

Abstract
The direct reductive amination of aromatic aldehydes has been realized using a photocatalyst under visible light irradiation. The single electron oxidation of an in situ formed aminal species generates the putative α-amino radical that eventually delivers the reductive amination product. This method is operationally simple, highly selective, and functional group tolerant, which allows the direct synthesis of benzylic amines by a unique mechanistic pathway.
Graphical Abstract
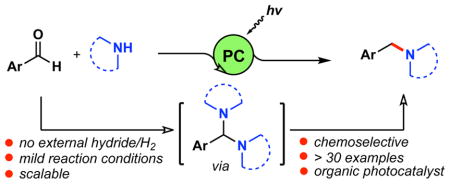
Amines and their derivatives are ubiquitous in pharmaceuticals, agrochemicals and biologically relevant natural products.1 Reductive amination represents one of the most convenient and versatile methods for their synthesis.2 In fact, a recent survey has revealed that reductive amination is the sixth most frequently used reaction in the pharmaceutical industry.3 Although various methods are known, direct reductive amination (DRA) is considered the most practical method for the synthesis of amines, and therefore, is the most widely used. DRA has been used mainly for synthesizing secondary amines; however, the synthesis of tertiary amines has been described as challenging, and few methods are reported in the literature.4 In addition to an amine and a carbonyl compound, the traditional reductive amination requires hydrogen (H2) or a hydride source such as borohydride or a hydrosilane reagent. Application of these reagents is often associated with toxicity issues, poor selectivity, air and moisture sensitivity, use of an external activator, and/or purification issues.4a, 5 Recently, several methods have been reported regarding metal-catalyzed reductive amination of aldehydes under external H2/hydride-free conditions.4b, 6 Despite the conceptual simplicity, these methods usually require the application of high pressure CO gas and high temperatures (120–140 °C).
Recently, visible light-mediated photoredox catalysis has emerged as an enabling platform to access new synthetic transformations through single-electron transfer (SET) events.7 In particular, the generation of carbon-centered α-amino radicals and their use in coupling reactions with various electrophilic partners has become a powerful strategy in organic synthesis.8 Such radical species have been generated from α-amino acids,9 α-amino trifluoroborates10 and silylamines,8b among others. Amine scaffolds have also been used to generate α-amino radicals through successive oxidation and α-deprotonation steps.7e Considering these recent advancements in this area, we questioned whether it might be feasible to use aldehydes and amines to generate α-amino radicals in situ, capable of undergoing reduction to give amines by a unique mechanistic pathway. Scouring the literature, we found DRA under photoredox catalysis is an underexplored area.11 Considering the prevalent role of reductive amination reactions, we conjectured that a photoredox-based DRA approach would offer utility in terms of mild reaction conditions, chemoselectivity, and functional group compatibility.
Herein, we report a novel method for direct reductive amination from aromatic aldehyde feedstocks and readily available secondary amines to synthesize tertiary benzylic amines under photoredox catalysis using iridium-based- or organic photocatalysts (Scheme 1).
Scheme 1.

Photoredox-catalyzed reductive amination of aldehydes
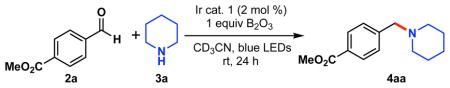
At the outset of our studies, methyl 4-formylbenzoate 2a and piperidine 3a were chosen as model substrates for the desired reductive amination (Table 1). A variety of photocatalysts were screened that possessed sufficiently high excited-state redox potentials to provide the desired amine compound 4aa. Although 4CzIPN (E*1/2= 1.35 V vs SCE)13 proved to be a viable photocatalyst, the Ir catalyst 1 (E*1/2= 1.32 V vs SCE)12 provided superior yields (entries 1–2, Table 1). The yield was decreased when the catalyst 1 was replaced by Ru(bpy)3 (PF6)2, which has a high reduction potential, but relatively low oxidation potential (E*1/2= 0.77 V vs SCE). Although MesAcr (mesitylacridinium) has a high oxidation potential in the excited state (E*1/2= 2.06 V vs SCE),14 no product was observed (entry 5), likely because of its low reduction potential. Other organophotocatalysts such as Eosin Y were also ineffective (entry 4) under these reaction conditions, most likely because of their low oxidation potentials (E*1/2= 0.79 V vs SCE).15 Importantly, the reaction was also unsuccessful in the absence of either photocatalyst 1 or light (entries 7–8).
