

Abstract
The design and synthesis of a library of forty novel 2-aminoazole analogues as well as their evaluation as antifungal compounds against Histoplasma capsulatum and Cryptococcus neoformans is described. These structures were derived from N-[5-(1-naphthalenylmethyl)-2-thiazolyl]cyclohexanecarboxamide (41F5), a fungistatic agent previously identified through phenotypic screening (Antimicrob Agents Chemother. 2013; 57:4349). Modifications to improve potency and water-solubility of 41F5 focused primarily on the 5-naphthalenyl group, the thiazole core, and the methylene linker between these two structural elements. In general, compounds with lipophilic [5+6] bicyclics ring systems, such as the 7-benzothiophenyl- and 4-indanyl groups, at the 5-position were 2–3 times more active against both fungal species as compard to 41F5. Also, introduction of a carbonyl group at the methylene linker of 41F5 resulted in a 2–3 fold increase in potency. These highly active compounds also showed generally low toxicities against murine P388D1 macrophages resulting in selectivity indices ranging from 63 to >200. Compounds that were highly active against fluconazole-sensitive C. neoformans strains had almost identical activity against fluconazole-resistant variants of this fungus indicating that 14α-demethylase is not their molecular target. Highly active compounds also retained activity against H. capsulatum phagocytosed into P388D1 macrophages.
Keywords: Aminothiazoles, Antifungal activity, Structure-Activity-Relationship, Histoplasma capsulatum, Cryptococcus neoformans
Graphical abstract
1. Introduction
Histoplasma capsulatum, the fungal pathogen causing histoplasmosis, is endemic to the Ohio- and Mississippi River valleys of North America as well as parts of Latin American and Africa, where up to 90% of the residents display signs of prior infection.1 Up to 50 million people have had contracted H. capsulatum and an estimated 500,000 cases of new infections occur annually in the US.2,3 Fortunately, most infections are subclinical but over 3,000 hospitalizations occur per year.4 Histoplasmosis is the most common cause for hospitalization among endemic fungal disease.4 The cost for each hospitalization is estimated to be $20,300 for children and $17,000 for adults.4 Cryptococcus neoformans, the culprit of cryptococcosis, affects primarily the immunocompromised population in sub-Saharan Africa, where the human immunodeficiency virus (HIV) burden is the highest in the world. In sub-Saharan Africa, the approximate number of cases of cryptococcal meningitis is 720,000 and the 90-day case fatality is 70%.5–8
Major classes of current antifungal drugs include allylamines, azoles, pyrimidine analogues, polyenes, echinocandins, and oxaboroles.9,10 These agents have different molecular targets found in the fungal cell membrane or in fungal biochemical processes.11 All clinically utilized antifungals suffer from side effects and limited activity spectra.9,10,12–20 In particular, echinocandins, which have the least host toxicity potential, are ineffective against H. capsulatum and C. neoformans.12–14 Therefore, it is important to explore novel structural classes for the development of antifungal agents.
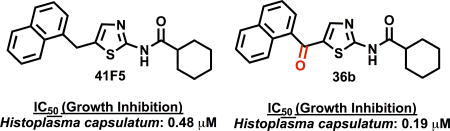
Edwards et al. performed a phenotypic screening of a purinome-focused library of 3600 commercially available compounds against H. capsulatum to find novel antifungal candidates and identified 41F5, which has a thiazole core structure, as the most active compound of this library (Table 1).21 This agent displayed fungistatic activity with an IC50 value of 0.87 µM against H. capsulatum and a selectivity index of ~63 over macrophages. It was also found that 41F5 is active against C. neoformans with an IC50 of 1.25 µM but 41F5 was inactive against the fungi Candida albicans, Aspergillus fumigatus and Blastomyces dermatitidis.21 41F5 did not enhance sensitivity of H. capsulatum to fluconazole suggesting the target of 41F5 differs from the cytochrome P450 sterol 14α-demethylase inhibited by azole-class antifungals.22
Table 1.
