

Abstract
Myeloid derived suppressor cells (MDSCs) are innate immune cells that potently inhibit T cells. In cancer, novel therapies aimed to activate T cells can be rendered ineffective due to the activity of MDSCs. Thus, targeted inhibition of MDSCs may greatly enhance T cell-mediated anti-tumor immunity, but targeted mechanisms remain obscure. Here we show, for the first time, that scavenger receptor type B-1 (SCARB1), a high-affinity receptor for spherical high-density lipoprotein (HDL), is expressed by MDSCs. Furthermore, we demonstrate that SCARB1 is specifically targeted by synthetic high-density lipoprotein-like nanoparticles (HDL NP), which reduces MDSC activity. Using in vitro T cell proliferation assays, data show that HDL NPs specifically bind SCARB1 to inhibit MDSC activity. In murine cancer models, HDL NP treatment significantly reduces tumor growth, metastatic tumor burden, and increases survival due to enhanced adaptive immunity. Flow cytometry and immunohistochemistry demonstrate that HDL NP-mediated suppression of MDSCs increased CD8+ T cells and reduced Treg cells in the metastatic tumor microenvironment. Using transgenic mice lacking SCARB1, in vivo data clearly show that the HDL NPs specifically target this receptor for suppressing MDSCs. Ultimately, our data provide a new mechanism and targeted therapy, HDL NPs, to modulate a critical innate immune cell checkpoint to enhance the immune response to cancer.
Keywords: Nanotechnology, Immunotherapy, Lipoprotein
INTRODUCTION
Modulating the immune system can be an effective mechanism to address numerous pathologies. In cancer, recent therapies including checkpoint blockade, cancer vaccines, and chimeric antigen receptor T cells aim to activate the adaptive immune response (1–3) and have shown success. However, despite their promise, they are only effective in sub-sets of patients (4, 5). As with the natural immune response, efficacy of these therapies is often drastically reduced by the action of specialized innate immune cells called myeloid derived suppressor cells (MDSCs), which potently inhibit adaptive immune cells (6–8). As such, novel mechanisms, targets, and therapies are required to better harness the immune system as a cancer therapy with a specific focus on MDSCs.
The size, shape, and composition of natural high-density lipoproteins (HDL) are constantly changing as the particles acquire and deliver cholesterol and cholesteryl ester to target cells (9). The main protein constituent of HDLs, apolipoprotein A-I (apo A-I), mediates this dynamic biological process whereby cholesterol-poor discoidal forms of HDL become progressively more spherical upon cholesterol uptake (10). Cholesterol-rich, spherical HDLs containing apo A-I bind with high affinity to scavenger receptor type B-1 (SCARB1) (11–13). HDL binding SCARB1 has been most studied in atherosclerosis where SCARB1 is expressed by innate myeloid cells such as macrophages and neutrophils (14–16). The interaction of apo A-I and HDLs with myeloid cells may have critical importance in cardiovascular disease (17, 18). Furthermore, in oncology, human apo A-I has been shown to generate an anti-tumor response in mouse models of cancer; however, the mechanisms remain obscure (19, 20). Importantly, increased plasma apo A-I and HDL cholesterol are associated with reduced cancer risk in patients (21).
Given the expression of SCARB1 on myeloid cells, the role of MDSCs in bridging innate and adaptive immunity, and the involvement of apo A-I and HDL in modulating the adaptive immune response, we hypothesized that MDSCs express SCARB1 that can be targeted to reduce MDSC function and generate a potent adaptive anti-tumor immune response. To explore this hypothesis, we used spherical, functional high-density lipoprotein-like nanoparticles (HDL NPs) developed by our lab (22–24). The HDL NPs have comparable size, shape, surface charge, and surface chemistry (e.g. contain apo A-I) to natural, spherical HDLs (24, 25), and more tightly bind SCARB1 when compared to native HDL (26, 27). Additionally, in vivo studies with HDL NPs have demonstrated a general lack of toxicity and selective targeting of cells that express SCARB1 (26, 28). Our data show that SCARB1, a high-affinity receptor for spherical HDL (29–32), is expressed by MDSCs. Targeting SCARB1 with HDL NP reduces MDSC activity in vitro. In murine models of melanoma, data show that HDL NP treatment significantly reduces tumor size, metastatic tumor burden, and increases survival due to an enhanced adaptive immune response through a mechanism requiring SCARB1. Additionally, data obtained in a murine model of lung cancer confirmed that HDL NP treatment reduced tumor volume and mass. As such, HDL NPs and MDSC expression of SCARB1 enable therapeutic modulation of a critical innate immune cell checkpoint on adaptive immunity.
MATERIALS AND METHODS
Cell Culture
B16F10 and Lewis Lung Carcinoma (LLC) cells were purchased from American Type Culture Collection (ATCC), and cultured in DMEM containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Life Technologies). Cells were passaged every 48-72 hours. B16F10 cells were purchased ~2 years ago, frozen at passage 3, and used for experiments prior to passage 8. The LLC cell line was purchased ~3 years ago, frozen at passage 3, and experiments were conducted prior to passage 8. Both of the cell lines are murine and were not authenticated. Mycoplasma testing was conducted using PlasmoTest Mycoplasma Detection Kit (Invivogen), and the cells were found to be negative (10/01/2017). Mouse T cells isolated for MDSC suppression assays were cultured in RPMI-1640 containing 10% FBS, 2.5% HEPES, 1% penicillin/streptomycin, 1% sodium pyruvate, 1% non-essential amino acids, and 0.1% 2-mercaptoethanol. Mouse MDSCs were grown in RPMI containing 10% FBS, 1% penicillin/streptomycin, IL-6 (10 ng/ml, Biolegend) and GM-CSF (20 ng/ml, Biolegend). Cell cultures were maintained in 5% CO2 at 37°C.
Experimental Animals
Mice were housed at the Northwestern University Center for Comparative Medicine according to NIH guidelines, following protocols approved by the Northwestern University Animal Care and Use Committee. Mixed male and female C57Bl/6 mice were purchased from Jackson Laboratories at 6-8 weeks of age. All animals were used soon after purchase. For SCARB1 knockout experiments, male and female heterozygous SCARB1Tm1Kri were purchased from Jackson Laboratory and bred to generate homozygous SCARB1 −/− mice. Animals were allowed to reach 6 weeks of age whereupon they were genotyped and utilized for experiments. Mice were randomized for all studies and researchers were blinded during analysis.
HDL NP Synthesis
High-density lipoprotein-like nanoparticles (HDL NPs) were synthesized and characterized as previously described (22, 25, 26). Citrate stabilized 5 nm diameter gold nanoparticles (AuNP, Ted Pella) were used as a template for surface modification. Purified human apolipoprotein AI (apo A-I, MyBioSource) was incubated with AuNPs (80 nM) at 5 molar excess (400 nM, final) overnight at room temperature (RT) with gentle stirring. Next, 1-2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[3-(2-pyridyldithio)propionate] (PDP-PE) were added at 250 molar excess relative to [AuNP] in a mixture of ethanol and water (1:4), and allowed to incubate at RT for 4 hours with gentle stirring. The HDL NPs were then purified and concentrated using tangential flow filtration. The HDL NP concentration and final conjugate size were determined using UV-Vis spectrophotometry (εAuNP = 9.696 × 106 M−1cm−1 at λmax = 520 nm) and dynamic light scattering (DLS, Malvern Zetasizer), respectively.