Table 1.
Optimization of the Reaction Conditionsa
| ||
|---|---|---|
| entry | deviation from standard condition | yield (%)b |
| 1 | no change | 78 |
| 2 | 5 mol % 4CzIPN instead of Ir cat. | 53 |
| 3 | 2 mol % Ru(bpy)3 instead of Ir cat. | 29 |
| 4 | 2 mol % Eosin Y instead of Ir cat. | 0 |
| 5 | 2 mol % MesAcr instead of Ir cat. | 0 |
| 6 | in absence of photo catalyst | 0 |
| 7 | in absence of light | 0 |
| 8 | in absence of B2O3 | 50 |
| 9 | 1 equiv B(OH)3 instead of B2O3 | 67 |
| 10 | 1 equiv of B(OMe)3 instead of B3O3 | 73 |
| 11c | SiO2 instead of B2O3 | 48 |
| 12c | 4 A MS instead of B2O3 | 24 |
| 13 | DME instead of MeCN | 22 |
| 14 | 1,4-dioxane instead of MeCN | <5 |
| 15 | THF instead of MeCN | <5 |
| 16 | DMF-d7 instead of MeCN | 56 |
| ||
General reaction conditions: 2a (1.0 equiv, 0.1 mmol), piperidine 3a (3.0 equiv, 0.3 mmol), Ir catalyst 1 (2 mol %), MeCN-d3 (0.20 M), rt, 24 h.
Yield was determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard.
50 mg of SiO2 and molecular sieves (4 Å) were used.
Although the reaction worked smoothly in the absence of B2O3, (entry 8), this additive was beneficial for affording high yields. Most likely, the boron trioxide acts as a weak Lewis acid, as well as a desiccant.16 B(OMe)3 was as efficient as B2O3 for the reaction, but boron trioxide was chosen, given its cost efficiency and ease of handling. Other dehydrating agents17 such as SiO2 and molecular sieves (MS) gave lower yields (entries 11–12). Although different aprotic solvents were examined for the amination reaction, acetonitrile (MeCN) was found to be superior to any other explored solvents (entries 13–16).
During the reaction optimization (entries 6–7, Table 1), we observed the clean formation of an aminal species in the reaction mixture. Intrigued by this result and its mechanistic implications, we set out to synthesize the corresponding aminal 5 (Figure 1). It has been reported that aminals can be used for reductive alkylation and allylation in the presence of Zn and strong Lewis acids, although the mechanism is not fully understood.18
Figure 1.

Synthesis of aminal compound 5
We anticipated that aminal 5 could be an intermediate in the amination reaction. To gain further insight, we measured the redox potential of 5 (Eox = +0.95 V vs SCE), which indicated the viability of oxidation with the excited state of Ir photocatalyst 1 (E* 1/2= 1.32 V vs SCE). Next, we subjected aminal 5 to the photocatalyst and light to examine the reactivity of this species. As outlined in Scheme 2, compound 5 afforded the amine 4aa in the presence of photocatalyst 1, although the yield was low (eq 1). Surprisingly, in the presence of piperidine (1 equiv), a three-fold increase of the yield was observed (eq 2). Different bases other than piperidine have also been tested. For example, the reaction also worked smoothly in the presence of diisopropylethylamine (DIPEA), with no cross-amination product (eq 3).19 Furthermore, as anticipated, aminal 5 was fully recovered, and no product was observed, in the absence of either photocatalyst or light.
Scheme 2. Reactivity of intermediate 5 under various conditions.a.

a5 (1.0 equiv, 0.1 mmol), piperidine 3a (1.0 equiv), DIPEA (1.0 equiv), Ir catalyst 1 (2 mol %), MeCN-d3 (0.20 M), rt, 24 h. bYield was determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard.
These results suggest that the aminal is most likely an active intermediate for the amination via generation of an α-aminobenzyl radical upon oxidation by the photoexcited catalyst (See Supporting Information (SI) for more experimental support).
