

SUMMARY
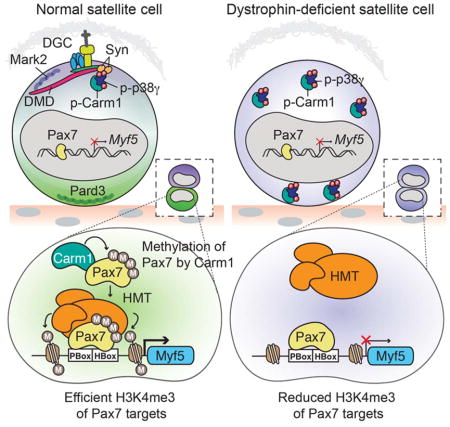
Asymmetrically dividing muscle stem cells in skeletal muscle give rise to committed cells, where the myogenic determination factor Myf5 is transcriptionally activated by Pax7. This activation is dependent on Carm1, which methylates Pax7 on multiple arginine residues, to recruit the ASH2L:MLL1/2:WDR5:RBBP5 histone methyltransferase complex to the proximal promoter of Myf5. Here we found that Carm1 is a specific substrate of p38γ/MAPK12, and that phosphorylation of Carm1 prevents its nuclear translocation. Basal localization of the p38γ/p-Carm1 complex in muscle stem cells occurs via binding to the dystrophin-glycoprotein complex (DGC) through β1-syntrophin. In dystrophin-deficient muscle stem cells undergoing asymmetric division, p38γ/β1-syntrophin interactions are abrogated resulting in enhanced Carm1 phosphorylation. The resulting progenitors exhibit reduced Carm1 binding to Pax7, reduced H3K4-methylation of chromatin, and reduced transcription of Myf5 and other Pax7 target genes. Therefore, our experiments suggest that dysregulation of p38γ/Carm1 results in altered epigenetic gene regulation in Duchenne Muscular Dystrophy.
eTOC blurb
Establishment of cell polarity by the dystrophin complex is required for muscle stem cell asymmetric divisions. Chang and colleagues identify p38γ MAPK as a critical downstream regulator of satellite stem cell commitment providing a link between dystrophin and epigenetic gene regulation to mediate asymmetric fates of daughter satellite cells.
INTRODUCTION
Satellite cells, the adult stem cells of skeletal muscle located between the basal lamina and sarcolemma of the muscle fiber (Mauro, 1961), are responsible for postnatal muscle growth and are indispensable for muscle regeneration in response to injury (Kuang et al., 2006; Lepper et al., 2011; Sambasivan et al., 2011; Seale et al., 2000; von Maltzahn et al., 2013). In addition to generating committed myogenic progenitors to repair muscle tissue after trauma, a small subset of the satellite cell population, termed satellite stem cells, retains long-term self-renewal capacity to ensure homeostatic muscle maintenance and manage muscle repair over the lifetime of the organism (Collins et al., 2005; Kuang et al., 2007; Sacco et al., 2008). For self-renewal, activated satellite stem cells can undergo either asymmetric division to generate one committed daughter cell while the other daughter cell retains stem cell characteristics, or symmetric division to generate two identical daughter stem cells to expand the stem cell pool (Gurevich et al., 2016; Kuang et al., 2007; Rocheteau et al., 2012). A balance between symmetric and asymmetric satellite stem cell division is critical for effective muscle regeneration, and modulation of this balance can have positive or detrimental consequences on muscle health (Bernet et al., 2014; Chang et al., 2016; Cosgrove et al., 2014; Dumont et al., 2015; Le Grand et al., 2009; Price et al., 2014).
Dystrophin protein, whose loss of expression underlies the etiology of Duchenne Muscular Dystrophy (DMD), was previously thought to primarily contribute to muscle fiber stability as an essential component of the large oligomeric dystrophin glycoprotein complex (DGC) at the muscle fiber membrane (Campbell and Kahl, 1989). In addition to its structural role in myofiber stability, dystrophin is expressed in satellite cells where it plays an essential role in regulating the establishment of satellite cell polarity and hence efficient asymmetric division (Chang et al., 2016; Dumont et al., 2015). Upon satellite cell activation, polarized distribution of dystrophin restricts localization of the polarity effector kinase Mark2 (also known as Par1b) to the same surface, which drives Pard3 to the opposite side of the cell (Dumont et al., 2015). The segregation of Mark2 and Pard3 to opposite ends of the cell is a well-conserved process for the establishment of polarity cues prior to asymmetric cell division (Neumuller and Knoblich, 2009). In dystrophin-deficient mdx mice, a mouse model for DMD, reduced numbers of asymmetric divisions strongly diminish the generation of myogenic progenitors needed for proper muscle regeneration (Dumont et al., 2015). However, the molecular effectors that control asymmetric satellite cell fate decisions downstream of polarity establishment have remained elusive.
Satellite cells express the paired box transcription factor Pax7, a master transcriptional regulator of genes required for myogenic commitment and myogenic progenitor proliferation (Seale et al., 2000; Soleimani et al., 2012). Commitment of satellite stem cells to the muscle lineage requires upregulation of the basic helix-loop-helix (bHLH) myogenic regulatory transcription factors Myf5 and MyoD (Kuang et al., 2007; Rudnicki et al., 1993; Troy et al., 2012). The majority of satellite cells have expressed Myf5 and are effectively primed for myogenic differentiation, however, a subpopulation of satellite cells (around 10%) have never expressed Myf5, representing a reservoir of genuine satellite stem cells (Crist et al., 2012; Kuang et al., 2007). Importantly, these satellite stem cells that are negative for Myf5 expression are capable of giving rise to committed Myf5 expressing satellite cells through asymmetric cell divisions and are superior in their ability to engraft into the satellite cell niche upon transplantation into muscle (Kuang et al., 2007).
The arginine methyltransferase Carm1 regulates epigenetic induction of Myf5 expression during asymmetric satellite stem cell division (Kawabe et al., 2012). Carm1 specifically methylates Pax7 at multiple arginine residues in the N-terminus of Pax7, facilitating the recruitment of the ASH2L:MLL1/2:WDR5:RBBP5 histone H3 lysine 4 (H3K4) methyltransferase complex to the proximal promoter of Myf5 resulting in permissive H3K4 tri-methylation (H3K4me3) of the surrounding chromatin (Kawabe et al., 2012; McKinnell et al., 2008). Transcription of Myf5 is thus activated in the committed daughter cell following an asymmetric division. Depletion of Carm1 in satellite cell-derived primary myoblasts results in reduced levels of H3K4me3 at Myf5 and deficient Myf5 gene expression (Kawabe et al., 2012), thus highlighting the importance of Carm1 in regulating this switch into the myogenic program. The differential regulation of Carm1 activity in the stem and committed daughter cells through an asymmetric division, however, has yet to be elucidated.
Here, we identify a role for the mitogen-activated protein kinase (MAPK) p38γ/MAPK12 as a critical regulator of satellite stem cell fate through phosphorylation of Carm1. Previous work has described a role for p38γ in preventing premature and precocious differentiation of myogenic progenitors (Gillespie et al., 2009). Intriguingly, we find that in satellite stem cells, p38γ functions oppose the activity of p38α, and stimulates self-renewal rather than differentiation. p38γ is regulated by its interaction with the DGC, via binding to β1-syntrophin, during asymmetric satellite stem cell divisions, which leads to polarized restriction of Carm1 phosphorylation within the stem cell. Importantly, in dystrophin-deficient satellite cells from mdx mice, p38γ-mediated regulation of Carm1 is impaired. Thus, we have defined an epigenetic regulatory pathway that mediates commitment during asymmetric division acting downstream of the DGC and polarity establishment. We suggest that these signaling pathways represent an additional deficit owing to the lack of dystrophin expression in satellite cells, thus impairing the function of satellite cells in DMD.
RESULTS
Carm1 is Directly Phosphorylated by p38γ MAPK
To identify regulators of Carm1 during asymmetric satellite stem cell division, we screened over 450 kinases for the ability to phosphorylate Carm1 and identified one kinase, p38γ MAPK as a Carm1 regulatory kinase. In vitro kinase assays combining Carm1 with active p38γ kinase demonstrated that p38γ directly phosphorylates Carm1 (Figures 1A and S1A). Phosphorylation of Carm1 was undetected in kinase assays with activated p38α MAPK, thus establishing Carm1 as a specific substrate for the p38γ isoform. A kinase-inactive mutant of p38γ (p38γ AF), containing amino acid substitutions at the kinase activation motif (T183A, Y185F), failed to phosphorylate Carm1, confirming that phosphorylation requires the canonical activation of p38γ (Figures 1B and S1B). However, co-immunoprecipitation experiments between Carm1 and both wild type p38γ and p38γ AF demonstrated that p38γ kinase activity is not required for binding with Carm1 (Figure 1C).
Figure 1. Carm1 is Directly Phosphorylated by p38γ MAPK.

(A) In vitro kinase assays between GST-Carm1 with either FLAG-p38γ or FLAG-p38α kinases. p38 kinases were activated by co-expression of MKK6EE, as indicated. The asterisk denotes phosphorylated Carm1. Arrowheads denote p38γ (γ) and p38α (α) kinases.
(B) In vitro kinase assays between GST-Carm1 with either wild type Myc-p38γ or kinase-inactive Myc-p38γ AF. The asterisk denotes phosphorylated Carm1 and the arrowhead denotes active Myc-p38γ kinase.
(C) HEK 293T cells were transfected with FLAG-Carm1 and either Myc-p38γ, Myc-p38γ AF, or pcDNA3. Immunoprecipitation (IP) and immunoblotting were performed with the indicated antibodies.