For fluorescence-tagged HDL NPs, the lipophilic fluorophore dialkylcarbocyanine (DiD, Life Technologies) was added to 1 ml of 1 μM HDL NPs at a 2.5 molar excess (2.5 μM) and incubated at 37 °C for 1 hour with mild shaking. Non-conjugated dye was removed from the HDL NP solution by centrifugation at 14,000 × g for 1 hour, decanting of the supernatant, and then the particle pellet was re-suspended in water.
Melanoma Lung Colonization
HDL NP pre-treatment studies: C57Bl/6 mice were treated intravenously with 100 μl HDL NPs (1 μM) every 48 hours for a total of 3 doses. 72 hours following the final dose, mice were inoculated with 1 × 105 B16F10 melanoma cells via tail vein and metastatic lung tumors were allowed to form over the course of 2 weeks at which point mice were sacrificed, lungs dissected and the metastatic tumors were counted. Lungs were then fixed and processed for IHC (see below). For the survival study, mice were treated as outlined above but removed from the study upon reaching the endpoint.
Established melanoma metastasis models: C57Bl/6 mice were inoculated with 1 × 105 B16F10 melanoma cells via tail vein and metastatic lung tumors were allowed to initiate for 5 days prior to initiating HDL NP dosing. At day 5, mice were treated with HDL NP at a concentration of 1 μM (200 μL) every 48 hours for 2 weeks. Mice were then sacrificed, lungs and lymph nodes dissected, the metastatic tumors were counted, and samples were processed for IHC.
Isolation of Bone Marrow cells
Mouse bone marrow (BM) cells were isolated as described previously (33). C57Bl/6 mice were sacrificed and the hind limbs removed using aseptic technique, leaving the femur and tibia intact. The excess muscle was removed from the bones with a razor blade. The bones were then cut at the joints and the bone marrow flushed with sterile DPBS (calcium and magnesium free) using a 1 ml syringe with a 27-gauge needle. Bone marrow cells were centrifuged at 300 × g and re-suspended in MACS buffer prior to MDSC isolation.
Isolation of Lymph Node Cells
C57Bl/6 mice were sacrificed and inguinal, axillary, cervical, and mesenteric lymph nodes were harvested using aseptic technique. Lymph nodes were then pressed through a 70 μm cell strainer to rupture the capsule and flushed using sterile PBS to generate a single cell suspension. Isolated cells were centrifuged at 300 × g for 10 minutes and re-suspended in MACS buffer for T cell isolation or PBS for analysis by flow cytometry.
Isolation of Spleen Cells
C57Bl/6 mice were sacrificed and the spleens harvested using aseptic technique. Spleens were finely cut and pressed through a 70 μm cell strainer with repeated flushes of sterile PBS to obtain single cell suspension. RBCs were then separated and discarded from the rest of the contents through density gradient centrifugation using Histopaque-1077 (Sigma-Aldrich) at room temperature. The remaining cells were re-suspended in PBS for analysis by flow cytometry.
Isolation of Lung Cells for Flow Cytometry
C57Bl/6 mice were euthanized, lungs removed and immune cells characterized as described previously (34). Briefly, lungs were minced in RPMI and subsequently incubated in digestion buffer containing 250 units of collagenase type IV and 200 units of Type IV DNase I in 8 mL of RPMI for 1 hour at 37 °C with gentle shaking. The digestion was quenched by adding 5% FBS. The lungs were subsequently filtered through a 70 μm cell strainer and rinsed in 5 ml of RPMI with 5% FBS. Cells were centrifuged at 300 × g for 10 minutes, re-suspended in 2 ml of ACK red blood cell lysis buffer and allowed to incubate for 10 minutes at 4 °C. Cells were again centrifuged at 300 × g for 10 minutes and washed in PBS. Cells were then counted and re-suspended in PBS at 1 × 107 cells/ml prior to flow cytometry.
Orthotopic Melanoma Survival Study
C57Bl/6 mice were inoculated with 5 × 104 B16F10 melanoma cells injected intradermally into the flank. Once the tumors became palpable, ~10 days following tumor cell inoculation, mice were treated every 48 hours with HDL NPs (100 μl, 1 μM). Survival of HDL NP and PBS treated mice was recorded. The study was terminated at Day 40, one week following the death of the final PBS-treated control mouse.
Lethal Irradiation and Bone Marrow Transplant
C57Bl/6 mice were treated with two doses of 500 rads of whole body irradiation separated by a two-hour break. Six hours following the final dose of radiation, mice were transplanted with 1 × 106 bone marrow cells from SCARB1 −/− mice. Mice were monitored for 1 month, allowing time for reconstitution of the immune system, at which point mice were subjected to the melanoma lung colonization experiment, as described previously.
MTS Assay
B16F10 cells were plated at a concentration of 20,000 cells/ml (2,000 cells/well) in 100 μl of DMEM, 10% FBS. Cells were allowed 24 hours to adhere at which point fresh media was added containing 50 nM HDL NP or natural human HDL (hHDL) (MyBiosource) and allowed to incubate for 24 or 48 hours. Then, the media was aspirated and the cells were washed with PBS. 80 μl of fresh serum free DMEM was added, as well as 20 μl of [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] (MTS) (Promega). A baseline absorbance was measured at 490 nm on a Synergy 2 plate reader (Biotek) and the cells incubated for 1 hour, at which point a second absorbance measurement was taken. Normalized metabolic activity was calculated by subtracting the baseline reading and normalizing to the untreated control.
Flow Cytometry
Isolated cells were washed with PBS and stained with 0.25 μl of LIVE/DEAD Fixable Aqua Dead Cell Stain (Thermo Fisher Scientific) in 500 μl of PBS for 20 min at room temperature. After washing in FACS buffer, cells were blocked with anti-CD16/CD32 (BD Biosciences) for 10 min and then stained in 100 μl of antibody mixture in FACS buffer (as detailed in Table S1) for 30 min at 4˚C in the dark. Cells were then washed in FACS buffer and fixed in 4% paraformaldehyde. Samples were run on an LSRII flow cytometer (BD Biosciences). Compensation on samples collected by the LSRII was performed in FlowJo or FCS Express post-collection.
MDSC HDL NP Treatment and Toxicity Analysis
Bone marrow of mice with B16F10 metastatic melanoma tumors established over the course of 2 weeks was isolated as outlined above and Myeloid Derived Suppressor Cells (MDSC) were separated using the magnetic activated cell sorting (MACS) MDSC isolation kit (Miltenyi Biotech). Cells were plated in a 24-well plate at 1 × 106 cells/well and cultured as described above. Cells were treated in triplicate with 50 nm HDL NP for either 24 or 48 hours. At which point, cells were harvested, washed to clear HDL NP and either analyzed using flow cytometry for percent live cell analysis or used in the MDSC mediated T cell suppression assay.
T Cell Suppression Assay
T cells were gathered from lymph node isolates (described above) employing EasySep Mouse T cell isolation kit (Stemcell). T cells were stained with CFSE (Thermo Fisher Scientific), as previously described (35). Briefly, T Cells were re-suspended in PBS with 5% FBS to a cell concentration of 8 × 106 cells/ml. CFSE was added to a concentration of 5 μM for 7 minutes at room temperature. Cells were washed 3 times in PBS with 5% FBS. Next, T cells were exposed to MDSCs, as previously described (35). T cells were plated in a 96-well plate at 2 ×105 cells/well and co-cultured with MDSCs isolated from wild-type or SCARB1 −/− mice, at a 2:1, 4:1 and 8:1 ratio of T cells to MDSCs in triplicate. T cells were stimulated using anti-CD3/CD28 activation beads (Invitrogen) and sustained in culture for 48 hours after which they were harvested, flow cytometry was performed, and the data were analyzed using FlowJo V10.