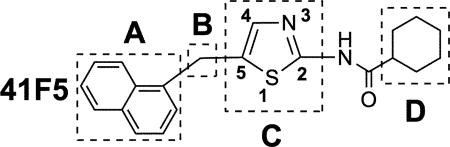
Alignment of research objectives with areas of structural modifications at the 41F5 structure and the numbering system for all target compounds
| ||
|---|---|---|
|
| ||
| Objective | Areas of chemical modifications |
Target compounds |
| 1 (Increased potency against H. capsulatum and C. neoformans) | A, B, C, and D | All the target compounds (14–33, 36, 38, 40) |
| 2 (Increased water solubility) | A and C | 28 (quinoline-substituted); 29, 31, 32 (pyridine-substituted); 40 (imidazole-substituted) |
| 3 (Molecular target identification) | A, B and D | 14, 15, 33 (ester-modified at 2- or 5-substituents) |
The thiazole core structure has been utilized as a pharmacophore in a number of biologically active agents.23 Examples are the anticancer agents tiazofurin and dasatinib, the anti-HIV agent ritonavir, the antiparasitic drug nitazoxanide, the anti-inflammatory agents fanetizole, meloxicam, and fentiazac, the antiulcer agent nizatidine, and the insecticide thiamethoxam.24–26 Abafungin is an antifungal drug that contains a thiazole ring systems but is otherwise structurally different from 41F5. This compound has inhibitory activity against sterol-C24-methyltransferase and can directly damage fungal cell membrane.27 Abafungin is fungicidal rather than fungistatic, and it is active against Candida albicans and Aspergillus fumigatus.24 Therefore, it was suggested that abafungin and 41F5 have different mechanisms of action.21
Khalil et al. synthesized 68 analogues of 41F5 to improve potency against H. capsulatum and C. neoformans and to develop a structure-activity relationship (SAR) for 41F5-derived antifungal compounds.22 Unfortunately, compounds with higher potency than 41F5 were not identified. Therefore, we synthesized and evaluated further analogues of 41F5 with the intention to improve potency against H. capsulatum and C. neoformans and to enhance water solubility. Another objective was the synthesis of 41F5 analogues that have the potential to be used for molecular target identification by unbiased affinity chromatography techniques. The results of these studies are presentend in this paper.
2. Results
2.1. Design strategies
Table 1 summarizes the primary objectives of our studies and aligns these with areas of structural modifications of the 41F5 structure and the numbering system for all target compounds. Analogues of 41F5 were synthesized to (1) improve potency against H. capsulatum and C. neoformans yeasts, (2) enhance water solubility, and (3) identify the molecular target(s) of 41F5-derived antigfungal compounds, which is(are) currently unknown. Additional related objectives were the evaluation of compound toxicities against fluconazole-resistant C. neoformans yeast, murine macrophages, and H. capsulatum phagocytosed into murine macrophages.
The “A” group was extensively modified to achieve all the three objectives (Table 1). In order to investigate the effect of the size on antifungal activity, the naphthalenyl group at the 5-position was replaced with tricyclic rings (fluorenyl), bicyclic rings (benzothiophenyl, benzofuranyl, 2,1,3-benzothiadiazolyl, indanyl, quinolinyl and tetralinyl), monocyclic rings (pyridinyl, cyclopentyl, and ester-modified phenyl) or the methyl group. Bioisosteric considerations played an important role in modifying this group (objective 1).28–30 To potentially improve water solubility (objective 2), pyridine- (pKa = 5.2) (29a/b, 31, 32a/b)31 and quinoline substituents (pKa = 4.85) (28a/b)32 were introduced at the 5-position. The phenyl ring at the 5-position of the thiazole core was substituted with an ester group either at the 3- or 4-position (objective 3). Using standard chemical methodology, such an ester function could be converted to an alkyne-containing amide group, which could be coupled to an azide-containing solid support matrix via click chemistry. A stationary phase modified in this way could potentially be used in an unbiased affinity chromatography approach for molecular target identification,33 which may have the potential to facilitate the structure-based design of 41F5-derived antifungal compounds. A crucial milestone in pursuing this methodololgy would be to initially evaluate the toxicity of ester containing compounds against H. capsulatum and C. neoformans. If such compounds would have significantly reduced activity as compared to 41F5, the fairly bulky ester group must interfere with binding to the target protein. In such a case, it would be reasonable to assume 5 that further increased steric hindrance by subsequent chemical modifications would be even more detrimental to binding, thus, rendering this affinity chromatography approach futile. In the “B” area, the methylene linker at the 5-position was replaced with a carbonyl group in order to explore the nature of the linker on potency (objective 1). In the “C” area, other heteroaromatic ring systems, such as oxazole (38a and 38b) and imidazole (40) were introduced to address objective 1. In addition, imidazole (pKa = 6.95)34 was also chosen as a core replacement to explore objective 2. Similar to the attachment of an ester group to a phenyl group at the 5-position (“A” area), modifications of the “D” area encompassed introduction of ester groups to a cyclohexane ring at the 2-position (33a, 33b and 33c) to potentially explore affinity chromatography approaches for molecular target identification (objective 3).
