reciprocal social interaction156, functionally defective neonatal social communication138, reduced working memory capacity156 and heightened thigmotaxis156. Sept5 deficiency impairs social interaction141 but has no effect on PPI or thigmotaxis141,152.
Mutant mice of other genes have been tested in various tasks listed, but no statistically significant phenotypes have been found when genetic background is controlled (Fig. 2). These genes include: Dgcr2 for aggression and adult vocalization152; Rtn4r for aggression and adult vocalization152; Ranbp1 for PPI, aggression and adult vocalization152; Arvcf for PPI and adult vocalization152; Comt for PPI, aggression, adult vocalization, social behaviors and anxiety-related behaviors152,157–160; Gp1bb for PPI, aggression and adult vocalization152; and Hira for adult vocalization152.
The presence and absence of behavioral phenotypes in these mutant mice provides a partial glimpse of how 22q11.2 CNV-encoded genes contribute to the overall behavioral profile. First, not all CNV-encoded genes are responsible for a given behavioral construct (i.e., non-contiguous gene effect)10–12. Out of seven genes tested for PPI, only Dgcr8 deficiency results in a deficit (see Fig. 2). This does not necessarily mean that this is the only 22q11.2 gene that contributes to PPI, however. As more genes are tested under rigorous experimental conditions, more are likely to be found to be required for normal expression of PPI. Out of eleven 22q11.2 genes tested for some aspects of social behaviors, the contribution of Prodh, Tbx1 and Sept5, but not other 8 genes, has been demonstrated.
The fact that many genes influence a specific dimensional aspect of mental illnesses also indicates that collective actions of more than one CNV-encoded gene determine the net behavioral phenotype (i.e., mass action)10–12. Various aspects of social behavior are reduced by the deletion of Prodh, Tbx1 and Sept5 (see Fig. 2). Moreover, those genes with no phenotypic consequence by themselves alone might cause a phenotype with other encoded genes (i.e., epistasis).
Further complicating the matter, mass action does not necessarily mean all genes involved act in the same direction. Some genes cause an antagonistic effect (i.e., antagonistic gene effect)10–12. Deletion of Comt improves working memory (see Fig. 2), although 22q11.2 hemizygous carriers are impaired in working memory. There might be more CNV-encoded genes that exert such antagonistic actions that are hidden against the mass action of other genes.
The non-contiguous gene and antagonistic actions pose a potential interpretative caveat of analysis for CNV models. Even if a CNV-encoded gene has no role or an antagonistic role in a particular behavioral phenotype, almost any gene within a CNV is expected to be involved in some molecular, cellular and neuronal functions, thereby causing some phenotypes in the brain without causing a behavioral phenotype. In such a case, that neuronal phenotype is an epi-phenomenon for a behavioral phenotype (i.e., epi-phenotype, see Fig. 3, C-c-1). Such a neuronal phenotype nonetheless could be a genuine neuronal phenotype of a different behavioral phenotype that has not been explored, however (i.e., misguided focus on a behavioral phenotype, Fig. 3, C-c-2). Such behavioral phenotypes might be relevant or irrelevant to dimensional aspects of mental disorders.
Figure 3.
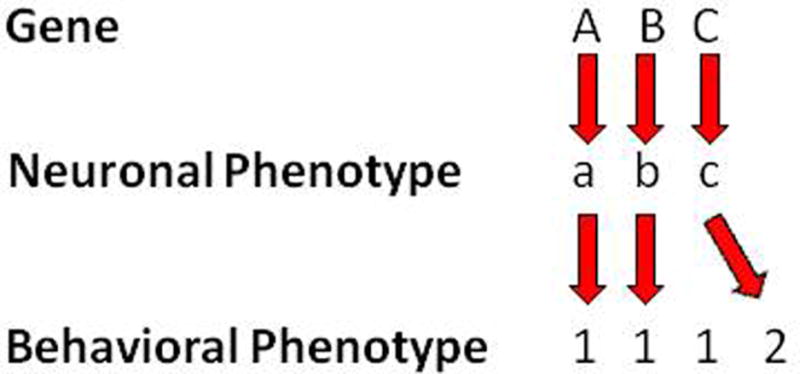
Hypothetical links between genes, neuronal phenotypes and behavioral phenotypes of a CNV. Hypothetical genes and neuronal phenotypes are indicated as letters in upper and lower cases, respectively. Behavioral phenotypes are indicated as numbers. The causal link is indicated by red arrows.
Neuronal phenotypes examined are often based on the consensus that they are relevant to mental disorders. However, there might be other neuronal phenotypes that have not been found yet which genuinely or more robustly contribute to a behavioral phenotype (i.e., unexplored neuronal phenotypes).
Existing primary evidence suggests that neuronal phenotypes caused by gene dose reductions of Prodh, Dgcr8, COMT, Tbx1 and Sept5 contribute to various behavioral constructs of mental illnesses (see Fig. 2). The brain functions of these genes suggest several potential neuronal phenotypes relevant to the behavioral phenotypes of their single gene mutant mice. Prodh is critical for mitochondrial respiration161 and dopaminergic and GABArgic transmission and gamma oscillation153,162. Dgcr8 deficiency alters embryonic neurogenesis163,164 and adult neurogenesis155 in mice. Moreover, neuronal proliferation, neural differentiation efficiency, neurite outgrowth, cellular migration and neurogenic-to-gliogenic competence ratio are reduced in cells derived from individuals with 22q11.2 deletion and schizophrenia diagnosis;altered neurogenesis and gliogenesis are partially restored by modulating a molecular target of Dgcr8165. Tbx1 expression is enriched in postnatal and adult neural progenitor cells107,156. When Tbx1 or Comt is over-expressed in adult neural progenitor cells in the hippocampal granule cell layer, the developmental maturation of working memory and the migration of newly generated neurons are eqaully blunted107. These observations suggest novel hypothetical genetic and cellular mechanisms for cognitive deficits seen in individuals with 22q11.2 duplication and triplication. Sept5 is functionally involved in cell death and synaptic transmission166–171. These neuronal phenotypes remain candidate mechanisms for specific dimensions of 22q11.2 CNV-associated psychiatric disorders.
Neuronal phenotypes of CNV mouse models
As some neuronal phenotypes are not technically tractable in humans, mouse models provide a complementary means of study. Neuronal phenotypes, including cellular and molecular phenotypes, detected in non-congenic mouse models of CNVs are not interpretable in terms of the impact of a CNV (see Mouse models of CNVs). Here I discuss primary evidence of mouse CNV models whose genetic background is controlled.
Some anatomic phenotypes are similarly observed in humans with many CNVs and their mouse models, but others might be distinct to some CNV cases. Altered tissue volumes are seen in the hippocampus CA3 of a mouse model of 7q11.23 deletion116, granular layer of the hippocampus and other regions of a mouse model of 15q11–13 duplication172, and cortex and other regions of a mouse model of 22q11.2 deletion173. Consistent with these anatomic features, humans with 7q11.23 deletion show decreased brain size, in particular, in gray matter density in the superior parietal lobe areas174 or altered shape, but not size, of the hippocampus175. Global brain volume reductions have been observed in various brain regions176–179 as well as increases in the corpus callosum180,181 of 22q11.2 hemizygous deletion carriers; however, the corpus callous volume is smaller among 22q11 deletion carriers with ADHD than without ADHD181. Heterotopia, dysplasias or malformations of the hippocampus in the alveus, CA4, and dentate gyrus and dysplasia in the dentate gyrus were detected in carriers of 15q11–13 duplication and individuals with autism diagnosis and hypoplasia of the corpus callosum182,183.
