

Abstract
The gut microbiome plays a key role in animal health, and perturbing it can have detrimental effects. One major source of perturbation to microbiomes, in humans and human-associated animals, is exposure to antibiotics. Most studies of how antibiotics affect the microbiome have used amplicon sequencing of highly conserved 16S rRNA sequences, as in a recent study showing that antibiotic treatment severely alters the species-level composition of the honeybee gut microbiome. But because the standard 16S rRNA-based methods cannot resolve closely related strains, strain-level changes could not be evaluated. To address this gap, we used amplicon sequencing of protein-coding genes to assess effects of antibiotics on fine-scale genetic diversity of the honeybee gut microbiota. We followed the population dynamics of alleles within two dominant core species of the bee gut community, Gilliamella apicola and Snodgrassella alvi, following antibiotic perturbation. Whereas we observed a large reduction of genetic diversity in G. apicola, S. alvi diversity was mostly unaffected. The reduction of G. apicola diversity accompanied an increase in the frequency of several alleles, suggesting resistance to antibiotic treatment. We find that antibiotic perturbation can cause major shifts in diversity, and that the extent of these shifts can vary substantially across species. Thus, antibiotics impact not only species composition, but also allelic diversity within species, potentially affecting hosts if variants with particular functions are reduced or eliminated. Overall, we show that amplicon sequencing of protein-coding genes, without clustering into operational taxonomic units (OTUs), provides an accurate picture of the fine-scale dynamics of microbial communities over time.
Keywords: antibiotics, genetic diversity, honeybee, microbiome, population dynamics
Introduction
Shifts in the gut microbiota, such as those following antibiotic exposure, can impact health of animal hosts. Thus, the resilience of the microbiota following perturbation is of major interest. However, characterizing shifts in microbiota composition is hampered by the high level of species and strain diversity within gut communities (Lozupone et al. 2012). In many hosts, each bacterial species residing in the gut contains fine-scale genetic variation that is not resolved using common methods, which rely on PCR amplification of regions of the highly conserved 16S rRNA molecule (Degnan & Ochman 2012). Bacterial gut inhabitants may have identical or nearly identical 16S rRNA but very different gene repertoires conferring different metabolic capabilities (Ellegaard & Engel 2016). Older methods, such as multilocus sequence typing (MLST) of several protein-coding genes could discriminate related strains (Ibarz Pavón & Maiden 2009) but were designed to characterize single isolates and are not practical for characterizing whole communities. Simple shotgun metagenomics, that is, random sequencing of pooled DNA from a community, is extremely cumbersome and expensive as an approach for sampling shifts in strain composition of communities over time. Methods based on deep sequencing of short amplicons, referred to as “phylotags” (Caro-Quintero & Ochman 2015) provide the possibility of deep sampling and strain-level discrimination for targeted bacterial species (Moeller et al. 2016). However, for complex communities such as those of humans and other mammals, these methods are limited by the need to assay each bacterial species separately, due to the high divergence rate of protein-coding genes.
In contrast to mammals, honeybees have a relatively simple gut microbiota, in which eight bacterial species comprise 95–99% of the community (Cox-Foster et al. 2007; Moran et al. 2012), making it feasible to study fine-scale changes in the community (Engel et al. 2016). Despite this simplicity, strain diversity within each of these species is high (Engel et al. 2014; Powell et al. 2016), and the honeybee gut community is spatially organized, with most of the bacteria residing in the hindgut (Kwong & Moran 2015). The bee gut community affects growth and metabolism (Kesnerova et al. 2017; Zheng et al. 2017), as well as immune function and susceptibility to pathogens (Kwong et al. 2017). Disrupting this community with antibiotics increases mortality and susceptibility to pathogens (Raymann et al. 2017).Two Gram-negative members of the honeybee gut microbiome, Gilliamella apicola and Snograssella alvi, dominate in the ileum region of the hindgut, where they form a biofilm on the gut wall, with S. alvi in direct contact with the host epithelium and G. apicola on top of it. These two species have contrasting and complementary metabolic capabilities (Kwong et al. 2014; Kesnerova et al. 2017).
Antibiotic exposure is known to decrease gut microbial diversity (Sekirov et al. 2008; Dethlefsen et al. 2008; Dethlefsen & Relman 2011; Theriot et al. 2014), and in some cases can permanently change community composition (Blaser 2016). The effects of antibiotic treatment vary across individual communities, with recovery being dependent on the starting composition (Dethlefsen & Relman 2011; Pérez-Cobas et al. 2013). Most studies on effects of antibiotics use 16S rRNA amplicons to evaluate shifts in community composition, and thus fail to discern strain-level changes. In a recent study of bee gut microbiomes based on 16S rRNA sequences, antibiotic exposure led to a decrease in the abundance of S. alvi but did not alter the abundance of G. apicola (Raymann et al. 2017). However, the impact of antibiotic treatment on the genetic diversity within these two species could not be characterized with this method.
Here, we applied deep amplicon sequencing of protein-coding gene loci to follow the fine-scale dynamics of two closely associated and abundant members of the honeybee core gut microbiome, S. alvi and G. apicola, after antibiotic perturbation. Although amplicon sequencing of protein-coding genes has been applied in recent studies to evaluate microbial species diversity (Caro-Quintero & Ochman 2015; Powell et al. 2016; Moeller et al. 2016), to our knowledge, it has not been used to study the dynamics of community members over time. We found that exposure to the antibiotic tetracycline (hereafter referred to as antibiotic treatment/exposure) differentially affected the genetic diversity of G. apicola and S. alvi. In particular, antibiotic exposure caused a decrease in genetic diversity in G. apicola whereas it had less effect on the genetic diversity of S. alvi. These results show that antibiotic perturbation can result in substantial shifts in genetic diversity within a community, due to variation in resistance among strains; the extent of these shifts can vary among bacterial species.
