

Abstract
Generation of conditional mutants in Trypanosoma brucei can be done by the use of RNA interference (RNAi). However, RNAi frequently produces off target effects. Here we present an alternative strategy in which the glmS ribozyme is inserted in the C-terminal region of one allele of a GOI and effectively knocks it down in response to the presence of glucosamine in the culture medium. Using several endogenous genes we show that the glmS ribozyme cleaves the mRNA in vivo leading to reduction in mRNA and protein expression following glucosamine treatment in both T. brucei procyclic and bloodstream forms. Glucosamine-induced ribozyme activation can be rapidly reversed by removing the inducer. In summary, the glmS ribozyme could be used as a tool to study essential genes in T. brucei.
Keywords: glucosamine, ribozyme, riboswitch, Trypanosoma brucei
Trypanosoma brucei belongs to the group of protist parasites that causes African trypanosomiasis or sleeping sickness, and is an excellent model for the study of trypanosomatids because of the availability of genetic tools for its manipulation (Clayton 1999, Docampo 2011). These tools, such as inducible gene expression, are essential for the identification and validation of new targets for drugs, vaccines or diagnostic purposes.
The fastest method for the generation of conditional mutants in T. brucei is the use of RNA interference (Alibu et al. 2005). This pathway was first reported in Caenorhabditis elegans (Fire et al. 1998) and soon after in T. brucei (Ngo et al. 1998), and is based on the ability of double stranded RNA (dsRNA) containing complementary sequences of interest to trigger downregulation of gene expression. Contrary to the results obtained in C. elegans (Fire et al. 1998), dsRNA transfection, although very effective in T. brucei RNAi, has only transient activity (Ngo et al. 1998). To overcome this limitation a tetracycline-regulated expression of dsRNA was established and T. brucei strains stably expressing the tet repressor and T7 RNA polymerase are used for this purpose (Wirtz and Clayton 1995, Wirtz et al. 1998). Strains expressing isopropyl β-D-1-thiogalactopyranoside (IPTG) (Tschopp et al. 2011), vanillic acid (Sunter 2016) and cumate (Li et al. 2017) switch systems have also been reported for inducible expression of tRNA (Tschopp et al. 2011) or proteins (Sunter 2016, Li et al. 2017) in T. brucei, although only the vanillic acid system (Sunter 2016) was appropriate for inducible RNAi.
There are many examples where RNAi has been shown to operate very effectively and specifically in T. brucei. However, in addition to the need to construct these strains, RNAi in T. brucei could trigger a number of unspecific events such as off target effects (Rusconi et al. 2005, Merritt and Stuart 2013), leakage (synthesis of dsRNA in the absence of tetracycline inducer (Motyka and Englund 2004)), loss of expression control (escape mutants (Motyka and Englund 2004)), or the incomplete reduction of the protein levels. Furthermore, the absence of a growth phenotype does not prove that the protein is not required for viability (Motyka and Englund 2004). Complementation with a tetracycline-regulated extra copy of the gene of interest to allow the knockout of the endogenous alleles in a cell line stably expressing a tet repressor and the T7 RNA polymerase (inducible knockout) has also been successfully employed although some of its drawbacks are the leakage of the system, the need to use a specific strain of T. brucei, and the trypanosome facility for mutations (estimated at 10−6 per cell per generation, (Valdés J 1996)) that could lead to loss of expression control (Clayton 1999).
An alternative method for the conditional loss of gene function mediated at the mRNA level is the use of riboswitches, which are naturally occurring self-cleaving RNAs (ribozymes) that can be modified to respond to ligands (Winkler et al. 2004). An example is the glmS gene from Bacilus subtilis, which has been shown to control reporter gene expression in response to exogenous glucosamine in other eukaryotes, such as Saccharomyces cerevisiae (Watson and Fedor 2011) and Plasmodium falciparum (Prommana et al. 2013). The glmS gene encodes the enzyme glutamine-fructose 6-phosphate amidotransferase that uses fructose 6-phosphate and glutamine to generate glucosamine 6-phosphate (GlcN6P). A conserved element in the 5′-unstranslated region of this gene acts, when transcribed into RNA, as a self-cleaving riboswitch stimulated by glucosamine 6-phosphate (GlcN6P), leading to specific scission of one internucleotide phosphodiester bond of the gene of interest (Winkler et al. 2004). When this element is inserted in the 5′-UTR or the 3′-UTR of a gene of interest the self-cleaving RNA will silence it when in the presence of GlcN6P produced within the cells. Addition of glucosamine to the culture medium will stimulate this activity through the endogenous generation of GlcN6P (Fig. S1). A mutant glmS element that has no self-cleaving activity (M9) can be used as negative control (Winkler et al. 2004).
In this work, we report the use of the glmS ribozyme as a valid alternative to the use of RNAi or inducible knockouts in T. brucei but lacking some of their limitations. Tagging of only one allele of the gene of interest (GOI) was sufficient to down-regulate gene expression at the mRNA level and, in some cases, produce phenotypic changes. Any strain of T. brucei can be used with this approach.
MATERIALS AND METHODS
Culture methods
T. brucei PCF and BSF trypanosomes (Lister strain 427) were used for the ribozyme experiments. PCF trypanosomes were grown at 27°C in SDM-79 medium (Cunningham 1977), with or without the addition of 5 mM D-glucosamine, and supplemented with hemin (7.5 μg/mL) and 10% heat-inactivated fetal bovine serum. BSF trypanosomes were grown at 37°C in HMI-9 medium (Hirumi and Hirumi 1989), supplemented with 10% fetal bovine serum (FBS), and 2.5 mM glucosamine where indicated. For the RNAi experiments PCF 29-13 and BSF single marker strains (gifts from Dr. George A.M. Cross, Rockefeller University, New York) were used as reported before (Huang et al. 2014). Growth was determined by counting cells in a Neubauer chamber.
