

Abstract
REV1 protein is a mutagenic DNA damage tolerance (DDT) mediator and encodes two ubiquitin-binding motifs (i.e., UBM1 and UBM2) that are essential for the DDT function. REV1 interacts with K164-monoubiquitinated PCNA (UbPCNA) in cells upon DNA-damaging stress. By using AlphaScreen assays to detect inhibition of REV1 and UbPCNA protein interactions along with an NMR-based strategy, we identified small-molecule compounds that inhibit the REV1/UbPCNA interaction and that directly bind to REV1 UBM2. In cells, one of the compound prevented recruitment of REV1 to PCNA foci on chromatin upon cisplatin treatment, delayed removal of UV-induced cyclopyrimidine dimers from nuclei, prevented UV-induced mutation of HPRT gene, and diminished clonogenic survival of cells that were challenged by cyclophosphamide or cisplatin. This study demonstrates the potential utility of a small-molecule REV1 UBM2 inhibitor for preventing DDT.
Keywords: REV1, ubiquitin-binding motif, PCNA, small-molecule, protein-protein interaction
ToC graphic
INTRODUCTION
Chemotherapy remains indispensable for frontline cancer therapy, but its efficacy is often partially limited by rapid and erroneous repair of DNA damage promoted by DNA replication irrespective of the damage1–2, leading to drug resistance and mutations. REV1 protein is indispensable for replicating over a variety of chemotherapeutic DNA damage types, including DNA crosslinks3–6. The mouse Rev1 is responsible for acquisition of resistance after repeated engraftment and cyclophosphamide treatment of lymphoma; depletion of Rev1 gene in the lymphoma dramatically improved the drug efficacy, suppressed the resistance, and inhibited the drug-induced mutations7. Carcinogen-induced adenomas developed in Rev1 transgenic mice approximately twice as often as in normal mice, with elevated DNA damage tolerance and mutations8, further indicating role of Rev1 in tumor generation. REV1 is also responsible for the resistance and mutations in ovarian cancer cells9 and for lung carcinogenesis10. Importantly, Rev1-knockout mice are healthy except for an approximate 10% reduction in body growth; Rev1−/− B cells undergo normal proliferation and apoptosis11. These findings suggest that REV1 inhibition is not likely to be mechanistically toxic unless a chemotherapy drug is co-administrated.
REV1 encodes the ubiquitin-binding motif (UBM) of REV1. When a DNA replication fork is stalled by DNA damage, PCNA on the fork is monoubiquitinated on K164 (UbPCNA) and recruits REV1, likely via its UBM12–13. REV1 coordinates Polζ, which extends over a variety of DNA lesions14, mediates most mutagenic DNA damage tolerance (DDT)15–16, and is composed of the regulatory subunit REV7 and the catalytic subunit REV3L. These observations suggest that the DDT processes may be inhibited by blocking one of the protein-protein interactions in the UbPCNA-REV1-REV7-REV3L complex (Figure 1). In accordance, we have reported small-molecules that inhibit UbPCNA-REV1 interaction by binding to PCNA17 and those that inhibit REV7 interaction with a partial structure of REV3L by binding to REV718. Another group has reported small-molecules that inhibit REV1 interactions with DNA polymerases19. Here, we focus on REV1 UBM as another potential target for inhibiting DDT.
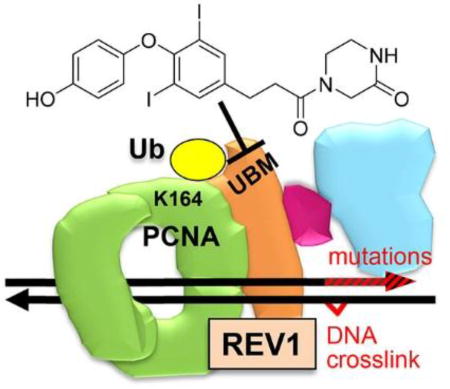
Figure 1.

The REV1 machinery for DNA damage tolerance and mutation. PCNA (homotrimer, green) is K164-monoubiquitinated (yellow) (UbPCNA) when a large DNA lesion such as a DNA crosslink is detected on the replication fork. REV1 (orange) has UBMs and binds to the UbPCNA. REV1 also binds to REV7 (wine red), recruiting REV3L (blue), which engages DNA replications that induce mutations (red arrowhead).
RESULTS
Small-molecule compounds inhibit REV1/UbPCNA interaction and directly bind to REV1 UBM
Our plan to identify inhibitor compounds was to screen a chemical library for UbPCNA-REV1 interaction inhibition and for direct REV1 protein binding. Previously, we used an immunoprecipitation assay using crude nuclear lysate that was harvested from cells challenged with DNA damage (i.e., ex vivo protocol using endogenous UbPCNA) to identify a UbPCNA inhibitor17, but this method is too tedious and not scalable for chemical library screening purposes. Therefore, we intended to use a scalable in vitro protocol. In our hands, full-length REV1 protein was very difficult to produce in large enough amounts for in vitro screening. Because studies have shown that BRCT domain12,20 and UBM6 may be responsible for REV1 interaction with UbPCNA, we alternatively generated REV1Δ, a truncated REV1 protein containing these domains (Figure 2A). Linear chimeras of PCNA-Ub fusion have been used as surrogates of UbPCNA for in vitro studies6,21. But in cells, PCNA is specifically monoubiquitinated at K164 upon DNA damage22. Therefore, to find compounds that are potentially active in cells, an assay should work with bona fide UbPCNA but not with the chimera. We have acquired an enzymatic protocol to produce UbPCNA in vitro23–24. This protocol ligated either non-tagged or N-terminus– tagged (hexahistidine, biotin-AviTag) ubiquitin to PCNA (Figure 2B).