Immunohistochemistry
C57Bl/6 mice were sacrificed and the lungs perfused with 10% formalin, excised and fixed in formalin for up to 48 hours. The lungs were paraffin embedded, sectioned and H&E staining was performed at the Robert H. Lurie Comprehensive Cancer Center Pathology Core. H&E staining was analyzed using SPOT software.
For immunostaining, 5 μm thick sections were de-paraffinized in two changes of xylene, 10 minutes each. Sections were hydrated in ethanol, 100% (2 times), 95%, 70% and 50%, 5 minutes each. Samples were then washed in deionized water and the antigens retrieved by heating in 1× Target Retrieval Solution, Citrate pH 6 (Dako) for 20 min at 95 °C followed by cooling to room temperature. For staining, sections were washed in PBS and blocked in PBS with 1% BSA, 1% donkey serum, 0.3% Triton-X 100 and 0.01% sodium azide for 1 hour at room temperature. Primary antibodies (Supplementary Table S1) were diluted in blocking buffer and incubated overnight at 4 °C. Samples were washed 3 × 10 minutes in PBS and incubated for 1 hour with fluorophore-conjugated secondary antibodies (Supplementary Table S1) in blocking buffer. Slides were then washed three times in PBS with 0.1% tween 20, counterstained with DAPI and mounted in Fluoromount-G (Southern Biotech). Fluorescence was visualized using a Nikon A1R confocal microscope at the Center for Advanced Microscopy at Northwestern University and images analyzed using NIS Elements (Nikon) and ImageJ/Fiji (National Institute of Health).
Lewis Lung Carcinoma Xenograft Model
5 × 105 LLC cells (ATCC) were injected subcutaneously into the flanks of C57Bl/6 mice. The tumors were allowed to develop for 8 days at which point the tumor dimensions were measured using calipers. Tumors were measured and volumes determined using the formula volume = [(width2 × length) × 0.5] with width being the smaller of the two measurements every 48 hours until the study was completed (36). Following the first tumor size measurement, mice were randomly placed in groups and treated with HDL NPs (5 doses over 10 days, 200 μl, 1 μM HDL NP) or left untreated. Following treatment, mice were sacrificed and the tumors were excised and weighed.
RT-PCR
MDSCs were isolated as detailed above and cultured at a concentration of 1 × 106 cells/ml. MDSCs were treated with HDL NP (50 nM) or left untreated for 48 hours at which point mRNA was isolated using an RNeasy kit (Qiagen). 200 ng of RNA was converted to cDNA using iScript cDNA synthesis kit (Bio-Rad). After cDNA synthesis samples were diluted 10 times in nuclease-free water. Quantitative RT-PCR was then used to amplify and measure the cDNA with PerfeCTa SYBR Green mastermix (Quanta) on a CFX Connect RT-PCR Detection System (Bio-Rad). (For primers see Supplementary Table S2).
Statistical Analysis
Data were expressed using ± standard deviation of at least triplicate experiments. GraphPad Prism version 6 was used to analyze data. Microscopy and flow cytometry were analyzed using ImageJ/Fiji and FlowJo V10 or FCS Express, respectively. P values were calculated using a two-tailed Student’s T-test of unpaired samples and statistical significance was considered significant if P ≤ 0.05. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
RESULTS
CD11b+Ly6G+ and CD11b+Ly6C+ cells express SCARB1 and are targeted by HDL NPs
Initially, we investigated immune cell expression of SCARB1 and if, by extension, HDL NPs targeted these cells after systemic administration. First, we isolated immune cells from the bone marrow, lymph nodes, and spleens of C57Bl/6 mice and measured SCARB1 expression. Flow cytometry revealed substantial SCARB1 expression in CD11b+Ly6C+ monocytic cells and moderate SCARB1 expression in CD11b+Ly6G+ granulocytic cells across all three immune foci (Fig. 1A and Supplementary Fig. S1A–C). With regard to adaptive immune cells, SCARB1 was not expressed by T or B cells (Fig. 1A). Next, HDL NPs were labeled with fluorescent dye (DiD) and administered to C57Bl/6 mice via tail vein injection to demonstrate specific targeting to SCARB1+ cells. At 24 hours, data showed that HDL NPs targeted CD11b+ cells (~86 ± 2%) in the bone marrow (Fig. 1B). Further, both CD11b+Ly6G+ and CD11b+Ly6C+ cells in the bone marrow were targeted, but with a high specificity toward CD11b+Ly6C+ cells (Fig. 1C and D). At three and five days following injection, labeled MDSCs were no longer detected in the bone marrow (Supplementary Fig. S2). Importantly, HDL NPs displayed no cytotoxic effects on CD11b+Ly6G+ and CD11b+Ly6C+ cells (Supplementary Fig. S3A and B). These data demonstrate that CD11b+Ly6G+ and CD11b+Ly6C+ cells express SCARB1 and are targeted by HDL NPs.
Figure 1.

SCARB1 expression and HDL NP targeting of immune cells. A, SCARB1 expression was quantified using flow cytometry of cells isolated from the lymph nodes, spleen, and bone marrow of WT mice. B, Flow cytometry of WT mouse bone marrow after treatment with HDL NP-DiD, first gating for HDL NP-DiD+ cells, to determine the percentage of HDL NP-DiD+ cells that are CD11b+. C, HDL NPs were labeled with the DiD fluorophore and administered to WT mice. 24-hours later, bone marrow was isolated and the labeled immune cell distribution was determined by flow cytometry. D, Quantification of HDL NP uptake (top) as percent of CD11b+, HDL NP-DiD+ cells that are Ly6C+ or Ly6G+ and the distribution (bottom) as percent of CD11b+ cells that are Ly6C+ or Ly6G+ cells; P values: **P < 0.01 by two-tailed T test.
HDL NPs inhibit T cell suppression by MDSCs
In order to determine if HDL NP targeting of SCARB1 on CD11b+Ly6G+ and CD11b+Ly6C+ cells impacts the adaptive immune cell balance in healthy mice, we systemically administered HDL NPs (3×/week for 1 week) to C57Bl/6 mice and then measured T cell distributions in lymph nodes. We focused on T cells because of their capacity for robust anti-tumor immune responses (6) and lymph nodes to sample multiple immune foci. Data show that after systemic administration there was a significant increase in total CD4+ and CD8+ T cells in the lymph nodes compared to control mice (Fig. 2A, Supplementary Fig. S4A and B). Additionally, there were increased memory CD4+ and CD8+ T cells and a significant increase in CD4+ naïve T cells (Fig. 2B and C). There was no significant change in the effector populations of either T cell subset (Fig. 2D). Interestingly, the increased naïve CD4+ T cells resulted from a relative reduction in CD4+CD62L−CD44− T cells after HDL NP treatment (Fig. 2E). As these data correlate with an established mechanism through which MDSCs suppress T cell function, cleaving CD62L to prevent T cell homing to lymphoid organs for activation (37), we went on to further characterize HDL NP effects on MDSCs. Additionally, there was no difference in either M-MDSC (CD11b+Ly6C+) or PMN-MDSC (CD11b+Ly6G+) cells in lymph nodes after treatment (Supplementary Fig. S5). These data, and SCARB1 expression in CD11b+Ly6G+ and CD11b+Ly6C+ cells, suggests that HDL NPs interact directly with MDSCs to reduce their suppressive functions on T cells.