Chemicals and reagents
The pMOTag4H vector was a gift from Dr. Thomas Seebeck (University of Bern, Bern, Switzerland). Monoclonal antibody L8C4 was a gift from Dr. Phillippe Bastin (Institute Pasteur, Paris). Wild type and M9 mutated Bacillus subtilis glmS ribozymes were gifts from Dr. Vasant Muralidharan (University of Georgia). Q5® Hot Start High-Fidelity DNA polymerase, BenchMark protein ladder, Alexa-conjugated secondary antibodies were purchased from Invitrogen (ThermoFisher Scientific, Waltham, MA). Purified HA.11 clone 16B12 monoclonal antibody against HA was purchased from Covance (Biolegend, San Diego, CA). The protein assay reagent, Magic Marker, Zeta-Probe GT Genomic Testing blotting and nitrocellulose membranes were from Bio-Rad Laboratories (Hercules, CA). AMAXA human T-cell Nucleofector kit was purchased from Lonza (Allendale, NJ). The primers were purchased from Integrated DNA Technologies (Coralville, IO). TRI® reagent and all other reagents of analytical grade were from Sigma-Aldrich (St. Louis, MO).
Construction of plasmids
The pMOTag4H vector designed for endogenous C-terminal tagging of T. brucei (Oberholzer et al. 2006) was used to construct the transfection plasmids. This vector contains a 3xHA tag and the hygromycin resistance marker (Fig. 1A). Wild-type and M9 mutated Bacillus subtilis glmS ribozyme sequences (Winkler et al. 2004) were used to generate the pMOTag-glmS-4H and pMOTag-M9-4H plasmids in one-step. PCR was performed with a primer set containing the SalI site (primers 1, 2 and 3, Table S1) to amplify the glmS ribozyme and the mutated M9 ribozyme using glmS-ribozyme-TOPO and M9-ribozyme-TOPO plasmids as templates. PCR was done in a reaction volume of 100 μl using Q5® Hot Start High-Fidelity DNA polymerase with ~100 ng of gDNA as follows: initial denaturation for 2 min at 95°C followed by 35 cycles of 30 s at 95°C, 30 s at 60°C, 15 s at 72°C and then a final extension for 2 min at 72°C. The amplified fragment was cloned into pMOTag4H, digested using SalI enzyme and subsequently the linear plasmid was dephosphorylated to prevent re-ligation. Insert correct orientation was determined by PCR and sequencing. For C-terminal tagging, the one-step epitope-tagging protocol reported by Oberholzer et al. (Oberholzer et al. 2006) was used to produce C-terminal tagging cassettes for transfection. Templates for homologous recombination were amplified by PCR. In brief, the PCR forward and reverse primers (primers 4 to 11, Table S1) included the terminal 100 nucleotides of each ORF before its stop codon and the reverse complement of the first 100 nucleotides of the 3′UTR, respectively, followed in frame by the 21 nucleotides of the backbone sequences of the modified pMOTag4H vector (Fig. 1A). Insert correct orientation was determined by PCR (primers 12 to 19) and sequencing.
Figure 1.

Schematic representation of the strategy used to generate endogenous C-terminal tagging of glmS/M9 in T. brucei and growth inhibition by glucosamine. A. Upper scheme, pMOTag-glmS/M9-HA vector map. The glmS or the M9 sequence is located between the 3xHA sequence and the tubulin intergenic region (IgR) and the gene that confers resistance to hygromycin (Hygro). HR1 Fw and HR2 Rv ultramers indicate oligonucleotides used to amplify DNA donor cassette. The annealing regions for ultramers to pMOTag and genomic DNA (gDNA) are indicated in filled and dotted line, respectively. Medium scheme, homologous recombination was induced by transfecting PCF trypanosomes with the DNA donor cassette, containing homologous regions to the GOI 3′end and to the GOI 3′UTR. Lower scheme, integration of 3xHA, glmS/M9, IgR, and antibiotic resistance gene at 3′ end of GOI by homologous recombination. Arrows indicate primers used for checking integration of donor DNA. B. Effect of different concentrations of glucosamine on PCF trypanosomes growth in SDM-79 medium. Values are means ± s.d. of n = 3.
RNA interference
We used RNA interference to generate knockdown of the gene Nudix hydrolase 2 (Tb427.05.4350) in PCF trypanosomes and TbVPS41 (Tb427.06.2770) in BSF trypanosomes and compare the efficacy of the mRNA deletion methods. A segment of the gene coding sequences was amplified by PCR (primers 20 and 21 for TbVPS41 and primers 22 and 23 for TbNUDIX2, Table S1) and cloned into the P2T7tiB vector (LaCount et al. 2002), which was linearized with NotI and transfected in T. brucei Lister 427 29-13 (PCF) or single marker (BSF) cell lines, respectively. To induce RNA interference, we added tetracycline (1 μg/ml) to cultures, and control un-induced cells were maintained in medium supplemented with tetracycline-free fetal bovine serum. RNA extraction, cDNA synthesis and quantitative Real Time PCR were performed as described below.
Cell transfection
Cell transfections were done as reported previously (Huang et al. 2014). In brief, For T. brucei mid-log phase PCF trypanosomes (5×106 cells/ml) were harvested by centrifugation at 1,000 × g for 7 min, washed with a Cytomix buffer (2 mM EGTA, 3 mM MgCl2, 120 mM KCl, 0.5% glucose, 0.15 mM CaCl2, 0.1 mg/ml BSA, 10 mM K2HPO4/KH2PO4, 1 mM hypoxanthine, 25 mM Hepes, pH 7.6) and resuspended in 0.4 ml of the same buffer at a cell density of 2.5×107 cells/ml. The washed cells were mixed with 50 μg of DNA or purified PCR products (10 μg) in a 0.4-cm electroporation cuvette and subjected to two pulses from a Bio-Rad Gene Pulser Xcell electroporator set at 1.5 kV and 25 μF. The stable transformants were obtained in SDM-79 glucosamine-free medium supplemented with 15% FBS and 50 μg/ml hygromycin. For BSF trypanosomes, 10 μg of NotI-linearized RNAi plasmid DNA or 10 μg of DNA donor cassette glmS/M9 amplicon (<10 μl) were used per 4×107 mid-log phase cells in 100 μl AMAXA Human T-cell Nucleofector solution. Electroporation was performed using 2 mm gap cuvettes with program X-001 of the AMAXA Nucleofector. Following each transfection, stable transformants were selected and cloned by limiting dilution in HMI-9 medium containing 15% FBS with 5 μg/ml of hygromycin for glmS/M9 transfectants and 2.5 μg/ml of phleomycin for RNAi tranfectants, in 24-well plates. The correct epitope-tagging of the target genes was confirmed by PCR followed by sequencing and western blot analyses.