A mouse model of 7q11.23 deletion shows a volume reduction in CA3, increased immature neurons in the subgranular zone of the hippocampal dentate gyrus and shorter dendrites reduced and spine density in the hippocampus, and reduced cell density in the amygdala, and a reduction in pyramidal cells in the cortex and CA1116. A congenic mouse model of 22q11.2 deletion has deficits in migration of cortical interneurons and hippocampal dentate precursor cells during the embryonic period164. The rate of spine formation and elimination is high, short-term depression in the prelimbic prefrontal region is greater, short-term potentiation and long-term potentiation are lower in a congenic mouse model of 22q11.2163. An enhanced turnover of spines, but not spine density, is found in the cortical regions of a 15q11–13 duplication model184.
A co-isogenic mouse model of 15q13.3 deletion exhibits a reduction in recruitment of neurons to fast steady-state gamma oscillatory activity and in the probability of interneurons to fire with low inter-spike intervals in response to auditory stimulation185. This mouse model also shows a reduction in the baseline firing rate and amplitude of fast-spiking PFC interneurons, but not pyramidal neurons and a delay in the onset of firing of both PFC pyramidal neurons and interneurons186.
A mouse model of 16p11.2 deletion exhibits normal synaptic transmission, normal paired pulse facilitation, but decreased CA1 long-term potentiation132.
Correlation between neuronal and behavioral phenotypes
Correlation needs to be first established between neuronal and behavioral phenotypes in a CNV model. However, this falls short of proof of casual relation between the two, given the possibility that some neuronal phenotypes might not be causally linked to a given behavioral phenotype (see Fig. 3, C-c-1) or act antagonistically against a behavioral phenotype (see Fig. 2, Comt, working memory).
Some neuronal and behavioral phenotypes co-exist, but others do not. Deficits in behavioral reversal are present together with reduced mature neurons in cortical 2/3 layers115,163 and reductions in parvalbumin interneurons in deep cortical layers of the medial anterior frontal cortex as well as in parvalbumin-positive perisomatic synaptic terminals onto projection neurons in lower layers in a congenic mouse model of 22q11.2 deletion187.
A co-isogenic mouse model of 22q11.2 deletion is impaired in PPI and shows increases in 3,4-Dihydroxyphenylacetic acid (DOPAC) in the prefrontal cortex and striatum and increased expression of GluR1 in the dorsal striatum56. However, this mouse model is normal in cortical layer composition, hippocampal structures, parvalbumin interneurons, myelination, levels of synaptic proteins (PSD-95, Syp, Syn1, Drebrin and geophysin), neurons and astrocytes (NeuN and GFAP), GABA (GABAA1, KCC2, VGAT, GAD65/67), glutamate (GluR2, NR2A, NR2B, VGluT1), dopamine, 5-HT and noradrenaline contents, baseline cortical low frequency oscillation and baseline spike frequency in the prefrontal cortex and GAD, thereby not lending strong support for a causal link between these latter neuronal phenotypes and PPI deficits56.
Although basal excitation and inhibition (E/I) balance is normal in the prefrontal cortex of a mouse model of 22q11.2 deletion, the E/I balance is more easily perturbed by dopaminergic stimulation in layer 5 prefrontal pyramidal neurons in this mouse model188. This perturbation depends on a potentiated excitation by D1R activation and reduced inhibition by D2R cells due to less effective GABAergic parvalbumin interneurons188. Low frequency oscillation in the cortex and elevation of pyramidal neuron spike frequency are normal in a co-isogenic 22q11.2 mouse model56.
A congenic mouse model of 22q11.2 deletion exhibits defective neural synchronization during pre-learning, training (acquisition) and maintenance of working memory110. Hippocampal-prefrontal synchrony, such as phase locking and theta coherence, is heightened when working memory demand is high. Baseline coherence in the delta (2–3 Hz), but not theta or gamma, range was lower before working memory training acquisition in mutant mice, compared to wild-type mice. Although mutant and wild-type mice were statistically indistinguishable in the speed to reach a criterion of working memory, individual mutant mice with low baseline coherence levels before acquisition took longer to reach a working memory criterion during training; such correlation was absent in wild-type mice. A gradual increase in theta coherence from the early to late stages of working memory training was correlated with the speed at which mice increased choice accuracy and mutant mice were slower in both the coherence increase and choice accuracy, compared to wild-type mice. After mutant and wild-type mice were trained to a level where their performance is indistinguishable, the strength of theta rhythm phase-locking was weaker and coherence was lower in the delta (1–4 Hz) and low theta (4–6 Hz), but not high theta (7–10 Hz) or gamma (30–80 Hz) range in mutant mice, compared to wild-type littermates. However, these neuronal phenotypes appeared in the absence of statistically significant deficits in the maintenance of working memory in mutant mice. The apparent dissociation might be due to a small sample size tested, as suggested by the authors110. Alternatively, basal delta coherence before training and a gradual increase in theta coherence during working memory training might be relevant to the pre-learning strategy and rapid acquisition of working memory, respectively, but not performance of working memory.
These studies provide many potential neuronal mechanisms underlying dimensional features of neuropsychiatric disorders. The challenge is to more tightly establish the causal link between these impressive neuronal phenotypes and precise behavioral phenotypes. The mere presence of a neuronal phenotype in a CNV mouse model does not automatically identify its causative behavioral targets. CNV models inherently suffer from a limitation in causally linking a neuronal phenotype and behavioral constructs of neuropsychiatric disorders because most CNV-encoded individual genes alter neuronal phenotypes but do not necessarily contribute to a specific behavioral phenotype (see Fig. 3, C-c-1) or some individual genes act antagonistically against a net behavioral phenotype (see Fig. 2, Comt deletion for working memory).
In this regard, some recent elegant studies have attempted to identify a tighter causal link between a neuronal phenotype and defective prepulse inhibition. A 22q11 deletion mouse model has a disrupted synaptic transmission at thalamocortical glutamatergic projections in the auditory cortex and defective prepulse inhibition due to elevation of D2 receptors in the thalamus; this D2R elevation is mediated by defective processing of miR-338–3p by Dgcr8, a 22q11.2-encoed gene55,154. These studies manipulate intermediate variables to predict the outcome, achieving a tighter degree of causative link between neuronal and behavioral phenotypes.
Concluding Remarks
Mouse models of CNVs have revealed anatomic and electrophysiologic phenotypes. One interpretative caveat is that any phenotype seen in the brains of CNV mouse models cannot be automatically taken as mechanistic bases of dimensions of mental disorders, because of the many interpretative pitfalls. The presence of behavioral and brain phenotypes is correlative, but not causative, just as these phenotypes are simply correlative in human subjects as well. Mouse models of single genes encoded in a CNV are being developed and tested to address this issue. As manipulation of gene doses in the living mouse brain and prediction of neuronal and behavioral phenotypes are technically feasible, mouse models provide a unique means to determine the causative link among genes, neuronal and behavioral phenotypes of CNVs.
A future challenge of mouse model analyses is to mechanistically deconstruct and reconstruct the causative genetic and neuronal mechanisms underlying behavioral dimensions of CNVs. Such efforts will not only provide hypothetical mechanistic bases for development of novel therapeutic options for mental illnesses, but also provide novel understanding of mental illnesses according to the RDoc criteria. It should be emphasized, however, that many brain structures and functions of a mouse might not be as developed as those of humans. Thus, there is an inherent limitation in the translational value of mouse models. The ultimate validation of hypothetical mechanisms found in mouse models depends on the effectiveness of therapeutic options, developed from mouse models, for human psychiatric disorders.