Figure 2.
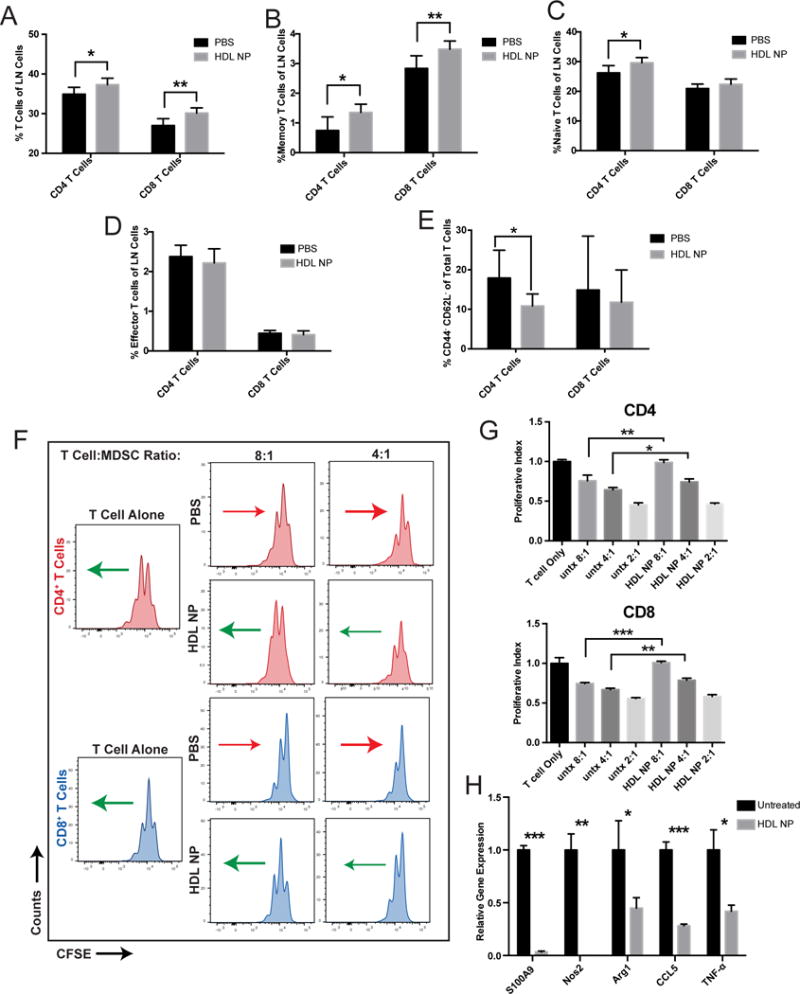
HDL NP treatment modulates systemic T cell distributions and inhibits MDSC suppression. A, After treating WT mice with either PBS or HDL NP (3×/week for 1 week), immune cell distributions in lymph nodes were analyzed by flow cytometry to quantify CD4+ and CD8+ T cells with regard to total T cells of each subtype, B, memory T cells, C, naïve T cells, D, effector T cells, E, and CD62L−CD44− naïve T cells. F, After CFSE staining, T cells were stimulated using anti-CD3/CD28 conjugated beads and co-cultured for 48 hours with isolated MDSCs at 4:1 and 8:1 ratios (T cells:MDSC) isolated from mice treated with HDL NP or PBS. Flow cytometry determined CFSE labeling in CD4+ and CD8+ T cells in order to evaluate suppressive effects of MDSCs. Arrows represent the general shift (red = less proliferation and green = more proliferation) in the CFSE signal of the cell population relative to its PBS or HDL NP treated counterpart. G, Quantification of the proliferative index, defined by the normalized number of T cell proliferations in F. H, After treating MDSCs with HDL NPs, gene expression was quantified by RT-PCR for S100A9, NOS2, Arg1, CCL5 and TNF-α with comparison made to PBS treated control MDSCs. A-H, P values: *P < 0.05, **P < 0.01 and ***P < 0.001 by two-tailed T test.
To test whether the measured differences in T cells were caused by an HDL NP-mediated reduction in MDSC activity, we first isolated MDSCs from tumor-free mice and matured them in vitro prior to measuring their functional capacity to suppress T cell proliferation. We measured the ability of the MDSCs to suppress CFSE-labeled T cell proliferation after stimulation with anti-CD3/CD28 conjugated beads. After 48 hours of culture in a 2:1 ratio of T cells to MDSCs, data reveal that HDL NPs significantly suppress the activity of MDSCs resulting in enhanced CD4+ and CD8+ T cell proliferation (Supplementary Fig. S6A). Next, we obtained similar data, but the MDSCs were isolated from mice with well-established melanoma lung metastases. Mice were treated 3 times with either PBS or HDL NPs (every 48 hours, 100 μl, 1 μM) whereupon MDSCs were isolated from the bone marrow, and then cultured with HDL NPs for 48 hours. As previously, CFSE-labeled T cells were stimulated with anti-CD3/CD28 conjugated beads. Data show that MDSCs only exposed to PBS considerably suppressed both CD4+ and CD8+ T cells at 2:1, 4:1, and 8:1 ratios (T cells:MDSCs), and HDL NP treatment substantially relieved the inhibitive effects and restored proliferation at each of the tested ratios (Fig. 2F, G and Supplementary Fig. S6B). Mechanistically, at the molecular level, HDL NPs caused a reduction in the expression of genes critical for MDSC-mediated T cell suppression including S100A9, inducible nitric oxide synthase (NOS2), Arginase-1 (ARG1), chemokine (C-C motif) ligand 5 (CCL5), and tumor necrosis factor alpha (TNF- α) as measured by quantitative RT-PCR (Fig. 2H). In order to verify that HDL NPs reduced MDSC suppression of T cell proliferation and the changes in MDSC gene expression were not caused by HDL NP cytotoxicity, we isolated MDSCs and treated them in vitro with HDL NPs at 50 nM, 10 nM and 2 nM concentrations for 48 hours. After the treatment period, cells were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain and the percentage of live cells was quantified using flow cytometry. Similar to in vivo results, the data show that HDL NPs did not cause a significant reduction in cell viability (Supplementary Fig. S7A, B).
HDL NPs reduce metastasis and increase survival in melanoma models
Given these data, we hypothesized that pre-treating a melanoma mouse model with HDL NPs would prime T cells to reduce melanoma lung metastasis. Accordingly, we systemically administered HDL NPs (3×/week for 1 week) to C57Bl/6 mice prior to systemic introduction of B16F10 melanoma cells (Fig. 3A). Two weeks later, mice were sacrificed and the metastatic burden was quantified in the lungs. Data demonstrated that HDL NP therapy drastically reduced lung metastasis with regard to the number and size of lesions (Fig. 3B–D). Notably, in vitro data demonstrate no direct cytotoxic effect of HDL or HDL NPs on B16F10 melanoma cells (Supplementary Fig. S8). To test if HDL NP pre-treatment translated to a significant increase in survival, the study was repeated and survival was measured. Data clearly show a significant survival advantage for mice treated with only three doses of HDL NPs prior to introduction of tumor cells (Fig. 3E). Finally, we utilized an orthotopic primary tumor model of melanoma to demonstrate that HDL NPs increased mouse survival as compared to PBS control treated mice. C57Bl/6 mice received intradermal injections of B16F10 melanoma cells. When the tumors became palpable, we treated the mice with intravenous injections of HDL NP (100 μl, 1 μM) or PBS (100 μl) every 48 hours. Data demonstrate a significant increase in survival of the HDL NP treated mice when compared to those treated with PBS (Supplementary Fig. S9). Together these data demonstrate that HDL NP treatment reduced melanoma metastasis and significantly increased survival.