Figure 2.

Proteins used to screen compounds for REV1/UbPCNA interaction inhibition. A: REV1 (1251 AA) and its truncated variant (REV1Δ, 695 AA), which was used to screen chemical compounds for inhibiting REV1/UbPCNA interaction. Dotted lines indicate the truncation to generate REV1Δ. B: In vitro production of K164-UbPCNA tagged with a biotin on the ubiquitin N-terminus. The reaction mixtures were analyzed by Coomassie-staining (left) or immunoblotting for pan-PCNA (right, PC-10 antibody) to confirm the UbPCNA bands.
To develop a scalable method to screen chemical compounds using these proteins, we used AlphaScreen that has been successfully applied to assay fairly weak protein-protein interactions25. This property is desirable for assaying ubiquitin interactions because affinities of ubiquitin-binding domains are weak or modest26. In addition, high sensitivity of the proximity signal generation by AlphaScreen is desirable for assaying compounds on proteins whose supply is limited such as UbPCNA by using relatively low protein concentrations. AlphaScreen cannot be used to determine the EC50 (surrogate of KD) of the REV1Δ/UbPCNA interaction because it is a sandwich assay that enforces the hooking effect in the direct binding curve. Despite, significant signals were generated by treating mixture of His-tagged REV1Δ and UbPCNA protein in which a biotin-AviTag is incorporated at N-terminus of the ubiquitin moiety by adding AlphaScreen beads (Figure 3A). We have screened ~50 chemical compounds that we have previously produced for another project27 to select ones that inhibit the signal generation in dose-dependent manner. Two structurally similar compounds, 1 and 2, appeared to be inhibitory for the REV1/UbPCNA interaction in this assay (Figure 3B). These compounds did not inhibit the signal generation from biotin-tagged polyhistidine peptide, indicating that they are not assay artifacts.
Figure 3.

Identification of small-molecule compounds that inhibit interaction of UbPCNA with REV1. A: The AlphaScreen assay for interaction of His-tagged REV1Δ and biotin-tagged UbPCNA that identified compounds that inhibit UbPCNA-REV1 interaction. B: Dose-response of the compounds for inhibiting the REV1Δ/UbPCNA interaction. The IC50 values of 1 and 2 were fitted as 3.4 µM and 9.7 µM, respectively.
We next used a protein NMR approach to show whether the compound binds to REV1Δ. Because REV1Δ and UbPCNA are still difficult to produce in large enough amounts for NMR experiments, we produced a small protein encoding only REV1 UBM1-UBM2 because they are potential site if the compounds inhibit the REV1/UbPCNA interaction by binding to REV1. In the saturation transfer difference (STD) NMR experiment28 of the compounds, they showed positive NOE peaks with the UBM1-UBM2 protein but no or negative NOE peaks without the protein (Figure 4A), indicative of direct binding to UBM1-UBM2. We next characterized the structural outcome of the REV1 interaction of compounds by 2D [1H, 15N]-HSQC NMR. Because we have found that HSQC peaks of UBM2 but not UBM1 of REV1 were perturbed by adding ubiquitin (manuscript under review), we anticipated that REV1/UbPCNA interaction is mediated by UBM2 and thus the compound should bind to UBM2. Compound 1 is not soluble enough to acquire clear HSQC spectra but 2 is soluble. In the 2D [1H, 15N]-HSQC spectra, peaks of several residues of REV1 UBM2 protein were shifted by adding 2 in dose-dependent manner (Figure 4B–C). By mapping the chemical shift perturbation (CSP) values (Figure 4D) on the NMR structure of REV1 UBM2, we found that all residues perturbed by 2 are on the α-helix structures (Figure 4E), which is similar to what we observed in the spectra of REV1 UBM2 with ubiquitin. This finding may explain the potential structural mechanism of Compound 2’s inhibition of UbPCNA- REV1Δ interaction (Figure 3).
Figure 4.

Compounds 1 and 2 bind to REV1 UBM2. A: One-dimensional proton spectra of 1 (upper) and 2 (lower) with REV1 UBM2 protein. Red trace indicates the 1D reference spectrum of each compound. Blue trace indicates the STD spectrum of each compound. B: Superposition of the 2D [1H, 15N]-HSQC spectra of REV1 UBM2 with 2 at different molar ratios: 1:0 (black), 1:2 (green), 1:4 (cyan), 1:6 (magenta), 1:8 (blue), and 1:10 (red). Arrows show the direction of perturbation. C: Selected peaks from the HSQC spectra are enlarged. D: Chemical shift perturbations (CSPs) are plotted as a function of the residue number for REV1 UBM2 with 2 at molar ratio 1:10. The secondary structure elements in the REV1 UBM2 are indicated at the top: α1-helices 1 and α2-helices 2. The CSP, with the average value of 0.006 displayed as a red dotted line. E: Assignment of CSP on the NMR structure of free REV1 UBM2 (6AXD.pdb) in a cartoon-embedded surface model. The secondary structure elements are indicated. Residues are colored on a blue (largest) to white (zero) gradient based on the CSP value.
We have attempted to accurately measure KD of the Compound 2 binding to REV1 UBM2. However, we could not observe saturation of the binding in either isothermal titration calorimetry (ITC) or surface plasmon resonance (SPR) (not shown), because compound 2 precipitated when the REV1 UBM2: 2 ratio exceeds 1:10. This suggests that the KD is significantly higher than the IC50 value observed in the AlphaScreen (Figure 3B). Therefore, we relied on NMR data to estimate the dose for the subsequent cellular experiments.
