

SUMMARY
PRC2 is a therapeutic target for several types of cancers currently undergoing clinical trials. Its activity is regulated by a positive feedback loop whereby its terminal enzymatic product, H3K27me3, is specifically recognized and bound by an aromatic cage present in its EED subunit. The ensuing allosteric activation of the complex stimulates H3K27me3 deposition on chromatin. Here, we report a step-wise feedback mechanism entailing key residues within distinctive interfacing motifs of EZH2 or EED that are found mutated in cancers and/or Weaver syndrome. PRC2 harboring these EZH2 or EED mutants manifest little activity in vivo but, unexpectedly, exhibited similar chromatin association as wild-type PRC2, indicating an uncoupling of PRC2 activity and recruitment. With genetic and chemical tools, we demonstrated that targeting allosteric activation overrode the gain-of-function effect of EZH2Y646X oncogenic mutations. These results revealed critical implications to the regulation and biology of PRC2 and a vulnerability in tackling PRC2-addicted cancers.
eTOC Blurb
Lee and Yu et al. dissect the stepwise mechanism of PRC2 activation involving the EZH2-SRM and EED-anchorage regions. Mutations in these regions diminish PRC2 activity in vivo and override the hyperactivity of oncogenic EZH2 mutations, but do not affect PRC2 association with chromatin. The α-helical mimetics against SRM abrogate PRC2 activation.
Introduction
Polycomb group (PcG) proteins are key epigenetic regulators that maintain transcriptional repression. During normal development, polycomb-mediated repression of lineage-specific genes contributes to cell identity stability and animal body plan formation (Aloia et al., 2013; Bracken and Helin, 2009; Bracken et al., 2006). Dysregulation of PcG proteins induces aberrant transcriptional programs that contribute to developmental diseases as well as cancer progression (Bachmann et al., 2006; Imagawa et al., 2017; Tan et al., 2014; Tatton-Brown et al., 2013).
Polycomb Repressive Complex 2 (PRC2) catalyzes the methylation of histone H3 lysine 27 (H3K27), with H3K27me2/3 being associated with repressive chromatin (Barski et al., 2007). The core complex of PRC2 comprises three subunits: EZH1/2, EED, and SUZ12, that associate with a histone binding protein RBAP46/48 (see review, Margueron et al., 2009). EZH1 and EZH2 are interchangeable subunits of PRC2 that harbor the histone lysine N-methyltransferase (HMT) activity through their respective SET domains, with the HMT activity of PRC2/EZH2 being markedly higher than that of PRC2/EZH1 (Margueron et al., 2008; Son et al., 2013, “Lee and Holder et al., Mol Cell, this issue”). The EZH2 catalytic SET domain exhibits an auto-inhibitory state (Jiao and Liu, 2015; Justin et al., 2016), which is relieved upon EZH2 association with EED and SUZ12, a prerequisite for HMT activity.
In addition, PRC2 exists in vivo as a holo-complex, associating with different accessory proteins that regulate its activity and recruitment to chromatin (see review, Holoch and Margueron, 2017). One such protein is JARID2 that assists in the recruitment of PRC2 to developmental loci in mouse embryonic stem cells (mESC) and more importantly, enhances its HMT enzymatic activity (Kaneko et al., 2014a; Li et al., 2010; Sanulli et al., 2015; Son et al., 2013). In addition, a recent study demonstrated that Polycomb-like proteins (PCLs), particularly MTF2/PCL2, facilitate PRC2 recruitment to CpG islands in mESC (Li et al., 2017a). Our previous work demonstrated that EED binds to H3K27me3 through its aromatic cage, leading to an allosteric activation of the complex, further assisting PRC2 in establishing repressive chromatin domains (Margueron et al., 2009). Of note, PRC2 association with JARID2 results in JARID2 being tri-methylated at lysine residue 116 (JARID2-K116me3), which also stimulates PRC2 in a manner similar to that of H3K27me3, and this function is likely required for the de novo deposition of H3K27me3 (Sanulli et al., 2015).
In human cancers, PRC2 plays a pleiotropic and highly context-dependent role. Loss-of-function mutations and deletions of EZH2, EED, or SUZ12 have been found in cancers including T cell acute lymphoblastic leukemia (T-ALLs) and malignant peripheral nerve sheath tumors (MPNSTs) (Kim and Roberts, 2016; Lee et al., 2014). On the other hand, EZH2 was found to be over-expressed in many other types of human cancers with an associated poor prognosis (Bachmann et al., 2006). Also, the “hotspot” gain-of-function mutations at residue Y646 (Y641 in human isoform c and in mouse EZH2) within the catalytic SET domain of EZH2 frequently were found in diffuse large B-cell lymphoma (DLBCL) (Bödör et al., 2011; Morin et al., 2010; Yap et al., 2011). The Y646X (X=S,N,F,C, or H) mutations within the SET domain of EZH2 specifically switches the substrate preference of EZH2, thereby facilitating the conversion of H3K27 di-methylation to the tri-methylation state in peptide-based HMT assays in vitro (Wigle et al., 2011; Yap et al., 2011). As EZH2Y646X mutant cancers often manifest a dependency on PRC2/EZH2 (Liu et al., 2010; McCabe et al., 2012a), oncogenic EZH2 has been implicated as a therapeutic target in cancers such as DLBCL that rely on its activity (Béguelin et al., 2013; McCabe et al., 2012a).
EZH2 inhibitors were developed as competitive inhibitors of S-adenosylmethionine (SAM), the methyl-donor essential for the HMT activity, several of which are currently in clinical trials. However, the therapeutic index of these SAM-competitive inhibitors is restricted by their imperfect pharmacological properties, including their short half-life, moderate to high clearance rate, and low permeability in cell-based assays (Bradley et al., 2014; Knutson et al., 2013; McCabe et al., 2012a). Of note, in cellular models of DLBCL, de novo mutations at residues I109, Y111, and Y666 of EZH2 were found to prevent inhibitor binding and led to an acquired resistance to these inhibitors (Baker et al., 2015; Gibaja et al., 2016). Therefore, new types of EZH2 inhibitors will be needed. New inhibitors targeting the cage of EED have been developed to abrogate the EED-H3K27me3 interaction and thereby prevent the initiation of H3K27me3-potentiated positive feedback activation of PRC2 (Curtin et al., 2017; He et al., 2017; Huang et al., 2017; Li et al., 2017c; Qi et al., 2017).
The structure of the fungal PRC2 was shown in its basal as well as allosterically stimulated states, and that of human PRC2 in a stimulated state with a JARID2-K116me3 peptide (Jiao and Liu, 2015; Justin et al., 2016). Additionally, the structure of a chimeric PRC2 consisting of EED and SUZ12 from human together with EZH2 from American chameleon (Anolis carolinensis) was solved in its basal state (Brooun et al., 2016). The structures of human and fungal PRC2 illustrated a major reorganization of the complex upon EED binding to JARID2-K116me3 or H3K27me3, namely the stabilization of the SET-I domain (an α-helix of the catalytic SET domain) through its interaction with an α-helix of the Stimulatory Responsive Motif (SRM) of EZH2.
Although structural studies have implicated the involvement of the SRM and SET-I domains of EZH2 in PRC2 allosteric activation, the structures of PRC2 were derived in complex with either the H3K27M oncohistone or a SAM competitive inhibitor which might conceal the dynamics of the SET domain during its allosteric activation (Brooun et al., 2016; Jiao and Liu, 2015; Justin et al., 2016). Yet, whether PRC2 acts in the same fashion while in its native state in vivo is unclear. To better understand the molecular mechanisms underlying the allosteric activation of PRC2, we revisited the structural data of PRC2 and examined the genetic mutations of PRC2 found in human diseases. We further analyzed the key regions that undergo conformational remodeling during allosteric activation of PRC2 and pinpointed the functional significance of key residues by introducing disease-associated point mutations. The biological impact of the allosteric activation of PRC2 was then investigated in a variety of cellular models, including mESC, melanoma and DLBCL cells.
Results
A stepwise model for allosteric activation of PRC2
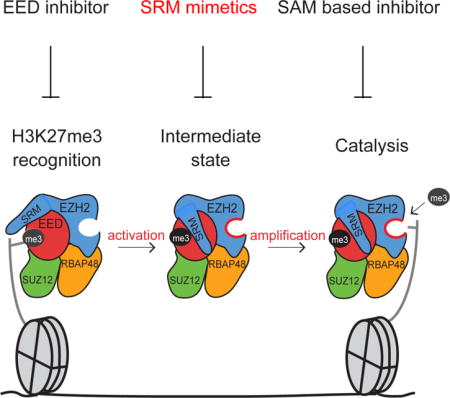
Previous studies demonstrated that EED, through its aromatic cage, binds to H3K27me3 or JARID2-K116me3, leading to the allosteric activation of PRC2 (Margueron et al., 2009; Sanulli et al., 2015). This enhanced activity entails a remodeling of the PRC2 subunits that orders the SRM domain of EZH2 (Justin et al., 2016). Together, these results favor a stepwise model for the allosteric activation of PRC2 (Justin et al., 2016; Margueron et al., 2009; Sanulli et al., 2015), which involves an initial recognition (EED aromatic cage binding to H3K27me3/JARID2-K116me3), an intermediate state (ordering of EZH2-SRM followed by the stabilization of the EZH2-SET-I domain) and a final step for substrate recognition and catalysis by a fully empowered SET domain (Figure 1A). Hereafter, we refer to the EED-H3K27me3 binding step as “recognition” and the deposition of PRC2 onto chromatin as “recruitment”.
Figure 1. Identification of key EZH2 and EED residues required for allosteric activation of PRC2.
