

Abstract
Glioblastoma multiforme is a malignant brain tumor noted for its extensive vascularity, aggressiveness, and highly invasive nature, suggesting that cell migration plays an important role in tumor progression. The poor prognosis in GBM is associated with a high rate of tumor recurrence, and resistance to the standard of care chemotherapy, temozolomide (TMZ). The novel compound NEO212, a conjugate of TMZ and perillyl alcohol (POH), has proven to be 10 fold more cytotoxic to glioma stem cells (GSCs) than TMZ, and is active against TMZ-resistant tumor cells. In this study, we show that NEO212 decreases migration and invasion of primary cultures of patient-derived GSCs, in both mesenchymal USC02 and proneural USC04 populations. The mechanism by which NEO212 reduces migration and invasion appears to be independent of its DNA alkylating effects, which cause cytotoxicity during the first hours of treatment, and is associated with a decrease in the FAK/Src signaling pathway, an effect not exhibited by TMZ. NEO212 also decreases the production of matrix metalloproteinases MMP2 and MMP9, crucial for GSC invasion. Gene expression analysis of epithelial and mesenchymal markers suggests that NEO212 increases the expression of epithelial-like characteristics, suggesting a reversion of the epithelial-to-mesenchymal transition (EMT) process. Furthermore, in an in vivo orthotopic glioma model, NEO212 decreases tumor progression by reducing invasion of GSCs, thereby increasing survival time of mice. These studies indicate that NEO212, in addition to cytotoxicity, can effectively reduce migration and invasion in GSCs, thus exhibiting significant clinical value in the reduction of invasion and malignant glioma progression.
Keywords: glioblastoma, glioma stem cells (GSCs), focal adhesion kinase (FAK), migration, invasion
Introduction
Glioblastoma multiforme (GBM) is a highly vascular and aggressive brain tumor, characterized by a high rate of recurrence and resistance to the standard of care chemotherapy, temozolomide (TMZ). Despite great progress made in radiotherapy, surgery and chemotherapy, there has been little improvement to patient outcome over the past decade[1]. The main problem with GBM is that once the tumor recurs, there are few treatment options available to patients; and repeated surgeries and radiation have a high potential for morbidity. Thus, chemotherapeutic agents with greater efficacy than TMZ are badly needed.
Over the past years, several groups have suggested that tumor recurrence is a result of a small sub-population of drug-resistant glioma stem cells (GSCs), also known as tumor-initiating cells[2]. This cell population shows capacity for self-renewal and in vivo tumor initiation, as well as resistance to antitumor drugs such as TMZ[2, 3]. Moreover, a recent study has demonstrated that GSCs are the first to proliferate and repopulate the tumor when TMZ treatment is discontinued in a spontaneous murine model of glioma[4]. These GSCs are highly resistant to TMZ, and able to differentiate into cells of different lineages[5]. At least two subtypes of GSCs have been reported: proneural and mesenchymal. The mesenchymal is the most aggressive and invasive of the two subtypes, with lower responsiveness to treatment, shorter median overall survival and worse overall patient outcome[6–9]. In the present study, we utilized two patient-derived primary cultures of GSCs to study the effects of NEO212 on these GSC phenotypes: mesenchymal-subtype USC02 cells and proneural-subtype USC04 cells[7].
Previous studies from this laboratory have shown that the novel drug, NEO212, a conjugate of TMZ to the antitumor agent perillyl alcohol (POH)[10] (structures depicted in Supplementary Figure 1), is more effective in reducing GBM progression than TMZ and/or POH[7]. Although TMZ is effective initially, GBMs have several mechanisms of resistance to this drug[11]. These mechanisms of resistance may result from mutations in the DNA repair mechanisms such as the base excision repair (BER) pathway, poly (ADP-ribose) polymerase (PARP)[12, 13], and mismatch repair (MMR) pathway[14]. However, TMZ exerts its cytotoxic effects mainly by methylating O6-guanine[12]. Hence, the most relevant mechanism of resistance to TMZ is the overexpression of the repair enzyme O6-methyl-guanine-DNA methyltransferase (MGMT)[15]. NEO212 is effective in inducing cytotoxicity in the presence of all of these mechanisms of resistance[11, 16].
Although GBMs do not metastasize outside the central nervous system, there is significant local infiltration of tumor cells in the brain, resulting in tumor invasion and progression[17]. Recent studies have shown that tumor progression can be associated with invasion of GSCs into the surrounding tumor microenvironment, and migration of these cells along blood vessels[18]. Thus for agents that regulate GSC activity, reducing their invasive potential would be essential for decreasing tumor progression. Since these are critical dimensions in tumor progression, we examined NEO212 for its effects on migration and invasion of GSCs.
Focal Adhesion Kinase (FAK) is a non-receptor protein tyrosine kinase activated by several agents including integrins, growth factor receptors, G protein-coupled receptors, and cytokine receptors[19, 20]. FAK activation triggers subsequent signaling cascades involved in various cell processes such as cancer stem cell renewal, cell survival (including drug resistance), proliferation, migration, invasion and epithelial-to-mesenchymal transition (EMT)[21]. Here, we show that NEO212 inhibits cell migration and invasion of proneural and mesenchymal GSCs, which correlates with a blockade of the activation of the FAK/Src signaling pathway.
Materials and Methods
Chemicals
Temozolomide (Merck), perillyl alcohol and NEO212 (NeOnc Technologies.) stocks were prepared in DMSO and used immediately or stored at -20 °C. NEO212 is accessible by the scientific community from NeOnc Technologies with appropriate non-disclosure agreement (NDA), and is subject to intellectual property (IP). In addition, the compound is commercially available from Axon Medchem.
Isolation of glioma stem cells
Human GBM tissues were obtained following written informed consent from the patients in accordance with Declaration of Helsinki guidelines and the Institutional Review Board (HS-09-00520), at Keck School of Medicine, University of Southern California.
The detailed protocol for GSC isolation has been published[7]. GSCs were cultured in cancer stem cell (CSC) culture medium containing DMEM-F12 with 1% penicillin-streptomycin, 1% B-27 (Life Technologies), and 20 ng/mL epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) (Peprotech).
Cell viability assays
Cytotoxicity was measured using the standard trypan blue and MTT assays. Results were reported as dose-dependent curves using data from three independent experiments carried out in triplicate.
Wound healing assays
USC02 cells were seeded in 96-well plates (1.5 × 104 cells/well) and cultured for 24 h, obtaining a 90-100% confluent monolayer. Wounds were made with a p10 pipette tip and washed with PBS to eliminate non-adherent cells and cell debris, and fresh medium with drug or DMSO was added. At 0 h and after 24 h, cells were photographed with an Eclipse TE300 Inverted Microscope (Nikon). For reversibility assays, photos were also taken after 72 h. Empty area in each wound was quantified using ImageJ software (NIH) and compared to the corresponding initial wound. The percentage of the areas from three independent experiments performed in quadruplicate was presented as mean ± standard error of the mean (SEM).
Boyden chamber invasion assays
Chemoinvasion of USC02 cells was tested with a Boyden chamber with 8 μm pore-size polycarbonate membranes coated with Matrigel (Corning). USC02 (2 × 104) cells were seeded in serum-free medium with the indicated concentration of drugs in the upper chamber, while medium containing 10% FBS as a chemoattractant, plus the appropriate concentration of drugs, was placed in the lower chamber. After 16 h at 37 °C, cells on the upper side of the membrane were removed with a cotton swab, and cells on the underside were fixed and stained with Diff-Quick (EMD Millipore). Photos were taken on an Eclipse 80i microscope (Nikon), and cells counted with ImageJ Software. Data from three independent experiments performed in triplicate were presented as mean ± SEM.
3D Spheroid migration and invasion assays
Spheroid-based migration and invasion assays of USC04 cells were performed as reported by Vinci et al.[22, 23]. Pictures at t = 0 h and t = 72 h were taken using an Eclipse TE300 Inverted Microscope (Nikon), and the area covered by the migrated or invaded cells was determined using ImageJ, and normalized to the original size of each corresponding spheroid at t = 0 h. Experiments were performed on three independent days in sextuplicate.
Western blot analysis
The detailed protocol has been published[24]. Primary antibodies used were rabbit anti-phospho-AKT (Ser473), rabbit anti-AKT, rabbit anti-phospho-FAK (Tyr397), rabbit anti-FAK, rabbit anti-eIF2α, rabbit anti-phospho-MEK1/2 (Ser217/221), rabbit anti-MEK1/2, rabbit anti-phospho-p38-MAPK (Thr180/Tyr182), rabbit anti-p38-MAPK, rabbit anti-phospho-Src (Tyr416), rabbit anti-Src (1:1000, Cell Signaling), goat anti-phospho-eIF2α (Ser51), rabbit anti-MMP2 (1:1000, Abgent), mouse anti-MMP9 (1:500, Abgent), rabbit anti-tubulin (1:1000, BioLegend). Secondary antibodies used were goat anti-mouse, goat anti-rabbit or donkey anti-goat IgG HRP conjugates (1:5000, Santa Cruz Biotechnology), accordingly. Relative levels from three independent experiments were presented as mean ± SEM.