Figure 3.
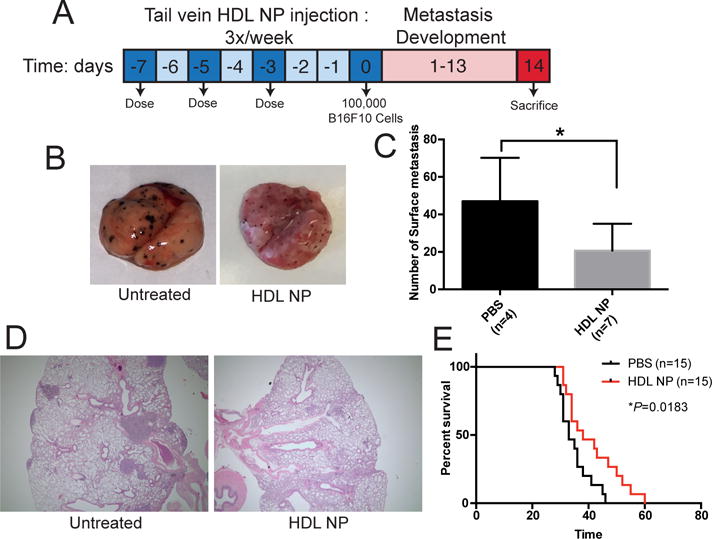
Metastasis and survival of mice pre-treated with HDL NPs prior to tumor cell administration. A, Time-line of C57Bl/6 metastatic lung colonization assay of mice treated with HDL NP (100 μl, 1 μM) or PBS (control) and inoculated with 1 × 105 B16F10 cells. B, Lung colonization 2-weeks after tumor inoculation. Surface metastases appear as darkly pigmented lesions. C, H&E staining of lungs of mice from B. D, Quantification of the number of surface metastases in B. E, Survival curve of PBS and HDL NP pre-treated mice after inoculation with B16F10 cells showing increased survival after HDL NP treatment (n=15 per group). *P = 0.0183 by Log-rank test. P values: *P < 0.05 by two-tailed T test.
SCARB1 is necessary for the anti-metastatic functions of HDL NPs
To unequivocally implicate SCARB1 targeting by HDL NPs, the in vitro suppression of SCARB1 −/− MDSCs on T cell proliferation was measured after HDL NP treatment compared to controls treated with PBS. SCARB1 −/− MDSCs inhibited T cell proliferation at all T cell:MDSC ratios (2:1, 4:1, 8:1), but, unlike MDSCs isolated from wild-type mice (Fig. 2F), HDL NP treatment did not inhibit MDSC activity and increase T cell proliferation when compared to controls (Fig. 4A, B and Supplementary Fig. S6B). These data demonstrate that HDL NPs function through SCARB1 to regulate MDSC function.
Figure 4.
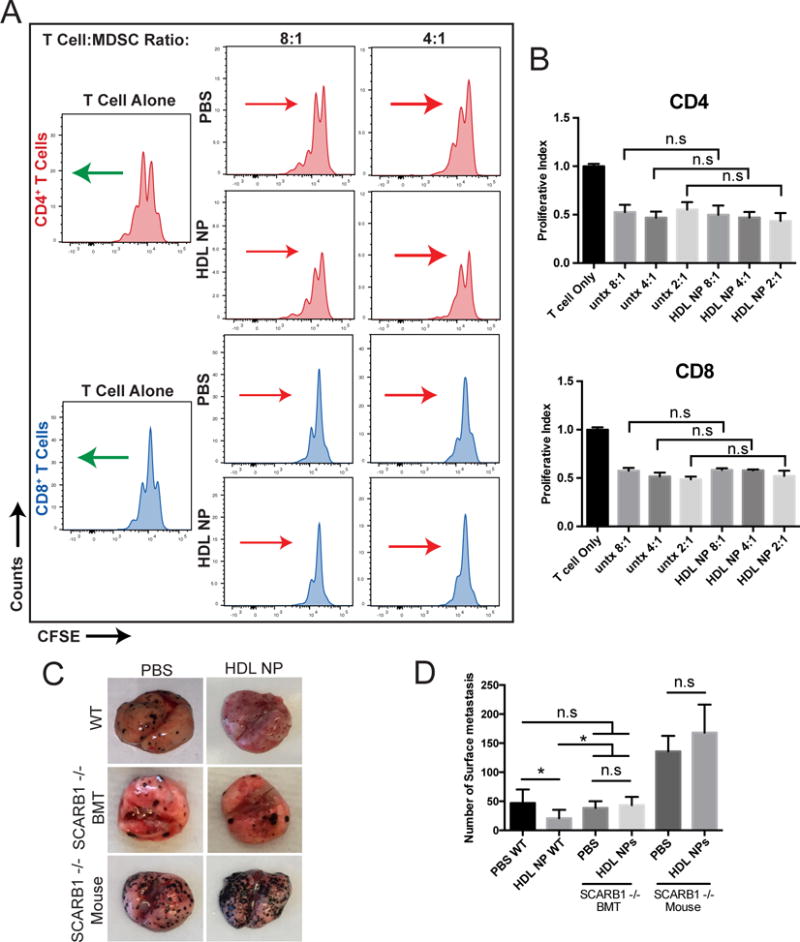
SCARB1 requirement for HDL NP activity. A, T cells stained with CFSE and stimulated with anti-CD3/CD28 conjugated beads were co-cultured for 48 hours with MDSCs isolated from tumor bearing SCARB1 −/− mice at ratios of 4:1 and 8:1 (T cell:MDSC). The mice had been treated with HDL NP or PBS. Flow cytometry determined CFSE labeling in CD4+ and CD8+ T cells in order to evaluate suppressive effects of SCARB1 −/− MDSCs. Arrows represent the general shift (red = less proliferation and green = more proliferation) in the CFSE signal of the cell population relative to its PBS or HDL NP treated counterpart. B, Quantification of the proliferative index, defined by the normalized number of T cell proliferations in A. C, B16F10 lung colonization in wild-type, SCARB1 −/− bone marrow transplant into wild-type mice (BMT), and global SCARB1 knockout mice with and without HDL NP pre-treatment. D, Quantification of the surface metastases in C. P values: **P < 0.01 and n.s.= not significant by two-tailed T test.