2.2. Chemistry
2.2.1. Precursor synthesis
Compound 2 (Scheme 1) was synthesized from commercially-available compound 1 as a precursor molecule according to a published procedure.35 This method was then adapted for the synthesis of 4 from commercially available compound 3. Briefly, the reactions of ethyl 4-aminobenzoate (1) and ethyl 3-aminobenzoate (3) with sodium nitrite resulted in the formations of corresponding diazonium salts, which were subsequently exposed to acrolein in the presence of copper(II) chloride dihydrate to yield the α-chloro aldehyde intermediates 1i and 3i. Cyclization of these intermediates with thiourea afforded products 2 and 4 in 21% and 22% over all yield, respectively. Intermediates 1i and 3i were not isolated and converted in situ to their respective end products 2 and 4.
Scheme 1.

Reagents and conditions: (a) NaNO2, 18% HCl, H2O, −5 °C, 24 h; (b) Acrolein, CuCl2 × 2 H2O, CaO, acetone, 5 °C, 2 h; (c) Thiourea, ethanol, reflux, 24 h.
Commercially available compound 5 was lithiated using lithium diisopropyl amide (LDA) followed by reaction with aldehydes to afford 6a–o in yields ranging from 9% to 81% (Scheme 2).36 Compounds 6a–l and 6o were converted to 7a–l and 7o by reduction of secondary alcohols and deprotection of t-butoxycarbonyl (Boc) with triethylsilane and trifluoroacetic acid (TFA), respectively, in yields ranging from 14% to 88%.22 Compounds 7m and 7n were prepared by simultaneous reduction of the hydroxyl group and removal of the Boc protective group of 6m and 6n using hydriodic acid in 10% and 43% yields, respectively.37 Boc-deprotection and deoxygenation of 6i afforded not only 7i but also 7i’ in a molar ratio of 84:16 as determined by 1H NMR and HR-ESI-MS (Scheme 2). It was not possible to separate this mixture by column chromatography. The deoxygenation of thiophene derivatives with triethylsilane in the presence of strong acids has been reported previously.38,39
Scheme 2.

Reagents and conditions: (a) LDA, THF, −78 °C, 30 min; (b) R1CHO, r.t., 16 h; (c) Et3SiH, TFA, DCM, r.t., 16 h; (d) 57% aq. HI, acetic acid, 100 °C, 4 h; *compound was purchased.
Aldehyde 9 was synthesized from commercially available compound 8 according to a published procedure (Scheme 3),40 which was adapted for the synthesis of 1241 from starting material 11. Briefly, 3- (8) and 4-pyridinepropanol (11) were oxidized with Dess-Martin periodinane to afford 9 and 12 in 43% and 44% yield, respectively. Compounds 9 and 12 were then α-brominated with t-butyl bromide and dimethyl sulfoxide (DMSO) followed by cyclization with thiourea to afford 1040 and 13 in 15% and 13% yield, respectively.