The small-molecule prevented cisplatin-mediated recruitment of REV1 that colocalized with PCNA on chromatin
To verify whether the inhibitors identified above in vitro can inhibit the full-length REV1/UbPCNA interaction in cells, we used a chromatin imaging approach that we have used to show that a compound that binds to and inhibits interactions of UbPCNA prevents chromatin localization of REV1 protein with PCNA foci in cells challenged with cisplatin (i.e., in situ generation of UbPCNA by DNA damage)17, indicating inhibition of REV1/UbPCNA interaction. The cells were treated with 1 in the concentration in which the NMR experiments (Figure 4A–D) indicated the UBM2 binding. Compound 2 was too toxic in this concentration. EGFP-tagged full-length REV1 protein was expressed in U2OS cells, and the cells were pulse-treated with cisplatin followed by Compound 1. The cells were pre-extracted to remove any non-chromatin proteins and the chromatin was immunostained for PCNA foci to image them with the EGFP-REV1. The recruitment of the EGFP-REV1 to chromatin was significantly suppressed by 1 (Figure 5).
Figure 5.

Compound 1 inhibits chromatin localization of REV1 following cisplatin treatment. A: Chromatin co-foci of full-length EGFP REV1/PCNA in U2OS cells that were pulse-treated with cisplatin (33 µM) followed by incubation with DMSO or 1 (150 µM). Pre-extracted cell nuclei were immunostained for PCNA. Confocal images of representative cells are shown. B: Colocalization of EGFP-REV1 and PCNA was determined by calculating Pearson’s correlation coefficients between red and green signals of all the pixels within a DAPI-positive area, n=32 (DMSO) and 37 (1). The dots in the graph indicate the correlation coefficient of individual cells. Data were analyzed by performing Student’s t-test (two-tailed). The bars in the graph indicate averages.
The small-molecule delayed removal of UV-induced DNA crosslinks from nuclei and prevented UV-induced mutation of the HPRT gene
REV1 supports DNA replication over various DNA lesions including crosslinks to prevent replication arrest and support the ultimate removal of the lesions5. Therefore, we assayed Compound 1 for inhibition of removal of DNA crosslinks. The cyclopyrimidine dimer (CPD) is a DNA crosslink that is generated by UVC and that can be reliably quantified on cell nuclei by using a specific antibody. The cells were UVC-irradiated to induce CPDs on their DNA and allowed to remove the CPDs while being incubated in Compound 1, which significantly suppressed removal of the CPD foci (Figure 6A–B). The incomplete inhibition regardless of the presence of 1 is presumably because the CPDs have been removed by Polη29 in addition to REV1.
Figure 6.

A: Compound 1 suppresses CPD removal. HeLa cells were irradiated by UVC (2.5 J/m2), and treated as indicated; chromatin was immunostained with a CPD antibody. (a) untreated cells; (b) cells <1 h after UVC irradiation; (c) cells 24 h after UVC irradiation and recovery under DMSO; and (d) ibid under 1 (150 µM). B: Dot blots quantify the CPD foci normalized with the DAPI staining for each panel in (A) as indicated. Data were analyzed by performing Student’s t-test (two-tailed). The bars in the graph indicate averages. C: Compound 1 (150 µM) suppresses HPRT gene mutations induced by UV. WTK-1 cells were UVC-challenged, treated as indicated, and cultured to allow HPRT protein expression. Mutation frequency was determined by culturing 480,000 cells with 6-TG for 10 days and calculated from the number of surviving colonies in each dish normalized to plating efficiency before adding 6-TG. Error bars represent standard deviation (n=2, 3, or 4).
REV1 supports DNA replication over a DNA crosslink by Polζ that is highly mutagenic (Figure 1). Therefore, we assayed 1 for inhibition of HPRT gene mutagenesis that is mediated by DNA crosslinking. WTK-1 human lymphoblastoid cells were given UVC with cisplatin followed by DMSO or 1 and cultured to induce mutation and express HPRT protein. Mutation of the UVC-crosslinked HPRT locus deactivates it; thus, the mutated clones are resistant against 6-thioguanine. Compound 1 significantly reduced the number of resistance clones (Figure 6C), indicating suppression of cisplatin-mediated mutagenesis. Few of the cells that received the highest UVC dose in this experiment (128 J/m2) survived, presumably because the amount of CPD generated by this UVC dose was too excessive to allow recovery under the presence of 1 and induced cell death. Given that CPD removal was suppressed incompletely by 1 (Figure 6A–B), we anticipated that the mutagenesis suppression by 1 would be more significant for DNA crosslinks other than CPD. However, the HPRT assays using cisplatin instead of UVC were unsuccessful because of enhanced cisplatin toxicity by the Compound 1 treatment (next section).
The small-molecule diminished clonogenic survival of cells that were challenged by chemotherapy drugs
We performed clonogenic cell survival assays to validate whether a small-molecule REV1 UBM2 inhibitor enhances the efficacy of chemotherapy drugs. U2OS cells received pulse treatment of 4-hydroxycyclophosphamide (active metabolite of cyclophosphamide) or cisplatin in doses not lethal to the cells, followed by Compound 1 or DMSO. Clonogenic survival of the cells that received only 1 was approximately 2-times lower than that of control cells (Figure 7), but survival of cells that received 1 in addition to non-lethal dose of either drug was approximately 10-times lower than that of control cells, indicating that 1 enhanced the drugs’ efficacy.
Figure 7.

Compound 1 enhances the efficacy of chemotherapy drugs. Clonogenic survival of U2OS cells after 4-hydroxycyclophosphamide (left) or cisplatin (right) treatment with or without 1 (75 µM). Error bars represent standard deviation (n=3).