When the cells were at a density of 2×106 PCF/ml or 1×105 BSF/ml, 5 mM (PCF) or 2.5 mM (BSF) of fresh D-glucosamine were added to induce ribozyme activity. Parasites were collected every 24 h for several days for further analysis.
Western blot analyses
Electrophoresed proteins were transferred to nitrocellulose membranes using a Bio-Rad transblot apparatus for 1 hour at 100 V at 4°C. Following transfer, the membrane blots were blocked with 5% non-fat dry milk in PBS containing 0.1% Tween-20 (PBS-T) at room temperature during 1 hour. Blots were probed with anti-HA mouse monoclonal antibody (1:1,000), anti-HA rat antibody (1:1,000), and anti-tubulin monoclonal antibody (1:40,000) for 1 h. After washing five times with PBS-T, the blots were incubated with anti-rat IgG antibody (1:20,000) or goat anti-mouse antibody (1:20,000). The membranes were washed three times with PBS-T, and western blot images were processed and analyzed using the Odyssey infrared system software (LICOR Biosciences, Lincoln, Nebraska). Density of the bands, as compared to controls considered as 1, was measured using an image processing software (FiJi, ImageJ, University of Wisconsin-Madison, WI).
Immunofluorescence analyses
PCF or BSF trypanosomes were washed with PBS and then fixed with cold methanol at room temperature for 2 min. The fixed parasites were washed twice with PBS for 10 min and then were allowed to adhere to poly-L-lysine-coated coverslips and permeabilized with 0.5% Triton X-100/PBS for 3 min for PCF and 0.1% Triton X-100/PBS for 5 min for BSF. After blocking with PBS containing 3% BSA, 1% fish gelatin, 50 mM NH4Cl and 5% goat serum for 1 h, trypanosomes were stained in 3% BSA/PBS with the monoclonal mouse antibody against TbL8C4 (1:25) or purified HA.11 clone 16B12 mouse monoclonal antibody against HA (1:50), or polyclonal rabbit TcHAf antibody (1:200), for 1 h. After thoroughly washing with PBS, cells were incubated with Alexa 488-conjugated goat anti-mouse or anti-rabbit antibody at 1:1,000 for 1 h. The cells were counterstained with DAPI before mounting with Gold ProLong Gold antifade reagent (Molecular Probes). Differential interference contrast and fluorescent optical images were taken with a 100× objective (1.35 aperture) under nonsaturating conditions with a Photometrix CoolSnapHQ charge-coupled device camera driven by DeltaVision software (Applied Precision, Issaquah, WA) and deconvolved for 15 cycles using Softwarx deconvolution software. For BSF trypanosomes, images were taken with a 100× oil immersion objective, a high-power solid-state 405 nm laser and EM-CCD camera (Andor iXon) under nonsaturating conditions in a Zeiss ELYRA S1 (SR-SIM) super resolution microscope. Images were acquired and processed with ZEN 2011 software with SIM analysis module.
Quantitative Real-time PCR
Total RNA was isolated from PCF and BSF trypanosomes using the TRI® reagent (Sigma) by following the manufacturer’s instructions. The total RNA was treated with DNase I to remove genomic DNA contamination. cDNA synthesis was accomplished using the iScript cDNA synthesis kit (Bio-Rad) for PCF trypanosomes and superscript III first-strand synthesis system (Invitrogen) for BSF trypanosomes with 100 ng of total RNA used per reaction. Real-time PCR was done using a CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad) and set up in white opaque polypropylene wells (LightCycler 480 Multiwell Plates 384) in a final volume of 10 μl per reaction. The primers for gene amplification are listed in Table S1. Reaction mixtures contained 2 μl of sample DNA (100 ng/μl), 5 μl of a master mix iQ™ SYBR® Green Supermix (Bio-Rad) and 4 μl of nuclease-free water with primers at a final concentration of 300 nM. Activation of polymerase was performed at 95°C for 2 min. PCR cycling conditions included 39 cycles of denaturation at 95°C for 10 s, and annealing and extension at 60°C for 30 s. SYBR Green fluorescent emission was measured at the end of the elongation step. Subsequently, a melting curve program was applied with a continuous fluorescent measurement starting at 65°C and ending at 95°C (ramping rate of 0.1°C/s). In order to normalize the expression of the genes, as housekeeping genes we used primers for actin, tubulin and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (primers 24 to 36). The samples were quantified according to the %Ct method and all the assays were performed at least three times.
Statistical analysis
All values are expressed as means ± s.d. Significant differences between treatments were compared using the tests indicated in the figure legends. Differences were considered statistically significant at P < 0.05, and n refers to the number of independent biological experiments performed. All statistical analyses were conducted using GraphPad Prism 5 (GraphPad Software, San Diego, CA).
RESULTS
The glmS ribozyme-mediated control of gene expression was first tested in T. brucei procyclic forms (PCF) by introducing the epitope tag sequence containing the active (glmS) or inactive (M9) ribozyme at the 3′end of two different genes: the T. brucei flagellar-adhesion glycoprotein 1 (TbFLA1, Tb427.08.4010) (Nozaki et al. 1996) and one of the seven T. brucei folate transporters (TbFT1, Tb427.08.3620).
TbFLA1 is a glycoprotein responsible for adhesion of the flagellum to the cell body originally discovered as an ortholog of T. cruzi glycoprotein 72 (GP72) (Nozaki et al. 1996). Attempts to delete both copies of TbFLA1 from the genome were unsuccessful suggesting that TbFLA1 is essential (Nozaki et al. 1996). RNAi of TbFLA1 in PCF trypanosomes results in flagellar detachment from the cell body and generation of multinucleated cells, suggesting that it is also important for cytokinesis (LaCount et al. 2002). TbFT1 is one of three folate transporter genes arranged in tandem identified in a genome-wide RNAi screen of T. brucei exposed to antifolates (Dewar et al. 2016). Knockdown of TbFT1-3 by RNAi does not affect growth (Dewar et al. 2016).