Materials and Methods
Marker identification and primer design
Genomes of G. apicola (n=9) and S. alvi (n=5) isolated from honeybees, available as of May 2015, were used to design species-specific protein-coding gene markers (see Table S1 for the list of genomes). Protein-coding gene families were defined using SiliX (Miele et al. 2011), with an identify threshold of 30% for proteins with 80% length conservation. In-house scripts were used to identify single copy protein-coding gene families present in 100% of the surveyed genomes. The nucleotide sequences corresponding to the single copy protein-coding gene families were aligned using Muscle v3 (Edgar 2004) and imported into Geneious 9.1.2 (Kearse et al. 2012). All genes smaller than 300 nucleotides were discarded as well as those that displayed 100% sequence identity between two or more strains. Primer3 (Untergasser et al. 2012) implemented in Geneious 9.1.2 (Kearse et al. 2012) was used to search for primer candidates. Candidate genes were considered if primers were identified that i) amplified a region of between 300–550 bp, ii) had a degeneracy of less than eight for both forward and reverse primers, and iii) did not hit any other species present in the honeybee gut microbiome (determined by BLASTn searches with default settings on NCBI, https://www.ncbi.nlm.nih.gov/). Based on these criteria, four candidate gene markers were selected for each species and experimentally tested. From these candidates, two gene markers (G. apicola: rimM and pflA, S. alvi: guaA and gluS) were selected for each species based on specificity (determined by preliminary Illumina sequencing trials).
Samples and sequencing
We used DNA samples from 60 control and 60 tetracycline-treated honeybees from (Raymann et al. 2017). All samples were from individual bees, and no bee was sampled multiple times. These bees were from a single colony that had not been treated with antibiotics for at least four years prior to our experiment. Thus, although many bee colonies in the United States contain gut bacteria with tetracycline resistance (tetR) genes (Tian et al. 2012), these colonies likely had low frequencies of tetR genes. Briefly, female workers with established microbiota were removed from the hive, given sucrose solution containing tetracycline for five days in the laboratory, sampled (Day 0), then marked, returned to their original colony, and sampled on Days 3, 5, and 7. Controls were given sucrose solution lacking antibiotic. Details are in (Raymann et al. 2017).
For each of our two target species, portions of two protein-coding genes were amplified for each bee (G. apicola: rimM and pflA corresponding to GAPWK_0415 and GAPWK_0528, S. alvi: guaA and gluS corresponding to SALWKB2_1196 and SALWKB2_0399) using the designed primers attached to Illumina adaptors (see Table S2 for primer sequences). Triplicate 25-μl reactions were carried out with 0.25 μl Phusion High-Fidelity DNA Polymerase (New England Biolabs), 5 μl of Phusion HF buffer (New England Biolabs), 1.25 μl of DMSO, 1 ul (each) 10μM primer, 14.5 μl H2O, and 1 μl of template DNA in buffer. The cycling conditions consisted of 98°C for 30 s, 25 cycles at 98°C for 10 s, 59°C for 30 s, 72°C for 30 s, with a final extension at 72°C for 7 min. Samples that did not amplify during the first round were rechecked by performing additional PCR assays (see Figure S1 for details).
The amplicons were pooled and cleaned using AMPure XP Beads (Beckman Coulter). The Genome Sequencing and Analysis Center at the University of Texas at Austin performed the barcoding and sequencing using Illumina MiSeq 2X300. Paired-end reads were assembled using FLASH v 1.2.11 (Magoč & Salzberg 2011) with default parameters. Reads were quality filtered with a minimum average Phred score of 30 (see Dataset S1 for final read numbers after quality filtering). In order to minimize the impact of sequencing errors on population genetics estimates, individual nucleotides were not considered (replaced by “N”) when their Phred quality score was lower than 30. Reads were checked for cross-contamination against the four reference genes (Table S2) using BOWTIE2 v2.2.8 (Langmead & Salzberg 2012) with default parameters. No cross-contamination was observed.
Population genetics analyses
For each gene marker, the nucleotide sequences from each bee were aligned with a reference sequence independently for each sample using MAFFT v7.310 (Katoh & Standley 2013) with the NW-NS-PartTree-1 setting. Due to the large sample size and the uneven depth of sequencing, the average nucleotide diversity (π) was estimated using a re-sampling method without replacement: for each alignment, 100 sequences were randomly sampled 100 times (excluding the reference sequence), and π was defined as the average across the 100 estimates (Dataset S1). For each pair of bees, we estimated the fixation index as , where is the average nucleotide diversity within each bee and is the average diversity between bees (Hudson et al. 1992). was computed by using the reference sequence present in each sample in order to retrieve the homologous positions of the alignment across samples. and were then estimated by a re-sampling strategy where 100 sequences were randomly selected at least 100 times (50 sequences of each sample were randomly selected for and 100 sequences for ) and averaged across all estimates. For each pair of bees A and B, the Fst was then defined as . Tajima’s D was estimated with the R package PEGAS v0.10 (Paradis 2010) for each sample. Haplotype networks were built with Fitchi (Matschiner 2015) with a minimal node size of 10. Due to the size of the data set, we were unable to represent all reads of each marker gene in the same haplotype network. Therefore, haplotype networks were built independently for each marker gene for each treatment day. For each bee, 100 reads were randomly selected twice. Haplotype networks were then built independently for each sub-sampling.