Microarray studies
The Human EMT RT2 Profiler PCR Array (Qiagen) was used to analyze the expression of 84 key genes related to the EMT process. Total RNA was isolated using the RNeasy Plus Mini Kit (Quiagen), including a genomic DNA elimination step with gDNA Eliminator columns (Quiagen). cDNA was subsequently obtained according to manufacturer’s instructions with iScript Advanced cDNA Synthesis kit (Bio-Rad). Real-time qPCR assay was performed using the Human EMT RT2 Profiler PCR Array in combination with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). Amplifications were run in a StepOnePlus cycler (Applied Biosystems) and data were analyzed using the SABiosciences PCR Array Data Analysis Template Excel (Qiagen).
In vivo experiments
All animal protocols were approved by the University of Southern California Institutional Animal Care and Use Committee and strictly adhered to. 100,000 USC02-luciferase labeled cells were implanted intracranially into the subcortical brain parenchyma of 8-10 week old male NOD/SCID mice (Envigo). The implantation coordinates were 1.0 mm posterior, 1.0 mm lateral (right) with respect to bregma, at a depth of -2.5 mm ventral. The implantation volume was 2 μL in PBS, and a 25G, 2.0 μL injection syringe was used (Hamilton). Tumor presence was confirmed by bioluminescent imaging 5 days post implantation, and treatment with either NEO212 or TMZ was initiated 6 days post implantation. NEO212 was administered subcutaneously in the neck scruff region at 5 mg/kg or 25 mg/kg in 10% DMSO, 45% ethanol, 45% glycerol. TMZ was administered via oral gavage using a 22G feeding needle, at 5 mg/kg or 25 mg/kg in water. Vehicle controls were subcutaneous injections of 10% DMSO, 45% ethanol, 45% glycerol. Treatment was administered in cycles of 5 days on, 2 days off. Bioluminescent imaging was performed at the University of Southern California Molecular Imaging Core. Mice were injected with 50 mg/kg D-Luciferin (Caliper Life Sciences) intravenously prior to imaging in the IVIS Spectrum (Xenogen Corporation). Living Image version 4.3 software (Perkin Elmer) was used for image analysis and quantification.
Mice were euthanized in compliance with the University of Southern California Institutional Animal Care and Use Committee’s general criteria for humane endpoint for tumor studies. The brains of euthanized mice were harvested and formalin-fixed for future staining.
Pharmacokinetic measurements of NEO212 in the brains were performed via HPLC, using a C18 column (50mm × 4.6mm, 3μm particle size) with 10 μL injection and flow rate of 1 mL/min. NEO212 was eluted with an isocratic flow of 40% acetonitrile over 10 minutes, and monitored at 316 nm.
Immunohistochemistry
Tissue sections were treated for antigen retrieval in a citrate buffer solution, and stained as previously described[25]. Primary antibodies used were mouse anti-humanTRA-1-85/CD147 (1:50, RD Systems), and goat anti-Sox2 (1:50, Santa Cruz Biotechnology). Secondary antibodies were biotinylated anti-mouse and biotinylated anti-goat (1:300; Vector Laboratories).
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 5.0 software. Statistical significance (P<0.05) was evaluated using 1-way ANOVA followed by Bonferroni’s or Dunnet’s Multiple Comparison Tests. Data were presented as mean ± SEM.
Results
NEO212 impairs migration of GSCs in vitro
Previous results from our laboratory showed that NEO212 reduces GSC proliferation in vitro, and increases survival in vivo[7]. Since migration and invasion play a significant role in GBM progression, our hypothesis was that NEO212 may also affect these processes in GBM cells, particularly in GSCs. To test this concept, we examined the effects of NEO212 on migration and invasion of GSCs. Using the wound healing assay, we showed that NEO212 decreases mesenchymal GSC migration. We observed that vehicle-treated USC02 cells were highly motile and almost completely repopulated the entire wound area within 24 h. By contrast, NEO212-treated GSCs exhibited impaired migration in a dose-dependent manner, and to a greater extent than cells treated with the individual components, TMZ and/or POH (Figure 1A,B). To rule out differences in cell viability or proliferation, the viable cells at the end of the experiment performed with 50 μM NEO212 were quantified, and showed no significant difference from vehicle-treated cells (Supplementary Figure 2A), indicating that there is little, if any, cell death at this dose of NEO212 after 24 h of treatment.
Figure 1. NEO212 reduces migration of GSCs.
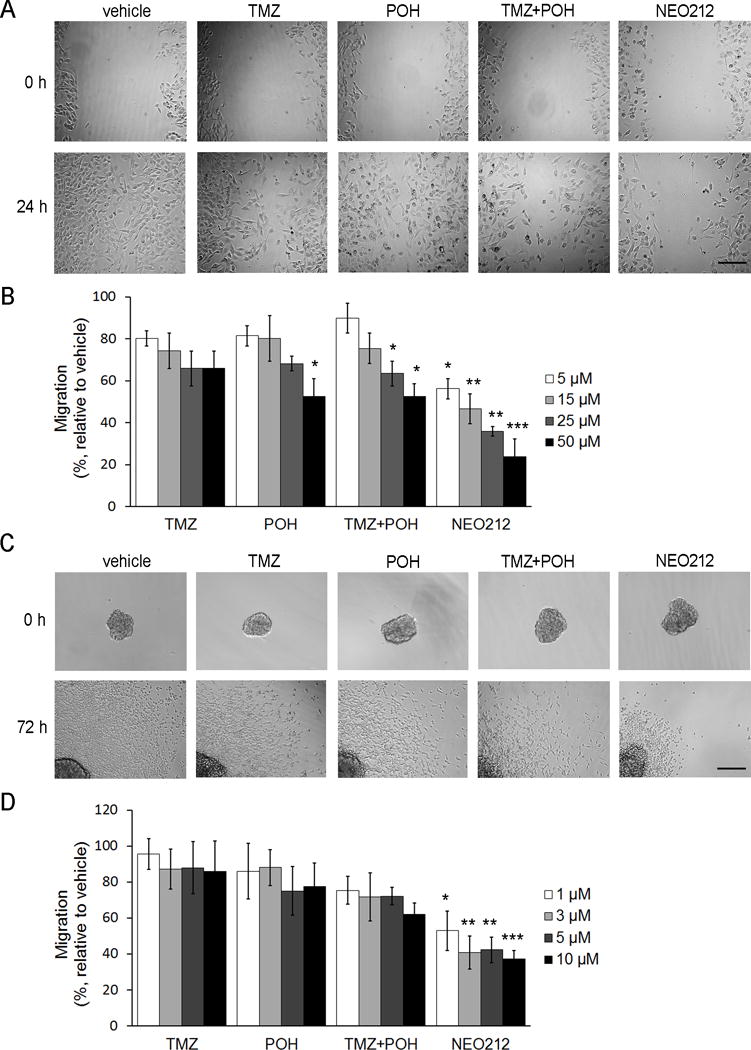
(A) Representative images of the wound healing assay performed with mesenchymal USC02 cells treated with 15 μM of each drug. Scale bar, 200 μm. (B) After 24 h of treatment, the remaining wound area was quantified and compared to the initial wound. The bar graph represents the average ± SEM of at least three independent experiments performed in quadruplicate. *, P<0.05; **, P<0.01; ***, P<0.001 (relative to vehicle-treated cells). (C) Representative images of the spheroid migration assay performed with proneural USC04 cells at 3 μM. Scale bar, 200 μm. (D) After 72 h, the area covered by migrated cells was quantified, and represented as relative to the initial sphere. The bar graph represents the average ± SEM of three independent experiments performed in sextuplicate. *, P<0.05; **, P<0.01; ***, P<0.001 (relative to vehicle-treated cells).