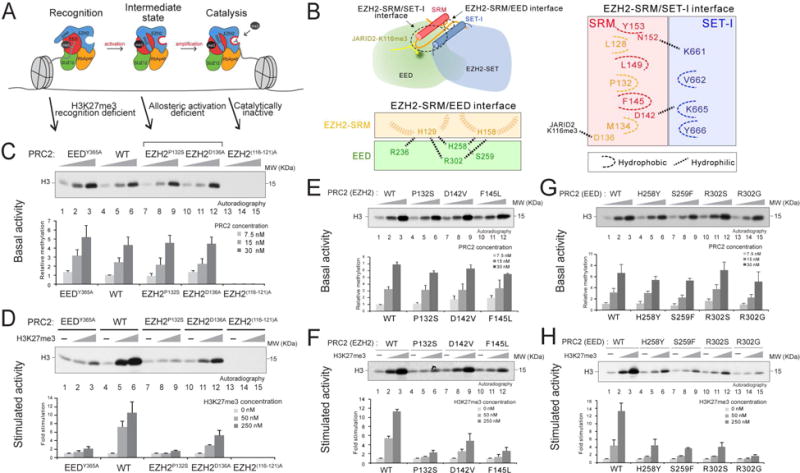
(A) Illustration of the stepwise model of H3K27me3-potentiated positive feedback activation of PRC2, including the initial recognition step (EED binding to H3K27me3), the intermediate state (EZH2-SRM and SET-I ordering), and the substrate binding step (catalysis). Arrows indicate the corresponding PRC2 mutants for the dissection of each step within the model.
(B) Illustration of the EZH2-SRM/SET-I and EZH2-SRM/EED interfaces (left-top). Residues that mediate the interaction within the EZH2-SRM/EED interface (left-bottom) or within the EZH2-SRM/SET-I interface (right) are highlighted. Note that the numbers of EZH2 residues are based on human EZH2 isoform a (751 amino acids). Hydrophobic interactions are marked as dashed curvy lines and hydrophilic interactions as dashed linear lines.
(C-H) Histone methyltransferase assays. Bar plots were presented as Mean±SD (n=3 for each data point).
(C and D) HMT assays using wild-type (WT) PRC2 or mutant PRC2, as indicated, with unmodified oligonucleosomes (150 nM) as substrate. (C) Top, a representative HMT assay measuring the basal activity of PRC2. HMT assays were performed with an increasing amount of PRC2 (7.5, 15, or 30 nM) in the absence of the H3K27me3 stimulatory peptide (amino acids 21-44), as illustrated in the panel. The levels of methylation on histone H3K27 are shown by autoradiography. (D) Top, representative assay measuring the stimulated activity of PRC2. HMT assays were performed using 15 nM PRC2 without or with an increasing amount of the H3K27me3 peptide (50 or 250 nM). (C-D) Bottom, Quantification of the relative amounts of 3H-SAM incorporated into histone H3 after 60 min incubation of the assays described in Top panel.
(E and F) HMT assays using WT PRC2 or PRC2 containing EZH2-SRM mutants. Top, Representative HMT assay measuring the basal activity (panel E) and the stimulated activity (panel F) of PRC2 with the experimental conditions described in panel (C-D). Bottom, Quantification of the assays described in Top panel.
(G and H) HMT assay using WT PRC2 or PRC2 containing EED anchoring mutants. Top, Representative HMT assay measuring the basal activity (panel G) and the stimulated activity (panel H) of PRC2 under the experimental conditions described in panel (C-D). Bottom, Quantification of the assays described in Top panel.
Specific mutations at the interface of EZH2-SRM/SET-I and that of EZH2-SRM/EED abrogate allosteric activation of PRC2
To test the model proposed above, we first scrutinized the structural features of the SRM domain of human EZH2. Upon EED binding to the JARID2-K116me3 peptide, the EZH2-SRM formed a loop-α-helix-loop structure, with the α-helix interacting with the SET-I domain of EZH2 and the loops interacting with JARID2-K116me3 and EED (Figure 1B, S1A and S1B). Several residues of the α-helices within the SRM and SET-I formed hydrophobic interactions, including P132 and F145 within the SRM, and V662 and Y666 within the SET-I. D142 of the SRM electrostatically interacted with K665 of the SET-I, as described previously (Justin et al., 2016). Further examination of the structural data revealed a potentially critical interaction between the SRM and EED during allosteric activation of PRC2. Moreover, several residues within the SRM, including H129 and H158, appeared to form an intercalated anchor at the EZH2-SRM/EED interface (Figure 1B, left-bottom, and S1B). Intriguingly, while searching for genetic alterations of PRC2 subunits in human diseases using the COSMIC cancer mutation dataset and literature search, we found a number of mutations in the SRM/SET-I and SRM/EED interfaces that are present in human cancers and Weaver syndrome (Table S1). Of note, the EED mutations found in patients affected by Weaver Syndrome were exclusively located at the SRM/EED interface, further highlighting the potential role of this region in regulating PRC2 activity (Figure 1B, S1B, and Table S1) (Cohen, A.S.A., Gibson, 2016; Cohen et al., 2015; Cooney et al., 2017).
Given the structural and human genetic data described above, we generated a series of point mutations within the SRM domain of EZH2, including P132S, D142V, and F145L which are located at the interface between the SRM/SET-I helices, as well as D136A that interacts with the JARID2-K116me3 peptide (Figures 1B and S1A). Additionally, residues on EED that anchor the SRM/EED interaction were mutated and we termed them “EED anchoring mutants”, including H258Y, S259F, R302S, and R302G (Figure 1B, left-bottom). Versions of PRC2 carrying each of the aforementioned mutations were produced using a baculovirus expression system and purified through tandem affinity-tag purification. The wild-type (WT) and mutant versions of PRC2 formed intact complexes (Figure S2A) and importantly, exhibited similar levels of basal HMT activity (Figures 1C, 1E, 1G, S2B, S2C and S2D). Note that EZH2D142V manifested an altered migration in SDS-PAGE likely due to a change in charge within the amino acid side chain (Figure S2A). As expected, the EEDY365A aromatic cage mutation specifically disrupted allosteric activation, and a catalytically inactive EZH2(116-121A) mutant had neither basal nor stimulated activity (Figures 1C, S1C and S2B). Among the SRM and EED anchoring mutants described above, EZH2P132S, EZH2F145L (within the SRM) and EEDR302G displayed a drastic reduction in allosteric activation, whereas EZH2D136A, EZH2D142V (within the SRM), and other EED mutants rendered a moderate effect (Figures 1D, 1F, 1H, S2C and S2D). Structural modeling of the EED anchoring mutants indicated a disturbance in anchorage formation involving residues H129 and H158 of the SRM (Figure S2E). Although the EEDY365A aromatic cage mutant impaired allosteric activation of PRC2, the defect was due to its deficiency in interacting with H3K27me3, the initial recognition step, whereas other SRM and EED anchorage mutants did not manifest such a recognition defect (Figure S3A), but were unable to be allosterically activated. Thus, two EZH2-SRM mutants (EZH2P132S and EZH2F145L) and an EED anchoring mutant (EEDR302G) are bona fide activation-deficient mutants of PRC2.
Allosteric activation of PRC2/EZH2 is critical for appropriate H3K27me2/3 levels in vivo
Having defined the key residues of EZH2 and EED important for the response to H3K27me3-potentiated PRC2 allosteric activation in vitro, we next examined the role of these residues in regulating PRC2 activity in vivo. In the case of EZH2, we generated EZH2-/- (EZH2-KO) mESCs and transduced them with lentiviruses containing either EZH2WT, or the activation-deficient SRM mutants (EZH2P132S, EZH2D142V or EZH2F145L). For the EED anchoring mutations, we generated a mESC line comprising the EED protein without its aromatic cage (EED ΔC-term mESCs) and transduced them with lentiviruses containing EEDWT or EED anchoring mutants (EEDH258Y, EEDS259F, EEDR302S, or EEDR302G) (Figure 2A and 2B). While ectopic expression of EZH2WT rescued H3K27me2 and H3K27me3 levels in the EZH2-KO mESC (Figure 2A, lane 4), EZH2P132S and EZH2F145L were ineffectual (Figure 2A, lanes 5 and 7). On the other hand, EZH2D142V displayed a partial rescue of H3K27me2, but H3K27me3 levels were severely compromised (Figure 2A, lane 6). In accordance, EEDWT and EED anchoring mutants regulated the endogenous levels of H3K27me2/3 levels in a manner similar to that observed in vitro, with EEDR302G (Figure 2B, lane 8) showing no rescue effect and EEDH258Y, EEDS259F, and EEDR302S showing a partial rescue of H3K27me2 levels, with levels of H3K27me3 being severely compromised (Figure 2B, lanes 5-7). Mass-spectrometry analyses of histone modifications further corroborated that EZH2P132S, EZH2F145L, and EEDR302G failed to catalyze any methylation on H3K27 in vivo (Figure S3B). These results were in full agreement with the biochemical studies for the stimulated activity, shown above (Figures 1D, 1F, and 1H). Unexpectedly, these results revealed that the basal state of PRC2 had little impact on endogenous H3K27me2/3 levels and underscored the predominant role of allosteric activation of PRC2 in attaining appropriate levels of H3K27me2/3 in vivo. Indeed, mESC expressing EEDR302G (Figure 2B, lane 8) phenocopied those expressing EED ΔC-term and failed to form embryoid bodies (EB), a well-established phenotype associated with PRC2-deficiency (Chou et al., 2011; Ezhkova et al., 2009; Obier et al., 2015; Pasini et al., 2007), as evidenced by the significantly low expression of the mesodermal marker, Brachyury (Figure S3C). Of note, immunoprecipitation (IP) experiments using anti-EZH2 antibody demonstrated that the interactions between each of the SRM mutants and core PRC2 subunits as well as accessory proteins JARID2 and AEBP2 were retained (Figure S3D), excluding the possibility that these mutations destabilized PRC2 complex formation in vivo. In addition to the above three SRM mutants, we further analyzed two other disease-associated (see Table S1) SRM mutants, L149Q and Y153C, that also participate in the hydrophobic interaction between the SRM and SET-I domains. In accordance, these two mutants formed intact PRC2 complex and manifested a defect in allosteric activation, while maintaining their basal activity (Figures S4A-S4C). Moreover, ectopic expression of these two mutants in EZH2-KO mESCs failed to rescue endogenous H3K27me2/3 levels (Figure S4D).
Figure 2. Point mutations of key residues at the EZH2-SRM/SET-I or EZH2-SRM/EED interface affect global H3K27me2/3 levels in vivo.