Further, in order to demonstrate the in vivo function of HDL NPs on MDSC activity using SCARB1 −/− mice, we applied the same pre-treatment regimen and melanoma model as was employed above for wild type mice. Convincingly, data showed that HDL NPs had no effect in SCARB1 −/− mice at reducing lung metastasis when compared to controls (Fig. 4C, D and Supplementary Fig. S10). In fact, both PBS control and HDL NP-treated SCARB1 −/− mice demonstrated increased metastasis compared to wild-type mice (Fig. 4C and D). The finding of increased metastasis may be due to the fact that SCARB1 −/− mice demonstrate high circulating HDL (38), which can bind SCARB1 expressed by the B16F10 cells and enhance metastasis and proliferation (39). To further explore, wild-type mice were lethally irradiated and then transplanted with bone marrow obtained from SCARB −/− mice. This allowed for the generation of wild-type mice with myeloid cells that do not express SCARB1 (BMT, Supplementary Fig. S11). Like in the SCARB1 −/− mice, HDL NP treatment did not reduce melanoma lung metastasis versus PBS in the BMT mice (Fig. 4C, D). Also, the number of lung metastases in both the PBS and HDL NP treated BMT groups were not statistically different from the numbers measured in wild-type mice treated with PBS, but were statistically different from the reduced numbers measured in the wild-type mice treated with HDL NP (Fig. 4D). Finally, transplanting the SCARB1 −/− bone marrow to wild-type mice reduced the number of metastases observed from the numbers measured in the mice with global SCARB1 knockout (Fig. 4D). In short, HDL NPs clearly and selectively target SCARB1 to release an innate immune checkpoint imparted by MDSCs.
Inhibition of MDSCs with HDL NPs leads to reduced metastasis and increased CD8+ T cell in the metastatic microenvironment
Next, we sought to demonstrate that HDL NP therapy administered after metastatic disease had been established would bolster T cell-mediated tumor clearance. To this end, we initiated melanoma lung metastasis by systemic administration of B16F10 cells, and waited 5 days to allow for development of florid lung metastases. We then treated the mice with HDL NPs (3×/week for two weeks) (Fig. 5A). After treatment, the mice were sacrificed and the lung metastatic burden was measured. In addition, immune cells in the tumor microenvironment were quantified using flow cytometry and immunohistochemistry. Data demonstrated reduced lung metastatic burden in mice treated with HDL NPs (Fig. 5B, C and Supplementary Fig. S12A–C). Furthermore, HDL NP treatment profoundly increased the number of cytotoxic CD8+ T cells, and decreased M-MDSCs both in the lung and in the metastatic tumor microenvironment as measured by flow cytometry and IHC, respectively (Fig. 5D–I). Furthermore, HDL NP treatment led to a reduction in CD4+ cells as measured by flow cytometry (Fig. 5E). Upon closer inspection by IHC, this was likely the result of a reduction in FOXP3+ T regulatory cells (Fig. 5H and J). Additionally, in the tumor microenvironment, we measured a decrease in the ratio of F4/80+ cells (macrophages) to CD3+ T cells after HDL NP treatment (Supplementary Fig. S13A and B). Collectively, these data demonstrate that HDL NPs potently enhance an adaptive immune response in the tumor microenvironment of established metastases and significantly reduce disease burden.
Figure 5.
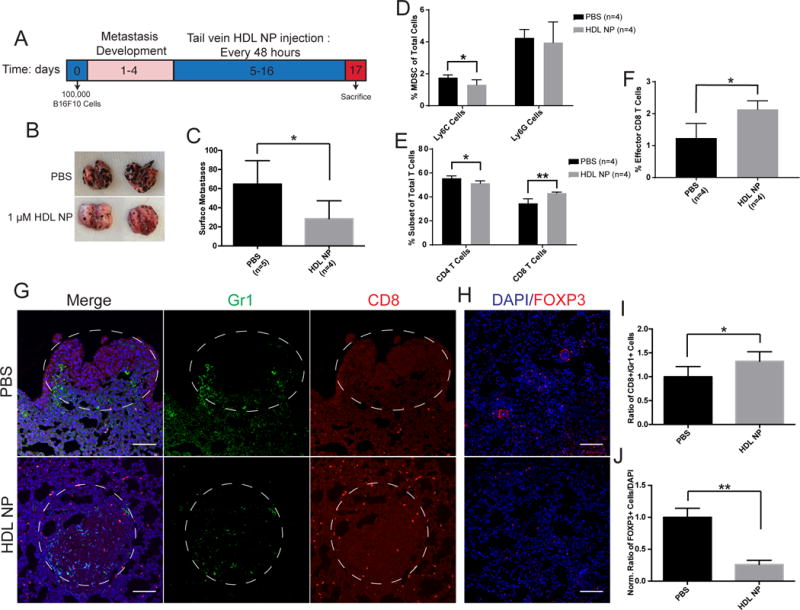
Effect of HDL NPs on established melanoma metastasis. A-J, B16F10 lung metastases were established for 5 days and then treatment was initiated with HDL NPs for two weeks. A, HDL NP treatment timeline. B, Representative images showing metastatic lung burden after HDL NP treatment. C, Quantification of lung metastases in B. D-F, Flow cytometry analysis of digested lungs of mice with B16F10 metastasis after HDL NP treatment D, Total percent PMN- and M-MDSCs. E, CD4+ and CD8+ T cell population subsets. F, Total effector CD8+ T cell percentage. G, IHC of lung sections for CD8+ T cell (red) and Gr-1 (green) tumor infiltration after HDL NP treatment in mice with metastases. H, IHC of lung sections showing T regulatory cells in the lungs of B16F10 metastasis bearing mice after HDL NP treatment. I, Quantification of CD8+:GR1+ cells infiltrating the metastatic tumor in G. J, Quantification of the number of T regulatory cells in H. C-J, P values: *P < 0.05 and **P < 0.01 by two-tailed T test.
HDL NPs significantly reduce Lewis lung carcinoma growth
In order to demonstrate that HDL NPs were effective in treating more than melanoma, we utilized another syngeneic murine model and measured tumor response with and without HDL NP treatment. Lewis lung carcinoma (LLC) xenografts were established in C57Bl/6 mice and the ability of systemically administered HDL NPs to slow LLC progression was tested. Tumors were initiated by injecting 5 × 105 LLC cells subcutaneously into the flank of each mouse. The tumors were measured 8 days following the initial injection and every 48 hours thereafter. On day 9, once the tumors reached ~50 mm3, the mice were treated intravenously with 200 μl of 1 μM HDL NPs for a total of 5 doses over the subsequent 10 days. Data show a significant reduction in the volume and weight of the tumors after treatment with HDL NPs (Fig. 6A–C). HDL NPs are not inherently toxic to cultured LLC cells (Supplementary Fig. S14). These data are noteworthy because they demonstrate that HDL NPs can effectively slow the progression of multiple cancer types.
Figure 6.
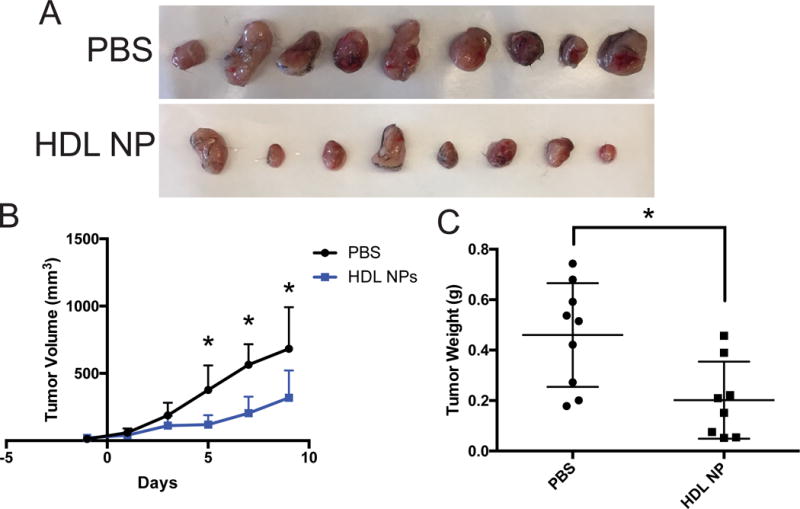
HDL NPs inhibit Lewis Lung Carcinoma tumor growth. A-C, LLC tumor xenograft study in C57Bl/6 mice. A, LLC tumors excised from C57Bl/6 mice following treatment with phosphate buffered saline (PBS) or HDL NPs. B, LLC growth curve as measured by volume (mm3) over the HDL NP treatment course. C, The weight (g) of the LLC tumors shown in figure A. B and C, P values: *P < 0.05 by two-tailed T test.