Acknowledgments
Research reported in this publication was supported by the National Institute of Mental Health of the National Institutes of Health under Award Numbers R01MH099660, R01DC015776, R21HD053114 and U54HD090260. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
I thank Dr. Yann Herault and Dr. Binnaz Yalcin of the IBGMC, France and members of my laboratory for insightful discussion.
Disclosure statement. I report a grant from Astellas.
Footnotes
Author Contributions. NH contributed to all aspects of the manuscript preparation.
References
- 1.Cnv, Schizophrenia Working Groups of the Psychiatric Genomics, C. & Psychosis Endophenotypes International, C. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49:27–35. doi: 10.1038/ng.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mosca SJ, et al. Copy-number variations are enriched for neurodevelopmental genes in children with developmental coordination disorder. J Med Genet. 2016;53:812–819. doi: 10.1136/jmedgenet-2016-103818. [DOI] [PubMed] [Google Scholar]
- 4.Girirajan S, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. 2012;367:1321–1331. doi: 10.1056/NEJMoa1200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Girirajan S, et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaugler T, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46:881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klei L, et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism. 2012;3:9. doi: 10.1186/2040-2392-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cross-Disorder Group of the Psychiatric Genomics, C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hiroi N, Hiramoto T, Harper KM, Suzuki G, Boku S. Mouse models of 22q11.2-associated autism spectrum disorder. Autism. 2012;S1:1–9. doi: 10.4172/2165-7890.S1-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiroi N, Nishi A. In: Modeling the Psychopathological Dimensions of Schizophrenia: From Molecules to Behavior Vol. 23 Handbook of Behavioral Neuroscience. Pletnikov MV, Waddington JL, editors. 2015. pp. 285–302. Ch. 17. [Google Scholar]
- 11.Hiroi N, et al. Copy Number Variation at 22q11.2: from rare variants to common mechanisms of developmental neuropsychiatric disorders. Mol Psychiatry. 2013;18:1153–1165. doi: 10.1038/mp.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishi A, Hiroi N. In: The Neurobiology of Schizophrenia. Abel T, Nickl-Jockschat T, editors. Academic Press/Elsevier; 2016. pp. 397–417. Ch 22. [Google Scholar]
- 13.Esposito G, Hiroi N, Scattoni ML. Cry, baby, cry: Expression of Distress as a Biomarker and Modulator in Autism Spectrum Disorder. Int J Neuropsychopharmacol. 2017 doi: 10.1093/ijnp/pyx014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kikusui T, Hiroi NA. Self-Generated Environmental Factor as a Potential Contributor to Atypical Early Social Communication in Autism. Neuropsychopharmacology. 2017;42:378. doi: 10.1038/npp.2016.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown AS, McGrath JJ. The prevention of schizophrenia. Schizophr Bull. 2011;37:257–261. doi: 10.1093/schbul/sbq122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim YS, Leventhal BL. Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol Psychiatry. 2015;77:66–74. doi: 10.1016/j.biopsych.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Insel T, et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167:748–751. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- 18.Morris SE, Cuthbert BN. Research Domain Criteria: cognitive systems, neural circuits, and dimensions of behavior. Dialogues Clin Neurosci. 2012;14:29–37. doi: 10.31887/DCNS.2012.14.1/smorris. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly MA, et al. Locomotor activity in D2 dopamine receptor-deficient mice is determined by gene dosage, genetic background, and developmental adaptations. J Neurosci. 1998;18:3470–3479. doi: 10.1523/JNEUROSCI.18-09-03470.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crusio WE. Flanking gene and genetic background problems in genetically manipulated mice. Biol Psychiatry. 2004;56:381–385. doi: 10.1016/j.biopsych.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 21.Flaherty L, Bolivar V. In: Neurobehavioral Genetics. Jones BC, Mormede P, editors. Taylor & Francis; 2007. pp. 115–127. Ch 8. [Google Scholar]
- 22.Gerlai R. Gene targeting: technical confounds and potential solutions in behavioral brain research. Behav Brain Res. 2001;125:13–21. doi: 10.1016/s0166-4328(01)00282-0. [DOI] [PubMed] [Google Scholar]
- 23.Wolfer DP, Crusio WE, Lipp HP. Knockout mice: simple solutions to the problems of genetic background and flanking genes. Trends Neurosci. 2002;25:336–340. doi: 10.1016/s0166-2236(02)02192-6. [DOI] [PubMed] [Google Scholar]
- 24.Kempermann G, Gage FH. Genetic influence on phenotypic differentiation in adult hippocampal neurogenesis. Brain Res Dev Brain Res. 2002;134:1–12. doi: 10.1016/s0165-3806(01)00224-3. [DOI] [PubMed] [Google Scholar]
- 25.Zilles K, Wu J, Crusio WE, Schwegler H. Water maze and radial maze learning and the density of binding sites of glutamate, GABA, and serotonin receptors in the hippocampus of inbred mouse strains. Hippocampus. 10:213–225. doi: 10.1002/1098-1063(2000)10:3<213::AID-HIPO2>3.0.CO;2-Q(2000). [DOI] [PubMed] [Google Scholar]
- 26.Crawley JN, et al. Behavioral phenotypes of inbred mouse strains: implications and recommendations for molecular studies. Psychopharmacology (Berl) 1997;132:107–124. doi: 10.1007/s002130050327. [DOI] [PubMed] [Google Scholar]
- 27.Faure A, et al. Social behaviors and acoustic vocalizations in different strains of mice. Behav Brain Res. 2017;320:383–390. doi: 10.1016/j.bbr.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 28.Scattoni ML, Gandhy SU, Ricceri L, Crawley JN. Unusual repertoire of vocalizations in the BTBR T+tf/J mouse model of autism. PLoS One. 2008;3:e3067. doi: 10.1371/journal.pone.0003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scattoni ML, Ricceri L, Crawley JN. Unusual repertoire of vocalizations in adult BTBR T+tf/J mice during three types of social encounters. Genes Brain Behav. 2011;10:44–56. doi: 10.1111/j.1601-183X.2010.00623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papandrea D, Anderson TM, Herron BJ, Ferland RJ. Dissociation of seizure traits in inbred strains of mice using the flurothyl kindling model of epileptogenesis. Exp Neurol. 2009;215:60–68. doi: 10.1016/j.expneurol.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen XJ, et al. Neuroanatomical differences between mouse strains as shown by high-resolution 3D MRI. Neuroimage. 2006;29:99–105. doi: 10.1016/j.neuroimage.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 32.Wahlsten D, Metten P, Crabbe JC. Survey of 21 inbred mouse strains in two laboratories reveals that BTBR T/+ tf/tf has severely reduced hippocampal commissure and absent corpus callosum. Brain Res. 2003;971:47–54. doi: 10.1016/s0006-8993(03)02354-0. [DOI] [PubMed] [Google Scholar]