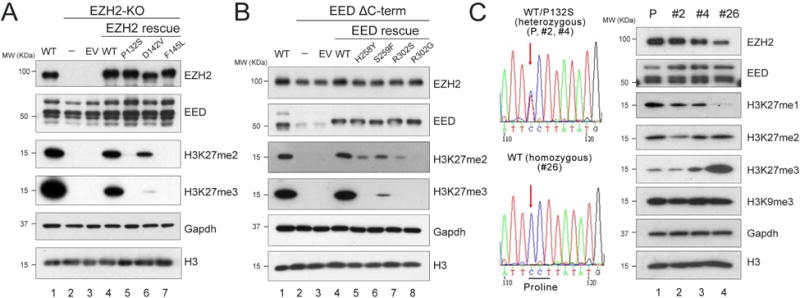
(A-B) Western blot analysis of EZH2, EED, H3K27me2, H3K27me3 and total histone H3 levels in E14 mESC cells, including WT, EZH2-KO, and EZH2 rescue conditions (A) or EEDΔC-term and EED rescue conditions (B). Gapdh was used as a loading control. mESC, mouse embryonic stem cells. EV, empty vector. EEDΔC-term, EED C-terminal deletion.
(C) CRISPR/Cas9-mediated reversal of EZH2P132S to EZH2WT in COLO-679 melanoma cells. Left, Sanger sequencing results for the parental (P) COLO-679 cell line (EZH2WT/P132S), the negative CRISPR clones (EZH2WT/P132S; clones #2 and #4), and the positive CRISPR clone (EZH2WT/WT; clone #26). Right, Western blot analysis of EZH2, EED, H3K27me1, H3K27me2, H3K27me3, H3K9me3 and total histone H3 levels in the parental COLO-679 cells as well as CRISPR clones #2, #4, and #26. Gapdh was used as a loading control.
To further explore the impact of this positive feedback-regulated allosteric activation of PRC2 in an independent system, we performed CRISPR/Cas9-mediated gene editing in a melanoma cell line, COLO-679, which carries a heterozygous EZH2WT/P132S mutation. We generated a sub-clone (clone #26) that underwent a genetic “correction” and thus expressed only EZH2WT from the parental line. This re-introduced EZH2WT allele in clone #26 (EZH2WT/WT) led to markedly increased levels of H3K27me3, unchanged levels of H3K27me2, and decreased levels of H3K27me1 (Figure 2C, lane 4), when compared to the parental line (P, lane 1) and other negative CRISPR clones that remained EZH2WT/P132S (clones #2 and #4, lanes 2 and 3, respectively). A PRC2-independent repressive histone modification, H3K9me3, was unaffected by the elimination of EZH2P132S. These results further corroborated the critical role of allosteric activation in driving the higher order methylation status of H3K27 by PRC2 in vivo.
PRC2/EZH2 recruitment to chromatin is independent of its allosteric activation
As the allosteric activation-deficient mutants failed to rescue H3K27me2/3 levels in vivo, we examined their respective PRC2 species for chromatin recruitment. We performed chromatin immunoprecipitation followed by next-generation deep sequencing (ChIP-seq) using anti-EZH2 or anti-H3K27me3 antibody in WT and EZH2-KO mESCs, either alone or rescued with EZH2WT, EZH2P132S or EZH2F145L. Ectopic expression of EZH2WT in the EZH2-null background rescued the majority of the EZH2 peaks across the genome and at the transcription start sites (TSS) of PRC2/EZH2 target genes (Figure 3A and 3B, left). Surprisingly, the allosteric activation deficient mutants, EZH2P132S and EZH2F145L, not only rescued the genome-wide occupancy, but also manifested a more pervasive association with chromatin in either the max-peak-centered or TSS-centered heatmap (Figure 3A and 3B, left, see Discussion). Yet, while each mutant bound to chromatin, H3K27me3 levels remained similar to the case of EZH2-KO cells (Figure 3A and 3B, right). Of note, we observed an incomplete rescue of the genome-wide H3K27me3 peaks upon ectopic expression of EZH2WT in EZH2-KO cells, possibly due to clonal variation, as our EZH2-KO line was a clonal line derived from the CRISPR/Cas9 approach (Figure 3A, right). However, in the same experiment, EZH2WT did rescue most of the H3K27me3 peaks at the TSS of PRC2/EZH2 targets, suggesting that H3K27me3 occupancy at gene promoters is more conserved and fluctuated less between the clonal and the parental lines (Figure 3B, right). Intriguingly, we detected a small number of H3K27me3 peaks in EZH2-KO cells (Figure 3A-C), likely due to the presence of EZH1 that functions similarly, but to a considerably lesser extent than EZH2 (Margueron et al., 2008; Son et al., 2013, “Lee and Holder et al., Mol Cell, this issue”). Regardless of the residual H3K27me3 peaks detected at a small number of loci (Figure 3C), EZH2WT, EZH2P132S, and EZH2F145L were still fully deposited to the same loci as endogenous EZH2 in control E14 mESC, suggesting that the chromatin landscape in EZH2-KO mESCs preserved an epigenetic memory that facilitated appropriate restoration of PRC2/EZH2 occupancy (Figure 3A-C). These results indicated that PRC2 recruitment to target genes in the chromatin context is uncoupled from its enzymatic activity.
Figure 3. The activity of PRC2 is independent of its recruitment to chromatin.
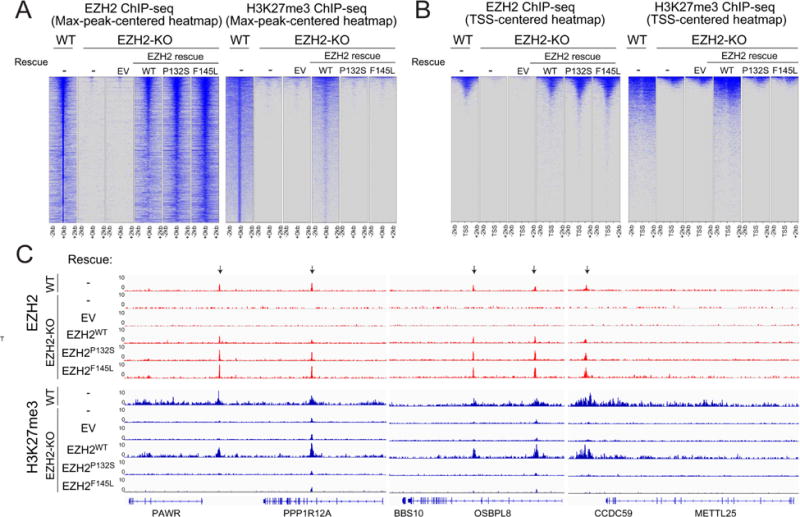
(A and B) Heatmaps representing EZH2 (left panels) or H3K27me3 (right panels) ChIP-seq peaks centered by maximum peak intensity at the genome-wide scale (A) or ~29000 UCSC annotated transcription start sites (TSS) and (B) in 4-kb windows within 200 bp bin size. The genotypes of E14 mESC for each experimental condition are indicated above each panel. WT, wild-type. KO, knockout. EV, empty vector.
(C) Representative track images for EZH2 (in red) and H3K27me3 (in blue) ChIP-seq experiments performed in panel (A and B) are shown for a select group of annotated genes. The UCSC annotations of exons and gene bodies are shown at the bottom. Arrows indicate the MACS peaks with FDR <5% in the respective windows. The scale of the peaks ranges from 0-10 reads per 10 million reads, with spike-in normalization using Drosophila chromatin and Drosophila H2A.X antibody in each ChIP reaction.
An allosteric activation-deficient mutation epistatically overrides a gain-of-function mutation in EZH2
As allosteric activation appears to be the predominant mechanism governing PRC2/EZH2 catalysis in vivo, we speculated that this mechanism might override the effect of cancer-associated, gain-of-function mutations at the Y646 residue within the SET domain of human EZH2 (Y641 in human isoform a and in mouse EZH2). Therefore, we generated a compound mutant of EZH2 that carried both the P132S activation deficient mutation (in the SRM) and the Y646N gain-of-function mutation (in the SET domain). We measured the HMT activity of PRC2 comprising EZH2WT, EZH2P132S, EZH2Y646N, or EZH2P132S;Y646N using mono-, di-, and un-methylated H3K27 oligonucleosomes as templates, as a function of the presence of the allosteric activator (H3K27me3). Consistent with the previous peptide-based HMT assay, PRC2 comprising EZH2Y646N exhibited no basal activity on un-methylated or mono-methylated H3K27 templates (Figure 4A, lane 4 and Figure 4B, lane 7). Moreover, EZH2Y646N had higher basal activity compared to EZH2WT on di-methylated templates (Figure 4C, compare lanes 1 and 7). This finding is consistent with the the gain-of-function mutation (EZH2Y646X) exhibiting an altered substrate preference for H3K27me2 over H3K27me1 or unmethylated templates (Wigle et al., 2011; Yap et al., 2011). In accordance, the EZH2Y646X oncogenic mutations were always found to be heterozygous, requiring one EZH2WT allele to cooperatively acquire a gain-of-function state by providing the H3K27me2 substrate (Sneeringer et al., 2010). More importantly, EZH2Y646N was also regulated by allosteric activation as it catalyzed elevated levels of H3K27me3 in response to the H3K27me3 peptide (Figure 4C, lanes 7-9). However, the presence of this allosteric activator was ineffectual in the case of EZH2P132S;Y646N (Figure 4C, lanes 10-12), suggesting that the gain-of-function effect driven by EZH2Y646N was restrained at the basal level by disruption of the induced interaction between SRM/SET-I. We also tested whether EEDR302G resembles EZH2P132S in this in vitro system. Indeed, PRC2 containing EEDR302G and EZH2Y646N manifested an activation defect similar to that of the EZH2P132S;Y646N compound mutant, further corroborating the essential involvement of EED anchorage in this process (Figures S2A and S5A).
Figure 4. EZH2P132S-driven inhibition of the allosteric activation of PRC2 overrides the effect of the EZH2Y646N gain-of-function mutation.