As outlined in Figure 2, we propose that an in situ formed aminal intermediate, 5a, undergoes single-electron oxidation by photoexcited catalyst 1 to form cationic radical species 5b, which subsequently forms the α-amino radical 5c.20 Fragmentation of 5b is likely to be facilitated by the presence of a base (Scheme 2, eqs 2–3), with concomitant formation of 6a. Notably, when pyrrolidine was used for the amination reaction, the formation of 1-pyrroline was observed (See SI). Next, amino benzyl radical 5c can undergo a single electron reduction by the reduced photocatalyst 1b to form benzylic anion 5d,21 and the subsequent protonation would deliver the final amine 4.22
Figure 2.

Plausible mechanism for the reductive amination
Unfortunately, this reductive amination protocol was not uniformly generalizable. When we employed electron-rich aldehydes such as p-anisaldehyde, the amination did not work under the standard conditions described in Table 1. This discrepancy was unexpected and showed that the substituent on the aromatic ring plays a crucial role beyond the condensation step. It is well known that the single electron reduction of benzylic radicals (e.g., 5c) is heavily dependent on the substituent of the arene ring.23 For example, benzyl radicals bearing electron-withdrawing substituents on the aromatic ring have lower reduction potentials (e.g., −0.77 vs SCE for p-CN) than those bearing electron-donating substituents on the arene ring (e.g., −1.75 vs SCE for p-OMe). Therefore, we hypothesized that because of this, a p-methoxybenzyl radical would not be reduced by photocatalyst 1b (Epc/−pc = −1.37 V vs SCE).24 To resolve this issue, we envisioned an alternative pathway (blue arrow, Figure 2).
We rationalized that for those radicals 5c exhibiting a high reduction potential, hydrogen atom transfer (HAT) would provide a means to bypass the single electron reduction and give a direct mechanistic pathway to 4.25 To validate this idea, we briefly screened different thiols. We found that 2,6-dimethylbenzenethiol (7) was highly efficient for the reductive amination of electron-rich p-anisaldehyde. Of note, aliphatic thiols such as t-BuSH were completely ineffective in providing the reductive amination product, likely as a result of mismatched polar effects.26 Consequently, it appears that amino benzyl radical species 5c can undergo two different pathways (SET or HAT) to give the amine, depending on the reduction potentials of the benzyl radicals and the reaction conditions. 27
We next turned our attention to the scope of this amination reaction, as shown in Scheme 3. The reaction proceeded well for both electron-rich and electron-deficient aromatic aldehydes (4aa-4qa). Aldehyde 2a reacted cleanly with piperidine, affording a good yield of the desired product. Importantly, the reaction was also scalable. Performing the amination on a larger scale (6.1 mmol, 12-fold increase) proved viable without compromising the yield (4aa). Other electron-withdrawing functional groups such as a nitrile (4fa), sulfone (4ga), and a benzyl ester (4ba-ca) were also compatible under the standard reaction conditions. In particular, boronate esters were untouched under the reaction conditions (4ca), thereby permitting the possibility of further functionalization using Chan-Lam amination28 or cross-coupling.29 Electron-rich aromatic aldehydes could be used when catalytic amount of 2,6-dimethylbenzenethiol (7) was used to afford the desired amines. Various electron-donating groups such as methoxy (4ha), and phenoxy (4ia) were well suited. The latter could be prepared on gram scale (5.05 mmol) using organic photocatalyst 4CzIPN ($6/gram). This provided a 63% yield of 4ia, and the catalyst was recyclable. Allyloxy (4jl), propargyloxy (4kl), thiomethyl (4ma), and amino groups (4oa) were also well tolerated and afforded the corresponding products in moderate to good yields. Of note, we did not observe any hydroaminated product when the allyl- or propargyl ether functionalized aldehydes were used (4jl-4kl).25 Interestingly, an O-glycoside aldehyde could be employed to synthesize 4na, although in modest yields. Sterically demanding 2-chlorobenzaldehyde could be used to synthesize the amine 4qa in acceptable yield. Finally, trifluoromethylated heterocyclic amine 4ra was accessed in modest yield after a prolonged reaction time (48 h). In an attempt to expand the aldehyde scope further, we also used aliphatic aldehydes such as isobutyraldehyde and pivalaldehyde. Unfortunately, none of these gave the desired amines (4sa-ta). Spectroscopic analysis showed that a lack of aminal formation was the likely cause.