DISCUSSION
Previously, we have shown that a small-molecule compound that binds to PCNA adjacent to K164 (to be monoubiquitinated upon DTT) inhibits UbPCNA protein interactions and suppresses REV1 recruitment to chromatin17. Here, we identified another compound that binds to REV1 UBM2 and inhibits the REV1/UbPCNA interaction. Because we have found that REV1 UBM2 can interact with ubiquitin (manuscript under review), it is likely that REV1/UbPCNA interaction is mediated by the UBM2. Altogether, the REV1/UbPCNA interaction can be inhibited by targeting either UbPCNA or REV1 UBM2. A potential limitation for targeting REV1 UBM2 is that intrinsic affinity of the UBM2 to UbPCNA is likely to be modest given that affinities of ubiquitin binding domains are weak or modest26. At a glance, inhibiting a protein-protein interaction with a modest affinity sounds easy, but it may not be because the modest affinity is due to a native property of the protein; that is, REV1 UBM2 itself may modestly interact with either ubiquitin or small-molecules intrinsically, which may partially explain the modest UBM2-inhibition potencies of Compounds 1 and 2. To generate potent analogs of these compounds, a fragment-based strategy could be applied by using protein NMR to identify fragment compounds that bind to REV1 at a site other than UBM2 and then conjugating the fragment compounds to 1 or 2.
A concern for targeting REV1 UBM is non-selectivity among other UBMs such as those of DNA Polι30, although there are few studies indicating potential toxicities by inhibiting UBMs other than those of REV1. However, we have observed several structural differences between REV1 UBM2 (6AXD.pdb) and Polι UBM (2KHU.pdb), indicating that a structure-based strategy is possible to rationally modify Compounds 1/2 or their fragment-conjugates to generate REV1 UBM2–specific small-molecules.
Once potent and selective REV1 UBM2 inhibitors are identified, such compounds will be useful chemical tools with which to study specific REV1 functions that UBM2 modulates because such UBM2 inhibitors will not affect the pathways that other protein interaction domains of REV1 (e.g., BRCT12) mediate. A study has shown that cells expressing REV1 point mutants in UBM are hypersensitive to UV and cisplatin6, genetic evidence that disrupting REV1 UBM interactions will deactivate REV1 functions for DDT. Given that Rev1 knock-out mice are viable11, small-molecules that potently and specifically inhibit REV1 would not be mechanistically toxic. Such inhibitors would reserve potential as future therapeutic agents to improve existing chemotherapies that generate interstrand DNA crosslinking (ICL), such as cisplatin and cyclophosphamide (Figure 7), because REV1 is required to remove ICL4 and REV1 has been validated for resistance against ICL drugs in mice7. A recent study demonstrating utility of small-molecules that inhibit DDT by targeting REV1 interactions to DNA polymerases19 also supports this therapeutic rationale.
MATERIALS AND METHODS
Reagents
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. All oligonucleotides for plasmid constructions were synthesized by Integrated DNA Technologies (Coralville, IA). The In-Fusion cloning kit (Takara Bio USA, Mountain View, CA) was used according to the manufacturers’ recommendations. The AlphaScreen assay kit was purchased from PerkinElmer (Waltham, MA). Anti-PCNA PC10 mouse IgG was purchased from Cell Signaling Technology (Danvers, MA). All cells were obtained from American Type Culture Collection (Manassas, VA) and cultured in Dulbecco's Modified Eagle Medium containing 10% FBS in an incubator at 37°C in a humidified atmosphere of 5% carbon dioxide.
Chemical synthesis
Production of His-tagged REV1Δ protein
To obtain a plasmid expressing minimal components of human REV1 with a histidine tag (REV1Δ) in bacteria, two coding regions were retrieved by PCR from pLLEV- human REV1 plasmid and introduced into pET15b vector (Millipore, Billerica, MA) by an in vitro recombination system using In-Fusion HD Cloning (Takara Bio, Mountain View, CA). The first fragment encoding codons 31 through 90 was retrieved by primers 5’-AGG AGA TAT ACC ATG GAA CAG TTT CGA TCA GAT GC-3’ and 5’-ACT CAG AAG AAA AGC ATA TGT GTT GTT TTA GAT CTG G-3’. The second fragment encoding codons 730 through 1251 was retrieved by 2 consecutive rounds of PCR, which added a thrombin cleavage site and a stretch of 8 histidines at the C-terminus of the truncated protein. The primers for the first round of PCR were 5’-GCT TTT CTT CTG AGT CTT TCA G-3’ and 5’-GTG GCT GCT GCC GCT GCC GCG CGG CAC CAG GCC GCT GCT TGT AAC TTT TAA TGT GCT TCC-3’, the latter of which contained the thrombin site and a histidine. The product was served for the second round of PCR, with primers 5’-GCT TTT CTT CTG AGT CTT TCA G-3’ and 5’-GGA TCC TCG AGC ATA TCA ATG ATG GTG ATG ATG ATG ATG ATG GCT GCT GCC-3’. The latter primer was designed to extend the histidine stretch. The 2 fragments of 210 bp and 1640 bp were introduced into NcoI and NdeI sites of pET15b vector by In-Fusion. The resultant sequence encodes the REV1Δ protein (695 AA) consisting of BRCT, UBM1, and UBM2 domains, of which the C-terminus is tagged with the thrombin site and the histidines but not with most of the catalytic core domain. The underscored nucleotides in the primer sequences denote the sequence used for recombination.