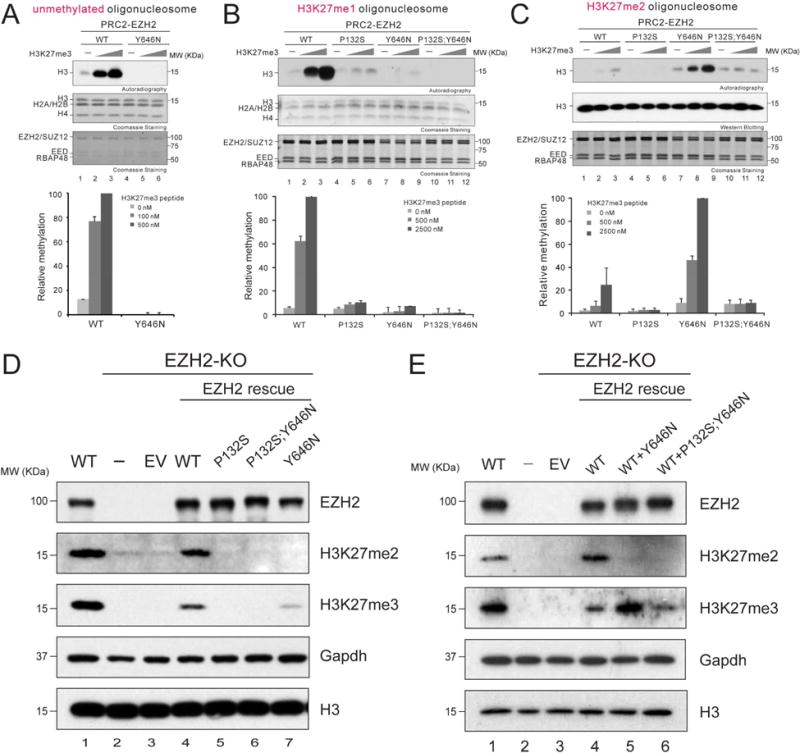
(A-C) Histone methyltransferase assays. Bar plots were presented as Mean±SD (n=3 for each data point).
(A) Top, a representative image of the HMT assays containing 100 nM WT PRC2 or PRC2/EZH2Y646N with unmodified oligonucleosomes (150 nM) as a substrate in the absence or presence of H3K27me3 peptide (100 or 500 nM). The levels of methylation on histone H3 are shown by autoradiography (top image). Coomassie blue staining of SDS-PAGE gels containing nucleosomes (middle image) or PRC2 components (bottom image) was used to visualize the relative concentration of each component present in each reaction. Bottom, quantification of the relative amounts of 3H-SAM incorporated into histone H3.
(B and C) Top, representative images for the HMT assays containing 500 nM WT PRC2 or mutant PRC2, as indicated, in the absence or presence of the H3K27me3 peptide (500 or 2500 nM) using 150 nM of H3K27me1 oligonucleosomes (B) or 150 nM of H3K27me2 oligonucleosomes (C) as substrate. The concentrations of PRC2 and H3K27me3 peptide were adjusted in these HMT reactions due to the less efficient catalysis on H3K27me1 or H3K27me2 oligonucleosomes. The middle/bottom gel images and the bottom panel for loading controls and signal quantifications, respectively, are as described in (A).
(D-E) Western blot analysis of EZH2, H3K27me2, H3K27me3, and total histone H3 in mESCs, either WT or under EZH2 rescue conditions, as indicated. Gapdh was used as a loading control. EV, empty vector.
To test if PRC2/EZH2Y646N catalysis of H3K27me3 in vivo was dependent on its allosteric activation, EZH2Y646N and EZH2P132S;Y646N were stably expressed in EZH2-KO mESC using a lentivirus approach. As expected, expression of EZH2Y646N alone in EZH2-null cells only partially rescued H3K27me3 due to a lack of EZH2WT that compromised the catalysis of mono- and di-methylation of H3K27 (Figure 4D, lane 7). Nevertheless, similar to EZH2P132S, EZH2P132S;Y646N was ineffectual in rescuing H3K27me3 levels, demonstrating that the activity of the hyperactive mutant was dependent on allosteric activation in vivo, and regulated in a manner similar to that of WT PRC2 (Figure 4D, lanes 5 and 6). To recapitulate the heterozygous nature of the EZH2Y646N mutation found in cancers, we co-expressed EZH2WT with either EZH2Y646N or EZH2P132S;Y646N in EZH2-null cells. In the presence of EZH2WT, EZH2Y646N manifested its expected hyperactivity for H3K27me3 catalysis (Figure 4E, lane 5), whereas EZH2P132S;Y646N was devoid of such hyperactivity (Figure 4E, lane 6). Thus, the P132S mutation epistatically overrode the effect of the Y646N gain-of-function mutation in catalyzing H3K27me3 in vivo.
Disrupting allosteric activation of PRC2/EZH2 effectively inhibits proliferation of PRC2/EZH2-addicted tumors
Next, we sought to evaluate the impact of allosteric activation of PRC2 on PRC2/EZH2-dependent human cancers. We focused on DLBCL cell lines that harbor EZH2Y646X mutations, as most EZH2Y646X-mutant cell lines showed a dependency on the activity of EZH2 and several SAM-competitive inhibitors are in clinical trials. We compared two EZH2Y646X-mutant DLBCL cell lines: KARPAS-422 (EZH2WT/Y646N), a known PRC2/EZH2-dependent cell line and SU-DHL-4 (EZH2WT/Y646S), an exceptional case without such EZH2-dependency. By using a lentivirus-based system, we first stably introduced an shRNA against the 3′ untranslated region (3′-UTR) of endogenous EZH2 mRNA transcripts in both cell lines. As expected, EZH2 knockdown led to a drastic reduction in H3K27me3 levels in both cell lines (Figure 5A, lanes 2 and 6), but only inhibited proliferation of KARPAS-422 cells (Figure 5B, black versus blue lines). Rescue by ectopic co-expression of EZH2WT and EZH2Y646N using lentiviruses restored H3K27me3 levels (Figure 5A, lane 7) and proliferation of KARPAS-422 cells (Figure 5B, right, red line), demonstrating that the effects of the EZH2 shRNA was on-target. However, rescue by co-expression of EZH2P132S and EZH2P132S;Y646N failed to negate the effects of EZH2 knockdown in these cells (Figure 5A, lane 8, and Figure 5B, right). On the other hand, co-expression of EZH2WT and EZH2Y646N partially rescued H3K27me3 levels in the SU-DHL-4 cells, likely due to the insufficient restoration of EZH2 levels (Figure 5A, lane 3). Nevertheless, concomitant expression of EZH2P132S and EZH2P132S;Y646N was ineffectual with respect to H3K27me3 levels and neither of the rescue conditions had any effect on SU-DHL-4 cell proliferation, due to their PRC2/EZH2-independency (Figure 5A, lane 4 and 5B, left). These proof-of-principle experiments highlight the requirement of PRC2 allosteric activation for H3K27me3 catalysis and the dependency on EZH2Y646X in DLBCL. These findings point to the importance of the SRM/SET-I interface as a potential target in a therapeutic strategy against cancers that rely on either WT or hyperactive PRC2/EZH2.
Figure 5. Disrupting allosteric activation of PRC2 impairs both H3K27me3 levels in EZH2Y646X-mutant DLBCLs and proliferation of PRC2-addicted cells.
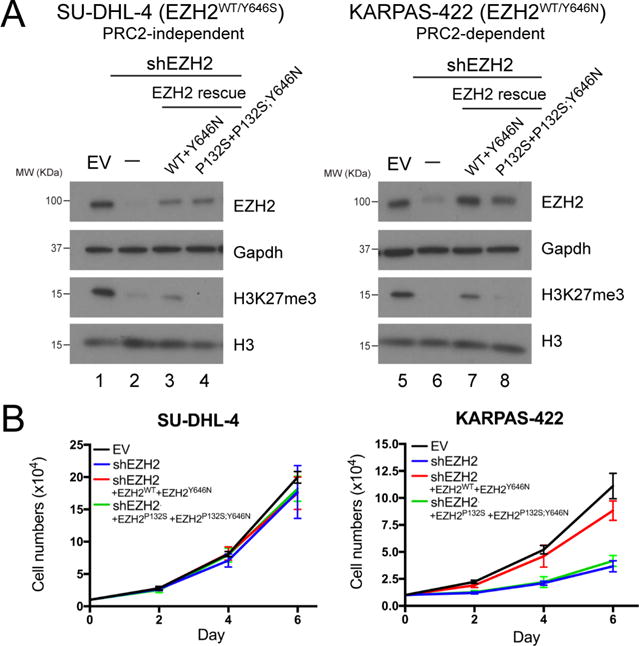
(A) Western blot analysis of EZH2, H3K27me3, and total histone H3 levels in SU-DHL-4 (left, PRC2-independent) and KARPAS-422 (right, PRC2-independent) DLBCL cells with control (EV), EZH2 knockdown (shEZH2), and EZH2 rescue conditions, as indicated on the right side. Gapdh was used as a loading control. EV, empty vector.
(B) Proliferation assays for SU-DHL-4 (left) and KARPAS-422 (right) cells under the abovementioned conditions. 10,000 cells were seeded in 6-well plates and cell numbers were counted after 2, 4, and 6 days. The culture medium was replenished every two days. Cell numbers are shown as Mean ± SD (n=3 for each data point).