Scheme 3.

Reagents and conditions: (a) Dess-Martin periodinane, 4Å molecular sieves, DCM, r.t., 16 h; (b) t-BuBr, DMSO, acetonitrile, 65 °C, 16 h; (c) Thiourea, ethanol, reflux, 16 h.
2.2.1. Target compound synthesis
Reactions of the precursor molecules 2, 4, 7a–n, 7p, 10 and 13 containing free amino groups with acid chlorides in the presence/absence of triethylamine (Scheme 4) yielded target compounds 14–32. As 7i was contaminated with 7i’ (vide supra), two target compounds 24 and 24’ were produced in molar ratio of 93:7 as determined by 1H NMR (SM-S.6.17.1) and HR-ESI-MS (SM-S.6.17.3). As in the case of the precursor molecules, it was not possible to separate this mixture by column chromatography. Precursor molecule 7o was coupled with commercially-available ester-substituted cyclohexane-1-carboxylic acids in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI)22 to afford 33a, 33b and 33c in 10%, 16% and 9% yield, respectively (Scheme 5). Carboxylic acids were used in these experiments because the corresponding acid chlorides were not commercially available.
Scheme 4.

Reagents and conditions: (a) R2COCl, Et3N, THF, r.t., 15 min; (b) R2COCl, THF, r.t., 15 min.
Scheme 5.

Reagents and conditions: (a) (1R/S,4R/S)-4-(methoxycarbonyl)cyclohexane-1-carboxylic acid or (1R,4R)-4-(methoxycarbonyl)cyclohexane-1-carboxylic acid or (1R,2S)-2-(methoxycarbonyl)cyclohexane-1-carboxylic acid, EDCI, DMAP, Et3N, DCM/DMF (3:1, v/v), r.t., 16 h.
The secondary alcohol of compound 6o was oxidized with Dess-Martin periodinane to a carbonyl group to afford 34 in 54% yield (Scheme 6)42 followed by the removal of the Boc protective group with trifluoroacetic acid (TFA) to yield 35 in 82% yield. Compounds 36a and 36b were synthesized from 35 in 53% and 76% yields, respectively, using the method described for the synthesis of 14–27, and 30a/b.
Scheme 6.

Reagents and conditions: (a) Dess-Martin periodinane, DCM, r.t., 16 h; (b) TFA, DCM, r.t., 16 h; (c) 3-Cyclohexylpropanoyl chloride or cyclohexanecarbonyl chloride, Et3N, THF, r.t., 15 min.
The oxazole derivatives 38a and 38b (Scheme 7) were prepared from a reported compound 3743 in 8% and 27% yields, respectively, using the method described for synthesis of 14–27, 30a/b, and 36a/b. Imidazole derivative 40 was synthesized in 65% yield from commercially available cyclohexanecarboxylic acid 39 by reaction with hydroxybenzotriazole (HOBt) in the presence of EDCI hydrochloride followed by reaction with commercially available 5-benzyl-1H-imidazol-2-amine hydrochloride (Scheme 8). Acylation occurred exclusively at the amino group under the applied reaction conditions.
Scheme 7.

Reagents and conditions: (a) 3-Cyclohexylpropanoyl chloride or cyclohexanecarbonyl chloride, Et3N, THF, r.t., 15 min.
Scheme 8.

Reagents and conditions: (a) HOBt, Et3N, EDCI hydrochloride, acetonitrile, r.t., 10 min; (b) 5-Benzyl-1H-imidazol-2-amine HCl, 80 °C, 16 h.
2.3. Biology
Compounds A–C and 41F5, previously reported by Khalil et. al,22 were used in our biological studies as general reference compounds because they showed significant antifungal activity and are structurally similar to the newly synthesized compounds (Table 2). The cytoxicities of target compounds (14–33, 36, 38 and 40) were evaluated in vitro against H. capsulatum and C. neoformans yeasts as well as in murine P388D1 macrophages and the obtained data are summarized in the Tables 3–5.