The resultant plasmid was used to transform E. coli strain BL21(DE3) (Millipore). A 10-L culture was grown at 37°C in LB media plus ampicillin to an OD600nm of 0.6. The culture was then shifted to 16°C, and REV1Δ protein expression was induced with 0.1 mM IPTG. Growth was continued for a further 16 h, at which point the cell pellet was collected by centrifugation. The pellet was resuspended in 20 mM HEPES, pH 7.4, 500 mM NaCl, 10% glycerol and 2 mM tris (2-carboxyethyl) phosphine hydrochloride (TCEP) and lysed by passage through a microfluidizer. The lysate was cleared by centrifugation and applied to a 5-mL bed volume Ni-NTA column (GE Healthcare). The REV1Δ protein was eluted by adding a linear gradient of imidazole in the same buffer. Fractions containing REV1Δ were pooled and dialyzed against 20 mM HEPES pH 7.4, 500 mM NaCl, 10% glycerol, and 2 mM TCEP overnight at 4°C.
Production of site-specifically biotin-tagged UbPCNA protein
Human Uba1 and UbcH5c S22R proteins were expressed in E. coli BL21(DE3) cells (Novagen). Cells were initially grown at 37°C and induced with 0.5 mM IPTG at 18°C overnight. Cells were harvested in a lysis buffer of 25 mM Tris, pH 8.0, 500 mM NaCl, and 10% glycerol. Affinity purification was initially done through a Ni-NTA column followed by a second purification using a 5-mL HiTrap Q HP column (GE Healthcare). Final purification was performed by size-exclusion chromatography using a HiPrep 16/60 Sephacryl S-200 HR prepacked column in 20 mM sodium phosphate, pH 7.0, 100 mM NaCl, and 2 mM TCEP. This expression and purification procedure is applicable to both Uba1 and UbcH5c S22R. Next, ubiquitination of PCNA was performed —initially in small-scale reactions that contained 80 nM Uba1 (E1 ubiquitin ligase), 16 µM UbcH5c S22R (E2 ubiquitin ligase), 32 µM biotin-AviTag-ubiquitin (Supporting Information), and 16 µM PCNA in a buffer containing 50 mM Tris or malic acid-MES-Tris (MMT) buffer, pH 8.0, 100 mM NaCl, 3 mM MgCl2, 0.5 mM TCEP, and 3 mM ATP. Experiments were performed for 16 h at 37°C in a 50 µL volume. Finally, the reaction was optimized for a 500 µL volume consisting of 100 µM Uba1, 128 µM UbcH5c S22R, 256 µM biotin-AviTag-ubiquitin, and 64 µM PCNA in a buffer containing 50 mM MMT pH 9.0, 25 mM NaCl, 10 mM MgCl2, 0.5 mM TCEP, and 3 mM ATP. Ubiquitinated PCNA was further purified by using a 1-mL HiTrap Q XL column (GE Healthcare) with 0 to 500 mM NaCl gradient in 20 mM HEPES, pH 8.0, and 0.5 mM TCEP.
AlphaScreen assays
The AlphaScreen assay was conducted in a solution consisting of 200 nM biotin-AviTag-ubiquitin-PCNA protein and 40 nM N-terminal His-tagged REV1Δ in the AlphaScreen buffer (1× PBS, pH 7.4, 0.1% BSA, and 0.05% Tween-20). Briefly, 5 µL of the assay solution containing the indicated serial dilution of indicated compound was transferred into each well of a white OptiPlate-384 (PerkinElmer, Waltham, MA). AlphaScreen nickel chelate acceptor beads (PerkinElmer) diluted 1:100 in 10 µL of the AlphaScreen buffer were added. After 1 h, streptavidin AlphaScreen donor beads (PerkinElmer) diluted 1:100 in 10 µL of the AlphaScreen buffer were added. The AlphaScreen signal was read an hour later by using an EnVision plate reader (PerkinElmer). The dose-response curve was fitted by using Prism 7 (GraphPad Software, La Jolla, CA) for final concentration of each serial dilution.
Production of REV1 UBM1-UBM2 and UBM2 proteins
The coding region of UBM1 and UBM2 domains (324 bp, 108 aa) was retrieved by PCR of pLLEV- human REV1 plasmid by using the primers 5’-AAG AGT TCC GGT CTG CCG TCA CCT TCC CAG CTG GAT C-3’ and 5’-TTA GCA GCC GGA TCC TAT TGT CTT TGA TCA TAC GCT GC -3’. The linear vector was generated by PCR of pET15b by using the primers 5’-CAG ACC GGA ACT CTT AAT GC-3’ and 5’-GGA TCC GGC TGC TAA CAA AGC C-3’. These fragments were recombined by In-Fusion. The resultant sequence encodes a protein (137 aa) with UBM1 and UBM2 domains, with the histidines and thrombin site at its N-terminus. The REV1 UBM1-UBM2 protein was produced using a method similar to that described for the REV1Δ protein followed by removal of the histidine-tag by trypsin digestion. Preparation of the REV1 UBM2 protein is reported elsewhere.
STD NMR experiments
STD experiments were performed at 280 K at a concentration of 10 µM protein and 200 µM of compound. For saturation transfer difference (STD) experiments, pre-saturation was achieved using a pulse train of 30 ms Gaussian shaped pulses for a 4-s duration and an offset of 0 ppm (on resonance) or −40 ppm (off resonance). To obtain STD spectra, 256 on and off resonance of free induction decays were recorded. Subtraction of the on-resonance from the off-resonance spectra yielded the STD signal.