To determine whether this effect of NEO212 on GSC migration was specific for mesenchymal GSCs, we tested NEO212 on USC04 proneural GSCs (Figure 1C,D). Since these cells grow as spheres, we used the spheroid-based migration assay, which takes into account tumor heterogeneity, cell-cell contacts and interactions with extracellular matrix proteins, leading to more accurate results than a two-dimensional (2D) migration assay[22]. In this assay, USC04 cells were allowed to migrate from a previously formed sphere over a Matrigel coating, and the area covered by migrating cells was evaluated. Recent studies from this laboratory showed that proneural USC04 cells are more sensitive to NEO212-induced cytotoxicity, when compared to mesenchymal USC02 cells (IC50 = 43 ± 9 μM for USC02 and IC50 = 8 ± 2 μM for USC04 cells, following 72 h treatment[7]). Hence, the doses of NEO212 used for USC04 cells were adjusted accordingly, making them equipotent to the doses used for USC02 cells. Similarly to what occurred with mesenchymal GSCs, NEO212-treated USC04 cells exhibited impaired migration in a dose dependent manner, and to a greater extent than cells treated with the individual components or the mixture of both (Figure 1C,D), suggesting a general effect of NEO212 on GSCs. To rule out cytotoxic effects, cell viability was examined and found not to be significantly affected at the highest concentration, indicating also that treating the cells once they have formed spheres confers resistance to the compound (Supplementary Figure 2B). In summary, these results show that NEO212 reduces proneural and mesenchymal GSC migration, at doses below cytotoxic concentrations.
NEO212 has several distinct mechanisms of activity
TMZ undergoes rapid chemical conversion at physiologic pH to the active compound, the highly reactive methyldiazonium cation, which causes cytotoxicity through methylation of DNA at the O6 position of guanine[26]. These cytotoxic effects of TMZ decay rapidly within the first few hours of treatment. Previous studies from this laboratory showed that when we incubate TMZ in medium at 37 °C, it loses its cytotoxicity after 2 h, while NEO212 loses its cytotoxicity after 24 h of incubation at 37 °C in the medium[16, 27, 28]. To determine whether there is a relationship between the cytotoxic effects of NEO212 and its ability to block the migration of GSCs, we treated USC02 cells with NEO212 for 24 h; then the supernatant was replaced with fresh medium without the drug in half of the cell cultures, or medium with NEO212 was not removed in the other half; and the cells were incubated for an additional 48 h-period. USC02 cells recovered their ability to migrate after removal of the compound, showing significant differences in the wound area after 72 h when compared to cells treated with NEO212 during the whole 72 h period (Figure 2A,B). There were no significant differences in the number of viable cells treated with 50 μM NEO212 at the end of the experiment (Figure 2C, P=0.4882). These data show that the effects of NEO212 on cell motility at these doses are reversible, and it is essentially affecting the process of cell migration and not simply inducing general cytotoxicity.
Figure 2. The cytotoxic and anti-migratory effects of NEO212 are independent.
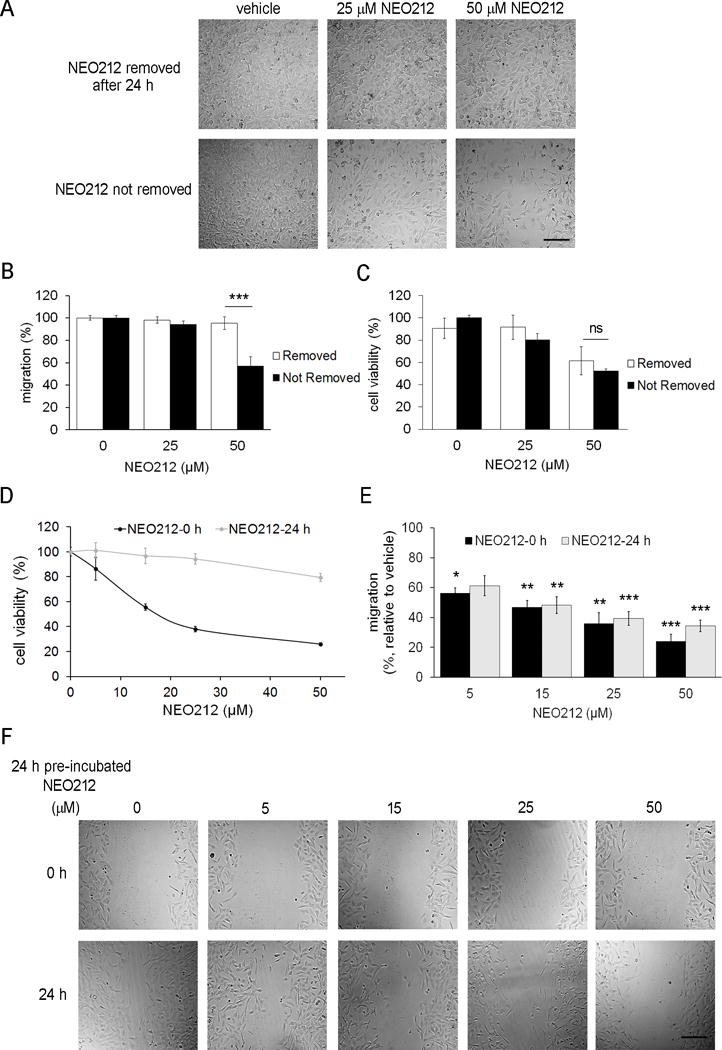
(A–C) USC02 cells recover their ability to migrate after removal of NEO212. (A) Representative images taken 72 h after performing the wound healing assay. Primary cultures of USC02 cells were treated with the indicated concentrations of NEO212 for 24 h, and medium was replaced with fresh medium without compound in half of the cells (upper panel), while in the other half the medium was not changed (lower panel). Scale bar, 200 μm. (B) After 72 h, the remaining wound area was quantified and compared to the corresponding initial area. (C) Cell viability after 72 h, relative to vehicle-treated cells whose medium was not changed after 24 h. (D-F) NEO212 losses its cytotoxic effects, but not its ability to block cell migration after 24 h of incubation in medium without cells. (D) Cell viability of USC02 after 120 h treatments with increasing concentrations of fresh NEO212 (black line) or NEO212 previously incubated for 24 h in medium at 37 °C and then added to the cells (grey line). (E) Wound healing assay performed with the indicated concentrations of fresh NEO212 (NEO212-0h, black bars) or NEO212 previously incubated in medium for 24 h at 37 °C (NEO212-24h, grey bars). After 24 h, the remaining wound area was quantified, and represented as relative to each corresponding initial area. (F) Representative images taken at 0 h (upper panel) and 24 h (lower panel), of the wound healing assay performed with USC02 cells at the indicated concentrations of NEO212 previously pre-incubated during 24 h at 37 °C. Scale bar, 200 μm. In all cases, the bar graphs represent the average ± SEM of at least three independent experiments performed in quadruplicate. *, P<0.05; **, P<0.01; ***, P<0.001; ns, not significant (relative to vehicle-treated cells).
To definitively discern whether there is a relationship between the cytotoxic effects of NEO212 and its ability to block migration of GSCs, we incubated the drug in medium without cells for 24 h at 37 °C (NEO212-24h), and this solution was added to the cells. Since both TMZ and NEO212 require several rounds of cell replication to cause cell death through the induction of DNA strand breaks[11], cell viability was analyzed after 120 h, and compared to that of cells treated with fresh NEO212 (NEO212-0h). The results show that the NEO212 pre-incubated for 24 h before being added to the cells lost most of its cytotoxic effects, when compared to fresh NEO212 (Figure 2D). By contrast, NEO212-24h exhibited a decrease in cell migration similar to that obtained with fresh NEO212-0h, as determined by wound healing assay (Figure 2E,F). In summary, the cytotoxic function of NEO212 was lost after 24 h, but the migration activity still remained. These outcomes indicate that NEO212 has at least two different and unique functions: an initial cytotoxic effect that is stable only within 24 h, and a migration inhibitory effect, stable beyond 24 h, which suggests that one or more of the breakdown products of NEO212 could be responsible for the blockade on GSC migration.
NEO212 reduces the invasive capacity of GSCs in vitro
We then tested whether NEO212 could affect cell invasion, a critical activity of stem cells during glioma progression where the degradation of extracellular matrix[29] by secreted enzymes is required. To investigate the invasiveness of USC02 cells, we used the Boyden chamber assay with Matrigel-coated polycarbonate membranes. NEO212 was tested at 15 μM, the concentration causing a significant decrease in the migration of USC02 cells (Figure 1A,B), with no apparent effect on cytotoxicity (Supplementary Figure 2A). We observed that at this dose, NEO212 significantly decreased the invasion rate of USC02 cells (Figure 3A,B), while TMZ and/or POH showed no significant effects on mesenchymal GSC invasion.
Figure 3. NEO212 suppresses invasion of mesenchymal (A, B, E) and proneural (C, D, F) GSCs.