(D) Carm1:p38γ PLA (red) performed on satellite cells cultured on myofibers. Satellite cells are marked by expression of Pax7 (green) and nuclei were counterstained with DAPI (blue). Scale bar represents 20 μm.
See also Figure S1.
To confirm that endogenous p38γ and Carm1 interact in satellite cells, we performed a proximity ligation assay (PLA) on isolated satellite cells associated with their myofiber either immediately after isolation (time 0) or after culturing in growth conditions for 42h, a time point by which most satellite cells will have undergone their first mitosis. PLA detection of p38γ and Carm1 generated a very prominent signal at the cell periphery of activated satellite cells (42h), while no signal was detected in quiescent satellite cells (0h) (Figures 1D and S1C). No PLA signal was detected in satellite cells transfected with p38γ or Carm1 siRNA, thus confirming the specificity of the assay (Figure S1D).
p38γ MAPK Phosphorylates Carm1 on S572
To identify the site of Carm1 phosphorylation by p38γ, we employed liquid chromatography-tandem mass spectrometry (LC-MS/MS) to analyze Carm1 protein treated with activated p38γ kinase (Figure 2A). A phosphopeptide was identified with a single phosphorylation at serine 572 (S572 in Carm1 isoform 2, S595 in Carm1 isoform 1) with 95% certainty (Figure 2B and Tables S1 and S2). S572 is located at the C-terminus of Carm1 (Figure 2C) and is a novel phosphorylation site that is highly conserved between vertebrate species (Figure 2D). This site is within a conserved p38 MAPK phosphorylation motif, which consists of a serine or threonine followed by a proline (S/T-P) (Clark-Lewis et al., 1991). We performed site-directed mutagenesis to substitute S572 with an alanine (S572A) to prevent phosphorylation at this site. p38γ was unable to phosphorylate mutant Carm1 S572A (Figures 2E and S2A), thus confirming that p38γ phosphorylates Carm1 specifically on S572.
Figure 2. p38γ MAPK Phosphorylates Carm1 on Ser 572.

(A) Coomassie blue staining of Carm1 treated with activated p38γ (+MKK6EE) or non-activated p38γ (+pcDNA3) for LC-MS/MS analysis. Asterisks denote Carm1 and arrowheads denote p38γ kinase.
(B) MS/MS spectrum of Carm1 peptide containing phosphorylated S572. Matched b and y ions and the position of oxidations (OX) and phosphorylations (PH) are indicated.
(C) Diagram of Carm1 ΔE15 protein with indicated phosphorylation site at S572.
(D) Multiple sequence alignment of the last 35 amino acids of Carm1 in 5 different vertebrate species. S572 is marked in red. An asterisk (*) represents identical amino acids, a colon (:) represents highly conserved amino acids, and a period (.) represents weakly conserved amino acids.
(E) In vitro kinase assays between active p38γ and wild type or S572A mutant Carm1. The asterisk denotes phosphorylated Carm1 and the arrowhead denotes active p38γ kinase.
(F) Carm1:p-S/T-P PLA (red) performed on satellite cells cultured on myofibers isolated from p38γ+/+ or p38γfl/fl mice. Satellite cells are marked by expression of Syndecan-4 (Sdc4, green) and nuclei were counterstained with DAPI (blue). Scale bar represents 10 μm.
See also Figure S2 and Tables S1 and S2.
To examine the phosphorylation status of Carm1 in satellite cells, we conducted PLA with antibodies recognizing Carm1 and the phosphorylated S/T-P motif (p-S/T-P). Isotype controls were used to confirm PLA specificity (Figure S2B). PLA was performed on cultured myofibers isolated from wild type and satellite cell specific p38γ-deleted (Pax7CreER/+:p38γfl/fl) mice. A PLA signal was detected in satellite cells from p38γ+/+ myofibers, strongly suggesting that Carm1 is phosphorylated on a S/T-P motif (Figures 2F, S2C and D). Importantly, no signal was detected in p38γfl/fl myofibers (Figure 2F), indicating that Carm1 phosphorylation at this motif is p38γ-dependent.
Immunoprecipitation with the anti-p-S/T-P antibody and immunoblot analysis with anti-Carm1 antibody provided further evidence that Carm1 is phosphorylated in satellite cell-derived primary myoblasts (Figure S2E). Additionally, we transfected Carm1 null primary myoblasts with either wild type Carm1 or Carm1 S572A, and subsequently performed the Carm1:p-S/T-P PLA. We observed a very robust cytoplasmic PLA signal in wild type Carm1-transfected cells and a very dim signal in a minor population of Carm1 S572A-transfected cells (Figures S2F and G). Thus, while we cannot exclude the possibility that the Carm1:p-S/T-P PLA signal is the result of Carm1 interacting with another phosphorylated protein, our results suggest that the majority of the signal is from phosphorylated Carm1 at this motif.
Carm1 Isoform 2 (Carm1 ΔE15) is Responsible for Pax7 Methylation
Carm1 protein exists as two isoforms in humans and mice (Figure 3A). Full length isoform 1 is comprised of 16 exons (referred to as Carm1 FL), while exclusion of exon 15 due to alternative splicing gives rise to isoform 2 (Carm1 ΔE15). In most tissues, isoform 2 is the predominant isoform (Wang et al., 2013), and we have utilized Carm1 ΔE15 in our past and present studies (Kawabe et al., 2012). S572 of Carm1 is encoded by exon 16, and is available within both isoforms for phosphorylation by p38γ. We subjected Carm1 ΔE15 and FL, as well as their respective S572/595A mutants, to in vitro kinase assays with activated p38γ. Both Carm1 isoforms were phosphorylated by p38γ whereas both mutants were not (Figure S3A).
Figure 3. Phosphorylation of Carm1 by p38γ Restricts Carm1 Subcellular Localization to the Cytoplasm.

(A) Schematic of Carm1 FL (isoform 1) and Carm1 ΔE15 (isoform 2).
(B) In vitro methylation assays with GST-Carm1 FL or GST-Carm1 ΔE15 with Pax7 or core histone proteins.
(C) Densitometric quantification of (B), represented as the mean (n = 3 replicates) ± SEM (*p ≤ 0.05).
(D) Immunostaining of Carm1-null primary myoblasts transfected with either wild type, S572A, or S572E Carm1, or pcDNA3. Cells were immunostained with α-tubulin (green), Carm1 (red), and nuclei were counterstained with DAPI (blue). Scale bar represents 10 μm.
(E) Quantification of the mean fluorescence intensity of Carm1 expression in the nucleus and cytoplasm from the immunostaining performed in (D) and expressed as a nucleus:cytoplasm expression ratio, represented as the mean (n = 11 WT, n = 13 S572A, n = 13 S572E cells) ± SEM (**p ≤ 0.01. NS, not significant).
See also Figure S3.
We subsequently compared the methyltransferase activities of Carm1 FL and ΔE15 using Pax7 and core histone proteins as substrates, and found that while both isoforms methylated histone H3 to the same extent, Pax7 was clearly a preferred substrate for Carm1 ΔE15 (Figures 3B and C and S3B). Based on these results, we conclude that Carm1 ΔE15 is the specific isoform responsible for Pax7 methylation.
p38γ Regulates the Subcellular Localization of Carm1 ΔE15
To determine how phosphorylation by p38γ regulates Carm1 ΔE15 function, we utilized the Carm1 S572A mutant and also generated an S572E mutant to mimic constitutive phosphorylation. In vitro methylation assays revealed that both Carm1 S572A and S572E mutants were able to methylate Pax7 and histone H3 to the same level as that of wild type (Figure S3C), demonstrating that phosphorylation does not affect Carm1 methyltransferase activity.
We examined the subcellular localization of the Carm1 phosphorylation mutants by expressing wild type Carm1 ΔE15, S572A or S572E mutants in Carm1 null primary myoblasts. Immunostaining for Carm1 protein revealed that cells transfected with the empty vector were negative for Carm1 expression, while wild type and S572A Carm1 protein localized mainly to the nucleus with weaker expression in the cytoplasm (Figure 3D). Interestingly, many of the cells expressing Carm1 S572E displayed predominantly cytoplasmic Carm1 localization. Determination of the ratio of average Carm1 fluorescence intensities between the nuclear and cytoplasmic subcellular compartments revealed that cells expressing the S572E mutant exhibited a significantly lower nucleus to cytoplasm expression ratio compared to wild type Carm1 (Figure 3E). Thus, phosphorylation of Carm1 by p38γ acts to enrich Carm1 subcellular localization to the cytoplasm and reduce its nuclear translocation.
Additionally, we employed subcellular fractionation to examine endogenous Carm1 localization in satellite cell-derived primary myoblasts derived from control Pax7CreER/+ and Pax7CreER/+:p38γfl/fl mice (Figure S3D). Densitometric analysis of Carm1 expression levels revealed that Pax7CreER/+:p38γfl/fl cells exhibited higher Carm1 nuclear to cytoplasm expression ratios (Figure S3E). Thus, consistent with our model, in p38γ-deleted primary myoblasts, Carm1 subcellular localization is preferentially nuclear compared to wild type control cells.
We previously demonstrated that Carm1 directly binds Pax7 in the nucleus of activated satellite cells (Kawabe et al., 2012). To examine the direct effects of p38γ on the function of Carm1 ΔE15, we performed Carm1:Pax7 PLA analysis on satellite cells cultured for 42h on myofibers treated with control, p38γ or Carm1 siRNA (Figure 4A). Enhanced Carm1 and Pax7 interactions were observed in satellite cells treated with p38γ siRNA compared to control, and interactions were abrogated in Carm1-depleted satellite cells (Figures 4B and C, and S4F). Taken together, these data provide compelling evidence that p38γ negatively regulates Carm1 ΔE15 translocation to the nucleus, where Carm1 binds and methylates Pax7 to epigenetically activate transcription of Myf5, thus inducing myogenic commitment of satellite stem cells.