Identification of selective sweeps
We defined “robust” populations of G. apicola or S. alvi as those that likely resisted antibiotic treatment based on a relative abundance of over 40% (for each species) of the total gut community following antibiotic treatment based on 16S rDNA gene profiling data from (Raymann et al. 2017). In contrast, all control bees and any treated bees with a relative abundance of G. apicola or S. alvi lower than 40% following treatment were considered “vulnerable” populations. We then looked for the presence of alleles for which the frequency was over-represented in the robust populations relative to the vulnerable populations by computing both a Wilcoxon test and a t-test with Bonferroni corrections.
Results
Antibiotic treatment decreases allelic diversity
In order to determine the effects of antibiotic treatment on the allelic diversity of S. alvi and G. apicola, we computed the average nucleotide diversity (π) for control and antibiotic-treated bees for the two gene markers from each species. Overall the two markers used for each species were very consistent with each other (Figure 1). We found that the overall allelic diversity was significantly lower in treated bees for both G. apicola gene markers (P<0.0001, Wilcoxon test), whereas S. alvi allelic diversity was not significantly different between control and treated bees for either gene marker (P>0.06, Wilcoxon test, Figure 1). Additionally, we compared allelic diversity for each sampling day post-treatment (Days 0, 3, 5, and 7). We saw no effect at Day 0 for either species. Starting at Day 3, we observed a reduction in the allelic diversity of G. apicola for both gene markers, with significant decreases in diversity observed at Days 5 and 7 post-treatment (P<0.001, Wilcoxon test, Figure 1). However, for S. alvi we saw a significant difference only for one gene maker (guaA) at Days 3 and 5 (P<0.05, Wilcoxon test). Although we did not see a significant decrease in allelic diversity for all genes at Days 3, 5, and 7, the median nucleotide diversity is consistently lower in treated bees for all gene markers (Figure 1). Thus, the antibiotic treatment caused an overall loss of genetic diversity for both S. alvi and G. apicola, although this effect is much more pronounced for G. apicola.
Figure 1.
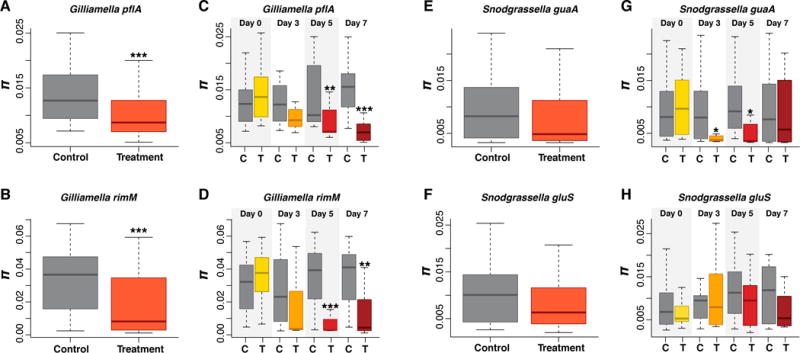
Effect of antibiotic treatment on allelic diversity in G. apicola (A–B) and S. alvi (E–F). The average nucleotide diversity (π) between all control and antibiotic treated bees (C–D and G–H) for each day post treatment (Days 0, 3, 5, and 7) for the two marker genes from each species. Box-and-whisker plots show high, low, and median values, with lower and upper edges of each box denoting first and third quartiles, respectively. The central vertical lines represent the data range, with a maximal distance of 1.5 interquartile ranges. * = P < 0.05, ** = P < 0.001, and *** = P < 0.0001, Wilcoxon test.
To further analyze allelic diversity of gut bacteria in control and antibiotic-treated bees we examined the variation in S. alvi and G. apicola nucleotide diversity for each individual bee and compared this to the nucleotide diversity present in the entire population of sampled bees (Figure 2). We found that most individual bees have lower allelic diversity than the entire population, indicating that individual bees possess distinct variants of G. apicola and S. alvi (Figure 2). At the individual bee level we observed a high level of variation in nucleotide diversity for these two species, even in control bees, but at Days 3, 5 and 7 polymorphism was substantially reduced in most treated bees (Figure 2). However, the allelic diversity was not reduced in some treated bees, which could indicate that the different variants of S. alvi and G. apicola were not equally impacted by the treatment (Figure 2).
Figure 2.
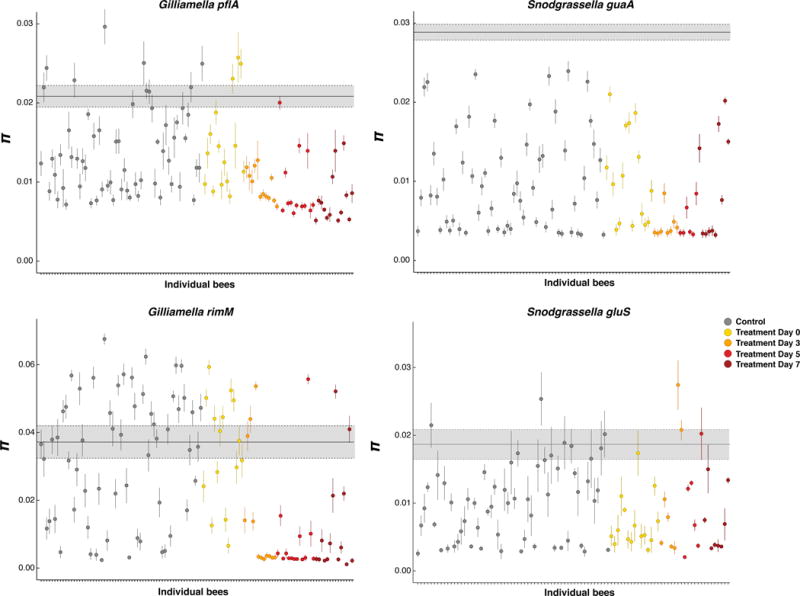
Allelic diversity of populations of G. apicola and S. alvi in individual bees compared to the entire population for each gene marker. The average nucleotide diversity (π) for each population within individual bee (dots) was compared to the average nucleotide diversity across all the populations found in all control and treated bees (gray box). For each bee bacterial population, the circle represents the average π estimate (100 sequences were randomly sampled 100 times, and π was defined as the average across the 100 estimates); vertical lines show the standard deviation. The bold gray horizontal line indicates the average nucleotide diversity of the entire population (average nucleotide diversity of G. apicola and S. alvi computed across all control and treated bees), with the gray shading representing the standard deviation. All the populations from control bees are shown in gray circles and the populations from treated bees are colored by sampling day post-treatment.