Scheme 3. Evaluation of Aldehydes and Amine Under Photoredox-Catalyzed Reductive Aminationa.

aGeneral reaction conditions: aldehyde 2 (1.0 equiv, 0.5 mmol), amine 3 (3.0 equiv, 1.5 mmol), B2O3 (1.0 equiv, 0.5 mmol), Ir-catalyst 1 (2 mol %), MeCN (0.20 M), rt, 24 h. bYield for the reaction using 2 equiv of amine. cYield for the 6.1 mmol scale reaction. dYield for the reaction (36 h) using 5 mol % 4CzIPN instead of 1. e10 mol % 2,6-dimethylbenzenethiol (7) was used. fYield for a 5.0 mmol scale reaction. gReaction time 48 h
Next, we turned our attention to explore the amine scope for the reductive amination (4ab-4fm). Morpholines are highly represented among small molecules that are biologically and therapeutically relevant, including many natural products.30 Consequently, we used morpholine (4ab) and thiomorpholine (4hc) for the amination, finding that both were well tolerated. Likewise, substituted piperidines were also well accommodated to afford tertiary amines (4ad-4ae and 4ge) in good yields. Compound 4ef bearing two carbonyl groups showcases the excellent chemoselectivity of this method. Phenyl- and nitrile-substituted piperidines (4gh, 4fg) could also be used. A spiro-piperidine reacted cleanly to afford 4aj. Furthermore, the Boc-protected piperazine could be used to synthesize 4ak, providing a direct method to access an important class of molecules31 from aldehyde feedstocks. Pyrrolidine also worked smoothly with 2a to give 4al in good yield. Moreover, an enantioenriched amine (4fm) was prepared without any stereochemical ablation, indicating the stereochemical fidelity of the method. Unprotected β-amino alcohols such as 4-piperidinol reacted with p-anisaldehyde to afford 4hi, demonstrating the high functional group compatibility and mildness of this method. Unfortunately, acyclic amines were unsuitable for the process (4hn). Spectroscopic analysis of the reaction between diethylamine (HNEt2) and p-anisaldehyde revealed the lack of aminal intermediate formation, which seems likely to be the cause of the unsuccessful reaction.
In summary, a novel method for the direct synthesis of benzylic, tertiary amines from aromatic aldehydes is reported. To the best of our knowledge, this reaction is the first example of the direct reductive amination of aromatic aldehydes to generate tertiary amines under photoredox catalysis. Furthermore, the excellent functional group compatibility and mild reaction conditions favor its implementation in late-stage functionalization of complex structural motifs. Notably, a new protocol for accessing α-amino radicals via aminal formation/oxidation/fragmentation has been reported, with demonstrated potential for the discovery of new transformations.
Supplementary Material
Acknowledgments
Funding Sources
The authors are grateful for the financial support provided by the NIGMS (R01 GM113878). R.A. is thankful to the Swedish Chemical Society for the Bengt Lundqvist postdoctoral fellowship.