DISCUSSION
High-density lipoprotein nanoparticles are capable of therapeutically activating an adaptive immune response by targeting SCARB1 expressed by MDSCs. Using HDL NPs, our data are the first to implicate SCARB1 expression by MDSCs as a target for therapy. Data in healthy mice reveal that HDL NP targeting of MDSCs may modulate the balance of the immune system to reduce immune suppression and activate T cell-mediated immunity. Further, in vitro data using MDSCs obtained from mice with existing melanoma lung metastases support that HDL NP targeting of SCARB1 reduces MDSC-mediated suppression and enhances CD8+ and CD4+ T cell proliferation, with a more pronounced effect on the CD8+ T cells (40). In vivo data collected in murine models of cancer clearly show that HDL NP binding to SCARB1 reduces the ability of MDSCs to inhibit T cell mediated immune responses to cancer through molecular mechanisms, summarized in Fig. S15, which include reduced MDSC expression of ARG1, NOS2, and CCL5. SCARB1 targeting data were confirmed using MDSCs isolated from SCARB1 −/− mice using in vitro and in vivo model systems. Ultimately, from a therapeutic standpoint, HDL NP treatment significantly reduced tumor growth, metastatic tumor burden, and increased survival by enhancing adaptive immunity.
With regard to HDL metabolism, our data are consistent with other studies that employed apoA-I knockout mice demonstrating that these mice were much more prone to tumor growth and metastasis. In this model, apo A-I infusions led to reduced metastasis and tumor burden (19, 20). However, the mechanism through which apo A-I achieved this response was not identified. Additional studies implicate the other two known transporters of cholesterol to HDLs, ATP binding cassette transporters A1 and G1 (ABCA1 and ABCG1), in the proliferative abilities of hematopoietic stem cells with an emphasis on myeloid progenitor cells (17). Data showed that proliferation of these cells was increased in mice lacking both ABCA1/ABCG1, and reduced myeloid progenitor cell proliferation was achieved by infusing HDL, but not apo A-I. Finally, data from humans suggest that serum levels of HDLs and apo A-I correlate with a reduced incidence of cancer, metastasis, and cancer-associated mortality (21). Collectively, these findings substantiate our data demonstrating that HDL NPs target SCARB1 expressed by MDSCs, reduce their function, and enhance anti-tumor immunity.
High-density lipoproteins function to transport cholesterol, and cholesterol metabolism is known to play a critical role in the proliferation of myeloid cells (41). For instance, patients receiving HMG-CoA reductase inhibitors (i.e. “statins”), which inhibit the rate-limiting step in de novo cholesterol synthesis, have reduced neutrophil and monocyte counts, further demonstrating the importance of cholesterol metabolism to myeloid cell homeostasis (42). Utilizing the HDL NP, a mimic of mature spherical HDL that binds SCARB1, enabled us to identify and implicate this receptor on MDSCs. The potency of the HDL NP at reducing MDSC functions is attributed to tight binding of SCARB1 imparted by the gold nanoparticle core that stabilizes the HDL NP size and shape, which has been shown to potently reduce cellular cholesterol (22, 26). Therefore, specifically reducing cellular cholesterol by targeting SCARB1 with HDL NPs has great potential to suppress the function of MDSCs.
Our data, and work by others (43), demonstrate that, in addition to MDSCs, macrophages express SCARB1. In the tumor microenvironment macrophages can be polarized to an M1, tumor inhibitive, or M2, tumor promoting, phenotype (44). The current work does not address differences in macrophage polarization in the tumor microenvironment due to HDL NP treatment. However, data obtained using the established melanoma tumor model show that HDL NP treatment reduced the F4/80+:CD3 cell ratio in the tumor microenvironment, and did not appear to change the number of F4/80+ cells in the tumor microenvironment. Further studies of HDL NPs on macrophages and macrophage polarization are underway.
Targeting MDSCs can be an extremely effective cancer treatment especially in combination with other drugs (45). Recently, researchers have shown that suppressing MDSCs with non-specific DNA methyltransferase and histone deacetylase inhibitors can eradicate tumors resistant to conventional checkpoint blockade (46). There has also been significant discussion focused on preventing the recruitment of MDSCs to the tumor site by targeting the chemokine receptors CXCR2 and CXCR4 and colony stimulating factor 1 receptor (CSF1R) (47). Ultimately, targeting MDSCs and inhibiting their suppressive functions is a promising treatment, especially alongside other immune activators, to treat cancer. Certainly, exquisite targeting of SCARB1 by HDL NPs provides a new strategy to reduce MDSC activity and enhance antitumor immunity.
In summary, our data conclusively demonstrate that SCARB1 is expressed by MDSCs. Engaging this receptor by HDL NPs inhibits MDSC function and activates an adaptive immune response to cancer. Interestingly, our data also point to the importance of cholesterol metabolism to MDSC biology. From a therapeutic standpoint, HDL NPs may augment the efficacy of existing immunotherapies whose function is drastically reduced by MDSCs. Therefore, combination therapies using HDL NPs with conventional checkpoint inhibitors or cell therapies, may provide significant therapeutic benefit. In short, SCARB1-targeted HDL NP therapy may be effective for multiple diseases, like cancer, potentiated by MDSCs.
Supplementary Material
Acknowledgments
CST thanks the Air Force Office of Scientific Research (FA95501310192) for grant funding; H Foundation NCI Stimulus Award from the Robert H. Lurie Comprehensive Center at Northwestern University; and, grant funding from the National Cancer Institute (R01CA167041). Also, we gratefully acknowledge the Robert H. Lurie Comprehensive Cancer Center Flow Cytometry Core and the Northwestern University Center for Advanced Microscopy for technical assistance. We kindly thank Praveen Thumbikat and Chonghui Cheng for reading the manuscript and offering important insight.
Footnotes
Disclosure of Potential Conflicts of Interest: CST is the founder of AuraSense, LLC that licensed the HDL NP technology from Northwestern University.