Table 2.
Highly active thiazole antigfungal compounds synthesized and evaluated by Khalil et al.22
| Cmpd |
|
H. capsulatum |
C. neoformans (H99 strain) |
|
|---|---|---|---|---|
|
| ||||
| R1 | R2 | IC50 (µM) | IC50 (µM) | |
| 41F5 |
|
|
0.4 | 0.4 |
| A |
|
|
0.4 | > 10 |
| B |
|
|
1.6 | 1.3 |
| C |
|
|
0.7 | > 10 |
Table 3.
Activity of 41F5-derived antifungals with bicyclic ring systems at the 5-position
|
H. capsulatum |
C. neoformans (H99 strain) |
P388D1 | SI (Hc) |
SI (Cn) |
||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Cmpd | R1 | R2 | IC50 (µM) [±SD] |
MIC (µM) |
IC50 (µM) [±SD] |
MIC (µM) |
IC50 (µM) [±SD] |
||
| A |
|
|
0.60 [±0.03] | 1.25 | > 20 | > 40 | 27.59 [±4.12] | 46 | nd |
| 41F5 |
|
|
0.48 [±0.01] | 1.25 | 0.67 [±0.06] | 1.25 | > 40 | > 83 | > 60 |
| 17a |
|
|
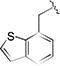
0.92 [±0.38] | 5.00 | > 20 | > 40 | 18.87 [±3.05] | 21 | nd |
| 17b |
|
|
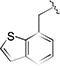
0.20 [±0.01] | 0.31 | 0.34 [±0.04] | 0.63 | > 40 | > 200 | > 118 |
| 20a |
|
|
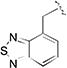
> 20 | > 40 | > 20 | > 40 | nd | nd | nd |
| 20b |
|
|
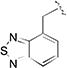
1.54 [±0.42] | 2.50 | 1.52 [±0.22] | 5.00 | > 40 | > 26 | > 26 |
| 21a |
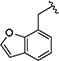
|
|
0.40 [±0.01] | 0.63 | > 20 | > 40 | 17.75 [±5.00] | 44 | nd |
| 21b |
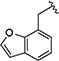
|
|
0.42 [±0.01] | 1.25 | 0.62 [±0.12] | 2.50 | > 40 | > 95 | > 65 |
| 22a |
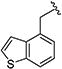
|
|
0.20 [±0.01] | 0.31 | > 20 | > 40 | > 40 | > 200 | nd |
| 22b |
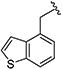
|
|
0.20 [±0.01] | 0.31 | 0.37 [±0.09] | 1.25 | > 40 | > 200 | > 108 |
| 23a |
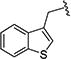
|
|
0.27 [±0.02] | 0.63 | > 20 | > 40 | 16.90 [±1.75] | 63 | nd |
| 23b |
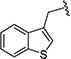
|
|
> 20 | > 40 | > 20 | > 40 | nd | nd | nd |
| 24 |
|
|
> 20 | > 40 | > 20 | > 40 | nd | nd | nd |
| 26a |
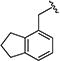
|
|
0.38 [±0.02] | 0.63 | > 20 | > 40 | > 40 | > 105 | nd |
| 26b |
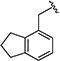
|
|
0.25 [±0.02] | 0.63 | 0.41 [±0.07] | 1.25 | > 40 | > 160 | > 98 |
| 27a |
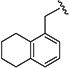
|
|
0.90 [±0.29] | 2.50 | > 20 | > 40 | 24.27 | 27 | nd |
| 27b |
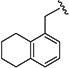
|
|
> 20 | > 40 | > 20 | > 40 | nd | nd | nd |
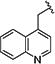
| 28a |
|
|
> 20 | > 40 | > 20 | > 40 | nd | nd | nd |
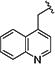
| 28b |
|
|
3.05 [±0.25] | 10.00 | 3.24 [±0.42] | 10.00 | 13.92 [±1.19] | 5 | 5 |
| 36a |
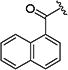
|
|
0.64 [±0.03] | 1.25 | > 20 | > 40 | > 40 | > 63 | nd |
| 36b |
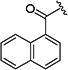
|
|
0.19 [±0.01] | 0.31 | 0.89 [±0.32] | 2.50 | > 40 | > 211 | > 45 |
SD: Standard deviation; SI: Selectivity index. Not determined
Table 5.