- 33.Routh BN, Johnston D, Harris K, Chitwood RA. Anatomical and electrophysiological comparison of CA1 pyramidal neurons of the rat and mouse. J Neurophysiol. 2009;102:2288–2302. doi: 10.1152/jn.00082.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwegler H, et al. Genetic variation in the morphology of the septo-hippocampal cholinergic and GABAergic systems in mice: II. Morpho-behavioral correlations. Hippocampus. 1996;6:535–545. doi: 10.1002/(SICI)1098-1063(1996)6:5<535::AID-HIPO6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 35.Schwegler H, et al. Genetic variation in the morphology of the septo-hippocampal cholinergic and GABAergic system in mice. I. Cholinergic and GABAergic markers. Hippocampus. 1996;6:136–148. doi: 10.1002/(SICI)1098-1063(1996)6:2<136::AID-HIPO5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 36.Yilmazer-Hanke DM, Roskoden T, Zilles K, Schwegler H. Anxiety-related behavior and densities of glutamate, GABAA, acetylcholine and serotonin receptors in the amygdala of seven inbred mouse strains. Behav Brain Res. 2003;145:145–159. doi: 10.1016/s0166-4328(03)00107-4. [DOI] [PubMed] [Google Scholar]
- 37.Restivo L, Roman FS, Ammassari-Teule M, Marchetti E. Simultaneous olfactory discrimination elicits a strain-specific increase in dendritic spines in the hippocampus of inbred mice. Hippocampus. 2006;16:472–479. doi: 10.1002/hipo.20174. [DOI] [PubMed] [Google Scholar]
- 38.Lee AW, Emsley JG, Brown RE, Hagg T. Marked differences in olfactory sensitivity and apparent speed of forebrain neuroblast migration in three inbred strains of mice. Neuroscience. 2003;118:263–270. doi: 10.1016/s0306-4522(02)00950-8. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen PV, Abel T, Kandel ER, Bourtchouladze R. Strain-dependent differences in LTP and hippocampus-dependent memory in inbred mice. Learn Mem. 2000;7:170–179. doi: 10.1101/lm.7.3.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen PV, Duffy SN, Young JZ. Differential maintenance and frequency-dependent tuning of LTP at hippocampal synapses of specific strains of inbred mice. J Neurophysiol. 2000;84:2484–2493. doi: 10.1152/jn.2000.84.5.2484. [DOI] [PubMed] [Google Scholar]
- 41.Moore SJ, Throesch BT, Murphy GG. Of mice and intrinsic excitability: genetic background affects the size of the postburst afterhyperpolarization in CA1 pyramidal neurons. J Neurophysiol. 2011;106:1570–1580. doi: 10.1152/jn.00257.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valor LM, Grant SG. Clustered gene expression changes flank targeted gene loci in knockout mice. PLoS One. 2007;2:e1303. doi: 10.1371/journal.pone.0001303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang S, et al. Biochemical, molecular and behavioral phenotypes of Rab3A mutations in the mouse. Genes Brain Behav. 2007;6:77–96. doi: 10.1111/j.1601-183X.2006.00235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ricard G, et al. Phenotypic consequences of copy number variation: insights from Smith-Magenis and Potocki-Lupski syndrome mouse models. PLoS Biol. 2010;8:e1000543. doi: 10.1371/journal.pbio.1000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Leary J, Osborne LR. Global analysis of gene expression in the developing brain of Gtf2ird1 knockout mice. PLoS One. 2011;6:e23868. doi: 10.1371/journal.pone.0023868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geyer MA. The family of sensorimotor gating disorders: comorbidities or diagnostic overlaps? Neurotox Res. 2006;10:211–220. doi: 10.1007/BF03033358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kohl S, et al. Prepulse inhibition of the acoustic startle reflex in high functioning autism. PLoS One. 2014;9:e92372. doi: 10.1371/journal.pone.0092372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McAlonan GM, et al. Brain anatomy and sensorimotor gating in Asperger’s syndrome. Brain. 2002;125:1594–1606. doi: 10.1093/brain/awf150. [DOI] [PubMed] [Google Scholar]
- 49.Madsen GF, Bilenberg N, Cantio C, Oranje B. Increased prepulse inhibition and sensitization of the startle reflex in autistic children. Autism Res. 2014;7:94–103. doi: 10.1002/aur.1337. [DOI] [PubMed] [Google Scholar]
- 50.Oranje B, Lahuis B, van Engeland H, Jan van der Gaag R, Kemner C. Sensory and sensorimotor gating in children with multiple complex developmental disorders (MCDD) and autism. Psychiatry Res. 2013;206:287–292. doi: 10.1016/j.psychres.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 51.Takahashi H, Nakahachi T, Stickley A, Ishitobi M, Kamio Y. Stability of the acoustic startle response and its modulation in children with typical development and those with autism spectrum disorders: A one-year follow-up. Autism Res. 2017;10:673–679. doi: 10.1002/aur.1710. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi H, Komatsu S, Nakahachi T, Ogino K, Kamio Y. Relationship of the Acoustic Startle Response and Its Modulation to Emotional and Behavioral Problems in Typical Development Children and Those with Autism Spectrum Disorders. J Autism Dev Disord. 2016;46:534–543. doi: 10.1007/s10803-015-2593-4. [DOI] [PubMed] [Google Scholar]
- 53.Ornitz EM, Lane SJ, Sugiyama T, d J. Startle modulation studies in autism. J Autism Dev Disord. 1993;23:619–637. doi: 10.1007/BF01046105. [DOI] [PubMed] [Google Scholar]
- 54.Yuhas J, et al. Brief Report: Sensorimotor Gating in Idiopathic Autism and Autism Associated with Fragile X Syndrome. J Autism Dev Disord. 2010;41:248–253. doi: 10.1007/s10803-010-1040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chun S, et al. Specific disruption of thalamic inputs to the auditory cortex in schizophrenia models. Science. 2014;344:1178–1182. doi: 10.1126/science.1253895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Didriksen M, et al. Persistent gating deficit and increased sensitivity to NMDA receptor antagonism after puberty in a new mouse model of the human 22q11.2 microdeletion syndrome: a study in male mice. J Psychiatry Neurosci. 2016;41:150381. doi: 10.1503/jpn.150381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sobin C, et al. Neuropsychological characteristics of children with the 22q11 Deletion Syndrome: a descriptive analysis. Child Neuropsychol. 2005;11:39–53. doi: 10.1080/09297040590911167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suzuki G, et al. Over-expression of a human chromosome 22q11.2 segment including TXNRD2, COMT and ARVCF developmentally affects incentive learning and working memory in mice. Human Molecular Genetics. 2009;18:3914–3925. doi: 10.1093/hmg/ddp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rees E, et al. Evidence that duplications of 22q11.2 protect against schizophrenia. Mol Psychiatry. 2014;19:37–40. doi: 10.1038/mp.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakatani J, et al. Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell. 2009;137:1235–1246. doi: 10.1016/j.cell.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tamada K, et al. Decreased exploratory activity in a mouse model of 15q duplication syndrome; implications for disturbance of serotonin signaling. PLoS One. 2010;5:e15126. doi: 10.1371/journal.pone.0015126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fejgin K, et al. A Mouse Model that Recapitulates Cardinal Features of the 15q13.3 Microdeletion Syndrome Including Schizophrenia- and Epilepsy-Related Alterations. Biol Psychiatry. 2013;76:128–137. doi: 10.1016/j.biopsych.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 63.Forsingdal A, Fejgin K, Nielsen V, Werge T, Nielsen J. 15q13.3 homozygous knockout mouse model display epilepsy-, autism- and schizophrenia-related phenotypes. Transl Psychiatry. 2016;6:e860. doi: 10.1038/tp.2016.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Luca CR, et al. Normative data from the CANTAB. I: development of executive function over the lifespan. J Clin Exp Neuropsychol. 2003;25:242–254. doi: 10.1076/jcen.25.2.242.13639. [DOI] [PubMed] [Google Scholar]