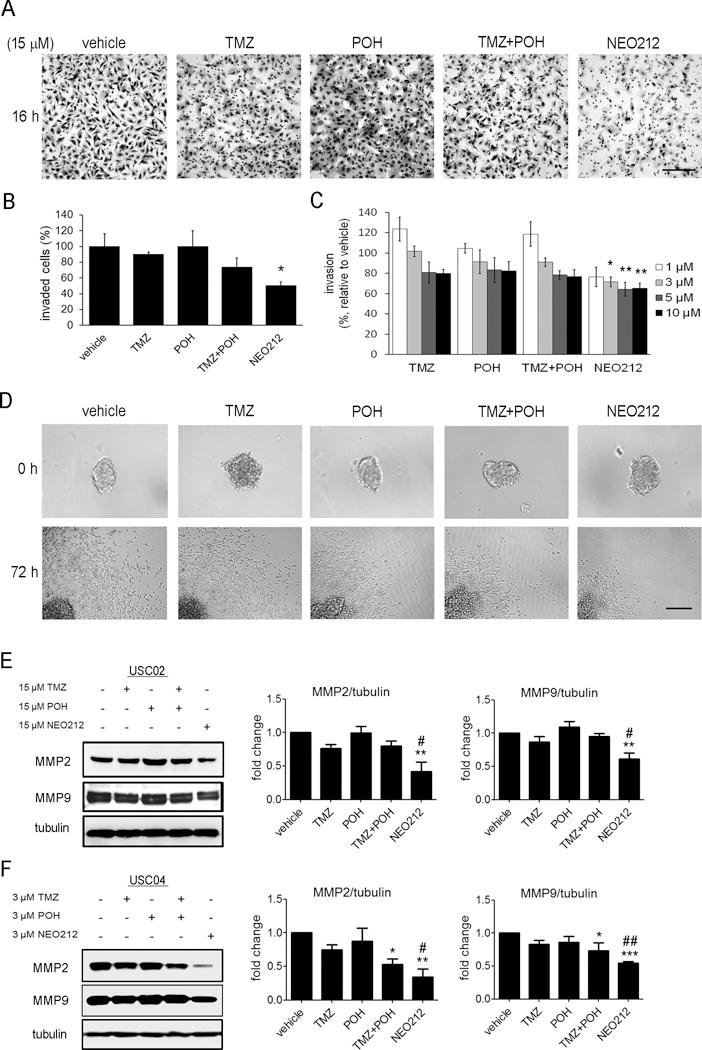
(A) Representative images of invasion assay through a Matrigel-coated Boyden Chamber, performed with USC02 cells and 15 μM treatments. Scale bar, 200 μm. (B) After overnight treatments with 15 μM NEO212, the number of invaded cells per field was counted. The bar graph represents the average ± SEM of at least three independent experiments performed in triplicate. *, P<0.05 (relative to vehicle-treated cells). (C) After 72 h, area covered by invaded USC04 cells in the spheroid invasion assay was quantified, and represented as relative to the initial sphere. The bar graph represents the average ± SEM of three independent experiments performed in sextuplicate. *, P<0.05; **, P<0.01 (relative to vehicle-treated cells). (D) Representative images of the spheroid migration assay performed with USC04 cells at 3 μM. Scale bar, 200 μm. (E-F) Western blot analysis of matrix metalloproteinases MMP2 and MMP9 in USC02 (E) and USC04 (F) cells. The bar graphs represent the average of three independent experiments. *, P<0.05; **, P<0.01, ***, P<0.001 (relative to vehicle-treated cells); #, P< 0.05; ##, P< 0.01 (relative to TMZ-treated cells).
To analyze this effect in primary cultures of proneural GSCs (USC04), we performed a spheroid-based invasion assay, which consists in embedding previously formed spheres into Matrigel as ECM, and allowing the cells to invade within this ECM for 72 h[23]. As shown in Figure 3C,D, treatment with NEO212 reduced USC04 cell invasion, with little effect caused by any of the other treatments.
To further explore the mechanism of reducing cell invasion, we analyzed the protein levels of two matrix metalloproteinases (MMPs), enzymes that degrade the ECM. We selected MMP2 and MMP9 as they are usually critical in tumor cell invasion and metastasis due to their specificity for type IV collagen, the principal component of the basement membrane[30]. Treatment with NEO212 significantly decreased the protein levels of MMP2 and MMP9 in the mesenchymal (USC02) and proneural (USC04) GSC populations, as compared to vehicle-treated cells (Figure 3E,F; asterisks). This effect is also significant when we compare NEO212-treated cells with TMZ-treated cells (Figure 3E,F; hash signs). Taken together, these data show that NEO212 decreases the invasion capacity of GSCs to a greater extent than TMZ.
Finally, to discern whether this blockade of the invasiveness occurs also in freshly derived glioma cells, we performed the Boyden chamber invasion assays with five different samples freshly obtained from patient tumors. Despite their different invasion rates, in all the cases treatment with 15 μM NEO212 decreased the invasion capacity of the glioma cells (Supplementary Figure 3A). Furthermore, we tested NEO212 in three more primary cultures of GSCs: USC08, with an intermediate phenotype between proneural and mesenchymal; mesenchymal USC10; and USC11, whose phenotype -mesenchymal or proneural- remains unknown. As observed in Supplementary Figure 3B, the results obtained were comparable, suggesting that this is a rather general effect of NEO212 in glioma cells.
NEO212 blocks the activation of the FAK/Src signaling pathway
The autophosphorylation of focal adhesion kinase (FAK) at Tyr397 has a central role in motility and invasion in a variety of solid cancers, including GBM[31, 32]. Previous data from our laboratory have shown that NEO212 reduces GSC renewal and proliferation[7]. We have reported here that NEO212 affects cell migration and invasion. Hence, we hypothesized that NEO212 might be regulating FAK signaling pathway.
Src binds to phosphorylated Tyr397 and further phosphorylates FAK, promoting its full kinase activity[20]. Therefore, we examined the status of FAK at its first phosphorylation site, Tyr397, as well as that of its activator, Src, upon exposure to NEO212. Western blot analysis indicated that NEO212 incubation decreased the phosphorylation levels of both FAK and Src, to a greater extent than treatment with the same concentrations of TMZ and/or POH (Figure 4A,B). To further confirm that NEO212 was affecting the FAK/Src signaling pathway, we analyzed the phosphorylation levels of the downstream kinases AKT, MEK1/2 and p38-MAPK in both USC02 and USC04 cells. As observed in Figure 4C, the levels of the phosphorylated kinases were decreased or even disappeared only when cells were treated with NEO212. Thus, the FAK/Src pathway showed different responses at equimolar concentrations of NEO212 and TMZ.
Figure 4. NEO212 blocks the activation of the FAK/Src signaling pathway.
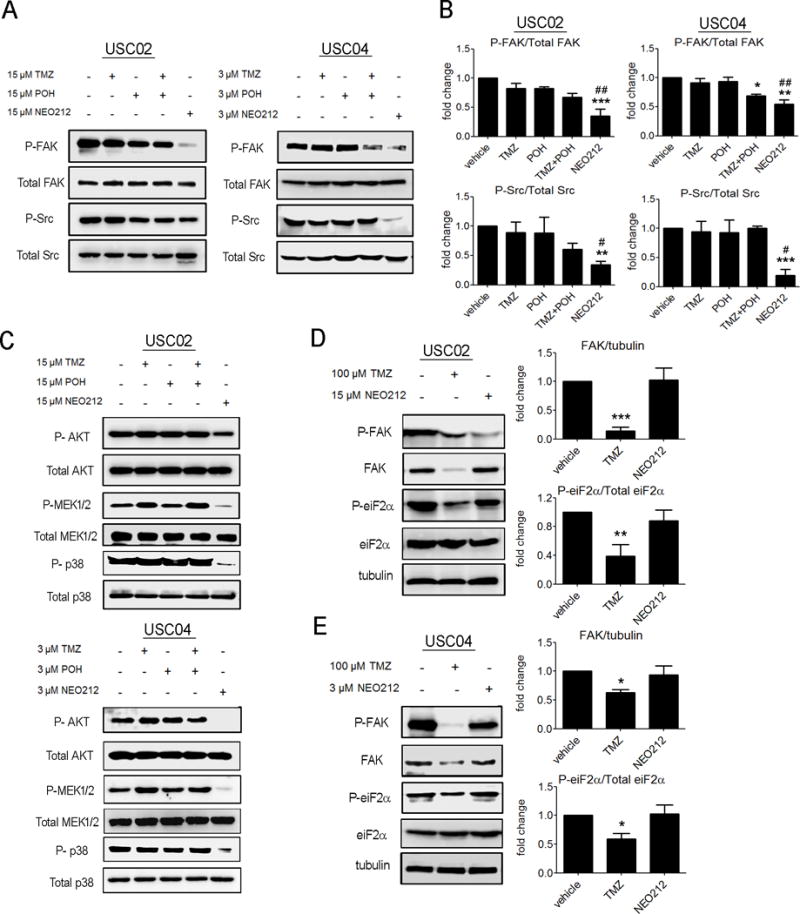
(A) Representative western blots of phosphorylated FAK (P-FAK), total FAK, phosphorylated Src (P-Src) and total Src, from experiments performed with equimolar concentrations of TMZ, POH, TMZ+POH and NEO212, in mesenchymal USC02 and proneural USC04 GSCs. (B) Data correspond to the average ± SEM of three independent experiments. *, P<0.05; **, P<0.01; ***, P<0.001 (relative to vehicle-treated cells); #, P< 0,05; ##, P< 0,01 (relative to TMZ-treated cells). (C) NEO212 reduces the activation of the downstream kinases AKT, MEK1/2 and p-38 MAPK. Representative western blots of phosphorylated (P-AKT) and total AKT, phosphorylated MEK1/2 (P-MEK1/2) and total MEK1/2, and phosphorylated p-38 MAPK (P-p38) and total p-38 MAPK. (D-E) NEO212 and TMZ at equipotent doses have different mechanisms of action. Representative western blots of phosphorylated FAK (P-FAK), total FAK, phosphorylated Src (P-Src), total Src, phosphorylated eiF2α (P-eiF2α) and total eiF2α, from experiments performed with equipotent concentrations of TMZ and NEO212 in USC02 (D) and USC04 (E) cells. Data from three independent experiments are represented with bar graphs as average ± SEM. *, P<0.05; **, P<0.01; ***, P<0.001 (relative to vehicle-treated cells).