Activity of antifungals with oxazole- and imidazole core structures
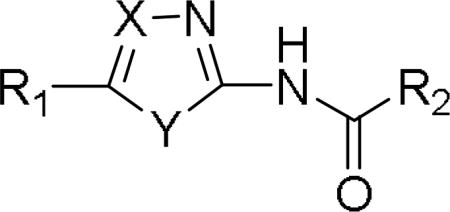
|
H. capsulatum |
C. neoformans (H99 strain) |
P388D1 | SI (Hc) |
SI (Cn) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Cmpd | X | Y | R1 | R2 | IC50 (µM) [±SD] |
MIC (µM) |
IC50 (µM) |
MIC (µM) |
IC50 (µM) |
||
| 38a | CH | O |
|
|
> 20 | > 40 | > 20 | > 40 | nd | nd | nd |
| 38b | CH | O |
|
|
> 20 | > 40 | > 20 | > 40 | nd | nd | nd |
| 40 | CH | NH |
|
|
8.11 [±0.33] | 20.00 | > 20 | > 40 | > 40 | > 5 | > 2 |
SD: Standard deviation; SI: Selectivity index, nd: not determined
Table 3 shows data of target compounds having a bicyclic ring system at the 5-position. Amongst the group of compounds with a cyclohexylamide substituent at the 2-position, compounds 17b, 22b and 26b, having a 7-benzothiophenyl-, 4-benzothiophenyl-, and indanyl group, respectively, at the 5-position, were ~2 times more active than 41F5 both against H. capsulatum and C. neoformans. Compound 36b, substituted at the 2-position with cyclohexylamide and having a carbonyl linked naphthalenyl group at the 5-position, was also ~ 2 times more active than 41F5 against H. capsulatum but appears to be slightly less active than 41F5 against C. neoformans. The activities of benzofuran-substituted 21a/b (Table 3) against H. capsulatum and C. neoformans were similar to that of 41F5 and compound A (Table 2).
Amongst the group of compounds with a cyclohexylethylamide substituent at the 2-position, compounds 22a and 23a with a 4- and 3-benzothiophenyl group, respectively, were ~ 2 times more active against H. capsulatum than 41F5. Compounds 21a and 26a, substituted with a 7-benzofuranyl- and an indanyl group, respectively, had similar activity against H. capsulatum as 41F5 whereas compound 36a, with a carboranyl linked naphthalenyl group, was slightly less active against this fungal species. A limited set of compounds with relatively small alicyclic (25a/b) and aliphatic (30a/b) substituents at the 5-position was evaluated in assays with H. capsulatum and C. neoformans (Table 4). Compound 25b, with a cyclohexylamide subsituents at the 2-position and a cyclopentylmethyl group at the 5-position, was active against H. capsulatum in the same range as reference compounds B and C.
Table 4.