Fig. 1A shows the strategy followed. We transfected a pMOTag4H vector that has a 3xHA tag, a tubulin intergenic region (IgR), and the hygromycin resistance marker, with the introduction, after a stop codon, of the active (glmS) or inactive (M9) ribozyme, to the C-terminus of the gene of interest (GOI), as described under Materials and Methods. We also tested whether different concentrations of glucosamine have any effect on parasite growth and found that concentrations between 2.5–5 mM glucosamine did not greatly change the doubling time (in hours) that went from 11.52 ± 0.10 (no glucosamine) to 12.15 ± 0.33 (2.5 mM) and 12.50 ± 0.23 (5 mM) (Fig. 1B). Doubling times went to 15.55 ± 0.61 and 17.15 ± 0.33 at 7.5 and 10 mM glucosamine, respectively. Values are means ± s.d. of 3 independent experiments.
Fig. 2A shows that TbFLA1-3xHA-glmS and TbFLA1-3xHA-M9 were correctly tagged at the endogenous locus, because the corresponding band amplified with a reverse primer annealing at the C-terminal tagged region (Fig. 1A, and S2A) is present in the hygromycin-resistant parasites (lanes M9 and glmS) but is absent in the wild type (WT) cells. Reverse primers annealing on the TbFLA1 gene demonstrated that, as expected, only one allele was tagged (Fig. 1A, and S2A). We analyzed the transfectants by western blotting, using commercial antibodies against the HA tag, and a 60 kDa band was detected in the TbFLA1-3xHA-M9- and TbFLA1-3xHA-glmS-transfectants. Addition of 5 mM glucosamine resulted in a decrease in intensity of the band after 48 h of culture of the TbFLA1-3xHA-glmS- but not of the TbFLA1-3xHA-M9-transfectants (Fig. 2B). No further decrease was observed after 72 h of culture with glucosamine (data not shown). Although only one allele was tagged (Fig. 1A), qRT-PCR showed decrease in the TbFLA1 mRNA with time of glucosamine exposure (Fig. 2C). Accordingly, differential interference contrast microscopy of the mutants revealed flagellar detachment in more than 50% of the cells when in the presence of medium containing 5 mM glucosamine (Fig. 2D). This effect was confirmed by immunofluorescence analysis (IFA) using monoclonal antibodies L8C4 against the paraflagellar rod 2 (PFR2) protein of T. brucei (Kohl et al. 1999) (Fig. 2D). To rule out the possibility of an effect of the addition of the glmS element to the 3′UTR of the GOI we followed, by qRT-PCR, the expression of the hygromycin resistance marker, which is present before the 3′UTR (Fig. 1A). No changes were observed (Fig. S3) in contrast to the changes observed in the expression of TbFLA1 (Fig. 1C).
Figure 2.

Integration of glmS/M9 ribozyme sequences into the TbFLA1 gene. A. PCR analysis using gDNA isolated from WT and TbFLA1-3xHA cell lines. A DNA fragment (band of 2079 bp) was amplified in 3xHA-glmS/M9-tagged PCF trypanosomes (indicated with arrow), whereas the band is absent in WT PCF trypanosomes. A band of 409 bp corresponds to the second allele maintained in the mutant and WT cells. L, ladder. B. Western blot analyses of TbFLA1-3xHA-expressing cell lines. Anti-HA antibodies detect TbFLA1-3xHA-glmS (glmS) and TbFLA1-3xHA-M9 (M9) (expected size 60 kDa). Anti-tubulin antibody was used as loading control. Antibodies are indicated on the right side of the blots, and molecular weights (MW) are on the left side. After 48 h of exposure to 5 mM glucosamine the band decreases in intensity in TbFLA1-3xHA-glmS- but not in TbFLA1-3xHA-M9-expressing cells. Values under the blots show relative adjusted density of the bands as compared to control ± s.d., n = 2. C. qRT-PCR shows down-regulation of TbFLA1 transcript in TbFLA1-3xHA-glmS- and TbFLA1-3xHA-M9-expressing cells after 0–3 days glucosamine addition. Values are means ± s.d. (n = 3). ****P < 0.0001. Two-way ANOVA with multiple comparisons. D. Fluorescence microscopy of TbFLA1-3xHA-M9-expressing PCF trypanosomes indicates localization of the endogenous tagged TbFLA1 (green) in the flagellum, which is detached in TbFLA1-3xHA-glmS-expressing cells. DIC, differential interference contrast microscopy. Scale bars = 5 μm.
Using a similar approach, we found that TbFT1-3xHA-glmS and TbFT1-3xHA-M9 were correctly tagged to one of the alleles at the endogenous locus, as detected by PCR (Fig. S2B). Western blot analysis showed that a 70 kDa band corresponding to TbFT1-3xHA greatly decreased 48 h after addition of 5 mM glucosamine to the medium in the TbFT1-3xHA-glmS- but not in the TbFT1-3xHA-M9-transfectants (Fig. 3A). qRT-PCR also showed decrease in the TbFT1 mRNA after 1–3 days of glucosamine exposure (Fig. 3B) but if the cells were transferred to a medium without glucosamine they recovered their mRNA levels (Fig. 3C). Immunofluorescence analysis of endogenously tagged cells (TbFT1-3xHA-M9-expressing cells) showed punctated staining close or co-localizing with a plasma membrane marker (antibody TcHAf against a T. cruzi P-type H+-ATPase that cross-reacts with T. brucei P-type H+-ATPase (Luo et al. 2006)) (Fig. 3D) in about 50% of the cells.
Figure 3.