To determine whether this mechanism of action was specific to NEO212, or higher doses of TMZ would lead to similar results, we compared FAK phosphorylation and expression levels when we treated the cells with NEO212 or TMZ at equipotent concentrations. Previous studies showed that GSCs are relatively resistant to TMZ and relatively sensitive to NEO212 after 72 h treatments[7]. The IC50 values of TMZ in USC02 and USC04 cells are not significantly different (317 ± 42 μM for USC02 and 323 ± 61 μM for USC04), whereas the IC50 values of NEO212 are different (43 ± 9 μM for USC02 and 8 ± 2 μM for USC04)[7]. Hence, GSCs treated with equipotent (IC17) doses of TMZ (100 μM for both USC02 and USC04) or NEO212 (15 μM for USC02 and 3 μM for USC04), were tested for FAK phosphorylation and expression. Cells treated with 100 μM TMZ showed relatively little phospho-FAK due to low FAK expression, which correlated with a significant decrease in the phosphorylation levels of eukaryotic Initiation Factor 2 (eIF2α), an essential factor for protein translation (Figure 4D,E). This is in contrast to the effects of NEO212 in these cells, where FAK and phospho-eIF2α protein levels were not affected, and only the phosphorylated levels of FAK were decreased. These data demonstrate that treatment with functionally equivalent doses of TMZ causes a non-specific drop in protein synthesis, while treatment with NEO212 is selective, and carefully regulated. Taken together, these studies suggest that the mechanisms of action of these two drugs are likely to be different.
NEO212 reverses the epithelial-to-mesenchymal transition (EMT) phenotype of GSCs in vitro
Overexpression of FAK has been associated with EMT, a prerequisite for cell migration and invasion, and for metastatic processes in several tumor types[21]. To test whether the FAK inhibition caused by NEO212 was regulating the EMT process, we analyzed the expression of 84 key genes involved in this phenotype transition. Only genes showing at least 4 fold differences in expression were considered significantly affected by NEO212. As expected, several genes whose expression is generally downregulated upon EMT induction, were upregulated in response to NEO212 (Figure 5A,B). Among these, we found typical epithelial markers critical for the maintenance of cell-cell adhesion such as E-cadherin (CDH1), desmocollin 2 (DSC2), desmoplakin (DSP) and epithelial cytokeratins KRT7 and KRT19[33, 34]. In addition, secreted factors that promote an epithelial-like phenotype[34] were also upregulated by NEO212: fibroblast growth factor binding protein 1 (FGFBP1), interleukin 1 receptor antagonist (IL1RN/IRAP), secreted phosphoprotein 1 (SPP1); and tissue factor pathway inhibitor 2 (TFPI-2), whose expression levels in tumors are inversely related to their malignancy as it blocks tumor growth and cell invasion through MMP inhibition[35]. Finally, the expression of two cell surface receptors that are classically downregulated during EMT[34], was upregulated with NEO212 treatment: estrogen receptor alpha (ESR1/ERα), whose activity is associated with differentiation and promotion of an epithelial phenotype[36]; and macrophage stimulating 1 receptor (MST1R), which inhibits cell migration and proliferation in vivo[37]. Furthermore, the transcription levels of other genes related to cell differentiation and development[34, 38] were increased by treatment with NEO212, such as the bone morphogenetic protein 2 (BMP2), described to induce differentiation in human mesenchymal stem cells[39]. Consistent with previous results from our laboratory showing that NEO212 triggers apoptosis[28], we observed an upregulation in the expression of Wnt11, which inhibits the canonical Wnt pathway and promotes cell differentiation via caspase activation[40]. Additionally, the expression levels of the mesenchymal marker CDH2 (N-cadherin) were downregulated by more than 4 fold in mesenchymal USC02 cells, while there was no change in proneural USC04 (Figure 5C,D). Surprisingly, we did observe that some genes related to the mesenchymal phenotype[34] were upregulated by treatment with NEO212, including GNG11, SNAI3, SPARC and MMP3 (Figure 5C,D).
Figure 5. NEO212 reverses the EMT phenotype of GSCs in vitro.
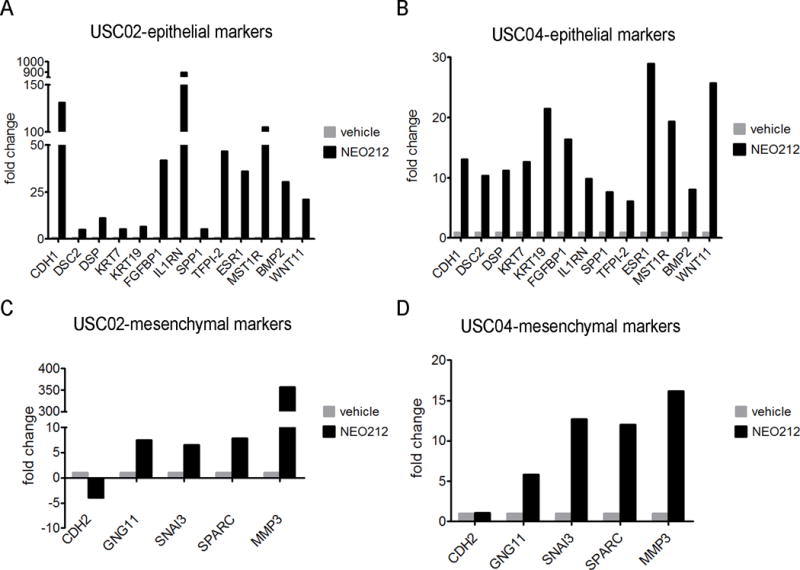
An EMT PCR array was performed in mesenchymal USC02 and proneural USC04 cells treated with NEO212 or vehicle. The graphs show those genes that were modulated (threshold = 4-fold change) in NEO212-treated cells vs vehicle-treated cells. (A,B) Genes related to an epithelial phenotype upregulated with NEO212 treatment in USC02 (A) and USC04 (B) cells. (C,D) Genes related to a mesenchymal phenotype whose expression was more than 4 fold regulated by NEO212 treatment in USC02 (C) and USC04 (D) cells. Results are expressed as fold change, relative to vehicle-treated cells.
NEO212 blocks invasion of mesenchymal GSCs in vivo
Recent studies from this laboratory have demonstrated that NEO212 at 25 mg/kg significantly prolonged the survival of mice bearing GSC tumors when low numbers of cells (1,000–5,000) were implanted[7]. To demonstrate the potential of NEO212 decreasing GSC invasion in vivo in resistant gliomas, we first determined whether NEO212 was active in blocking tumor growth in an orthotopic murine model. We implanted 100,000 mesenchymal USC02 cells intracranially into the brain parenchyma of immune-incompetent NOD/SCID mice, and compared treatment with NEO212 to TMZ in vivo at doses of 5 mg/kg and 25 mg/kg. The Kaplan-Meier survival curve shows that NEO212 at both doses significantly prolonged the survival of mice bearing mesenchymal GSCs (Figure 6A). No significant effects were observed in mice treated with TMZ. Using bioluminescence imaging, we confirmed the highly significant delay in tumor progression with these two doses of NEO212 (Figure 6B), much more evident than the one obtained with TMZ treatment.
Figure 6. NEO212 decreases tumor progression and GSC invasion in an in vivo mesenchymal-GSC (USC02) tumor model.