Disrupting EZH2-SRM/SET-I interaction by alpha-helical mimetics selectively inhibits allosteric activation of PRC2
To thwart the α-helical-mediated interaction between the SRM and SET-I domain of EZH2, we utilized an oligopyridylamide scaffold-based α-helix mimetic approach. The oligopyridylamides form a network of intramolecular hydrogen bonds that stabilize a single conformation and project side chain functionalities from one face that correspond to the side chain residues of an α-helical conformation at i, i+3 (i+4), and i+7 (Davis et al., 2007). The use of these oligopyridylamides effectively disrupts α-helix-mediated protein-protein interactions (Kumar and Hamilton, 2017; Kumar et al., 2015). To disrupt the SRM/SET-I helical interaction, we synthesized oligopyridylamides by designing the surface functionalities complementary to the side chain residues of the SRM helical interface (Figure 6A and 6B). For example, to mimic side chain residues of the SRM helix (residues D142, F145, and L149), we designed oligopyridylamides ADH-61 (side chain groups: Amine, Benzyl, Benzyl). We conducted a structural activity relationship study to gauge the mode of action of the oligopyridylamides against the SRM/SET-I interface and ADH-61 was predicted to be one of the most potent antagonists. We postulated that the amine group on ADH-61 forms a salt bridge with the carboxylate group of D142 and that the two benzyl groups on ADH-61 interact with the hydrophobic side chains (F145 and L149) of the SRM helix (Figure 6A and 6B, left). ADH-24 served as a negative control in which the amine group was replaced with carboxylate and the central planar benzyl group was replaced with a non-planar cyclohexane in order to diminish its hydrophobicity as well as the salt bridge interaction with the SRM helix (Figure 6A and 6B, right). We applied these α-helix mimetics to the HMT assays, in the absence or presence of the allosteric activator (H3K27me3 peptide). As predicted, incubation with ADH-61 selectively inhibited the response of reconstituted WT PRC2 to the stimulatory H3K27me3 peptide, whereas ADH-24 was ineffectual (Figure 6C). Note that the PRC2 complex remained stable in the presence of ADH-61 during allosteric activation (Figure S5B). The half maximal inhibitory concentration (IC50) of ADH-61 was 0.766 ± 0.710 μM for inhibition of PRC2 HMT activity in vitro (Figure 6D). Moreover and importantly, neither ADH-61 nor ADH-24 exhibited an overt effect on the basal activity of either WT or mutant PRC2 (Figure 6E and S5C). The data indicate that ADH-61 likely inhibited PRC2 HMT activity by targeting the SRM/SET-I interface. We also investigated the effect of ADH-61 on the gain-of-function mutant, PRC2/EZH2Y646N. Using H3K27me2 oligonucleosomes as template, the gain-of-function of PRC2/EZH2Y646N towards H3K27 tri-methylation was efficiently inhibited by ADH-61 in a dose-dependent manner with an IC50 of 0.891 ± 0.096 μM, while ADH-24 had no significant effect (Figures 6F and 6G). Together, these results provided the proof-of-principle for the concept that ordering of the SRM and the ensuing SRM/SET-I interaction are central to PRC2 allosteric activation.
Figure 6. Oligopyridylamide-mediated selective inhibition of the allosteric activation of PRC2.
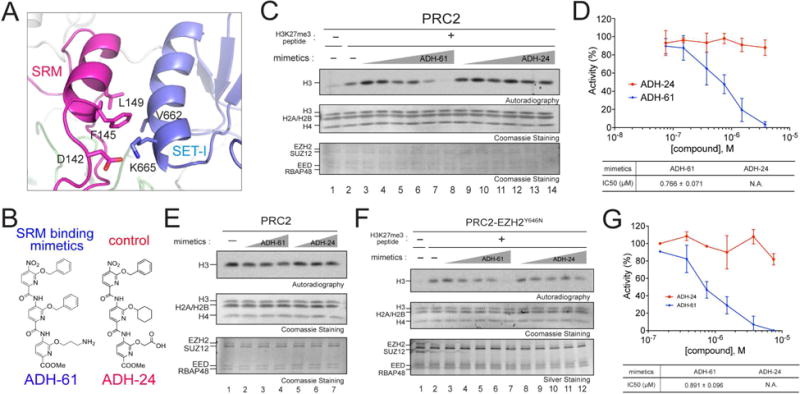
(A) An enhanced image depicting molecular features of the EZH2-SRM/SET-I interface. Key residues on the interface of SRM and SET-I α-helices are highlighted (modified from PDB:5HYN). Note that the numbers of EZH2 residues are based on human EZH2 isoform a (751 amino acids).
(B) Chemical structures of the α-helix mimetics used in the study.
(C) Representative HMT assays containing 7.5 nM PRC2 in the absence or presence of the H3K27me3 peptide (150 nM), with increasing amounts (0.075, 0.15, 0.375, 0.75, 1.5 or 3.75 μM) of the α-helix mimetic compounds using unmodified oligonucleosomes (150 nM) as substrate. Autoradiography shows the levels of methylation on histone H3 (top) and Coomassie blue staining of SDS-PAGE gels shows histones (middle) or PRC2 components (bottom).
(D) Top, Quantification of the HMT assays described in (C) Data were plotted as Mean±SD (n=3 for each data point). Bottom, IC50 was extracted for ADH-61 using a dose dependent study.
(E) Representative images of the HMT assays containing 7.5 nM PRC2 with increasing amounts (0.75, 1.5 or 3.75 μM) of the α-helix mimetic compounds in the absence of the H3K27me3 peptide using unmodified oligonucleosomes (150 nM) as substrate. Other panels are as described in (C).
(F) Top, Representative images of HMT assays containing 10 nM PRC2/EZH2Y646N with increasing amounts (0.15, 0.375, 0.75, 1.5, or 3.75 μM) of ADH-61 or ADH-24 in the absence or presence of the H3K27me3 peptide (300 nM) using H3K27me2 oligonucleosomes (150 nM) as substrate. Other panels are as described in (C).
(G) Quantification of the HMT assays described in (F). Data were plotted as Mean±SD (n=3 for each data point). Bottom, IC50 was extracted for ADH-61 using a dose dependent study.
Discussion
Mammalian heterochromatin contains large repressive chromatin domains, including H3K9me2/3-decorated constitutive heterchromatin at pericentromeric and telomeric regions and H3K27me2/3-decorated facultative heterochromatin with a more context-dependent deposition (Margueron and Reinberg, 2010; Shilatifard, 2006). The writers of these two classes of histone modifications include Suv39h1/2 and PRC2, respectively. These enzymes are regulated by a “read-and-write” mechanism whereby they recognize their own catalytic product through a chromodomain (CD) and an aromatic cage of EED, respectively. In turn, such recognition stimulates their enzymatic activity as a positive feedback loop that is determinant for the spreading and the formation of the respective repressive chromatin domain (Liu et al., 2015; Margueron et al., 2009; Müller et al., 2016; Zhang et al., 2015). Although the exact mechanism of allosteric activation of the enzymes that catalyze H3K9 methylation are not clearly defined, the studies presented here provide a detailed mechanism of H3K27me2/3-mediated allosteric activation by PRC2.
By identifying PRC2 mutants that specifically abrogate its allosteric activation, we demonstrated that this effect is crucial for its activity in vivo and the requirement for its basal activity observed in vitro may not be met in vivo, at least in mESCs (Figures 1 and 2). Our findings also highlight an unprecedented role of EZH2-SRM/EED anchorage in addition to the EZH2-SRM/SET-I interface in mediating the allosteric effect on H3K27 methylation. This notion is further supported by the EED anchorage mutations found in patients affected by Weaver Syndrome (Table S1 and Figure S2E) (Cohen, A.S.A., Gibson, 2016; Cohen et al., 2015; Cooney et al., 2017). Unlike the initial discovery of EED cage mutants, the EZH2-SRM and EED anchoring mutants are not defective in H3K27me3 binding (Figure S3A), allowing us to specifically segregate the allosteric effect and dissect the stepwise action of PRC2. An interesting observation from our study is that the EZH2-SRM and EED anchoring mutants manifested a spectrum of inhibitory effects on the allosteric activation of PRC2, with several of them retaining the ability to catalyze H3K27me2 in vivo (Figure 2A and 2B). These results implicate the depletion of H3K27me3, but not that of H3K27me2, as being key to the development of those diseases associated with these respective mutations. Further analysis of mutants specifically defective in H3K27me3 catalysis should be performed to elucidate the potentially unique role of H3K27me3 in these contexts.
Inherently, EZH2 catalyzes H3K27me2 more efficiently than H3K27me3 (Figure 4A–4C), as the Y646 residue within the SET domain likely restricts the transition from H3K27me2 to H3K27me3 (McCabe et al., 2012b). Intriguingly, the gain-of-function EZH2Y646X oncogenic mutations were shown to break this rate-limiting step by switching substrate preference and therefore, specifically promoting the conversion of H3K27me2 to H3K27me3. Such a deregulation of the catalytic kinetics is central to the oncogenesis in DLBCL, underscoring the importance of the rate-limiting control of the feed-forward mechanism and balancing endogenous H3K27me3 levels at undesired loci underlying disease pathogenesis (Bödör et al., 2011; McCabe et al., 2012a; Morin et al., 2010; Wigle et al., 2011; Yap et al., 2011). Importantly, our results demonstrated that the allosteric effect not only governs the activity of WT PRC2 in vivo, but also surpassed the hyperactivity of PRC2 containing the EZH2Y646N mutation (Figures 4D and 4E). In PRC2/EZH2-addicted DLBCL cells, genetic inactivation of the allosteric effect suppressed their proliferation as well as their endogenous H3K27me3 levels (Figure 5). Together with the proof-of-principle experiments exploiting the α-helix mimetics against the EZH2-SRM/SET-I interaction (Figure 6), these results provide a molecular and functional basis for future development of a different class of allosteric inhibitor in addition to the recently developed one that binds to the aromatic cage of EED (He et al., 2017; Li et al., 2017c; Qi et al., 2017). Albeit challenging, a number of protein-protein interaction (PPI) inhibitors have been developed and are under clinical testing, such as the BCL2-BH3 domain inhibitor, ABT-199 and the MDM2/TP53 interaction inhibitor, RG7112 (Andreeff et al., 2016; Arkin et al., 2014; Sheng et al., 2015; Souers et al., 2013; Tovar et al., 2013). In addition, the interfaces of EZH2-SRM/SET-I and EZH2-SRM/EED contain a groove surface feature (Figure S6), which may offer ideal target sites for the screening or structural design of potential small molecular inhibitors against the allosteric activation of PRC2.
To date, a number of factors have been implicated in regulating PRC2 recruitment to chromatin, perhaps reflective of the distinct types of genes targeted by PRC2 and that PRC2, by default, functions generally to maintain a repressed transcriptional state (Comet et al., 2016; Margueron and Reinberg, 2011). At imprinted genes or at active protein coding genes, the main PRC2 recruiter appears to be non-coding RNA (ncRNA) or the nascent transcript itself, respectively (Inoue et al., 2017; Kaneko et al., 2014b; Plath et al., 2003; Zhao et al., 2008). We suggest that at these genes, PRC2-mediated repression occurs passively, as silencing of gene expression is intially established by other factors (such as DNA metylation or sequence-specific repressors, respectively) prior to PRC2 recruitment to target DNA sequences. However, at other genes, such as those that regulate lineage commitment, the role of PRC2 is much more active. Upon initial PRC2 recruitment, the deposition of H3K27me2/3 is critical and highly dependent on its allosteric activation. Recruitment can be mediated by accessory factors known to associate with PRC2, such as JARID2, AEBP2, and PCLs, as well as DNA itself (see review (Holoch and Margueron, 2017), and “Lee and Holder et al., Mol Cell, this issue”). Among these factors, MTF2 (PCL2) and JARID2 might be pivotal (Li et al., 2017b; Oksuz et al., 2018). Indeed, MTF2 binds to CpG containing DNA and its absence in mESCs revealed reduced deposition of PRC2 on chromatin. Given that PRC2 methylates JARID2 at lysine 116 (JARID2-K116me3), which functions similarly to H3K27me3, we speculate that JARID2-K116me3 is important in igniting the allosteric mechanism upon PRC2 recruitment to its target genes for de novo H3K27me3 deposition.