References
- 1.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalos M, et al. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Science Translational Medicine. 2011;3(95) doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief AJM. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. 2016;16(4):219–233. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- 4.Snyder A, et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. New Engl J Med. 2014;371(23):2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 Blockade: New Immunotherapeutic Modalities with Durable Clinical Benefit in Melanoma Patients. Clin Cancer Res. 2013;19(19):5300–5309. doi: 10.1158/1078-0432.CCR-13-0143. [DOI] [PubMed] [Google Scholar]
- 6.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016;37(3):208–220. doi: 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu C, Workman CJ, Vignali DAA. Targeting regulatory T cells in tumors. Febs J. 2016;283(14):2731–2748. doi: 10.1111/febs.13656. [DOI] [PubMed] [Google Scholar]
- 9.Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circulation Research. 2005;96(12):1221–1232. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 10.Rosenson RS, et al. Cholesterol Efflux and Atheroprotection Advancing the Concept of Reverse Cholesterol Transport. Circulation. 2012;125(15):1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nieland TJF, Penman M, Dori L, Krieger M, Kirchhausen T. Discovery of chemical inhibitors of the selective transfer of lipids mediated by the HDL receptor SR-BI. P Natl Acad Sci USA. 2002;99(24):15422–15427. doi: 10.1073/pnas.222421399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shih AY, Sligar SG, Schulten K. Maturation of high-density lipoproteins. J R Soc Interface. 2009;6(39):863–871. doi: 10.1098/rsif.2009.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kozarsky KF, et al. Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol levels. Nature. 1997;387(6631):414–417. doi: 10.1038/387414a0. [DOI] [PubMed] [Google Scholar]
- 14.Murphy AJ, et al. Neutrophil activation is attenuated by high-density lipoprotein and apolipoprotein A-I in in vitro and in vivo models of inflammation. Arterioscler Thromb Vasc Biol. 2011;31(6):1333–1341. doi: 10.1161/ATVBAHA.111.226258. [DOI] [PubMed] [Google Scholar]
- 15.Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12(1):10–17. doi: 10.1038/nrcardio.2014.173. [DOI] [PubMed] [Google Scholar]
- 16.Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15(2):104–116. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yvan-Charvet L, et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328(5986):1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang J, et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci Adv. 2015;1(3) doi: 10.1126/sciadv.1400223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zamanian-Daryoush M, et al. The cardioprotective protein apolipoprotein A1 promotes potent anti-tumorigenic effects. The Journal of biological chemistry. 2013;288(29):21237–21252. doi: 10.1074/jbc.M113.468967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su F, et al. Apolipoprotein A-I (apoA-I) and apoA-I mimetic peptides inhibit tumor development in a mouse model of ovarian cancer. Proc Natl Acad Sci U S A. 2010;107(46):19997–20002. doi: 10.1073/pnas.1009010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandler PD, et al. Lipid biomarkers and long-term risk of cancer in the Women’s Health Study. Am J Clin Nutr. 2016;103(6):1397–1407. doi: 10.3945/ajcn.115.124321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plebanek MP, et al. Nanoparticle Targeting and Cholesterol Flux Through Scavenger Receptor Type B-1 Inhibits Cellular Exosome Uptake. Sci Rep-Uk. 2015;5 doi: 10.1038/srep15724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Angeloni NL, et al. Pathways for Modulating Exosome Lipids Identified By High-Density Lipoprotein-Like Nanoparticle Binding to Scavenger Receptor Type B-1. Sci Rep-Uk. 2016;6 doi: 10.1038/srep22915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luthi AJ, et al. Tailoring of Biomimetic High-Density Lipoprotein Nanostructures Changes Cholesterol Binding and Efflux. Acs Nano. 2012;6(1):276–285. doi: 10.1021/nn2035457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thaxton CS, Daniel WL, Giljohann DA, Thomas AD, Mirkin CA. Templated Spherical High Density Lipoprotein Nanoparticles. J Am Chem Soc. 2009;131(4):1384–+. doi: 10.1021/ja808856z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang S, et al. Biomimetic, synthetic HDL nanostructures for lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(7):2511–2516. doi: 10.1073/pnas.1213657110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McMahon KM, et al. Synthetic high-density lipoproteins as targeted monotherapy for chronic lymphocytic leukemia. Oncotarget. 2017;8(7):11219–11227. doi: 10.18632/oncotarget.14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mieszawska AJ, Mulder WJ, Fayad ZA, Cormode DP. Multifunctional gold nanoparticles for diagnosis and therapy of disease. Mol Pharm. 2013;10(3):831–847. doi: 10.1021/mp3005885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Eck M, Pennings M, Hoekstra M, Out R, Van Berkel TJC. Scavenger receptor BI and ATP-binding cassette transporter A1 in reverse cholesterol transport and atherosclerosis. Curr Opin Lipidol. 2005;16(3):307–315. doi: 10.1097/01.mol.0000169351.28019.04. [DOI] [PubMed] [Google Scholar]
- 30.Rigotti A, et al. Scavenger receptor BI - a cell surface receptor for high density lipoprotein. Curr Opin Lipidol. 1997;8(3):181–188. doi: 10.1097/00041433-199706000-00009. [DOI] [PubMed] [Google Scholar]
- 31.Acton S, et al. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271(5248):518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 32.Mooberry LK, Sabnis NA, Panchoo M, Nagarajan B, Lacko AG. Targeting the SR-B1 Receptor as a Gateway for Cancer Therapy and Imaging. Front Pharmacol. 2016;7 doi: 10.3389/fphar.2016.00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008 doi: 10.1002/0471142735.im1401s83. Chapter 14: Unit 14 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bantikassegn A, Song X, Politi K. Isolation of epithelial, endothelial, and immune cells from lungs of transgenic mice with oncogene-induced lung adenocarcinomas. Am J Respir Cell Mol Biol. 2015;52(4):409–417. doi: 10.1165/rcmb.2014-0312MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quah BJ, Warren HS, Parish CR. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat Protoc. 2007;2(9):2049–2056. doi: 10.1038/nprot.2007.296. [DOI] [PubMed] [Google Scholar]
- 36.McMahon KM, Plebanek MP, Thaxton CS. Properties of Native High-Density Lipoproteins Inspire Synthesis of Actively Targeted In Vivo siRNA Delivery Vehicles. Adv Funct Mater. 2016;26(43):7824–7835. doi: 10.1002/adfm.201602600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J Immunol. 2009;183(2):937–944. doi: 10.4049/jimmunol.0804253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yesilaltay A, et al. Excess cholesterol induces mouse egg activation and may cause female infertility. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(46):E4972–E4980. doi: 10.1073/pnas.1418954111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jani JP, et al. Metastasis of B16F10 mouse melanoma inhibited by lovastatin, an inhibitor of cholesterol biosynthesis. Invasion Metastasis. 1993;13(6):314–324. [PubMed] [Google Scholar]
- 40.Nagaraj S, Gabrilovich DI. Regulation of suppressive function of myeloid-derived suppressor cells by CD4+ T cells. Semin Cancer Biol. 2012;22(4):282–288. doi: 10.1016/j.semcancer.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sica A, Strauss L. Energy metabolism drives myeloid-derived suppressor cell differentiation and functions in pathology. J Leukocyte Biol. 2017;102(2):325–334. doi: 10.1189/jlb.4MR1116-476R. [DOI] [PubMed] [Google Scholar]
- 42.Tani S, et al. Association of Leukocyte Subtype Counts With Coronary Atherosclerotic Regression Following Pravastatin Treatment. Am J Cardiol. 2009;104(4):464–469. doi: 10.1016/j.amjcard.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 43.Ji A, et al. Scavenger receptor SR-BI in macrophage lipid metabolism. Atherosclerosis. 2011;217(1):106–112. doi: 10.1016/j.atherosclerosis.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dominguez GA, et al. Selective Targeting of Myeloid-Derived Suppressor Cells in Cancer Patients Using DS-8273a, an Agonistic TRAIL-R2 Antibody. Clin Cancer Res. 2017;23(12):2942–2950. doi: 10.1158/1078-0432.CCR-16-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim K, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A. 2014;111(32):11774–11779. doi: 10.1073/pnas.1410626111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.