Decreased allelic diversity is correlated to increased relative abundance
In a previous study, we showed that antibiotic treatment alters either the relative or absolute abundance of all core species in the honeybee gut microbiota (Raymann et al. 2017). Here we looked at the relationship between shifts in relative and absolute abundance of G. apicola and S. alvi and shifts in allelic diversity. In our previous study we found that antibiotic treatment resulted in an increase in the relative abundance of G. apicola (with little to no effect on absolute abundance) and a decrease in absolute abundance of S. alvi (with no effect on relative abundance) (Raymann et al. 2017) (Figure 3, Figure S2). Based on the nucleotide diversity of our four gene markers, we observed a negative correlation between relative abundance and allelic diversity (Figure 3). Antibiotic treatment resulted in an increase in relative abundance, which was accompanied by a decrease in allelic diversity, specifically for Days 3, 5 and 7 post-treatment. This effect was more prominent for G. apicola than for S. alvi. An increase in relative abundance coupled with a loss of polymorphism suggests a clonal expansion of some resistant clones (selective sweeps). Conversely, we only observed a slight correlation between absolute abundance and allelic diversity for G. apicola or S. alvi (Figure S2), likely due to a high level of natural variation in absolute abundance in both control and treated bees.
Figure 3.
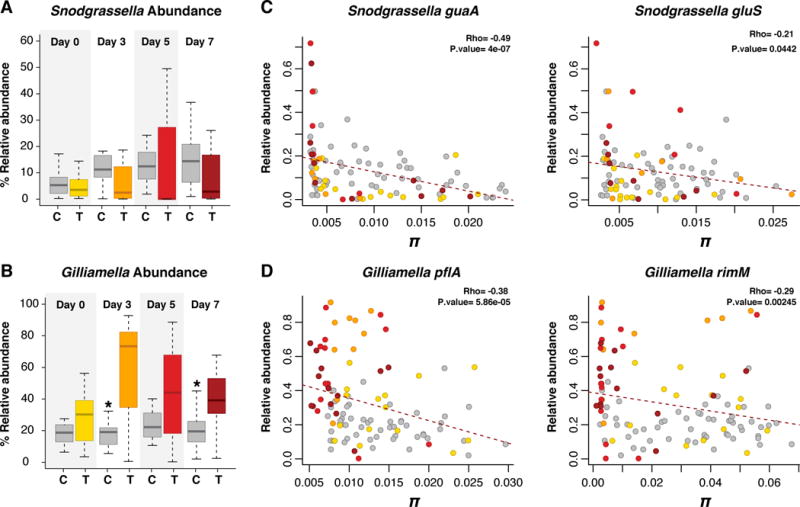
Correlation between allelic diversity and relative abundance for each gene marker. Relative abundance of S. alvi (A) and G. apicola (B) in control (C) and treated (T) bees at Days 0, 3, 5, and 7 post-treatment (data from Raymann et al. 2017). Box-and-whisker plots show high, low, and median values, with lower and upper edges of each box denoting first and third quartiles, respectively. The central vertical lines represent the data range, with a maximal distance of 1.5 interquartile ranges. * = P < 0.05, Wilcoxon test. Spearman correlation between relative abundance and π (C–D). All control bees are shown in gray dots. Treated bees are colored by day as in A–B.
Antibiotic treatment also affected both relative and absolute abundances of the other dominant members of the honeybee gut microbiome, based on 16S rDNA profiling. For example, antibiotic treatment caused a reduction in the relative and absolute abundance of Lactobacillus Firm-4, Lactobacillus Firm-5, Bartonella apis and Bifidobacterium; in contrast, Frischella perrara and Alpha 2.1 were only reduced in absolute abundance (Raymann et al. 2017). We evaluated whether changes in the relative or absolute abundance of other dominant members of the bee gut microbiome correlated with changes in genetic diversity of G. apicola or S. alvi (Table S3). We found that both relative and absolute abundance of B. apis were strongly correlated with allelic diversity of the two marker genes of G. apicola (Table S3), suggesting complex cross-species interactions between variants of these two members of the gut microbiome.
Antibiotic treatment causes changes in allele frequencies
We measured the impact of antibiotic treatment on allele frequencies using Tajima’s D for all control and treated bees as well as for control and treated bees at each day post-treatment. Overall we observed an excess of rare variants in control and treatment bees (Tajima’s D<0, Figure S3). This apparent large excess of rare alleles could potentially be attributed to sequencing errors that were not filtered despite the stringency of our procedure (see Methods). If so, Tajima’s D estimates should be equally affected by sequencing errors across our samples. However, we did not see any significant effect of treatment on the presence of low frequency alleles at Days 0, 3 and 5, but we observed a significant increase of Tajima’s D for G. apicola at Day 7 (P<0.001, Wilcoxon test). This indicates a loss of low-frequency alleles at Day 7 in G. apicola, which is consistent with the scenario of loss of diversity due to clonal expansion following treatment (i.e. a selective sweep).