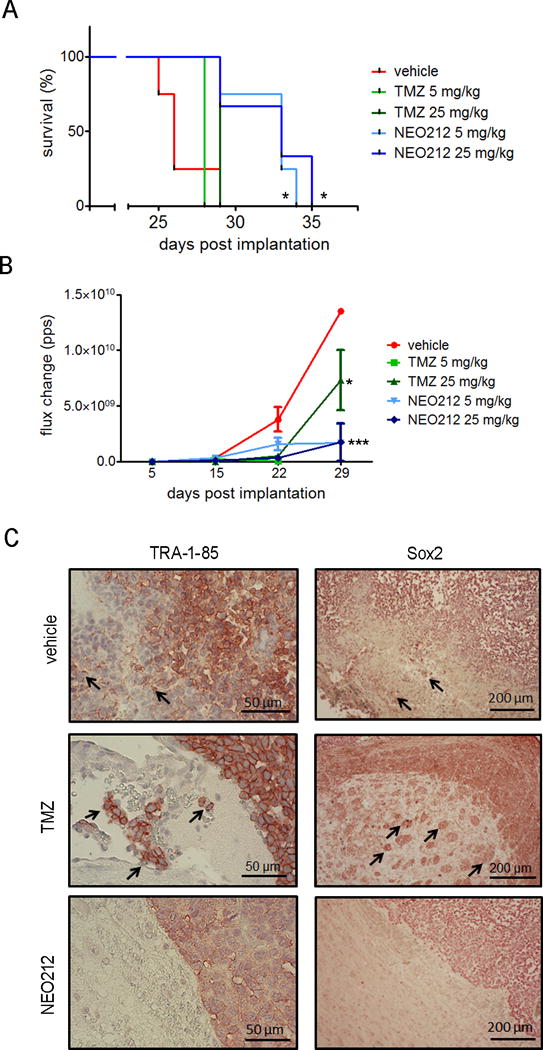
(A) Kaplan-Meier survival analysis of mice treated with vehicle (red line), TMZ 5 mg/kg (light green line), TMZ 25 mg/kg (dark green line), NEO212 5 mg/kg (light blue line) and NEO212 25 mg/kg (dark blue line). *, P<0.05 (relative to vehicle-treated mice). (B) Tumor growth curve of mice treated with vehicle (red line), TMZ 5 mg/kg (light green line), TMZ 25 mg/kg (dark green line), NEO212 5 mg/kg (light blue line) and NEO212 25 mg/kg (dark blue line). *, P<0.05; ***, P<0.001 (relative to vehicle-treated mice). (C) Representative images of brain sections stained for human cell marker TRA-1-85 (left panel, scale bar = 50 μm) and stem cell marker Sox2 (right panel, scale bar = 200 μm). Red denotes positive staining and arrows indicate invasive nature of GSCs.
To determine whether NEO212 had an effect on cell migration and invasion of GSCs in vivo, we stained the USC02-tumor bearing brains with anti-TRA-1-85 antibody, a marker of human cells; or anti-Sox2 antibody, which recognizes stem cells. In both cases, immunostaining showed that treatment with NEO212 decreased the invasion of human tumor cells into the brain, as demonstrated by a clear and distinct border between the tumor and normal brain parenchyma regions. By contrast, the brains of vehicle and TMZ-treated mice exhibited invasive cells from the tumor into the normal brain parenchyma (Figure 6C). These results suggest that NEO212 is effective in reducing migration and invasion of mesenchymal GSC populations in vivo.
To obtain the maximum tolerated dose (MTD) information, mice were treated daily for 10 days at doses of 60, 90, 120, 150 mg/kg. At the time of euthanasia, the following organs were retrieved: liver, heart, intestine, lungs, and kidneys. Specimens were formalin fixed and stained with hematoxylin/eosin. Bone marrow specimens were also stained and evaluated. The results showed no significant pathological changes to the organs examined. However, significant bone marrow toxicity was observed at 150 mg/kg but not at 120 mg/kg (Supplementary Figure 4A). Hence, we consider the 120 mg/kg as the MTD, being the doses used for the experiments (5 and 25 mg/kg) below the toxic doses.
Finally, through HPLC analysis of brains from both non-tumor-bearing and tumor-bearing mice, we characterized the pharmacokinetics (PKs) of NEO212 (Supplementary Figure 4B,C). At 5 minutes after drug administration, there was no detectable drug in either group. At 15 minutes post drug administration, there was negligible amounts of NEO212 in the non-tumor bearing brain, but significant levels of drug in the tumor-bearing brain. At 60 minutes post administration of drug, there was NEO212 detectable in both tumor-bearing and non-tumor bearing animals. By 120 minutes, the tumor-bearing brains accumulated and retained NEO212 while in the normal brain the drug was being washed out. These data, in conjunction with the in vivo drug efficacy data, confirm that following subcutaneous administration NEO212 is delivered to the brain.
Discussion
In this study, we explored the mechanism of action of the novel perillyl alcohol (POH)-conjugated analog of temozolomide (TMZ), NEO212, which has been shown to be active against TMZ-resistant cells as well as glioma stem cells (GSCs)[11, 16]. The current FDA-approved standard of therapy, TMZ, spontaneously converts to 5-(3-methyltriazen-1-yl)imidazole-4-carboxamide (MTIC), which then degrades to the 5-aminoimidazole-4-carboxamide (AIC), and the reactive methylating agent, methyldiazonium cation. This ion is the active component and has a very short half-life, as it rapidly reacts with DNA to form methyl adducts, such as N3-methyl-adenine, N7-methyl-guanine, and O6-methyl-guanine, resulting in DNA strand breaks and subsequent cell death[11]. Previous results from our laboratory demonstrated that NEO212 exerts a DNA alkylating activity similar to TMZ, but with a greater potency[7, 28].
Our previous studies showed that the cytotoxicity of NEO212 was independent of the status of the DNA repair protein MGMT, the main mechanism confering resistance to TMZ in about 50% of glioma patients[7, 41]. The results presented here support the hypothesis that NEO212 has additional therapeutic effects not present in TMZ. These novel effects are the anti-invasion properties of NEO212, which are likely due to one or more of the stable breakdown products of NEO212. This observation may explain why in the in vivo glioma model tumors progress more slowly when treated with NEO212 at doses that are not cytotoxic for these GSCs (Figure 6B). The decrease in tumor progression with NEO212 may be the result of inhibition of migration and invasion of GSCs, and decrease in MMP2 and MMP9 (Figures 1,3), essential for the degradation of the ECM[30]. In addition, we have shown differences between the cytotoxic effects of NEO212, which occur within the first 24 h of treatment, and the impaired cell migration and invasion, which is still maintained after 24 h of incubation of the compound in cell-free medium at 37 °C (Figure 2). Further studies regarding the breakdown products of NEO212 and their biological effects are in progress.
FAK is a key factor that regulates cell migration and invasion[21]. Since the FAK/Src route drives several tumor growth and metastasis-promoting signaling pathways, many small molecule FAK inhibitors have emerged as promising chemotherapeutics, several of which reaching the clinical trial phase[31]. FAK autophosphorylation at Tyr397 leads to Src recruitment, and subsequent phosphorylation of both proteins lead to a stimulation of the PI3K/Akt/mTOR, Ras/Raf/MEK/Erk and p-38 MAPKs signaling pathways[42–44]. The FAK/PI3K/AKT/mTOR and FAK/Ras/Raf/MEK/Erk cascades are involved in cell survival and proliferation, as well as in cell migration and invasion through the regulation of MMP2 and MMP9 activity[45]. The activation of p38-MAPKs has been reported to contribute to the acquisition of invasion and migrating capabilities, to the extravasations of migrating tumor cells, and to acquisition of EMT characteristics in cancer[46]. In the present study, we show that NEO212 blocks the activation of the FAK/Src pathway (Figure 4), as shown by a decrease in the phosphorylation levels of FAK, Src, and the downstream kinases AKT, MEK1/2 and p38-MAPK. In both mesenchymal and proneural GSCs, the effects of NEO212 were due to the TMZ and POH conjugate, and not simply to the additive effects of the components of NEO212. Furthermore, this mechanism of action differs from that shown by the reference compound TMZ, which at equipotent concentrations causes a general blockade in the protein synthesis (Figure 4D,E).
The overexpression of FAK has been associated with EMT, a prerequisite for cell migration, invasion, and metastasis[21]. During EMT, epithelial cells lose many of their epithelial cell-specific phenotypes and develop mesenchymal cell features[47]. This includes a loss of cell-cell binding proteins and polarity; and gain of cell motility and invasiveness, and of the ability to digest the surrounding ECM[48]. The association between EMT and cell invasion has been demonstrated in cancer progression and metastasis[49]. It has also been demonstrated that the FAK/Src route regulates the EMT process[21], so a decrease in the activation levels of this signaling pathway should translate into an increase of the epithelial markers and a decrease of the mesenchymal ones. Here, we show that NEO212 effectively reverses the EMT process, mainly by upregulating several genes that are normally downregulated during EMT (Figure 5), such as cell-cell adhesion molecules CDH1, DSC2, DSP; epithelial cytokeratins KRT7 and KRT19[33, 34]; secreted factors that promote an epithelial-like phenotype[34] like FGFBP1, IL1RN, SPP1, TFPI-2, ESR1 and MST1R; as well as BMP2 and Wnt11, both related to cell differentiation and development[34, 38].