Figure 4. p38γ Negatively Regulates Asymmetric Satellite Stem Cell Division and is Required for Symmetric Self-Renewal.

(A) Schematic of siRNA-treated myofiber culture experiments performed in Figure 4. (EDL, extensor digitorum longus. PLA, proximity ligation assay. IF, immunofluorescence).
(B) Carm1:Pax7 PLA (red) performed on satellite cells cultured for 42h on myofibers that were treated with indicated siRNAs. Satellite cells are marked by expression of Sdc4 (green) and nuclei were counterstained with DAPI (blue). Scale bar represents 10 μm.
(C) Quantification of PLA signal from (B), represented as the mean (n = 39 si-Ctrl, n = 54 si-p38γ, n = 47 siCarm1 cells from 3 mice) ± SEM (*p ≤ 0.05). The PLA was quantified by counting the number of nuclear PLA puncta for each satellite cell.
(D) Immunofluorescence of satellite cells cultured for 42h on myofibers that were treated with either control (si-Ctrl) or p38γ (si-p38γ) siRNA. Cells were immunostained for Sdc4 (green), and Myf5 (red). Nuclei were counterstained with DAPI (blue). Scale bar represents 10 μm.
(E) Quantification of mean fluorescence intensity of Myf5 staining from (D), represented as the mean (n = 67 si-Ctrl, n = 69 si-p38γ cells from 3 mice) ± SEM (***p ≤ 0.001).
(F) Quantification of the number of asymmetric satellite stem cell (Pax7+/YFP−) divisions per myofiber, represented as the mean (n = 9 mice) ± SEM (**p ≤ 0.01. NS, not significant).
(G) Quantification of the number of symmetric satellite stem cell (Pax7+/YFP−) divisions per myofiber, represented as the mean (n = 9 mice) ± SEM (*p ≤ 0.05. NS, not significant).
(H) Quantification of the number of committed satellite progenitor cell (Pax7+/YFP+) divisions per myofiber, represented as the mean (n = 9 mice) ± SEM (NS, not significant).
See also Figure S4.
Symmetric Satellite Cell Self-Renewal Requires p38γ
Enhanced Carm:Pax7 interactions in satellite cells depleted of p38γ would suggest that Myf5 expression levels are also augmented in these cells. To test this hypothesis, p38γ and control siRNA-treated satellite cells (Figure 4A) were immunostained with anti-Myf5 antibody (Figure S4G). Consequently, the average mean fluorescence intensity of Myf5 was significantly increased in si-p38γ treated satellite cells compared to control (Figure 4D and E). These data together suggest that p38γ is negatively regulating Carm1 and its ability to initiate commitment into the myogenic program.
To further elucidate a role for p38γ in regulating satellite stem cell fate, we treated myofibers from Myf5-Cre/R26R-EYFP mice (Kuang et al., 2007) with control, p38γ or p38α siRNA. Efficient knock-down and specificity of p38 isoform-specific siRNAs were confirmed by immunostaining and immunoblotting (Figures S4A, B and C). Employing Cre-mediated lineage tracing with Myf5-Cre and R26R-EYFP alleles allow for the discrimination of satellite stem cells that have never expressed Myf5, which remain negative for YFP expression (YFP−, < 10% of total satellite cells), and committed satellite cells that express YFP (Kuang et al., 2007). Upon culturing the siRNA-treated myofibers for 42h (Figure 4A), we found a significant increase in the number of asymmetric satellite stem cell (YFP−) divisions per myofiber when treated with p38γ siRNA (Figure 4F), which is consistent with the observation that Pax7:Carm1 interactions are enhanced upon p38γ depletion. Accordingly, we observed a significant diminution in the number of symmetric satellite stem cell (YFP−) divisions in p38γ siRNA-treated fibers (Figure 4G), suggesting that p38γ is required for symmetric stem cell maintenance. Thus, loss of p38γ greatly skewed the ratio of asymmetric to symmetric satellite stem cell divisions to favor asymmetric divisions and myogenic commitment at the expense of self-renewal (Figures S4D and E). No effect on committed satellite cell (YFP+) divisions were observed (Figure 4H). Notably, the p38γ-mediated alterations observed in satellite stem cell divisions is only attributable to the p38γ isoform, as we observed no significant difference in p38α-depleted myofibers (Figures 4F and G, S4D and E). All together, these results demonstrate that p38γ promotes symmetric self-renewal of satellite stem cells and negatively regulates myogenic commitment.
p38γ is Required for Satellite Cell Maintenance During Muscle Regeneration
To study the in vivo contribution of p38γ specifically in satellite cells, we generated mice allowing tamoxifen (TMX)-inducible genetic deletion of p38γ in Pax7-expressing cells (Figure S5A). TMX was administered to Pax7+/+:p38γfl/fl (wild type for p38γ expression, designated as p38γ+/+), Pax7CreER/+:p38γfl/+ (p38γfl/+) and Pax7CreER/+:p38γfl/fl (p38γfl/fl) mice (Figure 5A), and the efficiency of p38γ deletion was determined by both gene and protein expression analyses (Figures S5B and C).
Figure 5. Satellite Cell-Specific Genetic Deletion of p38γ Impairs Muscle Regeneration.

(A) Description of mice and schematic of muscle regeneration experiment performed in Figure 5 (CTX, cardiotoxin).
(B) Representative H&E stained sections of CTX-injured TA muscle from mice as described in (A). Scale bar represents 50 μm.
(C) Quantification of Pax7+ satellite cells normalized to TA cross-sectional area, represented as the mean (n = 4 p38γ+/+, n = 3 p38γfl/+, n = 3 p38γfl/fl mice) ± SEM (*p ≤ 0.05, **p ≤ 0.01).
(D) Ratio of the weight of injured TA muscle to total body weight, represented as the mean ± SEM (*p ≤ 0.05, **p ≤ 0.01).
See also Figure S5.
Based on our data showing a clear dependence on p38γ within the self-renewing satellite stem cell subpopulation (which make up approximately 10% of the satellite cell population), we performed a multiple muscle injury experiment in order to assess long term effects on satellite stem cell function upon challenging the muscle to repetitive regeneration. Thus following TMX administration, mice were subjected to 3 consecutive cardiotoxin (CTX)-induced injuries directly to the tibialis anterior (TA) muscle with 21 days between injuries to allow the muscle to regenerate. To prevent an overtake of satellite cells that have escaped TMX-induced deletion of p38γ, the mice were maintained on a TMX-based diet for the duration of the experiment (Figure 5A). Hematoxylin and eosin (H&E) staining of regenerated TA muscle cross sections revealed no gross histological differences between p38γ+/+, p38γfl/+ and p38γfl/fl mice (Figure 5B). However, enumeration of Pax7+ cells revealed a significant reduction in satellite cell numbers in p38γfl/fl regenerated muscles compared to both p38γfl/+ and p38γ+/+ (Figures 5C and S5D). Accordingly, upon examination of the weight of the injured TA muscle we found a significant reduction in the weight of the regenerated muscle in both p38γfl/+ and p38γfl/fl mice as compared to p38γ+/+ (Figure 5D). Despite the reduction in muscle mass, we observed increased numbers of myofibers in p38γfl/+ and p38γfl/fl regenerated muscles (Figure S5E). Consistent with this, a significant decrease in the percentage of large myofibers (minimum fiber Feret of 40–70 μm) and a significant shift towards smaller myofiber Feret was observed in p38γfl/fl fibers compared to p38γ+/+ (Figures S5F and G). The reduction in fiber size is consistent with the role of p38γ in myogenic progenitors, where it is required to prevent premature expression of Myogenin and precocious differentiation (Gillespie et al., 2009). Thus, satellite cell-specific deletion of p38γ results in a depletion of the satellite cell pool following multiple rounds of muscle regeneration and impairs the regeneration program, resulting in reduced muscle weight and increased numbers of small myofibers.
p38γ/Carm1 Interacts with the Dystrophin Glycoprotein Complex via β1-Syntrophin
In differentiated skeletal muscle tissue, p38γ has been reported to interact with the DGC via interaction with α1-syntrophin (Hasegawa et al., 1999). We therefore asked if p38γ in satellite cells could interact with syntrophin and be subject to regulation by the DGC complex. Gene expression analysis of satellite cells revealed little to no expression of α1-syntrophin and high expression of β1-syntrophin (Dumont et al., 2015), which was confirmed by immunofluorescence (Figure S6A). Immunostaining also indicated that p38γ co-localizes with β1-syntrophin and α7 integrin (Figures 6A, S6B and C), which localizes to the basal surface with the DGC in asymmetrically dividing satellite cells (Dumont et al., 2015).
Figure 6. p38γ/Carm1 interacts with β1-Syntrophin in Satellite Cells.

(A) Immunofluorescence of satellite cells cultured for 36h on myofibers isolated from wild type (WT) or mdx mice. Cells were immunostained for α7 integrin (white), β1-syntrophin (green), and p38γ (red). Nuclei were counterstained with Hoechst (blue). Scale bar represents 10 μm.
(B, C, and D) PLA with indicated antibodies (red) performed on satellite cells cultured for 36h on single EDL myofibers. Satellite cells were identified by their expression of α7 integrin (white) and nuclei were counterstained with DAPI (blue). Scale bar represents 10 μm.
(E, and F) PLA with indicated antibodies (red) performed on satellite cells cultured for 36h on single myofibers isolated from either WT or mdx mice. Satellite cells were identified by their expression of Sdc4 (green) and nuclei were counterstained with DAPI (blue). Scale bar represents 10 μm.