NMR titrations for interaction of compound with REV1 UBM2
A series of two-dimensional [1H, 15N]-HSQC spectra were collected on a Bruker 600 spectrometer at 298K. The REV1 UBM2 concentration for the entire titration was 50 µM, and the procedure was performed in a 20 mM sodium phosphate buffer (pH 7.0) and 100 mM NaCl. Chemical shifts (Δ) of individual resonances from 15N-labeled REV1 UBM2 were monitored as a function of increasing compound 2 concentration (REV1 UBM2: Compound 2 ratios: 1:2, 1:4, 1:6, 1:8, and 1:10). Compound 2 precipitated when the ratio exceeds 1:10 (not shown). 15N and 1H chemical shift values for the displaced peaks were determined for each of the successive titration points by using CARA 1.8.431. To determine the per-residue chemical shift perturbation upon binding and account for differences in spectral widths between 15N and 1H resonances32, weighted average chemical-shift differences [Δav(HN)] were calculated for the backbone amide 1H and 15N resonances by using the equation Δav (NH) = [(ΔH2+ (ΔN/5)2)/2]1/2, where ΔH and ΔN are chemical-shift differences for 1H and 15N, respectively33–34.
Chromatin co-localization studies
U2OS cells (4 × 105/well) were initially seeded onto coverslips in six-well cluster plates and allowed to attach overnight. Each sample was transfected with pEGFP-REV1 (2.5 µg) by using 7.5 µL of Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. Two days following transfection, cells were treated with 33 µM cisplatin for 3.5 h prior to release into media containing either DMSO vehicle or 150 µM of Compound 1 for 6 h. Samples were then pre-extracted with ice-cold CSK buffer (100 mM NaCl, 300 mM sucrose, 10 mM HEPES pH 7.4, 3 mM MgCl2, 1 × Halt Protease Inhibitor Cocktail [Thermo Fisher Scientific], 0.5% Triton X-100) for two minutes to remove non-chromatin bound protein and then fixed with 4% paraformaldehyde for 10 min at ambient temperature. Cells were exposed to 100% methanol for 15 min at −20°C prior to blocking in 3% FBS in PBS overnight at 4°C. The cells were then immunostained by probing with 1:2000 mouse α PCNA monoclonal antibody (PC10, Cell Signaling Technology) for 60 min followed by incubation with 1:200 donkey α mouse IgG Alexa 555 (Thermo Fisher Scientific) for 30 min. Cells were then washed extensively with PBS and mounted onto glass slides by using VectaShield containing 1 µg/mL DAPI. Immunostained cells were subsequently examined, and images were captured on a TE2000 microscope equipped with a C1Si confocal (Nikon, Tokyo, Japan). Nikon NIS-elements software (Nikon, Tokyo, Japan) was used to calculate the Pearson’s correlation coefficient of red (PCNA) and green (REV1) fluorescence signals for each nucleus, using DAPI staining to define the perimeter of each nuclei.
CPD immunofluorescence assay
HeLa cells (1.5 × 105 cells/dish) were seeded onto coverslips in 35-mm culture dishes and allowed to settle overnight. Cells were irradiated with 0 or 2.5 J/m2 UVC at 254 nm by using a Stratalinker UV Crosslinker 1800 (Stratagene, La Jolla, CA). Following irradiation, cells were released into media containing DMSO or 150 µM Compound 1. At designated time points, samples were rinsed with PBS, fixed with 3.7% formaldehyde in PBS for 15 min at room temperature, permeabilized with 0.5% triton X-100 for 5 min at room temperature, and stored at 4°C in PBS until all samples had been collected. Samples were subsequently denatured in 2 M HCl for 30 min and blocked in 3% FBS in PBS for 10 min at room temperature. Cells were probed with 1:400 mouse α CPD monoclonal antibody (clone TDM-2, Cosmo Bio USA) in 3% FBS in PBS for 70 min, thoroughly rinsed with PBS, and then exposed to 1:200 donkey anti-mouse IgG Alexa 488 (Thermo Fisher) in 3% FBS in PBS for 30 min at room temperature. Cells were then washed extensively by using 3 × PBS rinses and then mounted onto glass slides by using VectaShield containing 1 µg/mL DAPI. Samples were visualized by using an Olympus IX81 epi-fluorescence microscope (Olympus Corp., Tokyo, Japan), and Nikon NIS-elements software (Nikon, Tokyo, Japan) was used to calculate the mean green fluorescence intensity in each nucleus, using the DAPI staining pattern to define the perimeter of the nucleus.
HPRT gene mutagenesis assay
WTK-1 human lymphoblastoid cells were initially cleansed to minimize background levels of mutation in the HPRT gene by exposing cells to CHAT in RPMI media supplemented with 10% FBS for 2 days. Cells were allowed to recover in media containing THC for a further two days prior to conditioning in RPMI media containing 10% heat-inactivated horse serum. Conditioned WTK-1 cells (2 × 106) were subsequently exposed to UVC (0 – 128 J/m2) by using a UVP model UVGL-58 Handheld UV Lamp (Upland, CA) in the short-wavelength (254 nm) mode. Immediately following UVC treatment, cells were treated with either DMSO or 150 µM Compound 1 for 21 h. Following treatment, cells were resuspended in drug-free RPMI media supplemented with 10% horse serum, 1% penicillin/streptomycin, and 200 µg/mL sodium pyruvate. A portion of each suspension (0.5 mL) was used to evaluate survival (PE0) by immediate re-distribution into 96-well cluster plates at an average cell density of 1.6 cells per well. PE0 survival plates were incubated for 11 days and then scored for wells exhibiting colony growth. The remaining bulk of each cell suspension was cultured (4.8 × 106 cells per treatment) for a further five days to enable fixation of drug-induced mutations in the HPRT gene. Following fixation, cells were re-plated into 96-well cluster plates at a density of 5,000 cells per well in complete media containing 0.5 µg/mL 6-thioguanine (6TG) as a selective agent. Cells were also re-distributed in parallel into 96-well cluster plates without selective agent at an average density of 1.6 cells per well to evaluate survival (PE6). All 6TG and PE6 plates were incubated for a11 – 12 more days and then scored for colony formation.