It is worth noting that the higher expression levels of TFPI-2 (Figure 5A,B) could be a mechanistic support for the decrease in the levels of MMP2 and MMP9 (Figures 3,4), contributing to cell invasion impairment. Regarding the mesenchymal marker CDH2 (N-cadherin), which plays an essential role in stimulating cell migration, invasion and EMT process in cancers[50], its levels were more than 4 fold downregulated in USC02 cells, while there was no change in USC04 (Figure 5C,D). This could be explained by higher basal levels of N-cadherin in mesenchymal cells, when compared to proneural cells.
In contrast to these results, we observed that some genes that have been described to promote the EMT[34] were upregulated by NEO212 (Figure 5C,D). These seemingly contradictory results may be explained by different regulation at the transcriptional and translational levels, or by adaptive mechanisms activated by GSCs to compensate for the effects of NEO212. For example, in the case of MMP3, we had previously shown a decrease in the protein levels of two members of the same family of matrix metalloproteinases, MMP2 and MMP9 (Figure 3E,F). Either way, considering the number of genes affected in each case, that the fold change in the epithelial-like genes is much greater than in the mesenchymal-like ones, as well as the fact that the final phenotype shows a decrease in cell proliferation, migration and invasion, and a slower tumor progression, the overall results suggest that NEO212 drives a mesenchymal-to-epithelial transition in GSCs.
In vivo treatment with NEO212 increases the survival of mice and decreases tumor progression (Figure 6). Previous results demonstrated that NEO212 at high doses significantly prolong survival of mice bearing GSC-derived tumors[7]. Here we used higher numbers of cells and lower doses of NEO212 as compared to previous studies from our laboratory[7], which strengthens its potency as a chemotherapeutic agent. We have also incorporated TMZ treatment as a control, and performed a complete in vivo toxicity study of NEO212, demonstrating that the doses used were not toxic for the mice (Supplementary Figure 4A). Besides, the pharmacokinetic data from tumor-bearing and normal brains, in conjunction with the drug efficacy data, confirm that NEO212 is delivered to the brain, where it displays the expected antitumor effect. The observation that NEO212 is retained in the tumor-bearing brains while the drug is washed out in the normal brains suggests a possible binding or accumulation of NEO212 in tumor cells as compared to normal brain tissue (Supplementary Figure 4B,C). Finally, the immunostaining data that shows that the borders of the tumors in mice treated with NEO212 are much more defined than those of mice treated with vehicle or TMZ, reinforces the hypothesis that this compound is blocking GSC migration and invasion.
Clinically, we envision that NEO212 may be used both in upfront and recurrent malignant gliomas. In upfront gliomas, NEO212 can be active at lower concentrations than TMZ and show no toxicity in vivo at the proposed concentrations (Supplementary Figure 4A). This drug may be used in clinic at low doses in conjunction with radiation, similar to the Stupp protocol, to inhibit GSC invasion. As local invasion is the key to glioma recurrence, the ability of NEO212 to inhibit migration is unique in its action as an anti-tumor agent. Although the inhibition of migration is reversible in these studies, we are not sure whether long term usage would result in more permanent inhibition of migration or invasion. Moreover, it has implications in terms of how NEO212 would be administered in clinical settings. If it is administered at similar time schedule to TMZ (5 days on, 23 days off), lower doses of NEO212 could be administered for anti-migration/invasion action, and not solely for cytotoxicity. Subsequent cycles of NEO212 may then be applied at higher concentrations to induce cytotoxicity of the glioma cells themselves. In a recurrent glioma, application of NEO212 may be used to inhibit GSC invasion, and tumor recurrence and growth. As NEO212 works in both MGMT positive and negative glioma cells, it could be applied to all patients with malignant gliomas.
In summary, in addition to the higher alkylating potency of NEO212 compared to TMZ[11, 16], we have demonstrated that this novel compound significantly inhibits the migration and invasion capacities of GSCs, to a greater extent than TMZ and/or POH. This reduction in migration and invasion is associated with a decrease in the activation of the FAK-Src signaling pathway, although this may not necessarily be the cause of the inhibition of invasion. Furthermore, we have demonstrated that this mechanism of action is different from what occurs with TMZ, and is independent of its DNA alkylating activity. These data suggest that NEO212 acts as a multi-target drug, which resulting in an increased efficacy against tumor cells and lower risk of drug resistance and recurrence. Thus, the ability to block GSC migration and invasion in vivo as well as its selective cytotoxicity of tumor cells[11], make NEO212 an ideal candidate for the treatment of newly diagnosed and TMZ-resistant gliomas.
Supplementary Material
Acknowledgments
We thank Drs. Daniel Levin and Satish Puppali (Norac Pharma, Azusa, CA) for NEO212 conjugation and API manufacturing. We thank NeOnc Technologies, Inc. (T.C. Chen) and National Cancer Institute (NIH) (F.M. Hofman) for supporting this research.
Financial Support: Funding for these studies was provided by NeOnc Technologies, Inc. (T.C. Chen) and by the National Cancer Institute (NCI, NIH) award (R41CA192419) (F.M. Hofman).
Footnotes
Conflict of interest:
T.C. Chen has ownership interest in NEO212. No potential conflict of interest was disclosed by the other authors.
References
- 1.von Neubeck C, Seidlitz A, Kitzler HH, Beuthien-Baumann B, Krause M. Glioblastoma multiforme: Emerging treatments and stratification markers beyond new drugs. Br J Radiol. 2015;88:20150354. doi: 10.1259/bjr.20150354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 3.Beier D, Schulz JB, Beier CP. Chemoresistance of glioblastoma cancer stem cells - much more complex than expected. Mol Cancer. 2011;10:128. doi: 10.1186/1476-4598-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalkan R. Glioblastoma stem cells as a new therapeutic target for glioblastoma. Clin Med Insights Oncol. 2015;9:95–103. doi: 10.4137/CMO.S30271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lottaz C, Beier D, Meyer K, Kumar P, Hermann A, Schwarz J, Junker M, Oefner PJ, Bogdahn U, Wischhusen J, Spang R, Storch A, Beier CP. Transcriptional profiles of cd133+ and cd133− glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res. 2010;70:2030–2040. doi: 10.1158/0008-5472.CAN-09-1707. [DOI] [PubMed] [Google Scholar]
- 7.Jhaveri N, Agasse F, Armstrong D, Peng L, Commins D, Wang W, Rosenstein-Sisson R, Vaikari VP, Santiago SV, Santos T, Chen L, Schönthal AH, Chen TC, Hofman FM. A novel drug conjugate, neo212, targeting proneural and mesenchymal subtypes of patient-derived glioma cancer stem cells. Cancer Lett. 2016;371:240–250. doi: 10.1016/j.canlet.2015.11.040. [DOI] [PubMed] [Google Scholar]
- 8.Ho IAW, Shim WSN. Contribution of the microenvironmental niche to glioblastoma heterogeneity. Biomed Res Int. 2017;2017:9634172. doi: 10.1155/2017/9634172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olar A, Aldape KD. Using the molecular classification of glioblastoma to inform personalized treatment. J Pathol. 2014;232:165–177. doi: 10.1002/path.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen TC, Fonseca COD, Schönthal AH. Preclinical development and clinical use of perillyl alcohol for chemoprevention and cancer therapy. Am J Cancer Res. 2015;5:1580–1593. [PMC free article] [PubMed] [Google Scholar]
- 11.Cho HY, Wang W, Jhaveri N, Lee DJ, Sharma N, Dubeau L, Schönthal AH, Hofman FM, Chen TC. Neo212, temozolomide conjugated to perillyl alcohol, is a novel drug for effective treatment of a broad range of temozolomide-resistant gliomas. Mol Cancer Ther. 2014;13:2004–2017. doi: 10.1158/1535-7163.MCT-13-0964. [DOI] [PubMed] [Google Scholar]
- 12.Johannessen TCA, Bjerkvig R. Molecular mechanisms of temozolomide resistance in glioblastoma multiforme. Expert Rev Anticancer Ther. 2012;12:635–642. doi: 10.1586/era.12.37. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Stevens MFG, Laughton CA, Madhusudan S, Bradshaw TD. Acquired resistance to temozolomide in glioma cell lines: Molecular mechanisms and potential translational applications. Oncology. 2010;78:103–114. doi: 10.1159/000306139. [DOI] [PubMed] [Google Scholar]