(G) Quantification of PLA in (F), represented as the relative mean (n = 21 WT, n = 26 mdx cells from 3 mice) ± SEM (**p ≤ 0.01). The PLA was quantified by measuring mean fluorescence intensity of the PLA signal for each satellite cell.
See also Figures S6 and S7.
To assess if p38γ physically interacts with β1-syntrophin in satellite cells, PLA was performed on myofibers cultured for 36h, a time point by which satellite cells have entered the cell cycle. In satellite cells, we detected p38γ:β1-syntrophin PLA signals that co-localized with α7 integrin (Figure 6B). We also noted that p38γ and β1-syntrophin interactions were polarized in a subset of satellite cells (Figures 6E and S7A), suggesting that during an asymmetric cell division, p38γ localization would be basally restricted by the DGC complex via its interaction with β1-syntrophin. Co-immunoprecipitation experiments confirmed that β1-syntrophin interacts directly with p38γ (Figure S6E).
PLA for Carm1 and β1-syntrophin was performed on satellite cells cultured on myofibers to determine if Carm1 is also present in the p38γ/β1-syntrophin complex. While most satellite cells exhibited a Carm1:β1-syntrophin PLA signal that localized to the cell periphery, a subset of satellite cells (< 5%) exhibited a polarized signal, which also co-localized with α7 integrin (Figure 6C). Carm1 and β1-syntrophin protein interactions were also validated by co-immunoprecipitation experiments (Figure S6F). Additionally, PLA with Carm1 and p-S/T-P antibodies demonstrated that phospho-Carm1 PLA signals also exhibited a polarized distribution that co-localized with α7 integrin (Figure 6D), demonstrating that Carm1 is phosphorylated within this complex. Taken together, these results strongly suggest that β1-syntrophin restricts p38γ localization during asymmetric satellite stem cell division to phosphorylate Carm1 and prevent Carm1 nuclear translocation in the daughter cell that will remain a stem cell.
Impaired Epigenetic Activation of Myf5 in Dystrophin-Deficient Satellite Cells
To examine the functional consequences of dystrophin loss on p38γ regulation by the DGC, we performed p38γ:β1-syntrophin PLA on myofibers isolated from wild type and mdx mice. Mdx mice contain a spontaneous mutation that generates a premature stop codon within exon 23 of the dystrophin gene, thus rendering mdx mice essentially void of dystrophin expression (Bulfield et al., 1984). β1-syntrophin protein expression is reduced in mdx myofibers (De Arcangelis et al., 2010), however, it remained expressed in mdx satellite cells, where it did not appear to co-localize with p38γ (Figures 6A, S6B and D). Strikingly, p38γ:β1-syntrophin PLA interactions were completely absent in mdx satellite cells (Figures 6E and S7A).
To determine the consequences of this loss on Carm1 phosphorylation, we performed Carm1:p-S/T-P PLA on wild type and mdx satellite cells. We observed enhanced PLA signal intensity and frequency in mdx satellite cells (Figures 6F and G, and S7B and D). Enhanced Carm1 phosphorylation would predict an inhibition in Carm1 translocation to the nucleus. Thus, PLA was used to assess Carm1/Pax7 binding in wild type and mdx satellite cells. While the majority of wild type satellite cells exhibited a Carm1:Pax7 PLA signal, around half of mdx satellite cells exhibited little or no signal (Figures 7A and B, and S7C and E).
Figure 7. Carm1-Mediated Epigenetic Regulation of Myf5 Expression is Altered in mdx Cells.
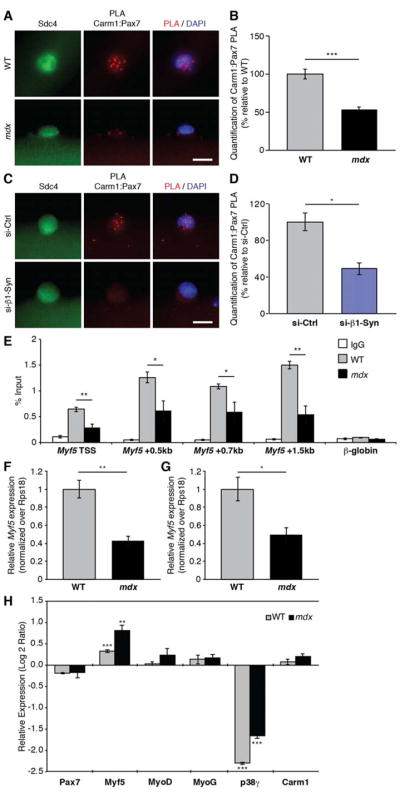
(A) Carm1:Pax7 PLA (red) performed on satellite cells cultured for 36h on single myofibers isolated from either wild type (WT) or mdx mice. Satellite cells were identified by their expression of Sdc4 (green) and nuclei were counterstained with DAPI (blue). Scale bar represents 10 μm.
(B) Quantification of PLA in (A), represented as the relative mean (n = 81 WT, n = 137 mdx cells from 3 mice) ± SEM (***p ≤ 0.001). The PLA was quantified by counting the number of nuclear PLA puncta for each satellite cell.
(C) Carm1:Pax7 PLA (red) performed on satellite cells cultured for 36h on myofibers that were treated with either control or β1-syntrophin siRNA. Satellite cells are marked by expression of Sdc4 (green) and nuclei were counterstained with DAPI (blue). Scale bar represents 10 μm.
(D) Quantification of PLA in (C), represented as the relative mean (n = 53 si-Ctrl, n = 73 si-SNTB1 cells from 3 mice) ± SEM (*p ≤ 0.05). The PLA was quantified by counting the number of nuclear PLA puncta for each satellite cell.
(E) Chromatin immunoprecipitation (ChIP)-qPCR analysis of H3K4me3 at Myf5 in WT and mdx primary myoblasts represented as the mean (n = 4 WT, n = 4 mdx mice) ± SEM (*p ≤ 0.05, **p ≤ 0.01).
(F) RT-qPCR analysis of Myf5 expression in freshly sorted satellite cells from WT and mdx mice represented as the mean (n = 3 WT, n = 4 mdx mice) ± SEM (**p ≤ 0.01).
(G) RT-qPCR analysis of Myf5 expression in WT and mdx primary myoblasts represented as the mean (n = 11 WT, n = 4 mdx mice) ± SEM (*p ≤ 0.05).
(H) RT-qPCR analysis of indicated genes in satellite cell-derived primary myoblasts isolated from WT and mdx mice that were treated with control and p38γ siRNA. Fold changes are shown relative to control siRNA-treated cells on a log base 2 axis and are represented as the mean (n = 3 replicates from 3 mice) ± SEM (**p ≤ 0.01, ***p ≤ 0.001).
See also Figure S7.
To demonstrate that β1-syntrophin directly mediates the ability of p38γ to regulate Carm1 interactions with Pax7, PLA for Pax7 and Carm1 were performed on satellite cells cultured on wild type EDL myofibers treated with siRNA against β1-syntrophin. Comparable to the 50% decrease in Pax7:Carm1 PLA signal observed in mdx satellite cells (Figure 7B), we similarly found a 50% reduction in Pax7:Carm1 PLA in satellite cells treated with si-β1-syntrophin compared to si-Ctrl treated cells (Figures 7C and D, and S7F). This finding strongly suggests that the ability of Carm1 to interact with Pax7 and promote myogenic commitment is directly controlled by β1-syntrophin and that defects in Carm1/Pax7 interactions in dystrophic satellite cells are a direct result of the loss of a functional DGC complex.
Carm1/Pax7 interactions result in methylation of Pax7 and recruitment of the ASH2L:MLL trithorax complex to the Myf5 promoter resulting in methylation of H3K4 to induce Myf5 expression (Kawabe et al., 2012). Notably, primary myoblasts lacking Carm1 exhibit reduced H3K4me3 over the Myf5 promoter and reduced levels of Myf5 mRNA (Kawabe et al., 2012). Thus, we compared levels of the H3K4me3 histone mark at the Myf5 locus in satellite cell-derived primary myoblasts isolated from wild type and mdx mice. A striking decrease in H3K4me3 at the transcriptional start site (TSS), +0.5kb, +0.7kb and +1.5kb regions of Myf5 were observed in mdx compared to wild type cells (Figure 7E). Accordingly, Myf5 mRNA was significantly reduced in both prospectively isolated mdx satellite cells (Figure 7F) and mdx satellite cell-derived primary myoblasts (Figure 7G) compared to wild type cells.
To demonstrate a direct relationship between p38γ and transcription of Myf5, we performed siRNA knockdown of p38γ in freshly isolated satellite cell-derived primary myoblasts isolated from both wild type and mdx mice followed by gene expression analysis of Myf5 transcript levels. In order to prevent the effects of precocious differentiation mediated by loss of p38γ and the consequent downregulation of Myf5 transcription (Gillespie et al., 2009), we cultured cells in the presence of the eIF2α phosphatase inhibitor, Sal003 (Zismanov et al., 2016). In wild type cells, knock-down of p38γ (downregulation of 4.9-fold) in the presence of Sal003 resulted in a modest (1.2-fold compared to si-Ctrl treated cells), but significant, increase in Myf5 mRNA levels (Figure 7H). Importantly, in mdx cells, where Myf5 expression is lower compared to wild type cells (Figures 7F and G), we observed a striking 1.8-fold increase in Myf5 when p38γ expression is diminished (downregulation of 3.2-fold) (Figure 7H). In established wild type primary myoblast cultures, where Myf5 expression is already epigenetically set, we did not observe any significant differences in H3K4me3 levels at Myf5 (Figure S7G). As well, in activated satellite cells isolated from Pax7-CreER/+ and Pax7-CreER/+:p38γfl/fl mice following CTX-injury, which are a heterogenous population of satellite stem cells and committed myogenic progenitors, we did not observe differences in Myf5 expression levels (Figure S7H).