- 65.Gur RC, et al. Age group and sex differences in performance on a computerized neurocognitive battery in children age 8-21. Neuropsychology. 2012;26:251–265. doi: 10.1037/a0026712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luna B. Developmental changes in cognitive control through adolescence. Adv Child Dev Behav. 2009;37:233–278. doi: 10.1016/s0065-2407(09)03706-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gathercole SE, Pickering SJ, Ambridge B, Wearing H. The structure of working memory from 4 to 15 years of age. Dev Psychol. 2004;40:177–190. doi: 10.1037/0012-1649.40.2.177. [DOI] [PubMed] [Google Scholar]
- 68.Luna B, Doll SK, Hegedus SJ, Minshew NJ, Sweeney JA. Maturation of executive function in autism. Biol Psychiatry. 2007;61:474–481. doi: 10.1016/j.biopsych.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 69.Rosenthal M, et al. Impairments in real-world executive function increase from childhood to adolescence in autism spectrum disorders. Neuropsychology. 2013;27:13–18. doi: 10.1037/a0031299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O’Hearn K, Asato M, Ordaz S, Luna B. Neurodevelopment and executive function in autism. Dev Psychopathol. 2008;20:1103–1132. doi: 10.1017/S0954579408000527. [DOI] [PubMed] [Google Scholar]
- 71.Bennett E, Heaton P. Is talent in autism spectrum disorders associated with a specific cognitive and behavioural phenotype? J Autism Dev Disord. 2012;42:2739–2753. doi: 10.1007/s10803-012-1533-9. [DOI] [PubMed] [Google Scholar]
- 72.Pennington BF, Ozonoff S. Executive functions and developmental psychopathology. J Child Psychol Psychiatry. 1996;37:51–87. doi: 10.1111/j.1469-7610.1996.tb01380.x. [DOI] [PubMed] [Google Scholar]
- 73.Russo N, et al. Deconstructing executive deficits among persons with autism: implications for cognitive neuroscience. Brain Cogn. 2007;65:77–86. doi: 10.1016/j.bandc.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 74.Goldenberg PC, et al. Computerized neurocognitive profile in young people with 22q11.2 deletion syndrome compared to youths with schizophrenia and at-risk for psychosis. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:87–93. doi: 10.1002/ajmg.b.32005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saykin AJ, et al. Neuropsychological function in schizophrenia. Selective impairment in memory and learning. Arch Gen Psychiatry. 1991;48:618–624. doi: 10.1001/archpsyc.1991.01810310036007. [DOI] [PubMed] [Google Scholar]
- 76.Schaefer J, Giangrande E, Weinberger DR, Dickinson D. The global cognitive impairment in schizophrenia: consistent over decades and around the world. Schizophr Res. 2013;150:42–50. doi: 10.1016/j.schres.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fioravanti M, Carlone O, Vitale B, Cinti ME, Clare L. A meta-analysis of cognitive deficits in adults with a diagnosis of schizophrenia. Neuropsychol Rev. 2005;15:73–95. doi: 10.1007/s11065-005-6254-9. [DOI] [PubMed] [Google Scholar]
- 78.Piskulic D, Olver JS, Norman TR, Maruff P. Behavioural studies of spatial working memory dysfunction in schizophrenia: a quantitative literature review. Psychiatry Res. 2007;150:111–121. doi: 10.1016/j.psychres.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 79.Gur RC, et al. Neurocognitive growth charting in psychosis spectrum youths. JAMA Psychiatry. 2014;71:366–374. doi: 10.1001/jamapsychiatry.2013.4190. [DOI] [PubMed] [Google Scholar]
- 80.Woodberry KA, Giuliano AJ, Seidman LJ. Premorbid IQ in schizophrenia: a meta-analytic review. Am J Psychiatry. 2008;165:579–587. doi: 10.1176/appi.ajp.2008.07081242. [DOI] [PubMed] [Google Scholar]
- 81.David AS, Malmberg A, Brandt L, Allebeck P, Lewis G. IQ and risk for schizophrenia: a population-based cohort study. Psychol Med. 1997;27:1311–1323. doi: 10.1017/s0033291797005680. [DOI] [PubMed] [Google Scholar]
- 82.Davidson M, et al. Behavioral and intellectual markers for schizophrenia in apparently healthy male adolescents. Am J Psychiatry. 1999;156:1328–1335. doi: 10.1176/ajp.156.9.1328. [DOI] [PubMed] [Google Scholar]
- 83.Fuller R, et al. Longitudinal assessment of premorbid cognitive functioning in patients with schizophrenia through examination of standardized scholastic test performance. Am J Psychiatry. 2002;159:1183–1189. doi: 10.1176/appi.ajp.159.7.1183. [DOI] [PubMed] [Google Scholar]
- 84.Reichenberg A, et al. Static and dynamic cognitive deficits in childhood preceding adult schizophrenia: a 30-year study. Am J Psychiatry. 2010;167:160–169. doi: 10.1176/appi.ajp.2009.09040574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meier MH, et al. Neuropsychological decline in schizophrenia from the premorbid to the postonset period: evidence from a population-representative longitudinal study. Am J Psychiatry. 2014;171:91–101. doi: 10.1176/appi.ajp.2013.12111438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bora E, et al. Cognitive deficits in youth with familial and clinical high risk to psychosis: a systematic review and meta-analysis. Acta Psychiatr Scand. 2014;130:1–15. doi: 10.1111/acps.12261. [DOI] [PubMed] [Google Scholar]
- 87.Bora E. Developmental trajectory of cognitive impairment in bipolar disorder: comparison with schizophrenia. Eur Neuropsychopharmacol. 2015;25:158–168. doi: 10.1016/j.euroneuro.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 88.Vorstman JA, et al. Cognitive Decline Preceding the Onset of Psychosis in Patients With 22q11.2 Deletion Syndrome. JAMA Psychiatry. 2015 doi: 10.1001/jamapsychiatry.2014.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kates WR, et al. White matter microstructural abnormalities of the cingulum bundle in youths with 22q11.2 deletion syndrome: associations with medication, neuropsychological function, and prodromal symptoms of psychosis. Schizophr Res. 2015;161:76–84. doi: 10.1016/j.schres.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Duijff SN, et al. Cognitive development in children with 22q11.2 deletion syndrome. Br J Psychiatry. 2012;200:462–468. doi: 10.1192/bjp.bp.111.097139. [DOI] [PubMed] [Google Scholar]
- 91.Gur RE, et al. Neurocognitive development in 22q11.2 deletion syndrome: comparison with youth having developmental delay and medical comorbidities. Mol Psychiatry. 2014;19:1205–1211. doi: 10.1038/mp.2013.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gothelf D, et al. COMT genotype predicts longitudinal cognitive decline and psychosis in 22q11.2 deletion syndrome. Nat Neurosci. 2005;8:1500–1502. doi: 10.1038/nn1572. [DOI] [PubMed] [Google Scholar]
- 93.Gothelf D, et al. Risk factors and the evolution of psychosis in 22q11.2 deletion syndrome: a longitudinal 2-site study. J Am Acad Child Adolesc Psychiatry. 2013;52:1192–1203. e1193. doi: 10.1016/j.jaac.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 94.Ackerman PL, Beier ME, Boyle MO. Working memory and intelligence: the same or different constructs? Psychol Bull. 2005;131:30–60. doi: 10.1037/0033-2909.131.1.30. [DOI] [PubMed] [Google Scholar]
- 95.Conway AR, Kane MJ, Engle RW. Working memory capacity and its relation to general intelligence. Trends Cogn Sci. 2003;7:547–552. doi: 10.1016/j.tics.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 96.Friedman NP, et al. Not all executive functions are related to intelligence. Psychol Sci. 2006;17:172–179. doi: 10.1111/j.1467-9280.2006.01681.x. [DOI] [PubMed] [Google Scholar]