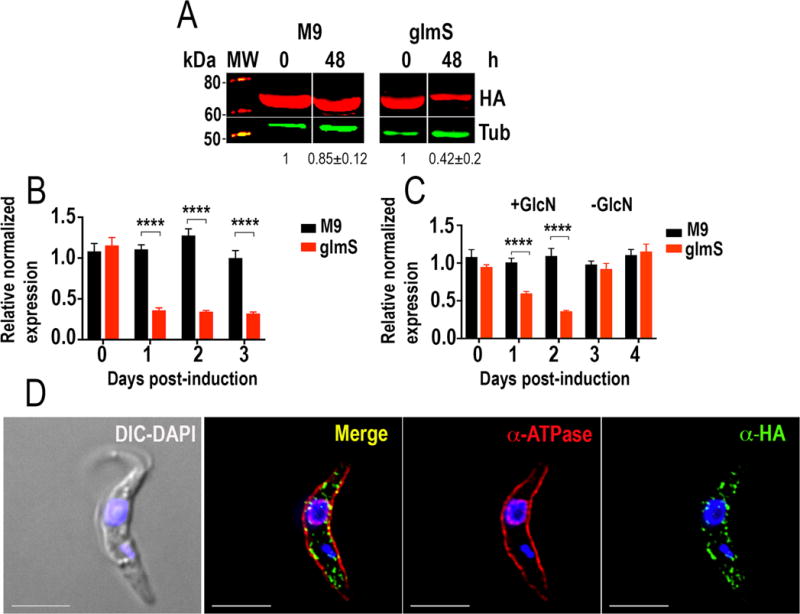
Integration of glmS/M9 ribozyme sequences into the TbFT1 gene. A. Western blot analyses of TbFT1-3xHA cell lines. Anti-HA antibodies detect TbFT1-3xHA-glmS and TbFT1-3xHA-M9 (expected size 70 kDa). Anti-tubulin antibody was used as loading control. Antibodies are indicated on the right side of the blots, and molecular weights (MW) are on the left side. After 48 h of exposure to 5 mM glucosamine the band decreases in intensity in TbFT1-3xHA-glmS- but not in TbFT1--3xHA-M9-expressing cells. Values under the blots are relative adjusted density of the bands as compared to control ± s.d., n = 2. B. qRT-PCR shows down-regulation of TbFT1 transcript in TbFT1-3xHA-glmS- and TbFT1-3xHA-M9-expressing cells after 0–3 days glucosamine addition. C. Ribozyme-mediated knockdown of TbFT1 expression is reversible. PCF trypanosomes were treated with 5 mM glucosamine (+GlcN) for 48 h and samples were grown in glucosamine-free medium (−GlcN) for another period of 48 h. Values in B and C are means ± s.d. (n = 3). ****P < 0.0001. Two-way ANOVA with multiple comparisons. D. Fluorescence microscopy of TbFT1-3xHA PCF trypanosomes indicates localization of the endogenous tagged TbFT1 (green) close to the plasma membrane with some co-localization with antibodies TcHAf (red) against the T. cruzi P-type H+-ATPase in the plasma membrane (see Merge in yellow). DIC, differential interference contrast microscopy. Bars = 5 μm.
We next compared the results of riboswitch-inducible downregulation of gene expression with those obtained with RNAi. We chose a gene encoding NUDIX hydrolase 2 (TbNUDIX2, Tb427.05.4350), an enzyme of proposed glycosomal localization (Guther et al. 2014). TbNUDIX2-3xHA-glmS and TbNUDIX-3xHA-M9 were correctly tagged to one allele at the endogenous locus as detected by PCR (Fig. S2C) and western blot analysis (Fig. 4A). qRT-PCR of both RNAi- and riboswitch-induced downregulation of TbNUDIX2 expression showed that the riboswitch reduction of gene expression was more efficient than RNAi in this case and with this individual RNAi construct (Fig. 4B). The values shown are the means of 3 independent experiments with the RNAi cell line that showed earlier changes in mRNA expression. No apparent phenotypic changes were observed in cells in which downregulation of TcNUDIX expression was generated using either method.
Figure 4.
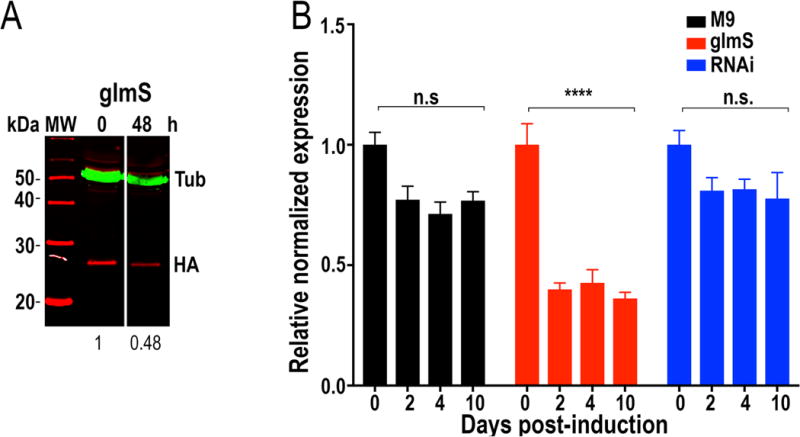
Integration of glmS/M9 ribozyme sequences into the TbNUDIX2 gene. A. Western blot analyses of TbNUDIX2-3xHA cell lines. Anti-HA antibodies detect TbNUDIX-3xHA-glmS. Anti-tubulin antibody was used as loading control. Antibodies are indicated on the right side of the blots, and molecular weights (MW) are on the left side. After 48 h of exposure to 5 mM glucosamine the band decreases in intensity in TbNUDIX2-3xHA-glmS-expressing cells. Values under the blots are relative adjusted density of the bands as compared to control of a representative blot. B. qRT-PCR shows down-regulation of TbNUDIX2 transcript at 2–10 days after addition of tetracycline (RNAi) and in TbNUDIX-3xHA-glmS-expressing cells (glmS) after 2–10 days glucosamine addition. Values are means ± s.d. (n = 3). ****P < 0.0001; n.s, no significant. Two-way ANOVA with multiple comparisons.