Clonogenic cell survival assay
After U2OS cells (300 cells/well) were settled in 6-well plates overnight, each indicated final concentration of cisplatin or 4-hydroxycyclophosphamide was added. After 24 h, Compound 1 or DMSO was added as indicated. After another 24 h, the culture medium was replaced with fresh medium, and cells were cultured for another 7 days. Each treatment was done in triplicate. The cells were gently rinsed twice with PBS, fixed with 3.7% formaldehyde, and stained by using 0.5% crystal violet. Colonies containing more than approximately 40 cells were counted. The survival rate was reported as 1 for the average of colony numbers in wells that received vehicle control (0.9% sodium chloride for cisplatin and DMSO for 4-hydroxycyclophosphamide).
Supplementary Material
Acknowledgments
We thank the Sanger DNA Sequencing Core for plasmid sequencing, Weixing Zhang for supervising the NMR, Jennifer Peters for assistance with microscopic foci quantification, Sivaraja Vaithiyalingam and Brett Waddell for attempting affinity measurements, Christine Canman for providing plasmids encoding human REV1, Hans Haecker for providing pBirA plasmid, Titia Sixma for providing pET3a- human Uba1 (E1) plasmid through Addgene (#63571, Cambridge, MA), Rachel Klevit for providing pET28a-Ubc5Hc S22R (E2) plasmid through Addgene (#12644), and Cherise Guess for editing the manuscript. The study was funded by ALSAC. The Cell & Tissue Imaging Center is supported by NCI P30 CA021765-34.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
MV produced proteins and performed protein NMR and SPR experiments. BJE performed the CPD removal assay, REV1 chromatin imaging, and HPRT mutagenesis assay. MLA performed chemical syntheses and AlphaScreen assays. AI generated protein expression constructs and performed AlphaScreen, HPRT mutagenesis, and cell survival assays. YS, RJH, and ETM produced proteins. NF designed the research and wrote the paper. All authors have approved the final version of the manuscript.
Conflict of interest statement
BJE is currently an employee of Noxopharm Ltd. The authors declare no other conflict of interest.
References
- 1.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nature reviews. Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 2.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nature reviews. Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ho TV, Scharer OD. Translesion DNA synthesis polymerases in DNA interstrand crosslink repair. Environmental and molecular mutagenesis. 2010;51:552–566. doi: 10.1002/em.20573. [DOI] [PubMed] [Google Scholar]
- 4.Sharma S, Canman CE. REV1 and DNA polymerase zeta in DNA interstrand crosslink repair. Environmental and molecular mutagenesis. 2012;53:725–740. doi: 10.1002/em.21736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hicks JK, Chute CL, Paulsen MT, Ragland RL, Howlett NG, Gueranger Q, Glover TW, Canman CE. Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA crosslinks. Molecular and cellular biology. 2010;30:1217–1230. doi: 10.1128/MCB.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo C, Tang TS, Bienko M, Parker JL, Bielen AB, Sonoda E, Takeda S, Ulrich HD, Dikic I, Friedberg EC. Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Molecular and cellular biology. 2006;26:8892–8900. doi: 10.1128/MCB.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie K, Doles J, Hemann MT, Walker GC. Error-prone translesion synthesis mediates acquired chemoresistance. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20792–20797. doi: 10.1073/pnas.1011412107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sasatani M, Xi Y, Kajimura J, Kawamura T, Piao J, Masuda Y, Honda H, Kubo K, Mikamoto T, Watanabe H, Xu Y, Kawai H, Shimura T, Noda A, Hamasaki K, Kusunoki Y, Zaharieva EK, Kamiya K. Overexpression of Rev1 promotes the development of carcinogen-induced intestinal adenomas via accumulation of point mutation and suppression of apoptosis proportionally to the Rev1 expression level. Carcinogenesis. 2017;38:570–578. doi: 10.1093/carcin/bgw208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin X, Okuda T, Trang J, Howell SB. Human REV1 modulates the cytotoxicity and mutagenicity of cisplatin in human ovarian carcinoma cells. Molecular pharmacology. 2006;69:1748–1754. doi: 10.1124/mol.105.020446. [DOI] [PubMed] [Google Scholar]
- 10.Dumstorf CA, Mukhopadhyay S, Krishnan E, Haribabu B, McGregor WG. REV1 is implicated in the development of carcinogen-induced lung cancer. Molecular cancer research : MCR. 2009;7:247–254. doi: 10.1158/1541-7786.MCR-08-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jansen JG, Langerak P, Tsaalbi-Shtylik A, van den Berk P, Jacobs H, de Wind N. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. The Journal of experimental medicine. 2006;203:319–323. doi: 10.1084/jem.20052227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo C, Sonoda E, Tang TS, Parker JL, Bielen AB, Takeda S, Ulrich HD, Friedberg EC. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol Cell. 2006;23:265–271. doi: 10.1016/j.molcel.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 13.Sharma NM, Kochenova OV, Shcherbakova PV. The non-canonical protein binding site at the monomer-monomer interface of yeast proliferating cell nuclear antigen (PCNA) regulates the Rev1-PCNA interaction and Polzeta/Rev1-dependent translesion DNA synthesis. J Biol Chem. 2011;286:33557–33566. doi: 10.1074/jbc.M110.206680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lawrence CW. Cellular functions of DNA polymerase zeta and Rev1 protein. Adv Protein Chem. 2004;69:167–203. doi: 10.1016/S0065-3233(04)69006-1. [DOI] [PubMed] [Google Scholar]