To further investigate the altered Carm1/Pax7-mediated epigenetic regulation in dystrophin-deficient satellite cells, we also interrogated expression levels of other Pax7 target genes. We found that in mdx satellite cells, a subset of Pax7 target genes (Soleimani et al., 2012) exhibited differential expression when compared to wild type cells (Figure S7I), suggesting that altered Pax7-mediated epigenetic regulation may extend to Pax7 target genes other than Myf5 and this may also contribute to satellite cell dysfunction in dystrophin-deficient satellite cells. All together, these findings provide compelling evidence that in the absence of dystrophin, syntrophin-mediated p38γ regulation is dysregulated resulting in a reduction in Carm1-dependent methylation of Pax7 and compromised epigenetic activation of Myf5 during satellite cell division.
DISCUSSION
The basal location of dystrophin and the polarity effector Mark2 set up the apical localization of Pard3 to establish stem cell polarity and facilitate the subsequent asymmetric division of satellite stem cells (Dumont et al., 2015). In this study, we place the p38γ MAPK signaling pathway downstream of the establishment of dystrophin-mediated cell polarity and demonstrate an important role of p38γ as an important determinant of satellite cell fate by regulating the nuclear localization of the epigenetic regulator Carm1.
Carm1 is ubiquitously expressed and has pleiotropic roles in many cellular processes including signal transduction, transcriptional regulation and mRNA splicing. Alternative splicing of Carm1 may provide a level of specificity with respect to its functions and it has been suggested that Carm1 isoforms differ in their substrate specificities (Wang et al., 2013). Here, we describe the first example of an isoform-specific role for Carm1 isoform 2 in the methylation of Pax7, a master transcriptional regulator of satellite cell function (Figure 3A). Moreover, we show that Carm1 translocation to the nucleus is prevented by phosphorylation on S572 by p38γ MAPK (Figure 3C). Of note, p38γ is the first Carm1 regulatory kinase identified that affects Carm1 subcellular localization. It was previously suggested that Carm1 localization could be regulated via phosphorylation at S217, however the biological relevance of this phosphorylation site and the kinase responsible remain to be determined (Feng et al., 2009).
In dystrophin-deficient satellite cells, Carm1 is hyper-phosphorylated, which retains Carm1 in the cytoplasm and prevents nuclear translocation (Figure 6F). In satellite cells and committed myoblasts lacking dystrophin, we observe reduced ability of Pax7 to recruit the trithorax complex to target genes such as Myf5, reduced levels of H3K4me3, and reduced transcription of Myf5 (Figures 7E–G). In accordance with the reduced frequency of asymmetric cell divisions observed in mdx satellite cells (Dumont et al., 2015), we observe around a 50% reduction in Pax7:Carm1 interactions (Figure 7B) and Myf5 mRNA levels in mdx satellite cells (Figure 7F). Thus, dystrophin-deficient satellite cells are capable of undergoing asymmetric divisions and generating committed progenitors, albeit at a much reduced rate. These observations suggest that perhaps the p38γ/Carm1 axis functions inefficiently in dystrophin-deficient satellite cells, or likely that other mechanisms that regulate muscle stem cell commitment are involved. Interestingly, consistent with reduced Myf5 expression, mdx myogenic progenitors display abnormal differentiation (Yablonka-Reuveni and Anderson, 2006). These findings indicate that dystrophin deficiency results in epigenetic alterations that perturb the expression of key regulators of the myogenic program. It will be interesting to fully document the epigenetic landscape in myogenic cells derived from dystrophin-deficient satellite cells in future experiments.
Of note, upon genetic depletion of p38γ in satellite cells in vivo, we did not observe an increase in H3K4me3 at the Myf5 locus (Figure S7G) or enhanced expression of Myf5 in satellite cells after injury (Figure S7H). As evidenced by the modest increase in Myf5 after knock-down of p38γ in wild type primary myoblasts (Figure 7H), we conclude that the majority of satellite cells (around 90% of the satellite cell population) are already epigenetically set for expression of Myf5, thus priming satellite cells for commitment into the myogenic program following their activation (Crist et al., 2012; Kuang et al., 2007). Thus, deletion of p38γ does not further enhance Myf5 levels or methylation in wild type satellite cells. In addition, previous studies have shown that p38γ-null satellite cells precociously activate Myogenin and differentiate, as well as downregulate Myf5 (Gillespie et al., 2009). Thus, genetic removal of p38γ in committed satellite cells results in precocious differentiation and downregulation of Myf5 thereby masking any increase in Myf5 expression within the satellite stem cell population. Addition of the eIF2α phosphatase inhibitor Sal003 during the culture of satellite cell-derived wild type and mdx myoblasts served to maintain cells in a more satellite cell-like state and prevented the translation of transcripts that would normally initiate differentiation (Zismanov et al., 2016). In the presence of Sal003, we observed significantly enhanced Myf5 transcript levels when p38γ was silenced (Figure 7H). Notably, the more pronounced augmentation of Myf5 mRNA in the dystrophin-deficient context, supports our view that in cells where Myf5 levels are not saturated, such as in mdx cells, removal of p38γ in the presence of Sal003 has the ability to rescue expression of Myf5.
Carm1 has recently been implicated in the regulation of autophagy, where it functions as a nuclear transcriptional co-activator to induce expression of genes required for autophagy and lysosome function (Shin et al., 2016). Intriguingly, studies have demonstrated that autophagy is impaired in dystrophic muscle of mdx mice and human DMD patients (De Palma et al., 2012; Spitali et al., 2013), and pharmacological stimulation of autophagy has been found to ameliorate the dystrophic phenotype of mdx mice (Bibee et al., 2014; Pauly et al., 2012). Thus, the consequence of enhanced Carm1 phosphorylation in mdx satellite cells described here may extend beyond regulation of Pax7 and influence other pathways that contribute to disease progression, such as the inability to activate autophagy in order to clear damaged mitochondria.
We have shown that in addition to loss of dystrophin/Mark2 signaling, that the p38γ/Carm1 pathway is also dysregulated in dystrophin-deficient satellite cells, ultimately affecting epigenetic-mediated gene transcription. Therapeutic strategies to restore cell polarity in dystrophic satellite cells should take into account the restoration of syntrophin-mediated p38γ regulation. Alternatively, alleviating the negative regulation of Carm1 may facilitate the generation of functional myogenic progenitors to help restore the normal myogenic function of dystrophic satellite cells. Translating this knowledge is thus an important goal towards developing appropriate personalized muscle regenerative therapies for DMD.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael A. Rudnicki (mrudnicki@ohri.ca).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Housing, husbandry and all experimental protocols for mice used in this study were performed in accordance with the guidelines established by the University of Ottawa Animal Care Committee, which is based on the guidelines of the Canadian Council on Animal Care. The health and immune status of all mice used were normal, and they were not involved in any previous procedures. Food and water were administered ad libitum. The Myf5-Cre:R26R-YFP mice (Kuang et al., 2007) were F1 progeny from Myf5tm3(cre)Sor (Myf5-Cre) x Gt(ROSA)26Sortm1(EYFP)Cos (R26R-YFP) breeding pairs, and were of mixed background (129SV and C57BL/6). For muscle regeneration experiments, an F2 cross between the offspring of Pax7tm1(cre/Esr1*)Cklr (Pax7CreER/+) mice (Nishijo et al., 2009) and p38γfl/fl mice (Ruiz-Bonilla et al., 2008) generated Pax7+/+:p38γfl/fl, Pax7CreER/+:p38γ fl/+, and Pax7CreER/+:p38γfl/fl mice (C57BL/6 genetic background). Six week-old male mice were used for muscle regeneration experiments.
Cell Lines
HEK293T cells were used for over-expression and purification of p38 kinases and for co-immunoprecipitation experiments. HEK293T cells were purchased from and authenticated by ATCC. These cells were verified to be free from mycoplasma contamination using the MycoSensor PCR Assay Kit (Agilent Technologies). HEK 293T cells were cultured at 37°C in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Sf9 insect cells (Gibco) were used to produce GST-Carm1 recombinant protein. Sf9 cells were cultured at 28°C in Sf-900 II Serum-Free Media (Gibco) supplemented with 1% penicillin/streptomycin.
Primary Cell Cultures
Satellite cells were cultured on extensor digitorum longus (EDL) myofibers isolated from the following mice (6–8 weeks old male and female mice were used): Myf5-Cre:R26R-EYFP (Kuang et al., 2007), Pax7CreER/+:p38γfl/fl, Pax7+/+:p38γfl/fl, C57BL/10ScSn (wild type), and C57BL/10ScSn-Dmdmdx/J (mdx) mice (homozygous Dmdmdx female mice and hemizygous DMDmdx males were used). EDL myofibers were cultured at 37°C in DMEM high glucose (Life Technologies) supplemented with 20% fetal bovine serum, 1% chick embryo extract, and 2.5 ng/ml of bFGF. Satellite cells were prospectively isolated by fluorescence-activated cell sorting (FACS) from 8-week old male C57BL/10ScSn and mdx mice. RNA was isolated from satellite cells immediately after isolation. Primary myoblasts were derived from either 3 week or 6–8 week-old female and male C57BL/10ScSn, mdx, and tamoxifen-treated Pax7CreER/+:Carm1fl/fl (Kawabe et al., 2012) mice by magnetic cell separation (MACS). Primary myoblasts were cultured at 37°C in Ham’s F-10 medium (Wisent) supplemented with 20% fetal bovine serum, 1% penicillin/streptomycin, and 2.5 ng/ml of bFGF.