- 97.Danielsson H, Henry L, Ronnberg J, Nilsson LG. Executive functions in individuals with intellectual disability. Res Dev Disabil. 2010;31:1299–1304. doi: 10.1016/j.ridd.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 98.Danielsson H, Henry L, Messer D, Ronnberg J. Strengths and weaknesses in executive functioning in children with intellectual disability. Res Dev Disabil. 2012;33:600–607. doi: 10.1016/j.ridd.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 99.Schuchardt K, Gebhardt M, Maehler C. Working memory functions in children with different degrees of intellectual disability. J Intellect Disabil Res. 2010;54:346–353. doi: 10.1111/j.1365-2788.2010.01265.x. [DOI] [PubMed] [Google Scholar]
- 100.Van der Molen MJ, Van Luit JE, Jongmans MJ, Van der Molen MW. Memory profiles in children with mild intellectual disabilities: strengths and weaknesses. Res Dev Disabil. 2009;30:1237–1247. doi: 10.1016/j.ridd.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 101.Westerberg H, Hirvikoski T, Forssberg H, Klingberg T. Visuo-spatial working memory span: a sensitive measure of cognitive deficits in children with ADHD. Child Neuropsychol. 2004;10:155–161. doi: 10.1080/09297040409609806. [DOI] [PubMed] [Google Scholar]
- 102.Castellanos FX, Tannock R. Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes. Nat Rev Neurosci. 2002;3:617–628. doi: 10.1038/nrn896. [DOI] [PubMed] [Google Scholar]
- 103.Martinussen R, Hayden J, Hogg-Johnson S, Tannock R. A meta-analysis of working memory impairments in children with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2005;44:377–384. doi: 10.1097/01.chi.0000153228.72591.73. [DOI] [PubMed] [Google Scholar]
- 104.Willcutt EG, Doyle AE, Nigg JT, Faraone SV, Pennington BF. Validity of the executive function theory of attention-deficit/hyperactivity disorder: a meta-analytic review. Biol Psychiatry. 2005;57:1336–1346. doi: 10.1016/j.biopsych.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 105.Nilsson SR, et al. Assessing the Cognitive Translational Potential of a Mouse Model of the 22q11.2 Microdeletion Syndrome. Cereb Cortex. 2016;26:3991–4003. doi: 10.1093/cercor/bhw229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Flurkey K, Currrer JM, Harrison DE. In: The Mouse in Biomedical Research. Fox JG, editor. Vol. 3. Elsevier; 2007. pp. 637–672. Ch. 20. [Google Scholar]
- 107.Boku S, et al. Copy number elevation of 22q11.2 genes arrests the developmental maturation of working memory capacity and adult neurogenesis. Molecular Psychiatry. 2017 doi: 10.1038/mp.2017.158. Online version published Aug 22, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dumontheil I, et al. Influence of the COMT genotype on working memory and brain activity changes during development. Biol Psychiatry. 2011;70:222–229. doi: 10.1016/j.biopsych.2011.02.027. [DOI] [PubMed] [Google Scholar]
- 109.Kogan JH, et al. Mouse Model of Chromosome 15q13.3 Microdeletion Syndrome Demonstrates Features Related to Autism Spectrum Disorder. J Neurosci. 2015;35:16282–16294. doi: 10.1523/JNEUROSCI.3967-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sigurdsson T, Stark KL, Karayiorgou M, Gogos JA, Gordon JA. Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature. 2010;464:763–767. doi: 10.1038/nature08855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.South M, Ozonoff S, McMahon WM. Repetitive behavior profiles in Asperger syndrome and high-functioning autism. J Autism Dev Disord. 2005;35:145–158. doi: 10.1007/s10803-004-1992-8. [DOI] [PubMed] [Google Scholar]
- 112.D’Cruz AM, et al. Reduced behavioral flexibility in autism spectrum disorders. Neuropsychology. 2013;27:152–160. doi: 10.1037/a0031721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Murray GK, et al. Reinforcement and reversal learning in first-episode psychosis. Schizophr Bull. 2008;34:848–855. doi: 10.1093/schbul/sbn078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Leeson VC, et al. Discrimination learning, reversal, and set-shifting in first-episode schizophrenia: stability over six years and specific associations with medication type and disorganization syndrome. Biol Psychiatry. 2009;66:586–593. doi: 10.1016/j.biopsych.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Meechan DW, et al. Cognitive ability is associated with altered medial frontal cortical circuits in the LgDel mouse model of 22q11.2DS. Cereb Cortex. 2015:1143–1151. doi: 10.1093/cercor/bht308. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Segura-Puimedon M, et al. Heterozygous deletion of the Williams-Beuren syndrome critical interval in mice recapitulates most features of the human disorder. Hum Mol Genet. 2014;23:6481–6494. doi: 10.1093/hmg/ddu368. [DOI] [PubMed] [Google Scholar]
- 117.Sturner R, Howard B, Bergmann P, Stewart L, Afarian TE. Comparison of Autism Screening in Younger and Older Toddlers. J Autism Dev Disord. 2017 doi: 10.1007/s10803-017-3230-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ozonoff S, et al. The broader autism phenotype in infancy: when does it emerge? J Am Acad Child Adolesc Psychiatry. 2014;53:398–407. doi: 10.1016/j.jaac.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zwaigenbaum L, Bryson S, Garon N. Early identification of autism spectrum disorders. Behav Brain Res. 2013;251:133–146. doi: 10.1016/j.bbr.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 120.Weigelt S, Koldewyn K, Kanwisher N. Face identity recognition in autism spectrum disorders: a review of behavioral studies. Neurosci Biobehav Rev. 2012;36:1060–1084. doi: 10.1016/j.neubiorev.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 121.Chawarska K, Macari S, Shic F. Decreased spontaneous attention to social scenes in 6-month-old infants later diagnosed with autism spectrum disorders. Biol Psychiatry. 2013;74:195–203. doi: 10.1016/j.biopsych.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zwaigenbaum L, et al. Behavioral manifestations of autism in the first year of life. Int J Dev Neurosci. 2005;23:143–152. doi: 10.1016/j.ijdevneu.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 123.Happe F, Frith U. Annual research review: Towards a developmental neuroscience of atypical social cognition. J Child Psychol Psychiatry. 2014;55:553–557. doi: 10.1111/jcpp.12162. [DOI] [PubMed] [Google Scholar]
- 124.Pellicano E. The development of core cognitive skills in autism: a 3-year prospective study. Child Dev. 2010;81:1400–1416. doi: 10.1111/j.1467-8624.2010.01481.x. [DOI] [PubMed] [Google Scholar]
- 125.Happe FG. The role of age and verbal ability in the theory of mind task performance of subjects with autism. Child Dev. 1995;66:843–855. [PubMed] [Google Scholar]
- 126.Senju A, Southgate V, White S, Frith U. Mindblind eyes: an absence of spontaneous theory of mind in Asperger syndrome. Science. 2009;325:883–885. doi: 10.1126/science.1176170. [DOI] [PubMed] [Google Scholar]
- 127.Sprong M, Schothorst P, Vos E, Hox J, van EH. Theory of mind in schizophrenia: meta-analysis. Br J Psychiatry. 2007;191:5–13. doi: 10.1192/bjp.bp.107.035899. [DOI] [PubMed] [Google Scholar]
- 128.Green MF, Horan WP, Lee J. Social cognition in schizophrenia. Nat Rev Neurosci. 2015;16:620–631. doi: 10.1038/nrn4005. [DOI] [PubMed] [Google Scholar]