We then tested the riboswitch system in T. brucei bloodstream forms (BSF) and compared the results with those obtained by downregulation of gene expression by RNAi. For these studies, we chose a gene encoding an ortholog to the Saccharomyces cerevisiae vacuolar protein sorting 41 (TbVPS41, Tb427.06.2770). This protein is important for PCF trypanosomes growth in low-iron medium (Lu et al. 2007). However, its significance in BSF trypanosomes has not been characterized yet. PCR analysis revealed correct tagging of one allele with either TbVPS41-3xHA-glmS or TbVPS41-3xHA-M9 (Fig. S2D). Using another primer pair designed to amplify the 3′UTR region of TbVPS41 we noticed a single 350 bp band corresponding to untagged copy in WT parasite line. However, for both TbVPS41-3xHA-glmS and TbVPS41-3xHA-M9 cell lines, we observed bands of 350 bp (corresponding to untagged copy) and 2,000 bp (corresponding to tagged copy). Therefore, only one allele was tagged as determined by PCR (Fig. S2D). For RNAi 5 clones were obtained and the clone that exhibited earliest regulation was selected for further analysis. qRT-PCR of both RNAi and riboswitch-induced downregulation of TbVPS41 showed that down-regulation was effective after 2 days culture. However, RNAi-treated parasites escaped downregulation after 4 days while riboswitch-induced downregulation remained stable (Fig. 5A). IFA of riboswitch-induced downregulation of TbVPS41 using high-resolution microscopy (Fig. 5B) showed morphological alterations similar to those produced by RNAi (data not shown) (Lu et al. 2007) with appearance of multinuclear cells 96 h after exposure to medium with 2.5 mM glucosamine in >70% of the cells.
Figure 5.

Integration of glmS/M9 ribozyme sequences into the TbVPS41 gene. A. qRT-PCR shows down-regulation of TbVPS41 transcript at 2 and 4 days after addition of tetracycline (RNAi) and in TbVPS41-3xHA-glmS-expressing cells (glmS) as compared to TbVPS41-3xHA-M9-expressing cells (M9) after 2 and 4 days glucosamine addition. Values are means ± s.d. (n = 3). *P < 0.05; ***P < 0.001; ****P < 0.0001. Two-way ANOVA with multiple comparisons. B. Super-resolution images of TbVPS41-3xHA-glmS-expressing BSF at 0, 2 and 4 days of cultivation in the presence of 2.5 mM glucosamine. Cells appear rounded and with multiple nuclei and kinetoplasts, as detected by DAPI staining (blue). The cell membrane was drawn to identify the cells. Scale bars = 5 μm.
DISCUSSION
In this work we adapted a C-terminal endogenous tagging method used in T. brucei (Oberholzer et al. 2006) to insert the glmS ribozyme and downregulate the expression of a number of genes by activating it by adding glucosamine to the medium in both PCF and BSF trypanosomes. The results were better than those using the RNAi method with the advantage that it can be used with any strain of T. brucei, there is no significant loss of expression control, and there is no risk of off target effects since only the mRNA of the gene adjacent to the ribozyme is cleaved. In this regard, previous high-throughput studies have shown that the expression of a significant number of genes was not downregulated using their RNAi experimental system (Alsford et al. 2011) (TriTrypDB.org).
As reported in S. cerevisiae (Meaux and Van Hoof 2006) and P. falciparum (Prommana et al. 2013) the ribozyme-cleaved mRNA 5′ fragment separated from its polyA tail could be rapidly degraded by the 3′exosome, which has been reported to be active in T. brucei (Estevez et al. 2001). It is also possible to position the glmS ribozyme in the 5′UTR, in which case mRNA cleavage would separate the mRNA from its 5′cap structure and the de-capped mRNA would be degraded by the 5′-3′-exonucleases, also present in trypanosomatids (Li et al. 2006).
A potential disadvantage of the method is the possible toxic effect of glucosamine on the parasite. However, glucosamine added to the tse tse fly food was shown to enhance T. brucei midgut infection (Peacock et al. 2006). In contrast to N-acetyl-glucosamine, which is not taken up, glucosamine is internalized by PCF trypanosomes in vitro (Ebikeme et al. 2008). Glucosamine could be toxic, apparently because of its acidity and ability to reduce the medium pH, only when used for prolonged periods and at very high concentrations (60 mM) (Peacock et al. 2006). Glucosamine is a component of the SDM-79 medium at a concentration of 1 mM, and we did not observe toxicity at the concentrations used to induce the glmS activity (2.5–5 mM). The use of the inactive ribozyme (M9) is also an appropriate control for potential side effects. Another potential disadvantage of this method is that, in contrast to RNAi method, it would not be useful for multi-copy genes. Interestingly, C-terminal tagging of the proteins encoded by only one allele of several genes resulted in downregulation of gene expression at the mRNA level demonstrating that both alleles were transcribed. In this regard, when targeting genes for deletion, several studies in T. cruzi (Campos et al. 2011, Cardoso et al. 2013, Perez Brandan et al. 2011) and Leishmania donovani (Dumas et al. 1997) have shown that disruption of a single allele results in a 40% (Cardoso et al. 2013) to 50% (Campos et al. 2011) reduction in the levels of the target mRNA in T. cruzi and up to 3- to 5-fold reduction in the target mRNA in L. donovani (Dumas et al. 1997) allowing the assessment of the phenotype of cells with this reduced expression. One possible reason for these dramatic effects could be the differential expression of different alleles.
Our method could be further improved to knockout both alleles of a non-essential gene using an additional plasmid with another antibiotic resistance marker.
Knockdown of TbFLA1 confirmed the role of the protein in flagellar attachment previously reported using RNAi (LaCount et al. 2002), while knockdown of TbFT1 did not result in any apparent phenotypic change. These latter results are in agreement with the presence of redundant copies of this transporter in T. brucei and previous reports using RNAi (Dewar et al. 2016). T. brucei possesses at least 5 NUDIX hydrolases (Ignatochkina et al. 2015) and this redundancy is probably also responsible for the lack of apparent phenotypic alterations in the cells in which TbNUDIX2 expression was downregulated although further experiments are needed to investigate other more cryptic phenotypic changes. However, as with the other genes, downregulation of TbNUDIX2 expression was obtained and the results were better than the results using RNAi in this case. Similar better results to those observed using RNAi were observed after downregulation of TbVPS41 expression with the riboswitch method but without the significant loss of expression regulation observed using RNAi. Work is in progress to investigate the role of this gene in BSF trypanosomes.