We then investigated the similarity of allelic diversity across samples by comparing the fixation index (Fst) across bees. We found a strong differentiation of S. alvi allelic diversity across bees (Fst = 0.7–0.8). In contrast, G. apicola allelic diversity is much less differentiated across bees (Fst = 0.2–0.3). These results indicate that G. apicola is more frequently exchanged across bees and/or that G. apicola engages in homologous recombination with other conspecifics more frequently than S. alvi (Moran et al. 2012; Kwong et al. 2014; Engel et al. 2014). Antibiotic treatment was associated with an increase in Fst for both markers of S. alvi and for the G. apicola marker rimM (P<0.0001, Wilcoxon test, not significant for the pflA marker), indicating that treated bees are more differentiated from one another than are the control bees (Figure 4). This finding suggests that the antibiotic treatment resulted in different allelic compositions, i.e. different variants survived and/or expanded following treatment.
Figure 4.
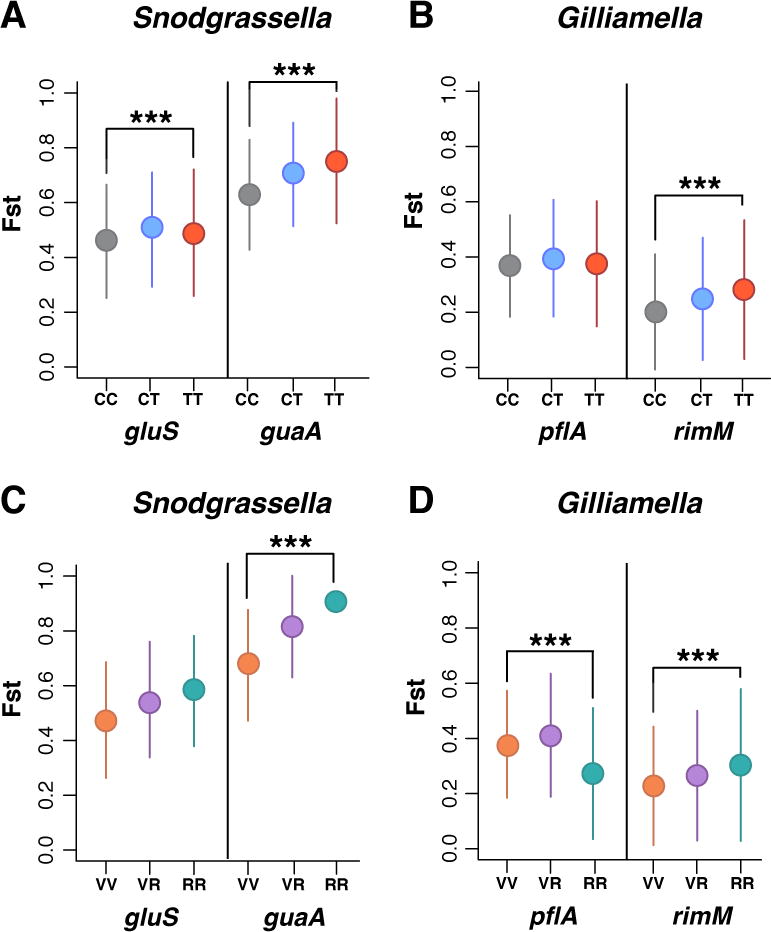
Effect of antibiotic treatment on population differentiation. Fixation index (Fst) between different populations of S. alvi and G. apicola from individual bees. (A–B) Fst between all control and treatment bees: Control/Control (CC), Control/Treatment (CT), and Treatment/Treatment (TT). (C–D) Fst between vulnerable and robust bees: Vulnerable/Vulnerable (VV), Vulnerable/Robust (VR), and Robust/Robust (RR). Circles represent median Fst values; vertical lines indicate standard deviation. *** = P < 0.0001, Wilcoxon test.
To analyze the disruptive effect of antibiotic treatment on allelic composition in more detail, we built a haplotype network for each gene marker for each treatment day by evenly sub-sampling bees. This analysis revealed that bee groups affected by treatment (S. alvi guaA at days 3 and 5 and both markers of G. apicola at days 5 and 7) presented more differentiated allelic profiles (Figure S4). This finding further suggests that the treatment resulted in heterogeneous allelic compositions through differential variant survival.
In some treated bees, S. alvi and G. apicola did not appear to be affected by antibiotic treatment. Therefore, we defined a subgroup of bees that we refer to as “robust” bees, based on their gut communities exhibiting an increase in relative abundance of S. alvi or G. apicola following antibiotic treatment. All control bees and any treated bees that did not have G. apicola or S. alvi present at greater than 40% relative abundance were categorized as “vulnerable”. We evaluated whether robust bees had a loss of rare variants by estimating Tajima’s D (Figure S4). Overall we did not see any change in rare variants in robust bees, except for a significant increase in Tajima’s D for one G. apicola marker (rimM) (Figures S5). We also compared Fst values between the vulnerable and robust bees. Consistent with our previous results, we found that robust bees were more differentiated from one another than were the control bees based on guaA and rimM (P<0.0001, Wilcoxon test, not significant for gluS, Figure 4). However, Fst estimates for pflA were significantly lower in robust bees, suggesting that this locus presents more similar allelic composition in robust bees relative to vulnerable bees. These results indicate that bees that were not affected by treatment (i.e. that did not show a decrease in allelic diversity) did not affect our overall results.