- 14.Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, Batchelor TT, Futreal PA, Stratton MR, Curry WT, Iafrate AJ, Louis DN. Loss of the mismatch repair protein msh6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res. 2007;13:2038–2045. doi: 10.1158/1078-0432.CCR-06-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W, Hegi ME. Mgmt promoter methylation in malignant gliomas: Ready for personalized medicine? Nat Rev Neurol. 2010;6:39–51. doi: 10.1038/nrneurol.2009.197. [DOI] [PubMed] [Google Scholar]
- 16.Chen TC, Cho HY, Wang W, Nguyen J, Jhaveri N, Rosenstein-Sisson R, Hofman FM, Schönthal AH. A novel temozolomide analog, neo212, with enhanced activity against mgmt-positive melanoma in vitro and in vivo. Cancer Lett. 2015;358:144–151. doi: 10.1016/j.canlet.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 17.Holland EC. Glioblastoma multiforme: The terminator. PNAS. 2000;97:6242–6244. doi: 10.1073/pnas.97.12.6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heddleston JM, Hitomi M, Venere M, Flavahan WA, Yan K, Kim Y, Minhas S, Rich JN, Hjelmeland AB. Glioma stem cell maintenance: The role of the microenvironment. Curr Pharm Des. 2011;17:2386–2401. doi: 10.2174/138161211797249260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. Fak integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 20.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: In command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 21.Yoon H, Dehart JP, Murphy JM, Lim STS. Understanding the roles of fak in cancer: Inhibitors, genetic models, and new insights. J Histochem Cytochem. 2015;63:114–128. doi: 10.1369/0022155414561498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vinci M, Box C, Zimmermann M, Eccles SA. Tumor spheroid-based migration assays for evaluation of therapeutic agents. In: Moll J, Colombo R, editors. Target identification and validation in drug discovery: Methods and protocols. Humana Press; Totowa, NJ: 2013. pp. 253–266. [DOI] [PubMed] [Google Scholar]
- 23.Vinci M, Box C, Eccles SA. Three-dimensional (3d) tumor spheroid invasion assay. J Vis Exp. 2015:e52686. doi: 10.3791/52686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marín-Ramos NI, Alonso D, Ortega-Gutiérrez S, Ortega-Nogales FJ, Balabasquer M, Vázquez-Villa H, Andradas C, Blasco-Benito S, Pérez-Gómez E, Canales Á, Jiménez-Barbero J, Marquina A, del Prado JM, Sánchez C, Martín-Fontecha M, López-Rodríguez ML. New inhibitors of angiogenesis with antitumor activity in vivo. J Med Chem. 2015;58:3757–3766. doi: 10.1021/jm5019252. [DOI] [PubMed] [Google Scholar]
- 25.Virrey JJ, Golden EB, Sivakumar W, Wang W, Pen L, Schönthal AH, Hofman FM, Chen TC. Glioma-associated endothelial cells are chemoresistant to temozolomide. J Neurooncol. 2009;95:13–22. doi: 10.1007/s11060-009-9891-7. [DOI] [PubMed] [Google Scholar]
- 26.Dresemann G. Temozolomide in malignant glioma. Onco Targets Ther. 2010;3:139–146. doi: 10.2147/ott.s5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen TC, Cho HY, Wang W, Barath M, Sharma N, Hofman FM, Schönthal AH. A novel temozolomide-perillyl alcohol conjugate exhibits superior activity against breast cancer cells in vitro and intracranial triple-negative tumor growth in vivo. Mol Cancer Ther. 2014;13:1181–1193. doi: 10.1158/1535-7163.MCT-13-0882. [DOI] [PubMed] [Google Scholar]
- 28.Chen TC, Cho HY, Wang W, Wetzel SJ, Singh A, Nguyen J, Hofman FM, Schönthal AH. Chemotherapeutic effect of a novel temozolomide analog on nasopharyngeal carcinoma in vitro and in vivo. J Biomed Sci. 2015;22:71. doi: 10.1186/s12929-015-0175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Citisli V, Dodurga Y, Eroglu C, Secme M, Avci CB, Satiroglu-Tufan NL. Temozolomide may induce cell cycle arrest by interacting with urg4/urgcp in sh-sy5y neuroblastoma cells. Tumour Biol. 2015;36:6765–6772. doi: 10.1007/s13277-015-3373-7. [DOI] [PubMed] [Google Scholar]
- 30.Komatsu K, Nakanishi Y, Nemoto N, Hori T, Sawada T, Kobayashi M. Expression and quantitative analysis of matrix metalloproteinase-2 and-9 in human gliomas. Brain Tumor Pathol. 2004;21:105–112. doi: 10.1007/BF02482184. [DOI] [PubMed] [Google Scholar]
- 31.Sulzmaier FJ, Jean C, Schlaepfer DD. Fak in cancer: Mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer-a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 33.Takaishi M, Tarutani M, Takeda J, Sano S. Mesenchymal to epithelial transition induced by reprogramming factors attenuates the malignancy of cancer cells. PLOS ONE. 2016;11:e0156904. doi: 10.1371/journal.pone.0156904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu S, Kanda T, Nakamoto S, Imazeki F, Yokosuka O. Knockdown of receptor-interacting serine/threonine protein kinase-2 (ripk2) affects emt-associated gene expression in human hepatoma cells. Anticancer Res. 2012;32:3775–3783. [PubMed] [Google Scholar]
- 35.Herman MP, Sukhova GK, Kisiel W, Foster D, Kehry MR, Libby P, Schönbeck U. Tissue factor pathway inhibitor-2 is a novel inhibitor of matrix metalloproteinases with implications for atherosclerosis. J Clin Invest. 2001;107:1117–1126. doi: 10.1172/JCI10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voutsadakis AI. Epithelial-mesenchymal transition (emt) and regulation of emt factors by steroid nuclear receptors in breast cancer: A review and in silico investigation. J Clin Med. 2016;5:11. doi: 10.3390/jcm5010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu X, Liu X, Koul S, Lee CY, Zhang Z, Halmos B. Axl kinase as a novel target for cancer therapy. Oncotarget. 2014;5:9546–9563. doi: 10.18632/oncotarget.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lavery K, Swain P, Falb D, Alaoui-Ismaili MH. Bmp-2/4 and bmp-6/7 differentially utilize cell surface receptors to induce osteoblastic differentiation of human bone marrow-derived mesenchymal stem cells. J Biol Chem. 2008;283:20948–20958. doi: 10.1074/jbc.M800850200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.An C, Cheng Y, Yuan Q, Li J. Igf-1 and bmp-2 induces differentiation of adipose-derived mesenchymal stem cells into chondrocytes-like cells. Ann Biomed Eng. 2010;38:1647–1654. doi: 10.1007/s10439-009-9892-x. [DOI] [PubMed] [Google Scholar]
- 40.Bisson JA, Mills B, Paul Helt J-C, Zwaka TP, Cohen ED. Wnt5a and wnt11 inhibit the canonical wnt pathway and promote cardiac progenitor development via the caspase-dependent degradation of akt. Dev Biol. 2015;398:80–96. doi: 10.1016/j.ydbio.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 41.Lee SY. Temozolomide resistance in glioblastoma multiforme. Genes & Diseases. 2016;3:198–210. doi: 10.1016/j.gendis.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zebda N, Dubrovskyi O, Birukov KG. Focal adhesion kinase regulation of mechanotransduction and its impact on endothelial cell functions. Microvasc Res. 2012;83:71–81. doi: 10.1016/j.mvr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parsons JT. Focal adhesion kinase: The first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 44.Stupack DG, Cheresh DA. Get a ligand, get a life: Integrins, signaling and cell survival. J Cell Sci. 2002;115:3729–3738. doi: 10.1242/jcs.00071. [DOI] [PubMed] [Google Scholar]
- 45.Chan KC, Ho HH, Huang CN, Lin MC, Chen HM, Wang CJ. Mulberry leaf extract inhibits vascular smooth muscle cell migration involving a block of small gtpase and akt/nf-κb signals. J Agric Food Chem. 2009;57:9147–9153. doi: 10.1021/jf902507k. [DOI] [PubMed] [Google Scholar]
- 46.Koul HK, Pal M, Koul S. Role of p38 map kinase signal transduction in solid tumors. Genes Cancer. 2013;4:342–359. doi: 10.1177/1947601913507951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4:657–665. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- 48.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Son H, Moon A. Epithelial-mesenchymal transition and cell invasion. Toxicol Res. 2010;26:245–252. doi: 10.5487/TR.2010.26.4.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X, Liu G, Kang Y, Dong Z, Qian Q, Ma X. N-cadherin expression is associated with acquisition of emt phenotype and with enhanced invasion in erlotinib-resistant lung cancer cell lines. PLOS ONE. 2013;8:e57692. doi: 10.1371/journal.pone.0057692. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.