In conclusion, the riboswitch method developed in this work will enable the downregulation of gene expression in any T. brucei strain without other limitations of current methods and could be potentially useful to generate expression knockdowns in T. cruzi, and in Leishmania spp. in which endogenous gene tagging using CRISPR/Cas9 has been possible (Lander et al. 2016b, Lander et al. 2016a).
Supplementary Material
Table S1. Primers used in this work
Fig. S1. Scheme of the mechanism of action of the ribozyme.
Fig. S2. PCR analysis and diagrams of primers used.
Fig. S3. Lack of downregulation of the hygromycin resistance marker.
Acknowledgments
We thank Philip Bastin for antibody L8C4, Thomas Seebeck for pMOTag plasmids, George A.M. Cross for the T. brucei cell lines to perform RNAi, Vasant Muralidharan for plasmids containing glmS and M9 ribozymes and useful discussions, and Muthugapatti Kandasamy and the Biomedical Microscopy Core of the University of Georgia for the use of microscopes. Funding for his work was provided by the U.S. National Institutes of Health (grant AI108222 to RD).
LITERATURE CITED
- Alibu VP, Storm L, Haile S, Clayton C, Horn D. A doubly inducible system for RNA interference and rapid RNAi plasmid construction in Trypanosoma brucei. Mol Biochem Parasitol. 2005;139:75–82. doi: 10.1016/j.molbiopara.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Alsford S, Turner DJ, Obado SO, Sanchez-Flores A, Glover L, Berriman M, Hertz-Fowler C, Horn D. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 2011;21:915–24. doi: 10.1101/gr.115089.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos PC, Silva VG, Furtado C, Machado-Silva A, Darocha WD, Peloso EF, Gadelha FR, Medeiros MH, Lana Gde C, Chen Y, Barnes RL, Passos-Silva DG, McCulloch R, Machado CR, Teixeira SM. Trypanosoma cruzi MSH2: Functional analyses on different parasite strains provide evidences for a role on the oxidative stress response. Mol Biochem Parasitol. 2011;176:8–16. doi: 10.1016/j.molbiopara.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso MS, Junqueira C, Trigueiro RC, Shams-Eldin H, Macedo CS, Araujo PR, Gomes DA, Martinelli PM, Kimmel J, Stahl P, Niehus S, Schwarz RT, Previato JO, Mendonca-Previato L, Gazzinelli RT, Teixeira SM. Identification and functional analysis of Trypanosoma cruzi genes that encode proteins of the glycosylphosphatidylinositol biosynthetic pathway. PLoS Negl Trop Dis. 2013;7:e2369. doi: 10.1371/journal.pntd.0002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton CE. Genetic manipulation of kinetoplastida. Parasitol Today. 1999;15:372–8. doi: 10.1016/s0169-4758(99)01498-2. [DOI] [PubMed] [Google Scholar]
- Cunningham I. New culture medium for maintenance of tsetse tissues and growth of trypanosomatids. J Protozool. 1977;24:325–9. doi: 10.1111/j.1550-7408.1977.tb00987.x. [DOI] [PubMed] [Google Scholar]
- Dewar S, Sienkiewicz N, Ong HB, Wall RJ, Horn D, Fairlamb AH. The role of folate transport in antifolate drug action in Trypanosoma brucei. J Biol Chem. 2016;291:24768–24778. doi: 10.1074/jbc.M116.750422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docampo R. Molecular parasitology in the 21st century. Essays Biochem. 2011;51:1–13. doi: 10.1042/bse0510001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas C, Ouellette M, Tovar J, Cunningham ML, Fairlamb AH, Tamar S, Olivier M, Papadopoulou B. Disruption of the trypanothione reductase gene of Leishmania decreases its ability to survive oxidative stress in macrophages. EMBO J. 1997;16:2590–8. doi: 10.1093/emboj/16.10.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebikeme CE, Peacock L, Coustou V, Riviere L, Bringaud F, Gibson WC, Barrett MP. N-acetyl D-glucosamine stimulates growth in procyclic forms of Trypanosoma brucei by inducing a metabolic shift. Parasitology. 2008;135:585–94. doi: 10.1017/S0031182008004241. [DOI] [PubMed] [Google Scholar]
- Estevez AM, Kempf T, Clayton C. The exosome of Trypanosoma brucei. EMBO J. 2001;20:3831–9. doi: 10.1093/emboj/20.14.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Guther ML, Urbaniak MD, Tavendale A, Prescott A, Ferguson MA. High-confidence glycosome proteome for procyclic form Trypanosoma brucei by epitope-tag organelle enrichment and SILAC proteomics. J Proteome Res. 2014;13:2796–806. doi: 10.1021/pr401209w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol. 1989;75:985–9. [PubMed] [Google Scholar]
- Huang G, Ulrich PN, Storey M, Johnson D, Tischer J, Tovar JA, Moreno SN, Orlando R, Docampo R. Proteomic analysis of the acidocalcisome, an organelle conserved from bacteria to human cells. PLoS Pathog. 2014;10:e1004555. doi: 10.1371/journal.ppat.1004555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatochkina AV, Takagi Y, Liu Y, Nagata K, Ho CK. The messenger RNA decapping and recapping pathway in Trypanosoma. Proc Natl Acad Sci U S A. 2015;112:6967–72. doi: 10.1073/pnas.1424909112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl L, Sherwin T, Gull K. Assembly of the paraflagellar rod and the flagellum attachment zone complex during the Trypanosoma brucei cell cycle. J Eukaryot Microbiol. 1999;46:105–9. doi: 10.1111/j.1550-7408.1999.tb04592.x. [DOI] [PubMed] [Google Scholar]
- LaCount DJ, Barrett B, Donelson JE. Trypanosoma brucei FLA1 is required for flagellum attachment and cytokinesis. J Biol Chem. 2002;277:17580–8. doi: 10.1074/jbc.M200873200. [DOI] [PubMed] [Google Scholar]