METHOD DETAILS
Muscle regeneration
6–8 week-old male mice were injected intraperitoneally with 100 μl of tamoxifen (TMX, 20 mg/ml in corn oil) on 5 consecutive days, and maintained on TMX containing chow (500 mg TMX/kg diet, Teklad, Envigo) for the remaining duration of the experiment. The diet provides approximately 80 mg TMX per kg of mouse body weight per day assuming an average intake of 3–4 g of diet for 20–25 g body weight. To perform muscle injury, mice were anesthetized with isoflurane and 50 μl of 10 μM cardiotoxin (CTX) from Naja mossambica (Sigma) was injected directly into the TA muscle. CTX was administered every 21 days for a total of 3 injections. Twenty-one days after the third injection, mice were euthanized and TA muscles were harvested and analyzed. Numbers of satellite cells, numbers of fibers and minimum fiber Feret were assessed by immunohistochemistry of TA sections with anti-Pax7 and anti-Laminin antibodies. Counting and measurements were performed with ImageJ software.
Immunohistochemistry
TA muscle cryosections were fixed with 2% PFA for 2 min and permeabilized (0.1% Triton X-100, 0.1M Glycine, PBS) for 5 min. Sections were washed and blocked with M.O.M. blocking reagent (Vector Laboratories) for 2–3 hours. Incubations with primary antibodies were performed overnight at 4°C. Incubation with secondary antibodies conjugated to Alexa Fluor (Life Technologies) was performed for 1 hour. After several washes with PBS, sections were stained with Hoechst, rinsed with water and mounted with PermaFluor mounting medium (Fisher).
Myofiber isolation and culture
Single myofibers were isolated from EDL muscles of 6–8 week old mice as previously described (Pasut et al., 2013). Briefly, EDL muscles were dissected and incubated in DMEM (Gibco) containing 0.2% collagenase I (Worthington) for 1 hour. Myofibers were detached using gentle trituration with a glass pipette and washed several times with DMEM. Myofibers were fixed with 2% PFA for 10 min either immediately after isolation, after 36 hours (to analyze when satellite cells are entering the cell cycle), or 42 hours (to analyze when satellite cells have undergone their first division) in culture. For culturing, myofibers were maintained in DMEM high glucose (Life Technologies) supplemented with 20% fetal bovine serum, 1% chick embryo extract and 2.5 ng/ml bFGF. siRNA transfections were performed at 4h and 16h post-culture using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer’s instructions. siRNAs were purchased from Dharmacon (Table S3) and used at a final concentration of 50 nM. For the assessment of symmetric and asymmetric satellite stem cell divisions, EDL myofibers were isolated from 9 Myf5-Cre:R26R-EYFP mice. Fibers from each mouse were subsequently treated with siRNA, and approximately 300 satellite cells from 30–35 myofibers were analyzed per condition.
Immunofluorescence
Fixed myofibers were permeabilized (0.1% Triton X-100, 0.1M Glycine, PBS) for 10 min and blocked (5% horse serum, 2% BSA, 0.1% Triton X-100 in PBS) for 2–3 hours. Incubation with primary antibodies diluted in blocking buffer was performed overnight at 4°C. Myofibers were subsequently washed 3 times with PBS and incubated with fluorophore-conjugated (Alexa Fluor 488, 546, or 647, Life Technologies, 1:1000 dilution) secondary antibodies for 1 hour at room temperature. After washing with PBS, myofibers were stained with Hoechst (1:1000 dilution) and mounted onto glass slides with PermaFluor (Fisher). For analysis of Carm1 fluorescence intensity in nuclear and cytoplasmic compartments, Z-stack images of transfected cells were acquired on an epifluorescent microscope equipped with a motorized stage (Zeiss AxioObserver Z1) with a step size of 0.2 μm to span the cell (25 slices in total) and images were deconvoluted using Zen Software (Zeiss). 3D sum intensity Z-projection was performed with ImageJ software and average fluorescence intensities were determined for both nuclear and cytoplasmic compartments (as determined by DAPI and tubulin markers). Carm1 localization was expressed as a ratio of nuclear to cytoplasmic expression.
Proximity ligation assay (PLA)
Fixed myofibers were permeabilized (0.1% Triton X-100, 0.1M Glycine, PBS) for 10 min and blocked with Duolink Blocking Solution (Sigma) for 2–3 hours. Incubation with primary antibodies diluted in Duolink Blocking Solution (Sigma) was performed overnight at 4°C. PLA reactions were subsequently carried out using Duolink PLA probes for mouse and rabbit and Duolink In Situ Detection Reagents Red (Sigma) following the manufacturer’s protocol. After the final wash steps, myofibers were mounted on glass slides with VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories). Quantification of PLA was performed either by counting PLA puncta per satellite cell (when distinct PLA signals were observed) or by measuring cell fluorescence intensity using ImageJ software (Burgess et al., 2010) (when the PLA signals were overly abundant). PLA experiments were performed in biological triplicate using EDL myofibers isolated from 3 different mice. For each individual experiment, a minimum of 20 myofibers were analyzed.
Co-immunoprecipitation
Indicated proteins were expressed in HEK293T cells using Lipofectamine 2000-mediated (Life Technologies) plasmid transfection. Cells were collected 2 days post-transfection and lysed in lysis buffer (50mM Tris pH 7.5, 150mM NaCl, 2mM MgCl2, 0.5mM EDTA, 0.5% Triton X-100, and protease inhibitors) for 30 min on ice. Cell lysates were cleared by centrifugation and were incubated with either anti-FLAG M2 Affinity Gel (Sigma) or c-Myc Monoclonal Antibody-Agarose Beads (Clontech) overnight at 4°C. Beads were washed 4 times with lysis buffer and eluted with Laemmli buffer. Immunoprecipitates were resolved by SDS-PAGE and analyzed by Western blot with the indicated antibodies.
Carm1 mutagenesis and protein purification
The Carm1 S595A/S572A mutant was generated by PCR site-directed mutagenesis using the following primers: 5′-CCATCTCTATGGCCGCGCCCATGTCC-3′ and 5′-GGATGGACATGGGCGCGGCCATAGAG-3′. For the Carm1 S572E mutant, the following primers were used: 5′-CATCTCTATGGCCGAGCCCATGTCCATCC-3′ and 5′-GGATGGACATGGGCTCGGCCATAGAGATG-3′. Carm1 S572A and S572E mutants were generated from Carm1 in pcDNA3 mammalian expression vector (Life Technologies) for transient expression in Carm1 null primary myoblasts. For Carm1 S595A/S572A mutant protein expression, GST-Carm1 in the baculovirus expression plasmid pFastBac1 (Life Technologies) was used to generate the mutation. GST-Carm1 WT, S595A, and S572A recombinant proteins were made from baculovirus-infected Sf9 cells and purified with glutathione sepharose (GE Healthcare) as previously described (Kawabe et al., 2012). Briefly, recombinant baculoviruses were generated using the Bac-to-Bac Baculovirus Expression System (Thermo Fisher Scientific) and used to infect Sf9 cells. 2–3 days post infection, cells were collected by centrifugation. Cells were lysed in lysis buffer (25mM Tris pH 7.6, 1% Triton X-100, 300mM NaCl, 2mM MgCl2, 1mM DTT) on ice for 20 min and cell lysates were cleared by centrifugation. Lysates were pre-cleared by incubation with protein G sepharose (GE Healthcare) for 30 min followed by incubation with glutathione sepharose (GE Healthcare) for 3 hours at 4°C. Beads were washed 3 times with lysis buffer followed by 2 washes with wash buffer (25mM Tris pH 7.6, 0.1% Triton X-100, 300mM NaCl, 2mM MgCl2, 1mM DTT). Protein was eluted from the resin with 50mM glutathione in wash buffer and eluted fractions were confirmed by SDS-PAGE followed by Coomassie blue staining.
In vitro kinase assays
FLAG and Myc-tagged p38 kinases were expressed either in the presence or absence of constitutively active upstream kinase MKK6EE in HEK293T cells and affinity purified with either anti-FLAG M2 Affinity Gel (Sigma) or c-Myc Monoclonal Antibody-Agarose Beads (Clontech). Active GST-p38γ kinase was purchased from EMD Millipore. GST-Carm1 recombinant proteins were affinity purified from Sf9 cells as described above. Kinases and Carm1 substrate were combined in the presence of ATP [γ-32P] (PerkinElmer) for 30 min at 30°C. In vitro kinase reactions were resolved by SDS-PAGE and analyzed by autoradiography and Coomassie blue staining.
In vitro methylation assays
GST-Carm1 recombinant proteins (isoform1 and 2) and Pax7-FLAG were affinity purified from Sf9 cells. GST-Carm1 and either Pax7-FLAG or core histone proteins (Upstate) were combined in the presence of S-adenosyl-L-[methyl-3H] methionine (PerkinElmer) for 1 hour at 30°C. In vitro methylation reactions were resolved by SDS-PAGE and analyzed by autoradiography and Coomassie blue staining.
Mass spectrometry
In vitro kinase assays were performed with either activated p38γ or non-activated p38γ together with GST-Carm1 (isoform 2) substrate. Reactions were resolved by SDS-PAGE and visualized by Coomassie blue staining. Protein bands corresponding to Carm1 protein were excised, digested with chymotrypsin (Promega) and peptides were analyzed by LC-MS/MS on an LTQ Orbitrap XL hybrid mass spectrometer with nanospray ionization source (Thermo Scientific) and an UltiMate 3000 RSLC Nano HPLC (Dionex). MASCOT 2.4 software (Matrix Science) was used to infer peptide and protein identities from the mass spectra.