We sincerely thank Dr. Alvaro Gutierrez-Bonet, Dr. Christopher B. Kelly and Jennifer K. Matsui of the University of Pennsylvania (UPenn) for useful discussions. We are grateful to Junchen Kang (UPenn) for synthesizing several aldehydes. We also thank Dr. Charles W. Ross III (UPenn) for assistance in obtaining HRMS and Simon Berritt (UPenn) for helping in SFC analysis. In addition, the authors thank Johnson Matthey for the donations of iridium(III) chloride and ruthenium(III) chloride trihydrate.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and characterization data for all compounds (PDF)
References
- 1.Lawrence SA. Amines: Synthesis, Properties and Applications. University Press; Cambridge: 2004. [Google Scholar]
- 2.(a) Margaretha P. Science of Synthesis. 40a. Thieme; Stuttgart: 2009. Reductive Amination of Carbonyl Compounds; pp. 65–89. [Google Scholar]; (b) Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. J Org Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]; (c) Abdel-Magid AF, Mehrman SJ. Org Process Res Dev. 2006;10:971–1031. [Google Scholar]
- 3.(a) Roughley SD, Jordan AM. J Med Chem. 2011;54:3451–3479. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]; (b) Brown DG, Boström J. J Med Chem. 2016;59:4443–4458. doi: 10.1021/acs.jmedchem.5b01409. [DOI] [PubMed] [Google Scholar]
- 4.(a) Varjosaari SE, Skrypai V, Suating P, Hurley JJM, Lio AMD, Gilbert TM, Adler MJ. Adv Synth Catal. 2017;359:1872–1878. [Google Scholar]; (b) Nayal OS, Thakur MS, Bhatt V, Kumar M, Kumar N, Singh B, Sharma U. Chem Commun. 2016;52:9648–9651. doi: 10.1039/c6cc04381j. [DOI] [PubMed] [Google Scholar]
- 5.Sapsford JS, Scott DJ, Allcock NJ, Fuchter MJ, Tighe CJ, Ashley AE. Adv Synth Catal. 2018;360:1066–1071. doi: 10.1002/adsc.201701418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Chusov D, List B. Angew Chem Int Ed. 2014;53:5199–5201. doi: 10.1002/anie.201400059. [DOI] [PubMed] [Google Scholar]; (b) Kolesnikov PN, Yagafarov NZ, Usanov DL, Maleev VI, Chusov D. Org Lett. 2015;17:173–175. doi: 10.1021/ol503595m. [DOI] [PubMed] [Google Scholar]; (c) Afanasyev OI, Usanov DL, Chusov D. Org Biomol Chem. 2017;15:10164–10166. doi: 10.1039/c7ob02795h. [DOI] [PubMed] [Google Scholar]; (d) Park JW, Chung YK. ACS Catal. 2015;5:4846–4850. [Google Scholar]
- 7.(a) Matsui JK, Lang SB, Heitz DR, Molander GA. ACS Catal. 2017;7:2563–2575. doi: 10.1021/acscatal.7b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tellis JC, Kelly CB, Primer DN, Jouffroy M, Patel NR, Molander GA. Acc Chem Res. 2016;49:1429–1439. doi: 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shaw MH, Twilton J, MacMillan DWC. J Org Chem. 2016;81:6898–6926. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Romero NA, Nicewicz DA. Chem Rev. 2016;116:10075–10166. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]; (e) Staveness D, Bosque I, Stephenson CRJ. Acc Chem Res. 2016;49:2295–2306. doi: 10.1021/acs.accounts.6b00270. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Marzo L, Pagire SK, Reiser O, König B. Angew Chem Int Ed. 2018. Early View. [DOI] [PubMed] [Google Scholar]
- 8.(a) Heinz C, Lutz JP, Simmons EM, Miller MM, Ewing WR, Doyle AG. J Am Chem Soc. 2018;140:2292–2300. doi: 10.1021/jacs.7b12212. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Remeur C, Kelly CB, Patel NR, Molander GA. ACS Catal. 2017;7:6065–6069. doi: 10.1021/acscatal.7b01973. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Patel NR, Kelly CB, Siegenfeld AP, Molander GA. ACS Catal. 2017:1766–1770. doi: 10.1021/acscatal.6b03665. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fuentes de Arriba AL, Urbitsch F, Dixon DJ. Chem Commun. 