- Lander N, Chiurillo MA, Docampo R. Genome editing by CRISPR/Cas9: A game change in the genetic manipulation of protists. J Eukaryot Microbiol. 2016a;63:679–90. doi: 10.1111/jeu.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander N, Chiurillo MA, Storey M, Vercesi AE, Docampo R. CRISPR/Cas9-mediated endogenous C-terminal tagging of Trypanosoma cruzi genes reveals the acidocalcisome localization of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 2016b;291:25505–25515. doi: 10.1074/jbc.M116.749655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CH, Irmer H, Gudjonsdottir-Planck D, Freese S, Salm H, Haile S, Estevez AM, Clayton C. Roles of a Trypanosoma brucei 5′->3′ exoribonuclease homolog in mRNA degradation. RNA. 2006;12:2171–86. doi: 10.1261/rna.291506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FJ, Xu ZS, Aye HM, Brasseur A, Lun ZR, Tan KSW, He CY. An efficient cumate-inducible system for procyclic and bloodstream form Trypanosoma brucei. Mol Biochem Parasitol. 2017;214:101–104. doi: 10.1016/j.molbiopara.2017.04.007. [DOI] [PubMed] [Google Scholar]
- Lu S, Suzuki T, Iizuka N, Ohshima S, Yabu Y, Suzuki M, Wen L, Ohta N. Trypanosoma brucei vacuolar protein sorting 41 (VPS41) is required for intracellular iron utilization and maintenance of normal cellular morphology. Parasitology. 2007;134:1639–47. doi: 10.1017/S0031182007003046. [DOI] [PubMed] [Google Scholar]
- Luo S, Fang J, Docampo R. Molecular characterization of Trypanosoma brucei P-type H+-ATPases. J Biol Chem. 2006;281:21963–73. doi: 10.1074/jbc.M601057200. [DOI] [PubMed] [Google Scholar]
- Meaux S, Van Hoof A. Yeast transcripts cleaved by an internal ribozyme provide new insight into the role of the cap and poly(A) tail in translation and mRNA decay. RNA. 2006;12:1323–37. doi: 10.1261/rna.46306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt C, Stuart K. Identification of essential and non-essential protein kinases by a fusion PCR method for efficient production of transgenic Trypanosoma brucei. Mol Biochem Parasitol. 2013;190:44–9. doi: 10.1016/j.molbiopara.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyka SA, Englund PT. RNA interference for analysis of gene function in trypanosomatids. Curr Opin Microbiol. 2004;7:362–8. doi: 10.1016/j.mib.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Ngo H, Tschudi C, Gull K, Ullu E. Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc Natl Acad Sci U S A. 1998;95:14687–92. doi: 10.1073/pnas.95.25.14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki T, Haynes PA, Cross GA. Characterization of the Trypanosoma brucei homologue of a Trypanosoma cruzi flagellum-adhesion glycoprotein. Mol Biochem Parasitol. 1996;82:245–55. doi: 10.1016/0166-6851(96)02741-7. [DOI] [PubMed] [Google Scholar]
- Oberholzer M, Morand S, Kunz S, Seebeck T. A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol Biochem Parasitol. 2006;145:117–20. doi: 10.1016/j.molbiopara.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Peacock L, Ferris V, Bailey M, Gibson W. Multiple effects of the lectin-inhibitory sugars D-glucosamine and N-acetyl-glucosamine on tsetse-trypanosome interactions. Parasitology. 2006;132:651–8. doi: 10.1017/S0031182005009571. [DOI] [PubMed] [Google Scholar]
- Perez Brandan C, Padilla AM, Xu D, Tarleton RL, Basombrio MA. Knockout of the dhfr-ts gene in Trypanosoma cruzi generates attenuated parasites able to confer protection against a virulent challenge. PLoS Negl Trop Dis. 2011;5:e1418. doi: 10.1371/journal.pntd.0001418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prommana P, Uthaipibull C, Wongsombat C, Kamchonwongpaisan S, Yuthavong Y, Knuepfer E, Holder AA, Shaw PJ. Inducible knockdown of Plasmodium gene expression using the glmS ribozyme. PLoS One. 2013;8:e73783. doi: 10.1371/journal.pone.0073783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusconi F, Durand-Dubief M, Bastin P. Functional complementation of RNA interference mutants in trypanosomes. BMC Biotechnol. 2005;5:6. doi: 10.1186/1472-6750-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunter JD. A vanillic acid inducible expression system for Trypanosoma brucei. Mol Biochem Parasitol. 2016;207:45–8. doi: 10.1016/j.molbiopara.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschopp F, Charriere F, Schneider A. In vivo study in Trypanosoma brucei links mitochondrial transfer RNA import to mitochondrial protein import. EMBO Rep. 2011;12:825–32. doi: 10.1038/embor.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdés J, T M, Cross MA, Ligtenberg MJ, RUdenko G, Borst P. The viral thymidine kinase gene as a tool for the study of mutagenesis in Trypanosoma brucei. Nuclei Acids Research. 1996;24:1809–1815. doi: 10.1093/nar/24.10.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PY, Fedor MJ. The glmS riboswitch integrates signals from activating and inhibitory metabolites in vivo. Nat Struct Mol Biol. 2011;18:359–63. doi: 10.1038/nsmb.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004;428:281–6. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- Wirtz E, Clayton C. Inducible gene expression in trypanosomes mediated by a prokaryotic repressor. Science. 1995;268:1179–83. doi: 10.1126/science.7761835. [DOI] [PubMed] [Google Scholar]
- Wirtz E, Hoek M, Cross GA. Regulated processive transcription of chromatin by T7 RNA polymerase in Trypanosoma brucei. Nucleic Acids Res. 1998;26:4626–34. doi: 10.1093/nar/26.20.4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers used in this work
Fig. S1. Scheme of the mechanism of action of the ribozyme.
Fig. S2. PCR analysis and diagrams of primers used.
Fig. S3. Lack of downregulation of the hygromycin resistance marker.