Our results suggested a selective sweep following antibiotic treatment; therefore, we looked for a signal of this sweep by examining the shifts in allele frequencies in robust bees relative to vulnerable bees. We identified three pflA alleles with a strong signal of sweep in G. apicola (Figure S6). We did not observe a similar signal of sweep in the G. apicola marker rimM. Consistent with this, the Fst for the robust bees significantly decreased for pflA but increased for rimM, suggesting that this was a local sweep.
Discussion
Antibiotic treatment has a major impact on the overall bee gut microbiota based on 16S rRNA gene profiling (Raymann et al. 2017). Here we investigated how antibiotic perturbation affects the fine-scale dynamics of two dominant and closely associated members of this community, G. apicola and S. alvi. Although none of the bee gut bacteria was completely eliminated following treatment, our results demonstrate that antibiotic exposure can have a severe impact on the genetic diversity within species, and arguably strain diversity. Because different strains have been shown to perform various functions in the bee gut (Kwong & Moran 2016; Kesnerova et al. 2017), this could have detrimental effects on honeybee health. For example, some strains of G. apicola secrete enzymes for pollen (pectin) degradation (Engel et al. 2012; Lee et al. 2015; Kesnerova et al. 2017) and some can metabolize sugars that are toxic to bees (Zheng et al. 2016). Overall, we found some variability in the response to antibiotic treatment across S. alvi and G. apicola within individual bees. This might be due to uneven treatment doses received by the bees (i.e. bees were fed antibiotics suspended in sugar syrup and might have consumed different amounts of the antibiotic). However, it could also reflect uneven strain compositions across bees. In fact, we found that the average nucleotide polymorphism of the bacterial populations in individual bees (including non-treated bees) was lower than the polymorphism calculated across all bees. Thus, individual bees, even from the same hive, naturally display distinct communities of S. alvi and G. apicola. Variation in strain composition among individuals from the same location (or hive) has been observed based on a single gene marker (minD) for S. alvi from honeybees (Powell et al. 2016). Due to this variation in strain profiles across individual hosts, some communities are likely to be more resilient than others to chemical perturbations. In humans, antibiotic treatment has individualized effects on gut microbial communities, and different individuals display various levels of microbiome recovery following treatment (Dethlefsen & Relman 2011; Pérez-Cobas et al. 2013). Our results provide additional evidence that these differences reflect variations in strain-level profiles among communities in different host individuals.
Despite their close association, S. alvi and G. apicola respond differently to antibiotic perturbation, suggesting that microbiota members undergo different population dynamics. Differences in the population dynamics of these two species have also been observed based on seasonal changes; G apicola abundance was highly impacted by seasonal changes whereas S. alvi showed little fluctuation (Ludvigsen et al. 2015). We observed a slight, non-significant reduction of allelic diversity in S. alvi in bees treated with antibiotics, despite the fact that this species was more negatively impacted by antibiotic treatment based on absolute abundance (Raymann et al. 2017). S. alvi was almost completely eliminated from the gut community of multiple bees following treatment (Figure S1) (Raymann et al. 2017). This indicates that S. alvi was, overall, not resistant to antibiotic perturbation and that no specific variants invaded S. alvi populations following treatment. The conservation of allelic diversity in the surviving populations could indicate that S. alvi will be able to return to (or retain) their original composition following antibiotic treatment. In contrast, treatment reduced allelic diversity of G. apicola far more, up to three-fold. If changes in allelic diversity represent strain variation, this reduction likely has substantial consequences for bee health, since different strains of G. apicola possess different metabolic capabilities (Engel et al. 2012; Lee et al. 2015; Zheng et al. 2016).
Interestingly, these results appear to contradict previous results based on 16S rDNA profiling using operational taxonomic unit (OTU) clustering. In Raymann et al. 2017, antibiotic treatment increased the number of G. apicola variants defined at 99% OTUs, suggesting that the diversity of G. apicola increased following treatment. Due to this apparent contradiction with our current results, we reanalyzed the previously defined 99% OTUs clusters. We found that the vast majority of the OTUs were present at very low frequencies; over 50% of the OTUs were supported by 3 reads or fewer across all samples. These results indicate that the diversity estimates based on OTU clustering were largely due to very rare variants, which could represent artifacts introduced by sequencing errors. The use of an arbitrary threshold (99% identity) to define strain-level diversity across large datasets can be very sensitive to sequencing errors; for this amplicon length, two reads would constitute different OTUs if they differed at only 3 nucleotide positions. Moreover, diversity measurements based on the number of OTUs have been shown to grossly overestimate diversity and to therefore lack biological relevance (Edgar 2017). Therefore, approaches based on OTU clustering are best reserved for measuring diversity at higher taxonomic levels (e.g. species, genus, etc.) and not at the strain level.
Unlike S. alvi, the absolute number of G. apicola cells did not decrease in the bee gut after treatment, and their relative abundance significantly increased (Raymann et al. 2017), implying that resistance mechanisms were at play. This pattern suggests that some resistant strains swept through the population during antibiotic exposure. Possibly, the different outcomes observed in the two species stem from a higher frequency of tetR genes in G. apicola. We found that 79.3% of the sequenced strains of G. apicola from honeybees (58 strains) contain at least one tetR gene, whereas 51.9% of previously sequenced S. alvi honeybee strains (27 strains) encode tetR genes (based on BLASTn search of the genomes available in NCBI as of July 2017).