In addition, a very interesting finding from our study is the uncoupling of PRC2 recruitment and catalysis in the context of chromatin (Figure 3). The EZH2-SRM mutants displayed increased and more pervasive association with chromatin as compared to WT EZH2, implying that the activated state of PRC2 might restrict its local occupancy independently of its initial recruitment. Is the actual deposition of H3K27me2/3 important in restricting/delineating PRC2-mediated repressed domains? If this were to be the case, as the data suggests, then H3K27me2/3 has yet an unknown function that perhaps requires the action of other factors.
In summary, our data defined the intermediate state during the allosteric activation of PRC2, which involved a structural remodeling and interplay between EZH2 and EED. We also demonstrated that the allosteric effect of PRC2 is a dominant feature in catalyzing H3K27 methylation in the context of chromatin comprising genes at which PRC2 is a major determinant in establishing an effective repressed state. Yet surprisingly, its allosteric activation can be uncoupled from PRC2 recruitment. Our findings that abrogating allosteric activation can suppress EZH2Y646X hyperactivity and DLBCL proliferation underscores the possibility of devising a novel class of allosteric inhibitors that target the SRM/SET-I interaction or SRM/EED anchorage against PRC2/EZH2-addicted cancers.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Danny Reinberg (Danny.Reinberg@nyumc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Embryonic Stem Cells (mESCs)
The mESCs used in this study were derived from ES-E14TG2a. These cells were grown in standard ESC medium containing Lif, 1 μM MEK1/2 inhibitor (PD0325901) and 3 μM GSK3 inhibitor (CHIR99021) at 27 °C.
Cancer cell lines
The COLO-679 cell line was derived from malignant melanoma. SU-DHL-4 and KARPAS-422 cell lines were derived from DLBCLs. These cells were grown in standard RPMI medium at 27 °C following the instruc tion from ATCC or Sigma.
METHOD DETAILS
Purification of protein using Baculovirus expression system
To purify human PRC2 core complexes, FLAG-tagged-EED, 6xHIS-tagged-EZH2, SUZ12, and RBAP48 were cloned independently into a baculovirus expression vector, pFASTBac1 (Invitrogen). EZH2 or EED mutant constructs were generated by site-directed mutagenesis and mutations were confirmed by Sanger DNA sequencing. All four core components of PRC2 (FLAG-EED, 6xHIS-EZH2, SUZ12, and RBAP48) were co-expressed in Sf9 cells by baculovirus infection. After 60 hr of infection, Sf9 cells were resuspended in BC150 buffer (25 mM Hepes-NaOH, pH 7.8, 1 mM EDTA, 150 mM NaCl, 10 % glycerol, 1 mM DTT, and 0.1 % NP-40) with protease inhibitors (1 mM phenylmethlysulfonyl fluoride (PMSF), 0.1 mM benzamidine, 1.25 mg/ml leupeptin and 0.625 mg/ml pepstatin A) and phosphatase inhibitors (20 mM NaF and 1 mM Na3VO4). Cells were lysed by sonication (Fisher Sonic Dismembrator model 100), and WT or mutant recombinant PRC2 was tandemly purified through Ni-NTA agarose beads (Qiagen), FLAG-M2 agarose beads (Sigma), Q sepharose beads (GE healthcare) and glycerol gradient (15-35%) sedimentation. The EZH2(116-121A) mutant consists of six alanine mutations on the SAL (SET-activation Loop) domain of EZH2, thereby preventing the establishment of a stable conformation of the SET domain (Jiao and Liu, 2015).
Nucleosome reconstitution
Recombinant histones were generated as previously described (Lee et al., 2013; Yun et al., 2012). Briefly, each core histone was expressed in Rosetta (DE3) cells (Novagen), extracted from inclusion bodies, and purified by sequential anion and cation chromatography. For refolding recombinant octamers, equal amounts of histones were mixed and dialyzed into refolding buffer (10 mM Tris-HCl, pH 7.5, 2 M NaCl, 1 mM EDTA, and 5 mM β-mercaptoethanol). Octamers were further purified by size exclusion chromatography on a 24-mL Superdex 200 column (GE healthcare) in refolding buffer. Recombinant oligonucleosomes were reconstituted by sequential salt dialysis of octamers and plasmid having 12 601-nucleosome positioning sequences. The methyl-lysine analogue production for the generation of pseudo-monomethyl- and pseudo-dimethyl-H3K27 nucleosomes was previously described in detail (Yun et al., 2012).
HMT assay
Standard HMT assays were performed in a total volume of 15 μL containing HMT buffer (50 mM Tris-HCl, pH 8.5, 5 mM MgCl2, and 4 mM DTT) with 500 nM of 3H-labeled S-Adenosylmethionine (SAM, Perkin Elmer), 10 nM (500 ng) of recombinant oligonucleosomes consisting of 12× repeat nucleosome arrays (120 nM of nucleosome), and recombinant human PRC2 complexes. The reaction mixture was incubated for 60 min at 30 °C and stopped by the addition of 4 μL SDS buffer (0.2 M Tris-HCl, pH 6.8, 20% glycerol, 10% SDS, 10 mM β-mercaptoethanol, and 0.05% Bromophenol blue). A titration of PRC2 (from 5 to 60 nM) was performed under these conditions to optimize the HMT reaction within a linear range, and the yield of each HMT reaction was measured using the following procedures. After HMT reactions, samples were incubated for 5 min at 95 °C and separated on SDS-P AGE gels. The gels were then subjected to Coomassie blue staining for protein visualization or wet transfer of proteins to 0.45 μm PVDF membranes (Millipore). The radioactive signals were detected by exposure on autoradiography films (Denville Scientific). To measure “basal” HMT activity of PRC2, a titration of WT PRC2 or PRC2 harboring EZH2 or EED mutations was used at a concentration of 7.5, 15, or 30 nM. To measure the “stimulated” HMT activity of PRC2, 15 nM of PRC2 or PRC2 harboring EZH2 or EED mutations was used in the presence of an H3K27me3 peptide (amino acids 21-44) at a concentration of 0, 50, or 250 nM. All HMT assays were performed with the above indicated concentrations of PRC2 and H3K27me3 peptide unless otherwise noted in the figure legends.
Cell culture and EB formation
Mouse embryonic stem cells (mESCs) were grown in standard ESC medium containing Lif, 1 μM MEK1/2 inhibitor (PD0325901) and 3 μM GSK3 inhibitor (CHIR99021). For EB formation, 600K mESCs were plated in suspension plates with medium containing DMEM, 20% FBS, 1% Pen/Strep, 2 mM L-Glutamine, and 0.1 mM 2-mercaptoethanol, 1 mM vitamin C. Cells were collected 3 days after the induction of EB differentiation.
CRISPR/Cas9-mediated genome editing
An optimal gRNA target sequence closest to the genomic target site was chosen using the http://crispr.mit.edu/ design tool. The gRNA was cloned into the SpCas9-2A GFP (Addgene: PX458) or SpCas9-2A Puro (Addgene:PX459) via BbsI digestion and insertion(Ran et al., 2013). mESC cells were seeded into 12-well plates at 100,000 cells per well, and transfected with 0.5 μg of the appropriate guide RNAs, template DNA for guide RNAs (listed in Table 2) and Cas9 endonuclease using Lipofectamine 2000 (Life Technologies). The transfection was performed according to the manufacturer’s recommended protocol, using a 3:1 ratio of Lipofectamine:DNA. After transfection, GFP-positive cells were sorted using the Sony SY3200 cell sorter and 20,000 cells were plated on a 15 cm dish. Puromycin resistant cells were selected in 2 μg/mL puromycin for 48 hr 7-10 days later, single ESC clones were picked, trypsinized in 0.25 % Trypsin-EDTA for 5 min, and plated into individual wells of a 96-well plate for genotyping. Genomic DNA was harvested via QuickExtract (Epicentre) DNA extraction, and genotyping PCRs were performed using primers surrounding the target site. The resulting PCR products were purified and sequenced to determine the presence of a deletion or a mutation event. mESCs with a C-terminal deletion of EED were generated by a frame-shift deletion resulting in a stop codon at residue D371 of EED.
Lentiviral production and delivery
WT or mutant EZH2 and EED constructs were subcloned into the pLV-EF1-alpha-IRES-mCherry vector (Clontech) for lentiviral production and delivery. For the production of viral particles, lentiviral vectors containing WT or mutant EZH2 and EED (10 μg) were co-transfected with pcREV (2.5 μg), BH-10 (3 μg), and pVSV-G (5 μg) packaging vectors into 293-FT cells. The virus-containing medium was collected 48 hr after transfection and the target cells were spin-infected. Polybrene was added to the viral medium at a concentration of 8 μg/mL. Infected cells were FACS-sorted for mCherry.
Synthesis of α-helix mimetics
The monomers for the preparation of the oligopyridylamides were synthesized according to a previously published method (Kumar and Hamilton, 2017). Following the monomer synthesis, chain elongation of pyridylamides was achieved using iterative amide coupling between oligo-pyridylamines and monomeric-pyridylacids using 2-chloro-1-methylpyridinium iodide (Mukaiyama’s reagent) followed by reduction of the nitro groups. The acid labile tert-butyl esters and NH-Boc groups were cleaved using a trifluoroacetic acid (TFA) cocktail (dichloromethane/TFA/triethylsilane, 80:15:5, v/v/v) in the final step to attain the series of oligopyridylamides.