2016;52:14434–14437. doi: 10.1039/c6cc09172e. [DOI] [PubMed] [Google Scholar]; (e) Beatty JW, Stephenson CRJ. Acc Chem Res. 2015;48:1474–1484. doi: 10.1021/acs.accounts.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Freeman DB, Furst L, Condie AG, Stephenson CRJ. Org Lett. 2012;14:94–97. doi: 10.1021/ol202883v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zuo Z, Cong H, Li W, Choi J, Fu GC, MacMillan DWC. J Am Chem Soc. 2016;138:1832–1835. doi: 10.1021/jacs.5b13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heitz DR, Rizwan K, Molander GA. J Org Chem. 2016;81:7308–7313. doi: 10.1021/acs.joc.6b01207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo X, Wenger OS. Angew Chem Int Ed. 2018;57:2469–2473. doi: 10.1002/anie.201711467. [DOI] [PubMed] [Google Scholar]
- 12.Koike T, Akita M. Inorg Chem Front. 2014;1:562–576. [Google Scholar]
- 13.Luo J, Zhang J. ACS Catal. 2016;6:873–877. [Google Scholar]
- 14.Ohkubo K, Mizushima K, Iwata R, Souma K, Suzuki N, Fukuzumi S. Chem Commun. 2010;46:601–603. doi: 10.1039/b920606j. [DOI] [PubMed] [Google Scholar]
- 15.Majek M, Jacobi von Wangelin A. Acc Chem Res. 2016;49:2316–2327. doi: 10.1021/acs.accounts.6b00293. [DOI] [PubMed] [Google Scholar]
- 16.Sekiya M, Sakai H. Chem Pharm Bull. 1969;17:32–35. [Google Scholar]
- 17.Williams DBG, Lawton M. J Org Chem. 2010;75:8351–8354. doi: 10.1021/jo101589h. [DOI] [PubMed] [Google Scholar]
- 18.Hatano B, Nagahashi K, Kijima T. J Org Chem. 2008;73:9188–9191. doi: 10.1021/jo8019797. [DOI] [PubMed] [Google Scholar]
- 19.Tararov VI, Kadyrov R, Riermeire TH, Börner A. Adv Synth Catal. 2002;344:200–208. [Google Scholar]
- 20.(a) Miyake Y, Nakajima K, Nishibayashi Y. J Am Chem Soc. 2012;134:3338–3341. doi: 10.1021/ja211770y. [DOI] [PubMed] [Google Scholar]; (b) Zhu S, Das A, Bui L, Zhou H, Curran DP, Rueping M. J Am Chem Soc. 2013;135:1823–1829. doi: 10.1021/ja309580a. [DOI] [PubMed] [Google Scholar]; (c) Ruiz Espelt L, Wiensch EM, Yoon TP. J Org Chem. 2013;78:4107–4114. doi: 10.1021/jo400428m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Huang H, Yu C, Zhang Y, Zhang Y, Mariano PS, Wang W. J Am Chem Soc. 2017;139:9799–9802. doi: 10.1021/jacs.7b05082. [DOI] [PubMed] [Google Scholar]; (b) Kohls P, Jadhav D, Pandey G, Reiser O. Org Lett. 2012;14:672–675. doi: 10.1021/ol202857t. [DOI] [PubMed] [Google Scholar]
- 22.Loh YY, Nagao K, Hoover AJ, Hesk D, Rivera NR, Colletti SL, Davies IW, MacMillan DWC. Science. 2017;358:1182–1187. doi: 10.1126/science.aap9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sim BA, Griller D, Wayner DDM. J Am Chem Soc. 1989;111:754–755. [Google Scholar]
- 24.Kelly CB, Patel NR, Primer DN, Jouffroy M, Tellis JC, Molander GA. Nat Protocols. 2017;12:472–492. doi: 10.1038/nprot.2016.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Musacchio AJ, Lainhart BC, Zhang X, Naguib SG, Sherwood TC, Knowles RR. Science. 2017;355:727–730. doi: 10.1126/science.aal3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts BP, Steel AJ. J Chem Soc, Perkin Trans 1. 1994:2155–2162. [Google Scholar]
- 27.Margrey KA, Nicewicz DA. Acc Chem Res. 2016;49:1997–2006. doi: 10.1021/acs.accounts.6b00304. [DOI] [PubMed] [Google Scholar]
- 28.Vantourout JC, Law RP, Isidro-Llobet A, Atkinson SJ, Watson AJB. J Org Chem. 2016;81:3942–3950. doi: 10.1021/acs.joc.6b00466. [DOI] [PubMed] [Google Scholar]
- 29.Yamashita Y, Tellis JC, Molander GA. Proc Natl Acad Sci USA. 2015;112:12026–12029. doi: 10.1073/pnas.1509715112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wijtmans R, Vink SKM, Schoemaker EH, Delft LF, Blaauw HR, Rutjes TJPF. Synthesis. 2004;5:641–662. [Google Scholar]
- 31.Patel VR, Park WS. Mini-Reviews in Medicinal Chemistry. 2013;13:1579–1601. doi: 10.2174/13895575113139990073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.