In agreement with a selective sweep scenario for G. apicola, we observed three alleles in pflA that reached fixation in nearly all treated bees that displayed a high relative abundance of G. apicola. However, this signal of sweep was not detected in the G. apicola gene marker rimM, suggesting that this was a local sweep, only affecting one or several loci of the chromosome close to pflA. Gene-specific sweeps have been shown to occur in natural populations of Vibrio cyclitrophicus (Shapiro et al. 2012) and in some freshwater lake bacterial populations (Bendall et al. 2016). In fact, genome-wide sweeps have rarely been observed in natural populations and are thought to only occur in populations with low recombination rates, whereas gene-specific sweeps are more likely in highly recombining species (Bendall et al. 2016), such as G. apicola (Moran et al. 2012; Kwong et al. 2014; Engel et al. 2014). Potentially, a local sweep would be more likely if rimM were located near a tetR gene, i.e., if rimM swept to high frequency through hitchhiking. However, we examined the 58 G. apicola genomes available and did not find a tetR gene within <50kb of rimM in these sequenced strains. Alternatively, it is possible that this pattern is due to the expansion of a single clone that was affected by recombination in rimM and not in pflA. Irrespective of the mechanisms at play, our results indicate that the resilience of G. apicola following antibiotic treatment was accompanied by a loss of allelic diversity, including a loss of rare variants. Therefore, it is possible that G. apicola, as opposed to S. alvi, will not be able return to its original allelic composition after antibiotic exposure.
Antibiotic treatment resulted in elevated mortality of bees in the hive; over 50% of the treated bees died by Day 3 post-treatment (Raymann et al. 2017). Because only surviving bees could be sampled, our profiles of community composition may represent an incomplete picture of the shifts that follow treatment. Additionally, we may have captured DNA from dead bacterial cells in our analysis, as noted previously (Raymann et al. 2017). This potential bias is especially likely for bees sampled at Day 0 post-treatment before return to the hive, as bees do not typically defecate while kept in the lab. In fact, we did not observe a significant difference in G. apicola or S alvi genetic diversity between control and treatment bees sampled on Day 0 post-treatment. A huge reduction in absolute numbers of bacterial cells was observed after bees were returned to the hive, but not on Day 0; this contrast suggests that DNA from dead bacterial cells was less problematic in samples after return to the hive (Days 3, 5, and 7). However, the problem of sequencing DNA from dead cells is inherent to metagenomic studies and could have caused some biases in our results.
Results showed some discordance between the two gene markers of G. apicola. Such discrepancies have been noted previously for the highly recombining species of Synechococcus (Rosen et al. 2015), suggesting that the population dynamics of highly recombinant species would be better elucidated by using additional markers. In S. alvi, the two gene markers gave similar results, consistent with a previous study suggesting that S. alvi recombines less frequently than does G. apicola (Moran et al. 2012). Thus, a single gene marker might be sufficient for following population dynamics of clonal or nearly clonal species, but multiple markers might be needed to accurately decipher the population dynamics of highly recombining species.
Conclusion
Using species-specific amplicon sequencing of protein-coding genes we documented a major impact of antibiotic treatment on the fine-scale diversity of two dominant species from the honeybee gut microbiota. Because distinct strains can have different roles in hosts; these shifts may impact bee health.
In addition, we show that avoiding OTU clustering and instead using population genetic metrics provides a more accurate picture of the fine-scale diversity within species of natural gut communities. OTU clustering using arbitrary thresholds will often either 1) overlook strain diversity by using very stringent thresholds or 2) overestimate strain diversity by overemphasizing rare variants, which cannot be reliably distinguished from sequencing errors.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health award 1R01GM108477-01 to N.A.M, the National Institute of Food and Agriculture award (2017-67012-26088) to K.R. L M.B. was supported through funding by National Institutes of Health award R35GM118038 to Howard Ochman.
We thank Kim Hammond for assistance with beekeeping, Zack Shaffer for research assistance, members of the Moran-Ochman lab group for helpful discussions, and the UT Genome Sequencing and Analysis Facility for sequencing services.
Footnotes
DR. KASIE RAYMANN (Orcid ID : 0000-0001-7008-2783)
DR. NANCY ANN MORAN (Orcid ID : 0000-0003-2983-9769)
Data Accessibility
All metagenomic sequence data was deposited on the Sequence Read Archive (SRA) on NCBI BioProject ID: PRJNA415093.
Author Contributions
K.R and LM.B. performed the research and analyzed data. K.R, LM.B and N.A.M designed the research and wrote the manuscript.