ChIP-seq and data analysis
Standard ChIP experiments were performed as previously described (Gao et al., 2012). 100 μg sonicated chromatin was used in each ChIP reaction with 2.5 μg of anti-EZH2 or anti-H3K27me3 antibody. 1 μg of Drosophila chromatin and 0.1 μg of anti-Drosophila H2A.X antibody were added in each ChIP reaction as spike-in references. For ChIP-seq library preparation, up to 30 ng of immunoprecipitated DNA was end-repaired, A-tailed, and ligated to custom barcode adapters with T4 ligase, as previously described (Gao et al., 2012). Libraries were sequenced on Illumina HiSeq. A custom barcoding system was employed. The ChIP-seq data were first mapped to the mouse genome (mm9 downloaded from UCSC genome browser) using the Bowtie2 software package (version 2.3.0). All reads that failed to align to the mouse genome were mapped to the fly genome (dm6 downloaded from UCSC genome browser) using the same software and criteria. The sum of the number of reads mapped to both genomes was used as the actual “library size”. The median of the library sizes across all the samples was used as the reference library size, which is about 20,000,000 reads. All samples were scaled to obtain the new library size equal to the reference library size. The input samples were normalized to the same total reference library size as the ChIP samples. After adjustment of the total library size, the number of reads mapped to the fly genome was used as the spike-in normalization factor. All samples were then scaled except for input samples in order to have the same total spike-in content. To obtain EZH2 binding sites, only reads with mapping quality greater than 30 were selected and removed as presumptive PCR duplicates with the same mapped coordinates. MACS2 software package was used (version 2.1.1) for calling significantly enriched peaks at a false discovery rate (FDR) less than 5% relative to the input samples. For H3K27me3 samples, SICER software package was used (version 1.1) to find broad peaks by setting the window size of 500 bp and allowing peaks within a distance of 500 bp to merge together. The significantly enriched peaks were filtered with the same criteria as above, FDR < 0.05.
Preparation of mESC whole cell extract
8×106 cells were harvested and lysed with standard RIPA buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, and 0.1% SDS) containing protease inhibitors (0.2 mM PMSF, 1 μg/mL Pepstatin A, 1 μg/mL Leupeptin, and 1 μg/mL Aprotinin) and phosphatase inhibitors (10 mM NaF and 1 mM Na3VO4). The cell suspension was briefly sonicated (40% amplitude, 5 strokes) and centrifuged at 20,000 × g at 4 °C for 20 min. The supernatant was collected as the whole cell extract.
Preparation of mESC nuclear extract
To prepare nuclear extract from mESC, approximately 4×107 cells were harvested and washed with PBS. Cells were lysed with intact nuclei in TMSD buffer (40 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 250 mM sucrose, 1 mM DTT, and 0.02% NP-40) containing protease inhibitors and phosphatase inhibitors, as indicated above. The cell suspension was centrifuged at 800 × g at 4 °C for 10 min. The pellets (nuclei) were resuspended in 20× cell pellet volume of BC400 buffer (20 mM Tris-HCl, pH 7.9, 400 mM KCl, 0.2 mM EDTA, 20% glycerol, 0.5 mM DTT, and 0.02% NP-40) with protease inhibitors and phosphatase inhibitors, and incubated at 4 °C for 1 hr. The cell suspension was centrifuged at 18,000 × g at 4 °C for 20 min. The supernatant was collected as the nuclear extract.
Immunoprecipitation
2 mg of mESC nuclear extract was incubated with 3 μg of corresponding antibody at 4 °C for 2 hr. 0.6 mg of Dynabeads Protein G (Thermo fisher) was added and incubated at 4 °C for 1 hr. The tubes were placed on magnets to separate the beads from the solution and unbound supernatant was removed. The beads were washed three times with BC400 buffer at 4 °C for 10 min. After the final wash, samples were incubated for 5 min at 95 °C to extract protein from the beads and separated on SDS-PAGE gels. The gels were then subjected to wet transfer of proteins to 0.45 μm PVDF membranes for Western Blotting.
Peptide binding assay
2 nmol of biotinylated H3K27(18-36) or H3K27me3(18-36) was immobilized to Streptavidin agarose resin (Millipore). 2 μg of recombinant PRC2 or PRC2 containing EED or EZH2 mutants was added and incubated at 4 °C for 1 hr. Unbound supernatants were removed and beads were washed three times with BC150 at 4 °C for 10 min. After the final wash, samples were incubated for 5 min at 95 °C to extract protein from the beads and the recovered protein was separated on SDS-PAGE gels. The gels were then subjected to Coomassie blue staining.
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphs display the mean ± SD from three independently performed experiments. GraphPad Prism 6.0 or Microsoft Excel was used for statistical analysis.
DATA AND SOFTWARE AVAILABILITY
The GEO number for ChIP-seq results in this study is GSE111433. Original gel images were uploaded to Mendeley Data: https://data.mendeley.com/datasets/7kpzswgdzf/2.
Supplementary Material
Highlights.
The EZH2-SRM and EED-anchorage specifically mediate the allosteric activation of PRC2
Activation-deficient PRC2 retain chromatin association despite minimal activity
SRM or EED anchorage mutations epistatically override the hyperactivity of EZH2Y646X
α-helical mimetics against SRM abrogate the allosteric activation of PRC2
Acknowledgments
We thank Drs. L. Vales, R. Margueron, K-J. Armache, and J. Wilson for critical reading of the manuscript as well as past and current Reinberg members for critical comments and discussion; D. Hernandez, Dr. O. Oksuz, K. Stafford, and Dr. J. Stafford for technical assistance. We also thank the NYU Flow Cytometry Core (grant: NIH/NCI P30CA016087) for cell purification. The work in D.R.’s lab is supported by NIH (R01CA199652) and the Howard Hughes Medical Institute (HHMI). The work in A.D.H’s lab is supported by New York University. The work in B.A.G’s lab is supported by NIH grants (R01GM110174) and (P01CA196539). J-R.Y. is supported by the American Cancer Society (PF-17-035-01). G.L. is supported by a grant from the Making Headway Foundation (189290).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
C-H.L., J-R.Y., and D.R. conceptualized and designed the study. C-H.L, J-R.Y., G.L., and S. Kaneko conducted the experiments. S. Kumar designed and synthesized the α-helical mimetics in A.D.H’s lab. Y.J. performed bioinformatics analyses. N.B. performed quantitative mass spectrometry analyses in B.A.G’s lab. J-R.Y, C-H.L, and S. Kumar wrote the manuscript with assistance from D.R.
Declaration of Interests
The authors declare no competing interests.
References
- Aloia L, Di Stefano B, Di Croce L. Polycomb complexes in stem cells and embryonic development. Development. 2013;140:2525–2534. doi: 10.1242/dev.091553. [DOI] [PubMed] [Google Scholar]
- Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, Bowen D, Martinelli G, Drummond MW, Vyas P, et al. Results of the phase i trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res. 2016;22:868–876. doi: 10.1158/1078-0432.CCR-15-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin MR, Tang Y, Wells JA. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, Salvesen HB, Otte AP, Akslen LA. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24:268–273. doi: 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- Baker T, Nerle S, Pritchard J, Zhao B, Rivera VM, Garner A, Gonzalvez F. Acquisition of a single EZH2 D1 domain mutation confers acquired resistance to EZH2-targeted inhibitors. Oncotarget. 2015;6 doi: 10.18632/oncotarget.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. EZH2 Is Required for Germinal Center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer Cell. 2013;23:677–692. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bödör C, O’Riain C, Wrench D, Matthews J, Iyengar S, Tayyib H, Calaminici M, Clear A, Iqbal S, Quentmeier H, et al. EZH2 Y641 mutations in follicular lymphoma. Leuk Off J Leuk Soc Am Leuk Res Fund, UK. 2011;25:726–729. doi: 10.1038/leu.2010.311. [DOI] [PubMed] [Google Scholar]
- Bracken AP, Helin K. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat Rev Cancer. 2009;9:773–784. doi: 10.1038/nrc2736. [DOI] [PubMed] [Google Scholar]
- Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley WD, Arora S, Busby J, Balasubramanian S, Gehling VS, Nasveschuk CG, Vaswani RG, Yuan CC, Hatton C, Zhao F, et al. EZH2 inhibitor efficacy in non-Hodgkin’s lymphoma does not require suppression of H3K27 monomethylation. Chem Biol. 2014;21:1463–1475. doi: 10.1016/j.chembiol.2014.09.017. [DOI] [PubMed] [Google Scholar]
- Brooun A, Gajiwala KS, Deng YL, Liu W, Bolaños B, Bingham P, He YA, Diehl W, Grable N, Kung PP, et al. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat Commun. 2016;7:11384. doi: 10.1038/ncomms11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou RH, Yu YL, Hung MC. The roles of EZH2 in cell lineage commitment. Am J Transl Res. 2011;3:243–250. [PMC free article] [PubMed] [Google Scholar]
- Cohen ASA, Tuysuz B, Shen Y, Bhalla SK, Jones SJM, Gibson WT. A novel mutation in EED associated with overgrowth. J Hum Genet. 2015;60:339–342. doi: 10.1038/jhg.2015.26. [DOI] [PubMed] [Google Scholar]
- Cohen ASA, Gibson WT. EED-associated overgrowth in a second male patient. J Hum Genet. 2016:1–4. doi: 10.1038/jhg.2016.51. [DOI] [PubMed] [Google Scholar]
- Comet I, Riising EM, Leblanc B, Helin K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat Rev Cancer. 2016;16:803–810. doi: 10.1038/nrc.2016.83. [DOI] [PubMed] [Google Scholar]
- Cooney E, Bi W, Schlesinger AE, Vinson S, Potocki L. Novel EED mutation in patient with Weaver syndrome. Am J Med Genet Part A. 2017;173:541–545. doi: 10.1002/ajmg.a.38055. [DOI] [PubMed] [Google Scholar]
- Curtin ML, Pliushchev MA, Li HQ, Torrent M, Dietrich JD, Jakob CG, Zhu H, Zhao H, Wang Y, Ji Z, et al. SAR of amino pyrrolidines as potent and novel protein-protein interaction inhibitors of the PRC2 complex through EED binding. Bioorganic Med Chem Lett. 2017;27:1576–1583. doi: 10.1016/j.bmcl.2017.02.030. [DOI] [PubMed] [Google Scholar]