- 129.Gur RC, et al. Neurocognitive growth charting in psychosis spectrum youths. JAMA Psychiatry. 2014;71:366–374. doi: 10.1001/jamapsychiatry.2013.4190. [DOI] [PubMed] [Google Scholar]
- 130.Penn DL, Sanna LJ, Roberts DL. Social cognition in schizophrenia: an overview. Schizophr Bull. 2008;34:408–411. doi: 10.1093/schbul/sbn014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Walcott CM, Landau S. The relation between disinhibition and emotion regulation in boys with attention deficit hyperactivity disorder. J Clin Child Adolesc Psychol. 2004;33:772–782. doi: 10.1207/s15374424jccp3304_12. [DOI] [PubMed] [Google Scholar]
- 132.Arbogast T, et al. Reciprocal Effects on Neurocognitive and Metabolic Phenotypes in Mouse Models of 16p11.2 Deletion and Duplication Syndromes. PLoS Genet. 2016;12:e1005709. doi: 10.1371/journal.pgen.1005709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hiroi N, et al. A 200-kb region of human chromosome 22q11.2 confers antipsychotic-responsive behavioral abnormalities in mice. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:19132–19137. doi: 10.1073/pnas.0509635102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Esposito G, Venuti P. Understanding early communication signals in autism: a study of the perception of infants’ cry. J Intellect Disabil Res. 2010;54:216–223. doi: 10.1111/j.1365-2788.2010.01252.x. [DOI] [PubMed] [Google Scholar]
- 135.Ozonoff S, et al. A prospective study of the emergence of early behavioral signs of autism. J Am Acad Child Adolesc Psychiatry. 2010;49:256–266. [PMC free article] [PubMed] [Google Scholar]
- 136.Lai JK, et al. Temporal and spectral differences in the ultrasonic vocalizations of fragile X knock out mice during postnatal development. Behav Brain Res. 2014;259:119–130. doi: 10.1016/j.bbr.2013.10.049. [DOI] [PubMed] [Google Scholar]
- 137.Lacaria M, Spencer C, Gu W, Paylor R, Lupski JR. Enriched rearing improves behavioral responses of an animal model for CNV-based autistic-like traits. Hum Mol Genet. 2012;21:3083–3096. doi: 10.1093/hmg/dds124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Takahashi T, et al. Structure and function of neonatal social communication in a genetic mouse model of autism. Mol Psychiatry. 2016;21:1208–1214. doi: 10.1038/mp.2015.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dickinson ME, et al. High-throughput discovery of novel developmental phenotypes. Nature. 2016;537:508–514. doi: 10.1038/nature19356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kirov G, et al. The Penetrance of Copy Number Variations for Schizophrenia and Developmental Delay. Biol Psychiatry. 2013;75:378–385. doi: 10.1016/j.biopsych.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Harper KM, et al. Alterations of social interaction through genetic and environmental manipulation of the 22q11.2 gene Sept5 in the mouse brain. Human Molecular Genetics. 2012;21:3489–3499. doi: 10.1093/hmg/dds180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Suzuki G, et al. Sept5 deficiency exerts pleiotropic influence on affective behaviors and cognitive functions in mice. Human Molecular Genetics. 2009;18:1652–1660. doi: 10.1093/hmg/ddp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Spencer CM, et al. Modifying behavioral phenotypes in Fmr1KO mice: genetic background differences reveal autistic-like responses. Autism Res. 2011;4:40–56. doi: 10.1002/aur.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Drapeau E, Dorr NP, Elder GA, Buxbaum JD. Absence of strong strain effects in behavioral analyses of Shank3-deficient mice. Dis Model Mech. 2014;7:667–681. doi: 10.1242/dmm.013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sittig LJ, et al. Genetic Background Limits Generalizability of Genotype-Phenotype Relationships. Neuron. 2016;91:1253–1259. doi: 10.1016/j.neuron.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yang M, Lewis F, Foley G, Crawley JN. Tribute to Bob Blanchard: Divergent Behavioral Phenotypes of 16p11.2 Deletion Mice Reared in Same-Genotype Versus Mixed-Genotype Cages. Physiol Behav. 2015 doi: 10.1016/j.physbeh.2015.04.023. [DOI] [PubMed] [Google Scholar]
- 147.Kalbassi S, Bachmann SO, Cross E, Roberton VH, Baudouin SJ. Male and Female Mice Lacking Neuroligin-3 Modify the Behavior of Their Wild-Type Littermates. eNeuro. 2017;4 doi: 10.1523/ENEURO.0145-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Karayiorgou M, Gogos JA. The molecular genetics of the 22q11-associated schizophrenia. Brain Res Mol Brain Res. 2004;132:95–104. doi: 10.1016/j.molbrainres.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 149.Jonas RK, Montojo CA, Bearden CE. The 22q11.2 deletion syndrome as a window into complex neuropsychiatric disorders over the lifespan. Biol Psychiatry. 2014;75:351–360. doi: 10.1016/j.biopsych.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Guna A, Butcher NJ, Bassett AS. Comparative mapping of the 22q11.2 deletion region and the potential of simple model organisms. J Neurodev Disord. 2015;7:18. doi: 10.1186/s11689-015-9113-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Brown SD, Moore MW. The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mamm Genome. 2012;23:632–640. doi: 10.1007/s00335-012-9427-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Koscielny G, et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 2014;42:D802–809. doi: 10.1093/nar/gkt977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Paterlini M, et al. Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nat Neurosci. 2005;8:1586–1594. doi: 10.1038/nn1562. [DOI] [PubMed] [Google Scholar]
- 154.Chun S, et al. Thalamic miR-338-3p mediates auditory thalamocortical disruption and its late onset in models of 22q11.2 microdeletion. Nat Med. 2017;23:39–48. doi: 10.1038/nm.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ouchi Y, et al. Reduced Adult Hippocampal Neurogenesis and Working Memory Deficits in the Dgcr8-Deficient Mouse Model of 22q11.2 Deletion-Associated Schizophrenia Can Be Rescued by IGF2. J Neurosci. 2013;33:9408–9419. doi: 10.1523/JNEUROSCI.2700-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hiramoto T, et al. Tbx1: identification of a 22q11.2 gene as a risk factor for autism spectrum disorder in a mouse model. Hum Mol Genet. 2011;20:4775–4785. doi: 10.1093/hmg/ddr404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.O’Tuathaigh CM, et al. Genetic vs pharmacological inactivation of COMT influences cannabinoid-induced expression of schizophrenia-related phenotypes. Int J Neuropsychopharmacol. 2012;15:1331–1342. doi: 10.1017/S1461145711001581. [DOI] [PubMed] [Google Scholar]