References
- Bendall ML, Stevens SL, Chan L-K, et al. Genome-wide selective sweeps and gene-specific sweeps in natural bacterial populations. The ISME journal. 2016;10:1589–1601. doi: 10.1038/ismej.2015.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaser MJ. Antibiotic use and its consequences for the normal microbiome. Science. 2016;352:544–545. doi: 10.1126/science.aad9358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Quintero A, Ochman H. Assessing the unseen bacterial diversity in microbial communities. Genome biology and evolution. 2015;7:3416–3425. doi: 10.1093/gbe/evv234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox-Foster DL, Conlan S, Holmes EC, et al. A metagenomic survey of microbes in honey bee colony collapse disorder. Science. 2007;318:283–287. doi: 10.1126/science.1146498. [DOI] [PubMed] [Google Scholar]
- Degnan PH, Ochman H. Illumina-based analysis of microbial community diversity. The ISME journal. 2012;6:183–194. doi: 10.1038/ismej.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proceedings of the National Academy of Sciences. 2011;108(Suppl 1):4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS biology. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Accuracy of microbial community diversity estimated by closed- and open-reference OTUs. PeerJ. 2017;5:e3889. doi: 10.7717/peerj.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegaard KM, Engel P. Beyond 16S rRNA community profiling: intra-species diversity in the gut microbiota. Frontiers in microbiology. 2016;7:1475. doi: 10.3389/fmicb.2016.01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel P, Kwong WK, McFrederick Q, et al. The bee microbiome: impact on bee health and model for evolution and ecology of host-microbe interactions. Mbio. 2016;7:e02164–15. doi: 10.1128/mBio.02164-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel P, Martinson VG, Moran NA. Functional diversity within the simple gut microbiota of the honey bee. Proceedings of the National Academy of Sciences. 2012;109:11002–11007. doi: 10.1073/pnas.1202970109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel P, Stepanauskas R, Moran NA. Hidden diversity in honey bee gut symbionts detected by single-cell genomics. PLoS genetics. 2014;10:e1004596. doi: 10.1371/journal.pgen.1004596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Slatkin M, Maddison WP. Estimation of levels of gene flow from DNA sequence data. Genetics. 1992;132:583–589. doi: 10.1093/genetics/132.2.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarz Pavón AB, Maiden MCJ. Multilocus sequence typing. Methods in molecular biology (Clifton, N.J.) 2009;551:129–140. doi: 10.1007/978-1-60327-999-4_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesnerova L, Mars R, Ellegaard KM, Troilo M. Disentangling metabolic functions of bacteria in the honey bee gut. bioRxiv. 2017 doi: 10.1371/journal.pbio.2003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong WK, Moran NA. Evolution of host specialization in gut microbes: the bee gut as a model. Gut microbes. 2015;6:214–220. doi: 10.1080/19490976.2015.1047129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong WK, Moran NA. Gut microbial communities of social bees. Nature reviews. Microbiology. 2016;14:374–384. doi: 10.1038/nrmicro.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong WK, Engel P, Koch H, Moran NA. Genomics and host specialization of honey bee and bumble bee gut symbionts. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:11509–11514. doi: 10.1073/pnas.1405838111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong WK, Mancenido AL, Moran NA. Immune system stimulation by the native gut microbiota of honey bees. Royal Society open science. 2017;4:170003. doi: 10.1098/rsos.170003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FJ, Rusch DB, Stewart FJ, Mattila HR, Newton ILG. Saccharide breakdown and fermentation by the honey bee gut microbiome. Environmental microbiology. 2015;17:796–815. doi: 10.1111/1462-2920.12526. [DOI] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludvigsen J, Rangberg A, Avershina E, et al. Shifts in the midgut/pyloric microbiota composition within a honey bee apiary throughout a season. Microbes and environments. 2015;30:235–244. doi: 10.1264/jsme2.ME15019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matschinder M. Fitchi: Haplotype genealogy graphs based on the Fitsch algorithm. Bioinformatics. 2015;32:1250–52. doi: 10.1093/bioinformatics/btv717. [DOI] [PubMed] [Google Scholar]
- Miele V, Penel S, Duret L. Ultra-fast sequence clustering from similarity networks with SiLiX. BMC bioinformatics. 2011;12:116. doi: 10.1186/1471-2105-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller AH, Caro-Quintero A, Mjungu D, et al. Cospeciation of gut microbiota with hominids. Science. 2016;353:380–382. doi: 10.1126/science.aaf3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, Hansen AK, Powell JE, Sabree ZL. Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS one. 2012;7:e36393. doi: 10.1371/journal.pone.0036393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E. pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics. 2010;26:419–420. doi: 10.1093/bioinformatics/btp696. [DOI] [PubMed] [Google Scholar]
- Pérez-Cobas AE, Artacho A, Knecht H, et al. Differential effects of antibiotic therapy on the structure and function of human gut microbiota. PLoS one. 2013;8:e80201. doi: 10.1371/journal.pone.0080201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JE, Ratnayeke N, Moran NA. Strain diversity and host specificity in a specialized gut symbiont of honey bees and bumble bees0. Molecular ecology. 2016 doi: 10.1111/mec.13787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymann K, Shaffer Z, Moran NA. Antibiotic exposure perturbs the gut microbiota and elevates mortality in honeybees. PLoS biology. 2017;15:e2001861. doi: 10.1371/journal.pbio.2001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MJ, Davison M, Bhaya D, Fisher DS. Microbial diversity. Fine-scale diversity and extensive recombination in a quasisexual bacterial population occupying a broad niche. Science. 2015;348:1019–1023. doi: 10.1126/science.aaa4456. [DOI] [PubMed] [Google Scholar]
- Sekirov I, Tam NM, Jogova M, et al. Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infection and immunity. 2008;76:4726–4736. doi: 10.1128/IAI.00319-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro BJ, Friedman J, Cordero OX, et al. Population genomics of early events in the ecological differentiation of bacteria. Science. 2012;336:48–51. doi: 10.1126/science.1218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot CM, Koenigsknecht MJ, Carlson PE, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nature communications. 2014;5:3114. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Fadhil NH, Powell JE, Kwong WK, Moran NA. Long-term exposure to antibiotics has caused accumulation of resistance determinants in the gut microbiota of honeybees. Mbio. 2012;3:e00377–12. doi: 10.1128/mBio.00377-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, et al. Primer3–new capabilities and interfaces. Nucleic acids research. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Nishida A, Kwong WK, et al. Metabolism of toxic sugars by strains of the bee gut symbiont Gilliamella apicola. Mbio. 2016;7:e01326–16. doi: 10.1128/mBio.01326-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Powell JE, Steele MI, Dietrich C, Moran NA. Honeybee gut microbiota promotes host weight gain via bacterial metabolism and hormonal signaling. Proceedings of the National Academy of Sciences. 2017;114:4775–4780. doi: 10.1073/pnas.1701819114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.