- Davis JM, Tsou LK, Hamilton AD. Synthetic non-peptide mimetics of α-helices. Chem Soc Rev. 2007;36:326–334. doi: 10.1039/b608043j. [DOI] [PubMed] [Google Scholar]
- Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su I hsin, Hannon G, Tarakhovsky A, Fuchs E. Ezh2 Orchestrates Gene Expression for the Stepwise Differentiation of Tissue-Specific Stem Cells. Cell. 2009;136:1122–1135. doi: 10.1016/j.cell.2008.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Zhang J, Bonasio R, Strino F, Sawai A, Parisi F, Kluger Y, Reinberg D. PCGF Homologs, CBX Proteins, and RYBP Define Functionally Distinct PRC1 Family Complexes. Mol Cell. 2012;45:344–356. doi: 10.1016/j.molcel.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibaja V, Shen F, Harari J, Korn J, Ruddy D, Saenz-Vash V, Zhai H, Rejtar T, Paris CG, Yu Z, et al. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene. 2016;35:558–566. doi: 10.1038/onc.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Selvaraju S, Curtin ML, Jakob CG, Zhu H, Comess KM, Shaw B, The J, Lima-Fernandes E, Szewczyk MM, et al. The EED protein–protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat Chem Biol. 2017;2:1–10. doi: 10.1038/nchembio.2306. [DOI] [PubMed] [Google Scholar]
- Holoch D, Margueron R. Mechanisms Regulating PRC2 Recruitment and Enzymatic Activity. Trends Biochem Sci. 2017;42:531–542. doi: 10.1016/j.tibs.2017.04.003. [DOI] [PubMed] [Google Scholar]
- Huang Y, Zhang J, Yu Z, Zhang H, Wang Y, Lingel A, Qi W, Gu J, Zhao K, Shultz MD, et al. Discovery of First-in-Class, Potent, and Orally Bioavailable Embryonic Ectoderm Development (EED) Inhibitor with Robust Anticancer Efficacy. J Med Chem. 2017;60:2215–2226. doi: 10.1021/acs.jmedchem.6b01576. [DOI] [PubMed] [Google Scholar]
- Imagawa E, Higashimoto K, Sakai Y, Numakura C, Okamoto N, Matsunaga S, Ryo A, Sato Y, Sanefuji M, Ihara K, et al. Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome. Hum Mutat. 2017;38:637–648. doi: 10.1002/humu.23200. [DOI] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, Suzuki T, Zhang Y. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature. 2017;547:419–424. doi: 10.1038/nature23262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao L, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science (80-) 2015;350:291. doi: 10.1126/science.aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. 2016;7:11316. doi: 10.1038/ncomms11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Bonasio R, Saldaña-Meyer R, Yoshida T, Son J, Nishino K, Umezawa A, Reinberg D. Interactions between JARID2 and Noncoding RNAs Regulate PRC2 Recruitment to Chromatin. Mol Cell. 2014a;53:290–300. doi: 10.1016/j.molcel.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Son J, Bonasio R, Shen SS, Reinberg D. Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev. 2014b;28:1983–1988. doi: 10.1101/gad.247940.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nat Med. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Hamilton AD. α-Helix Mimetics as Modulators of Aβ Self-Assembly. J Am Chem Soc. 2017;139:5744–5755. doi: 10.1021/jacs.6b09734. [DOI] [PubMed] [Google Scholar]
- Kumar S, Schlamadinger DE, Brown MA, Dunn JM, Mercado B, Hebda JA, Saraogi I, Rhoades E, Hamilton AD, Miranker AD. Islet amyloid-induced cell death and bilayer integrity loss share a molecular origin targetable with oligopyridylamide-based α-helical mimetics. Chem Biol. 2015;22:369–378. doi: 10.1016/j.chembiol.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Wu J, Li B. Chromatin remodelers fine-tune h3k36me-directed deacetylation of neighbor nucleosomes by Rpd3S. Mol Cell. 2013;52:255–263. doi: 10.1016/j.molcel.2013.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1227–1232. doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 2010;24:368–380. doi: 10.1101/gad.1886410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Liefke R, Jiang J, Kurland JV, Tian W, Deng P, Zhang W, He Q, Patel DJ, Bulyk ML, et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature. 2017a;549:287–291. doi: 10.1038/nature23881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Liefke R, Jiang J, Kurland JV, Tian W, Deng P, Zhang W, He Q, Patel DJ, Bulyk ML, et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature. 2017b;549:287–291. doi: 10.1038/nature23881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhang H, Zhang M, Zhao M, Feng L, Luo X, Gao Z, Huang Y, Ardayfio O, Zhang JH, et al. Discovery and molecular basis of a diverse set of polycomb repressive complex 2 inhibitors recognition by EED. PLoS One. 2017c;12 doi: 10.1371/journal.pone.0169855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Li L, Zhao K, Xu Q, Qi V, Teng L, Das R, Sheng Q, Gu J, Atadja P, et al. Tyrosine 646 mutation of EZH2 is neomorphic to promote H3K27 trimethylation. Nat Genet 2010 [Google Scholar]
- Liu N, Zhang Z, Wu H, Jiang Y, Meng L, Xiong J, Zhao Z, Zhou X, Li J, Li H, et al. Recognition of H3K9 methylation by GLP is required for efficient establishment of H3K9 methylation, rapid target gene repression, and mouse viability. Genes Dev. 2015;29:379–393. doi: 10.1101/gad.254425.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet. 2010;11:285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. Ezh1 and Ezh2 Maintain Repressive Chromatin through Different Mechanisms. Mol Cell. 2008;32:503–518. doi: 10.1016/j.molcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, Voigt P, Martin SR, Taylor WR, De Marco V, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012a;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27) Proc Natl Acad Sci. 2012b;109:2989–2994. doi: 10.1073/pnas.1116418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller MM, Fierz B, Bittova L, Liszczak G, Muir TW. A two-state activation mechanism controls the histone methyltransferase Suv39h1. Nat Chem Biol. 2016;12:188–193. doi: 10.1038/nchembio.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obier N, Lin Q, Cauchy P, Hornich V, Zenke M, Becker M, Müller AM. Polycomb Protein EED is Required for Silencing of Pluripotency Genes upon ESC Differentiation. Stem Cell Rev Reports. 2015;11:50–61. doi: 10.1007/s12015-014-9550-z. [DOI] [PubMed] [Google Scholar]
- Oksuz O, Narendra V, Lee CH, Descostes N, LeRoy G, Raviram R, Blumenberg L, Karch K, Rocha PR, Garcia BA, et al. Capturing the onset of PRC2-mediated repressive domain formation. bioRxiv. 2018 doi: 10.1016/j.molcel.2018.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol Cell Biol. 2007;27:3769–3779. doi: 10.1128/MCB.01432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300:131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- Qi W, Zhao K, Gu J, Huang Y, Wang Y, Zhang H, Zhang M, Zhang J, Yu Z, Li L, et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol. 2017;226:1–11. doi: 10.1038/nchembio.2304. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PDP, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanulli S, Justin N, Teissandier A, Ancelin K, Portoso M, Caron M, Michaud A, Lombard B, da Rocha ST, Offer J, et al. Jarid2 Methylation via the PRC2 Complex Regulates H3K27me3 Deposition during Cell Differentiation. Mol Cell. 2015;57:769–783. doi: 10.1016/j.molcel.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng C, Dong G, Miao Z, Zhang W, Wang W. State-of-the-art strategies for targeting protein–protein interactions by small-molecule inhibitors. Chem Soc Rev. 2015;44:8238–8259. doi: 10.1039/c5cs00252d. [DOI] [PubMed] [Google Scholar]
- Shilatifard A. Chromatin Modifications by Methylation and Ubiquitination: Implications in the Regulation of Gene Expression. Annu Rev Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010;107:20980–20985. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Shen SS, Margueron R, Reinberg D. Nucleosome-binding activities within JARID2 and EZH1 regulate the function of PRC2 on chromatin. Genes Dev. 2013;27:2663–2677. doi: 10.1101/gad.225888.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu RM, Zhu P, Li G. Cryo-EM Study of the Chromatin Fiber Reveals a Double Helix Twisted by Tetranucleosomal Units. Science (80-) 2014;344:376–380. doi: 10.1126/science.1251413. [DOI] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Tan JZ, Yan Y, Wang XX, Jiang Y, Xu HE. EZH2: biology, disease, and structure-based drug discovery. Acta Pharmacol Sin. 2014;35:161–174. doi: 10.1038/aps.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton-Brown K, Murray A, Hanks S, Douglas J, Armstrong R, Banka S, Bird LM, Clericuzio CL, Cormier-Daire V, Cushing T, et al. Weaver syndrome and EZH2 mutations: Clarifying the clinical phenotype. Am J Med Genet Part A. 2013;161:2972–2980. doi: 10.1002/ajmg.a.36229. [DOI] [PubMed] [Google Scholar]
- Tovar C, Graves B, Packman K, Filipovic Z, Xia BHM, Tardell C, Garrido R, Lee E, Kolinsky K, To KH, et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res. 2013;73:2587–2597. doi: 10.1158/0008-5472.CAN-12-2807. [DOI] [PubMed] [Google Scholar]
- Wigle TJ, Knutson SK, Jin L, Kuntz KW, Pollock RM, Richon VM, Copeland RA, Scott MP. The Y641C mutation of EZH2 alters substrate specificity for histone H3 lysine 27 methylation states. FEBS Lett. 2011;585:3011–3014. doi: 10.1016/j.febslet.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Xu S, Grullon S, Ge K, Peng W. Spatial clustering for identification of chip-enriched regions (SICER) to map regions of histone methylation patterns in embryonic stem cells. Methods Mol Biol. 2014;1150:97–111. doi: 10.1007/978-1-4939-0512-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap DB, Chu J, Berg T, Schapira M, Cheng SWG, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117:2451–2459. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun M, Ruan C, Huh JW, Li B. Reconstitution of modified chromatin templates for in vitro functional assays. Methods Mol Biol. 2012;833:237–253. doi: 10.1007/978-1-61779-477-3_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications – writers that read. EMBO Rep. 2015;16:1467–1481. doi: 10.15252/embr.201540945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9 doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–756. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.