Cell culture and transfection
For Carm1 subcellular localization experiments, Carm1 null primary myoblasts were cultured on a collagen-coated glass bottom 35mm μ-dish (Ibidi). Transfections with indicated plasmids were performed using Lipofectamine 3000 (Life Technologies) according to the manufacturer’s protocol. Cells were fixed 24 hours post-transfection and immunofluorescence staining was performed as described above.
Satellite cell and primary myoblast isolation
Satellite cells were prospectively isolated from C57BL/10 or mdx mice by flow cytometry using APC-conjugated anti-α7-integrin (Ablabs), and PE-conjugated anti-CD31 (BD Biosciences), anti-CD45 (BD Bioscences), anti-Sca1 (BD Biosciences) and anti-CD11b (BD Biosciences) antibodies (Pasut et al., 2012). In brief, dissected muscles from the hind limbs were minced in collagenase/dispase solution followed by dissociation using the gentleMACS Octo Dissociator with Heaters (Miltenyi Biotec). Cells were then filtered through a 70 μm nylon mesh and collected by centrifugation. Cell pellets were resuspended in FACS buffer (5% FBS, 1mM EDTA in PBS) and incubated with the antibodies listed above and Hoechst 33342. Unbound antibodies were washed with FACS buffer and cells were pelleted prior to resuspension in FACS buffer and a final filtration through a 30 μm filter. Satellite cells were sorted based on forward scatter and side scatter profiles, followed by negative lineage selection in PE (Cd11b−, Sca1−, Cd45−, Cd31−) and positive selection in APC (α7-integrin+). Flow cytometry was performed using a MoFlo XDP (Beckman Coulter) equipped with 5 excitation lasers (355 nm, 405 nm, 488 nm, 561 nm and 640 nm). Primary myoblasts were purified from C57BL/10 or mdx mice by magnetic cell separation (MACS) (Sincennes et al., 2017). Muscle dissociation and cell filtration was performed as described above for FACS. Cell pellets were resuspended in MACS buffer (0.5% BSA, 2mM EDTA in PBS), and then incubated with biotin-conjugated lineage antibodies (Cd11b−, Sca1−, Cd45−, Cd31−), followed by incubation with streptavidin microbeads for negative lineage selection. Cell suspensions were loaded onto a LD column (Miltenyi Biotec) in a magnetic field of a VarioMACS separator (Miltenyi Biotec) and rinsed with MACS buffer. Column flow-through (containing the lineage negative cells) was collected and stained with biotin-conjugated anti-a7-integrin antibody, followed by incubation with streptavidin microbeads, to purify the satellite cell/myoblast population. Cells were loaded onto a MS column (Miltenyi Biotec) and washed 3 times with MACS buffer. Flow-through containing unlabeled cells were discarded, and the column was flushed to collect positively labeled satellite cells and myoblasts. Cells were centrifuged and the cell pellets were resuspended in myoblast growth media and plated onto collagen-coated culture dishes.
Chromatin immunoprecipitation (ChIP)
Primary myoblasts were cross-linked using 1% formaldehyde in PBS for 10 minutes. Glycine was added to a final concentration of 0.125M for 5 minutes, followed by centrifugation. The cell pellet was washed in PBS and subsequently resuspended in ChIP lysis buffer (50mM Tris-HCl pH 8.0, 10mM EDTA, 0.5% SDS). Chromatin was fragmented using a Covaris sonicator. Before ChIP, equal volumes of ChIP assay buffer (20mM Tris-HCl pH 8.0, 1.2mM EDTA, 1.1% Triton X-100, 200mM NaCl) was added. Immunoprecipitation was performed using 0.5 mg of chromatin and 4 μg of rabbit anti-histone H3K4me3 antibody or rabbit IgG overnight at 4°C. Protein A/G mix magnetic beads (Millipore) were then added for 2 hours at 4°C. Antibody/protein/DNA complexes were collected, washed and eluted, and cross-links were reversed according to manufacturer’s instructions. DNA was purified by phenol/chloroform purification using linear acrylamide (Ambion) and GlycoBlue (Ambion) as carriers. ChIP enrichment was analyzed by quantitative PCR using the Mx3000P qPCR System (Stratagene). A list of the primers used can be found in Table S4.
RT-qPCR
For satellite cells, RNA was isolated using Arcturus Picopure RNA extraction kit (Thermo Fisher Scientific). For primary myoblasts, RNA was extracted using Trizol and isolated using the Nucleospin RNA II kit (Macherey-Nagel) according to the manufacturer’s instructions. cDNA was synthesized using SuperScript III First-Strand Synthesis System (Invitrogen) by following the manufacturer’s protocol. RT-qPCR reactions were performed using iQ SYBR Green Supermix (Bio-Rad) and the Mx3000P qPCR System (Stratagene). A list of the primers used can be found in Table S4.
Droplet digital PCR (ddPCR)
RNA was extracted with the PicoPure RNA Isolation Kit (Applied Biosystems) and cDNA was generated with the SuperScript III First-Strand Synthesis System (Invitrogen) according to manufacturer’s protocol. ddPCR was performed using ddPCR Supermix for Probes (no dUTP) (Biorad) according to manufacturer’s protocol and analyzed with the QX100 Droplet Digital PCR System (Biorad). p38γ expression was normalized to TATA-binding protein (TBP) levels and expressed as a percentage relative to control. Probes were purchased from Integrated DNA Technologies (Table S4).
Subcellular fractionation
Cells were collected by trypsin-EDTA digestion and centrifugation, and subsequently washed once with PBS. Cell pellets were lysed in Buffer A (10mM Tris-HCl, pH 7.9, 10mM KCl, 0.1mM EDTA, 0.1mM EGTA) with protease and phosphatase inhibitors on ice for 15 min. 10% NP40 was added to the cell lysis (1:16 volume) and lysates were vortexed vigorously followed by centrifugation at 14,500 rpm for 30 seconds. The supernatant was collected as the cytoplasm fraction. The cell pellet was resuspended in Buffer C (20mM Tris-HCl, pH 7.9, 400mM NaCl, 1mM EDTA, 1mM EGTA) with protease and phosphatase inhibitors and vortexed for 1 h at 4°C followed by centrifugation for 15 min at 14,500 rpm at 4°C. The supernatant was collected as the nuclear fraction. Protein concentrations were determined by Bradford assay (Biorad), and lysates were analyzed by SDS-PAGE and immunoblotting.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experiments involving mice were performed with a minimum of three biological replicates (as indicated) and results are presented as the mean ± SEM. Sample size or replicate number (designated as “n”) for each experiment are indicated in the figure legends. Only male mice were analyzed for the p38γfl/fl muscle regeneration experiment. Comparisons between two experimental groups were made using the two-tailed Student’s t test, and comparisons between three experimental groups were made using univariate ANOVA followed by Fisher’s Least Significant Difference post hoc tests where appropriate (SPSS). Dot density and box plots were generated using Graphpad software. P-values are indicated as *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and P-values <0.05 were considered to be statistically significant.
Supplementary Material
HIGHLIGHTS.