- 158.O’Tuathaigh CM, et al. Chronic adolescent exposure to Delta-9-tetrahydrocannabinol in COMT mutant mice: impact on psychosis-related and other phenotypes. Neuropsychopharmacology. 2010;35:2262–2273. doi: 10.1038/npp.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Papaleo F, et al. Genetic dissection of the role of catechol-O-methyltransferase in cognition and stress reactivity in mice. J Neurosci. 2008;28:8709–8723. doi: 10.1523/JNEUROSCI.2077-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Papaleo F, Burdick MC, Callicott JH, Weinberger DR. Epistatic interaction between COMT and DTNBP1 modulates prefrontal function in mice and in humans. Mol Psychiatry. 2014;19:311–316. doi: 10.1038/mp.2013.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hancock CN, Liu W, Alvord WG, Phang JM. Co-regulation of mitochondrial respiration by proline dehydrogenase/oxidase and succinate. Amino Acids. 2016;48:859–872. doi: 10.1007/s00726-015-2134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Crabtree GW, Park AJ, Gordon JA, Gogos JA. Cytosolic Accumulation of L-Proline Disrupts GABA-Ergic Transmission through GAD Blockade. Cell Rep. 2016;17:570–582. doi: 10.1016/j.celrep.2016.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fenelon K, et al. The pattern of cortical dysfunction in a mouse model of a schizophrenia-related microdeletion. J Neurosci. 2013;33:14825–14839. doi: 10.1523/JNEUROSCI.1611-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Toritsuka M, et al. Deficits in microRNA-mediated Cxcr4/Cxcl12 signaling in neurodevelopmental deficits in a 22q11 deletion syndrome mouse model. Proc Natl Acad Sci U S A. 2013;110:17552–17557. doi: 10.1073/pnas.1312661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Toyoshima M, et al. Analysis of induced pluripotent stem cells carrying 22q11.2 deletion. Transl Psychiatry. 2016;6:e934. doi: 10.1038/tp.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Amin ND, et al. Cyclin-dependent kinase 5 phosphorylation of human septin SEPT5 (hCDCrel-1) modulates exocytosis. J Neurosci. 2008;28:3631–3643. doi: 10.1523/JNEUROSCI.0453-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Beites CL, Campbell KA, Trimble WS. The septin Sept5/CDCrel-1 competes with alpha-SNAP for binding to the SNARE complex. Biochem J. 2005;385:347–353. doi: 10.1042/BJ20041090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dent J, et al. A prototypic platelet septin and its participation in secretion. Proc Natl Acad Sci U S A. 2002;99:3064–3069. doi: 10.1073/pnas.052715199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Dong Z, et al. Dopamine-dependent neurodegeneration in rats induced by viral vector-mediated overexpression of the parkin target protein, CDCrel-1. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12438–12443. doi: 10.1073/pnas.2132992100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang Y, et al. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci U S A. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Son JH, et al. Neurotoxicity and behavioral deficits associated with Septin 5 accumulation in dopaminergic neurons. J Neurochem. 2005;94:1040–1053. doi: 10.1111/j.1471-4159.2005.03257.x. [DOI] [PubMed] [Google Scholar]
- 172.Ellegood J, et al. Neuroanatomical Phenotypes Are Consistent With Autism-Like Behavioral Phenotypes in the 15q11-13 Duplication Mouse Model. Autism Res. 2015;8:545–555. doi: 10.1002/aur.1469. [DOI] [PubMed] [Google Scholar]
- 173.Ellegood J, et al. Neuroanatomical phenotypes in a mouse model of the 22q11.2 microdeletion. Mol Psychiatry. 2014;19:99–107. doi: 10.1038/mp.2013.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jackowski AP, et al. Brain abnormalities in Williams syndrome: a review of structural and functional magnetic resonance imaging findings. Eur J Paediatr Neurol. 2009;13:305–316. doi: 10.1016/j.ejpn.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 175.Meyer-Lindenberg A, et al. Functional, structural, and metabolic abnormalities of the hippocampal formation in Williams syndrome. J Clin Invest. 2005;115:1888–1895. doi: 10.1172/JCI24892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gothelf D, Schaer M, Eliez S. Genes, brain development and psychotiatric phenotypes in velo-cardio-facial syndrome. Dev Disabilities. 2008;14:59–68. doi: 10.1002/ddrr.9. [DOI] [PubMed] [Google Scholar]
- 177.Tan GM, Arnone D, McIntosh AM, Ebmeier KP. Meta-analysis of magnetic resonance imaging studies in chromosome 22q11.2 deletion syndrome (velocardiofacial syndrome) Schizophr Res. 2009;115:173–181. doi: 10.1016/j.schres.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 178.Kates WR, et al. Frontal and caudate alterations in velocardiofacial syndrome (deletion at chromosome 22q11.2) J Child Neurol. 2004;19:337–342. doi: 10.1177/088307380401900506. [DOI] [PubMed] [Google Scholar]
- 179.Bish JP, Nguyen V, Ding L, Ferrante S, Simon TJ. Thalamic reductions in children with chromosome 22q11.2 deletion syndrome. Neuroreport. 2004;15:1413–1415. doi: 10.1097/01.wnr.0000129855.50780.85. [DOI] [PubMed] [Google Scholar]
- 180.Machado AM, et al. Corpus callosum morphology and ventricular size in chromosome 22q11.2 deletion syndrome. Brain Res. 2007;1131:197–210. doi: 10.1016/j.brainres.2006.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Antshel KM, Conchelos J, Lanzetta G, Fremont W, Kates WR. Behavior and corpus callosum morphology relationships in velocardiofacial syndrome (22q11.2 deletion syndrome) Psychiatry Res. 2005;138:235–245. doi: 10.1016/j.pscychresns.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 182.Wegiel J, et al. Differences between the pattern of developmental abnormalities in autism associated with duplications 15q11.2-q13 and idiopathic autism. J Neuropathol Exp Neurol. 2012;71:382–397. doi: 10.1097/NEN.0b013e318251f537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Boronat S, Mehan WA, Shaaya EA, Thibert RL, Caruso P. Hippocampal abnormalities in magnetic resonance imaging (MRI) of 15q duplication syndromes. J Child Neurol. 2015;30:333–338. doi: 10.1177/0883073814538669. [DOI] [PubMed] [Google Scholar]
- 184.Isshiki M, et al. Enhanced synapse remodelling as a common phenotype in mouse models of autism. Nat Commun. 2014;5:4742. doi: 10.1038/ncomms5742. [DOI] [PubMed] [Google Scholar]
- 185.Thelin J, et al. The translationally relevant mouse model of the 15q13.3 microdeletion syndrome reveals deficits in neuronal spike firing matching clinical neurophysiological biomarkers seen in schizophrenia. Acta Physiol (Oxf) 2017;220:124–136. doi: 10.1111/apha.12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Nilsson SR, et al. A mouse model of the 15q13.3 microdeletion syndrome shows prefrontal neurophysiological dysfunctions and attentional impairment. Psychopharmacology (Berl) 2016;233:2151–2163. doi: 10.1007/s00213-016-4265-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Meechan DW, et al. Cognitive ability is associated with altered medial frontal cortical circuits in the LgDel mouse model of 22q11.2DS. Cereb Cortex. 2015;25:1143–1151. doi: 10.1093/cercor/bht308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Choi SJ, et al. A Schizophrenia-Related Deletion Leads to KCNQ2-Dependent Abnormal Dopaminergic Modulation of Prefrontal Cortical Interneuron Activity. Cereb Cortex. 2017:1–17. doi: 10.1093/cercor/bhx123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kumar V, et al. C57BL/6N mutation in cytoplasmic FMRP interacting protein 2 regulates cocaine response. Science. 2013;342:1508–1512. doi: 10.1126/science.1245503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Matsuo N, et al. Behavioral profiles of three C57BL/6 substrains. Front Behav Neurosci. 2010;4:29. doi: 10.3389/fnbeh.2010.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mouri A, et al. Mouse strain differences in phencyclidine-induced behavioural changes. Int J Neuropsychopharmacol. 2012;15:767–779. doi: 10.1017/S146114571100085X. [DOI] [PubMed] [Google Scholar]
- 192.Simon MM, et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013;14:R82. doi: 10.1186/gb-2013-14-7-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]