- 15.Sharma S, Shah NA, Joiner AM, Roberts KH, Canman CE. DNA Polymerase Zeta is a Major Determinant of Resistance to Platinum-based Chemotherapeutic Agents. Molecular pharmacology. 2012 doi: 10.1124/mol.111.076828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu F, Lin X, Okuda T, Howell SB. DNA polymerase zeta regulates cisplatin cytotoxicity, mutagenicity, and the rate of development of cisplatin resistance. Cancer Res. 2004;64:8029–8035. doi: 10.1158/0008-5472.CAN-03-3942. [DOI] [PubMed] [Google Scholar]
- 17.Inoue A, Kikuchi S, Hishiki A, Shao Y, Heath R, Evison BJ, Actis M, Canman CE, Hashimoto H, Fujii N. A small molecule inhibitor of monoubiquitinated Proliferating Cell Nuclear Antigen (PCNA) inhibits repair of interstrand DNA cross-link, enhances DNA double strand break, and sensitizes cancer cells to cisplatin. J Biol Chem. 2014;289:7109–7120. doi: 10.1074/jbc.M113.520429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Actis ML, Ambaye ND, Evison BJ, Shao Y, Vanarotti M, Inoue A, McDonald ET, Kikuchi S, Heath R, Hara K, Hashimoto H, Fujii N. Identification of the first small-molecule inhibitor of the REV7 DNA repair protein interaction. Bioorganic & medicinal chemistry. 2016;24:4339–4346. doi: 10.1016/j.bmc.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sail V, Rizzo AA, Chatterjee N, Dash RC, Ozen Z, Walker GC, Korzhnev DM, Hadden MK. Identification of Small Molecule Translesion Synthesis Inhibitors That Target the Rev1-CT/RIR Protein-Protein Interaction. ACS chemical biology. 2017;12:1903–1912. doi: 10.1021/acschembio.6b01144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pustovalova Y, Maciejewski MW, Korzhnev DM. NMR mapping of PCNA interaction with translesion synthesis DNA polymerase Rev1 mediated by Rev1-BRCT domain. Journal of molecular biology. 2013;425:3091–3105. doi: 10.1016/j.jmb.2013.05.029. [DOI] [PubMed] [Google Scholar]
- 21.Chen J, Ai Y, Wang J, Haracska L, Zhuang Z. Chemically ubiquitylated PCNA as a probe for eukaryotic translesion DNA synthesis. Nature chemical biology. 2010;6:270–272. doi: 10.1038/nchembio.316. [DOI] [PubMed] [Google Scholar]
- 22.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 23.Lau WC, Li Y, Zhang Q, Huen MS. Molecular architecture of the Ub-PCNA/Pol eta complex bound to DNA. Scientific reports. 2015;5:15759. doi: 10.1038/srep15759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hibbert RG, Sixma TK. Intrinsic flexibility of ubiquitin on proliferating cell nuclear antigen (PCNA) in translesion synthesis. J Biol Chem. 2012;287:39216–39223. doi: 10.1074/jbc.M112.389890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wigle TJ, Herold JM, Senisterra GA, Vedadi M, Kireev DB, Arrowsmith CH, Frye SV, Janzen WP. Screening for inhibitors of low-affinity epigenetic peptide-protein interactions: an AlphaScreen-based assay for antagonists of methyl-lysine binding proteins. Journal of biomolecular screening. 2010;15:62–71. doi: 10.1177/1087057109352902. [DOI] [PubMed] [Google Scholar]
- 26.Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. The Biochemical journal. 2006;399:361–372. doi: 10.1042/BJ20061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Actis M, Inoue A, Evison B, Perry S, Punchihewa C, Fujii N. Small molecule inhibitors of PCNA/PIP-box interaction suppress translesion DNA synthesis. Bioorganic & medicinal chemistry. 2013;21:1972–1977. doi: 10.1016/j.bmc.2013.01.022. [DOI] [PubMed] [Google Scholar]
- 28.Mayer M, Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. Journal of the American Chemical Society. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 29.McCulloch SD, Kokoska RJ, Masutani C, Iwai S, Hanaoka F, Kunkel TA. Preferential cis-syn thymine dimer bypass by DNA polymerase eta occurs with biased fidelity. Nature. 2004;428:97–100. doi: 10.1038/nature02352. [DOI] [PubMed] [Google Scholar]
- 30.Burschowsky D, Rudolf F, Rabut G, Herrmann T, Peter M, Wider G. Structural analysis of the conserved ubiquitin-binding motifs (UBMs) of the translesion polymerase iota in complex with ubiquitin. J Biol Chem. 2011;286:1364–1373. doi: 10.1074/jbc.M110.135038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keller R. The Computer Aided Resonance Assignment Tutorial. Verlag; Cantina, Switzerland: 2004. [Google Scholar]
- 32.Farmer BT, 2nd, Constantine KL, Goldfarb V, Friedrichs MS, Wittekind M, Yanchunas J, Jr, Robertson JG, Mueller L. Localizing the NADP+ binding site on the MurB enzyme by NMR. Nature structural biology. 1996;3:995–997. doi: 10.1038/nsb1296-995. [DOI] [PubMed] [Google Scholar]
- 33.Grzesiek S, Bax A, Clore GM, Gronenborn AM, Hu JS, Kaufman J, Palmer I, Stahl SJ, Wingfield PT. The solution structure of HIV-1 Nef reveals an unexpected fold and permits delineation of the binding surface for the SH3 domain of Hck tyrosine protein kinase. Nat. Struct. Biol. 1996;3:340–345. doi: 10.1038/nsb0496-340. [DOI] [PubMed] [Google Scholar]
- 34.Garrett DS, Seok YJ, Peterkofsky A, Clore GM, Gronenborn AM. Identification by NMR of the binding surface for the histidine-containing phosphocarrier protein HPr on the N-terminal domain of enzyme I of the Escherichia coli phosphotransferase system. Biochemistry. 1997;36:4393–4398. doi: 10.1021/bi970221q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.