p38γ MAPK positively regulates symmetric muscle stem cell expansion
p38γ phosphorylates Carm1 to prevent nuclear localization and interaction with Pax7
p38γ/Carm1 signaling is dysregulated in dystrophin-deficient mdx satellite cells
Carm1-mediated epigenetic induction of Myf5 is impaired in mdx satellite cells
Acknowledgments
We thank Drs. Jeffrey Dilworth and Lynn Megeney for careful reading of the manuscript. We also thank Jennifer Ritchie for animal husbandry, Dr. Lawrence Puente for mass spectrometry analysis, Dr. Chloë van Oostende for microscopy and imaging analysis, Paul Oleynik for FACS, and Fan Xiao, Natasha Mercier, and David Wilson for technical assistance. N.C.C. is a recipient of the Centre for Neuromuscular Disease Scholarship in Translational Research Award from the University of Ottawa Brain and Mind Research Institute, and was supported by Postdoctoral fellowships from the Canadian Institutes of Health Research (CIHR) and the Ontario Institute for Regenerative Medicine (OIRM). F.P.C. was supported by a Postdoctoral fellowship from the French Muscular Dystrophy Association (AFM)-Téléthon. C.E.B. is supported by a Postdoctoral fellowship from OIRM. M.L. was supported by a Postdoctoral fellowship from CIHR. P-M-C acknowledges support from ERC-STEM-AGING 741966 and SAF2015-67369-R. M.A.R. holds the Canada Research Chair in Molecular Genetics. These studies were carried out with support of grants to M.A.R. from the US National Institutes for Health [R01AR044031], the Canadian Institutes of Health Research [FDN-148387], the Muscular Dystrophy Association (USA), E-Rare-2: Canadian Institutes of Health Research/Muscular Dystrophy Canada [ERA-132935], and the Stem Cell Network.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Conceptualization, N.C.C. and M.A.R.; Methodology, N.C.C., M.C.S., F.P.C, C.E.B., M.L., and M.A.R.; Investigation, N.C.C., M.C.S., F.P.C., C.E.B., M.L., and H.M.; Writing – Original Draft, N.C.C., F.P.C. and M.A.R.; Writing – Review & Editing, N.C.C., M.C.S., F.P.C., M.L., and M.A.R.; Funding Acquisition, M.A.R.; Resources, J.S., P.M-C., and M.A.R.; Supervision, M.A.R.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, Olwin BB. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med. 2014;20:265–271. doi: 10.1038/nm.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibee KP, Cheng YJ, Ching JK, Marsh JN, Li AJ, Keeling RM, Connolly AM, Golumbek PT, Myerson JW, Hu G, et al. Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function. FASEB J. 2014;28:2047–2061. doi: 10.1096/fj.13-237388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess A, Vigneron S, Brioudes E, Labbe JC, Lorca T, Castro A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci U S A. 2010;107:12564–12569. doi: 10.1073/pnas.0914191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338:259–262. doi: 10.1038/338259a0. [DOI] [PubMed] [Google Scholar]
- Chang NC, Chevalier FP, Rudnicki MA. Satellite Cells in Muscular Dystrophy - Lost in Polarity. Trends Mol Med. 2016;22:479–496. doi: 10.1016/j.molmed.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark-Lewis I, Sanghera JS, Pelech SL. Definition of a consensus sequence for peptide substrate recognition by p44mpk, the meiosis-activated myelin basic protein kinase. J Biol Chem. 1991;266:15180–15184. [PubMed] [Google Scholar]
- Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA, Morgan JE. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. 2005;122:289–301. doi: 10.1016/j.cell.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, Blau HM. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med. 2014;20:255–264. doi: 10.1038/nm.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crist Colin G, Montarras D, Buckingham M. Muscle Satellite Cells Are Primed for Myogenesis but Maintain Quiescence with Sequestration of Myf5 mRNA Targeted by microRNA-31 in mRNP Granules. Cell Stem Cell. 2012;11:118–126. doi: 10.1016/j.stem.2012.03.011. [DOI] [PubMed] [Google Scholar]
- De Arcangelis V, Serra F, Cogoni C, Vivarelli E, Monaco L, Naro F. β1-Syntrophin Modulation by miR-222 in mdx Mice. PLoS One. 2010;5:e12098. doi: 10.1371/journal.pone.0012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma C, Morisi F, Cheli S, Pambianco S, Cappello V, Vezzoli M, Rovere-Querini P, Moggio M, Ripolone M, Francolini M, et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 2012;3:e418. doi: 10.1038/cddis.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont NA, Wang YX, von Maltzahn J, Pasut A, Bentzinger CF, Brun CE, Rudnicki MA. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat Med. 2015;21:1455–1463. doi: 10.1038/nm.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, He B, Jung SY, Song Y, Qin J, Tsai SY, Tsai MJ, O’Malley BW. Biochemical control of CARM1 enzymatic activity by phosphorylation. J Biol Chem. 2009;284:36167–36174. doi: 10.1074/jbc.M109.065524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie MA, Le Grand F, Scime A, Kuang S, von Maltzahn J, Seale V, Cuenda A, Ranish JA, Rudnicki MA. p38-{gamma}-dependent gene silencing restricts entry into the myogenic differentiation program. J Cell Biol. 2009;187:991–1005. doi: 10.1083/jcb.200907037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich DB, Nguyen PD, Siegel AL, Ehrlich OV, Sonntag C, Phan JM, Berger S, Ratnayake D, Hersey L, Berger J, et al. Asymmetric division of clonal muscle stem cells coordinates muscle regeneration in vivo. Science. 2016;353:aad9969. doi: 10.1126/science.aad9969. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Cuenda A, Spillantini MG, Thomas GM, Buee-Scherrer V, Cohen P, Goedert M. Stress-activated protein kinase-3 interacts with the PDZ domain of alpha1-syntrophin. A mechanism for specific substrate recognition. J Biol Chem. 1999;274:12626–12631. doi: 10.1074/jbc.274.18.12626. [DOI] [PubMed] [Google Scholar]
- Kawabe Y, Wang YX, McKinnell IW, Bedford MT, Rudnicki MA. Carm1 regulates Pax7 transcriptional activity through MLL1/2 recruitment during asymmetric satellite stem cell divisions. Cell Stem Cell. 2012;11:333–345. doi: 10.1016/j.stem.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang S, Charge SB, Seale P, Huh M, Rudnicki MA. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J Cell Biol. 2006;172:103–113. doi: 10.1083/jcb.200508001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand F, Jones AE, Seale V, Scime A, Rudnicki MA. Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell Stem Cell. 2009;4:535–547. doi: 10.1016/j.stem.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepper C, Partridge TA, Fan CM. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development. 2011;138:3639–3646. doi: 10.1242/dev.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnell IW, Ishibashi J, Le Grand F, Punch VG, Addicks GC, Greenblatt JF, Dilworth FJ, Rudnicki MA. Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat Cell Biol. 2008;10:77–84. doi: 10.1038/ncb1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumuller RA, Knoblich JA. Dividing cellular asymmetry: asymmetric cell division and its implications for stem cells and cancer. Genes Dev. 2009;23:2675–2699. doi: 10.1101/gad.1850809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijo K, Hosoyama T, Bjornson CR, Schaffer BS, Prajapati SI, Bahadur AN, Hansen MS, Blandford MC, McCleish AT, Rubin BP, et al. Biomarker system for studying muscle, stem cells, and cancer in vivo. FASEB J. 2009;23:2681–2690. doi: 10.1096/fj.08-128116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasut A, Jones AE, Rudnicki MA. Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J Vis Exp. 2013:e50074. doi: 10.3791/50074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasut A, Oleynik P, Rudnicki MA. Isolation of muscle stem cells by fluorescence activated cell sorting cytometry. Methods Mol Biol. 2012;798:53–64. doi: 10.1007/978-1-61779-343-1_3. [DOI] [PubMed] [Google Scholar]
- Pauly M, Daussin F, Burelle Y, Li T, Godin R, Fauconnier J, Koechlin-Ramonatxo C, Hugon G, Lacampagne A, Coisy-Quivy M, et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol. 2012;181:583–592. doi: 10.1016/j.ajpath.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Price FD, von Maltzahn J, Bentzinger CF, Dumont NA, Yin H, Chang NC, Wilson DH, Frenette J, Rudnicki MA. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat Med. 2014;20:1174–1181. doi: 10.1038/nm.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheteau P, Gayraud-Morel B, Siegl-Cachedenier I, Blasco MA, Tajbakhsh S. A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell. 2012;148:112–125. doi: 10.1016/j.cell.2011.11.049. [DOI] [PubMed] [Google Scholar]
- Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 1993;75:1351–1359. doi: 10.1016/0092-8674(93)90621-v. [DOI] [PubMed] [Google Scholar]
- Ruiz-Bonilla V, Perdiguero E, Gresh L, Serrano AL, Zamora M, Sousa-Victor P, Jardi M, Wagner EF, Munoz-Canoves P. Efficient adult skeletal muscle regeneration in mice deficient in p38beta, p38gamma and p38delta MAP kinases. Cell Cycle. 2008;7:2208–2214. doi: 10.4161/cc.7.14.6273. [DOI] [PubMed] [Google Scholar]
- Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM. Self-renewal and expansion of single transplanted muscle stem cells. Nature. 2008;456:502–506. doi: 10.1038/nature07384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud-Morel B, Guenou H, Malissen B, Tajbakhsh S, Galy A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138:3647–3656. doi: 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–786. doi: 10.1016/s0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- Shin HJ, Kim H, Oh S, Lee JG, Kee M, Ko HJ, Kweon MN, Won KJ, Baek SH. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature. 2016;534:553–557. doi: 10.1038/nature18014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sincennes MC, Wang YX, Rudnicki MA. Primary Mouse Myoblast Purification using Magnetic Cell Separation. Methods Mol Biol. 2017;1556:41–50. doi: 10.1007/978-1-4939-6771-1_3. [DOI] [PubMed] [Google Scholar]
- Soleimani VD, Punch VG, Kawabe Y, Jones AE, Palidwor GA, Porter CJ, Cross JW, Carvajal JJ, Kockx CE, van IWF, et al. Transcriptional dominance of Pax7 in adult myogenesis is due to high-affinity recognition of homeodomain motifs. Dev Cell. 2012;22:1208–1220. doi: 10.1016/j.devcel.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitali P, Grumati P, Hiller M, Chrisam M, Aartsma-Rus A, Bonaldo P. Autophagy is Impaired in the Tibialis Anterior of Dystrophin Null Mice. PLoS Curr. 2013 doi: 10.1371/currents.md.e1226cefa1851a1372f1079bbc1406c1370a1321e1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy A, Cadwallader AB, Fedorov Y, Tyner K, Tanaka KK, Olwin BB. Coordination of satellite cell activation and self-renewal by Par-complex-dependent asymmetric activation of p38alpha/beta MAPK. Cell Stem Cell. 2012;11:541–553. doi: 10.1016/j.stem.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Maltzahn J, Jones AE, Parks RJ, Rudnicki MA. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc Natl Acad Sci U S A. 2013;110:16474–16479. doi: 10.1073/pnas.1307680110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Charoensuksai P, Watson NJ, Wang X, Zhao Z, Coriano CG, Kerr LR, Xu W. CARM1 automethylation is controlled at the level of alternative splicing. Nucleic Acids Res. 2013;41:6870–6880. doi: 10.1093/nar/gkt415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yablonka-Reuveni Z, Anderson JE. Satellite cells from dystrophic (mdx) mice display accelerated differentiation in primary cultures and in isolated myofibers. Dev Dyn. 2006;235:203–212. doi: 10.1002/dvdy.20602. [DOI] [PubMed] [Google Scholar]
- Zismanov V, Chichkov V, Colangelo V, Jamet S, Wang S, Syme A, Koromilas AE, Crist C. Phosphorylation of eIF2alpha Is a Translational Control Mechanism Regulating Muscle Stem Cell Quiescence and Self-Renewal. Cell Stem Cell. 2016;18:79–90. doi: 10.1016/j.stem.2015.09.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.