Activity of 41F5-derived antifungals with monocyclic ring systems or an acyclic ethyl group at the 5-position
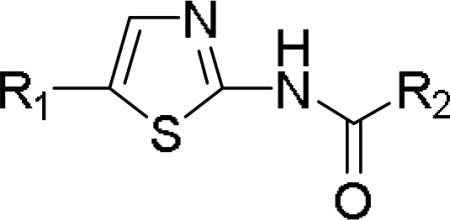
|
H. capsulatum |
C. neoformans (H99 strain) |
P388D1 | SI (Hc) |
SI (Cn) |
||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Comp | R1 | R2 | IC50 (µM) [±SD] |
MIC (µM) |
IC50 (µM) [±SD] |
MIC (µM) |
IC50 (µM) [±SD] |
||
| C |
|
|
0.80 [±0.04] | 1.25 | > 20 | > 40 | > 40 | > 50 | nd |
| B |
|
|
1.06 [±0.06] | 2.50 | 1.36 [±0.10] | 2.50 | > 40 | > 38 | > 29 |
| 25a |
|
|
> 20 | > 40 | > 20 | > 40 | nd | nd | nd |
| 25b |
|
|
0.76 [±0.05] | 1.25 | > 20 | > 40 | > 40 | > 53 | nd |
| 29a |
|
|
8.09 [±0.70] | 20.00 | > 20 | > 40 | > 40 | > 5 | nd |
| 29b |
|
|
18.30 [±1.06] | 40.00 | 7.17 [±0.84] | 20.00 | > 40 | > 2 | > 6 |
| 30a | C2H5 |
|
> 20 | > 40 | > 20 | > 40 | nd | nd | nd |
| 30b | C2H5 |
|
> 20 | > 40 | > 20 | > 40 | nd | nd | nd |
| 31 |
|
|
> 20 | > 40 | > 20 | > 40 | nd | nd | nd |
| 32a |
|
|
7.67 [±0.25] | 20.00 | > 20 | > 40 | > 40 | > 5 | nd |
| 32b |
|
|
> 20 | > 40 | > 20 | > 40 | nd | nd | nd |
SD: Standard deviation; SI: Selectivity index, nd: not determined
Khalil et. al.22 reported that 41F5-derived thiazole antifungal compounds with a cyclohexylethylamide group at the 2-position (e.g. A and C, Table 2) were highly active only against H. capsulatum whereas those having a cyclohexylamide group at the 2-position (e.g. 41F5 and B, Table 2) were highly active against both H. capsulatum and C. neoformans. Surprisingly, this SAR pattern was not observed for cyclohexylethylamide substituted compounds 20a and 28a, which were inactive against both H. capsulatum and C. neoformans, whereas their cyclohexylamide counterparts 20b and 28b were moderately active against both fungal species in accordance with the previously observed SAR pattern (Table 3). In contrast, the cyclohexylamide-substituted compounds 23b and 27b were inactive against both H. capsulatum and C. neoformans whereas their cyclohexylethylamide counterparts 23a and 27a adhered to the previously established SAR trend and were active/moderately active against H. capsulatum and inactive against C. neoformans (Table 3). Yet another SAR profile was observed for the 25a/b pair (Table 4). In this case, activity was observed only for cyclohexylamide-substituted compound 25b in H. capsulatum. Studies with compounds 20a/b, 23a/b, 25a/b, 27a/b, and 28a/b were repeated several times to exclude the possibility of experimental error. The obtained set of data indicates that differences in the SAR pattern between both fungal species are more complex than originally proposed by Khalil et al.22
Compounds 28a/b, 29a/b, 31 and 32a/b were moderately active or inactive against H. capsulatum and C. neoformans indicating that the presence of a nitrogen in the ring system at the 5-position negatively affects activity (Tables 3 and 4). No significant differences were observed when the activities of 20a/b, 28a/b, 29a/b, 31 and 32a/b against both fungal species were evaluated comparatively at pH 5 and and pH 7 (Supplementary Material [SM]-S.2). On the other hand, compounds with oxygen in the ring system at the 5-position (21a/b) had approximately similar activities as 41F5. These results are consistent with those reported by Khalil et al.,22 which indicates that polar atoms within the ring systems at the 2- and 5-positions generally have no positive effect on the activities of 41F5-derived antifungals. In addition, calculations with ACD/Percepta software (Version 14.1.0, Advanced Chemistry Development, Inc., Toronto, ON, Canada) indicated increased aqueous solubilities of compounds 28b, 29a/b, 31, 32a/b by factors ranging from 15 to 298 at pH 5 and from 10 to 88 at pH 7 as compared to that of 41F5 (SM-S.3), which suggests that improved water solubility may also have no marked effect on the activities of 41F5-derived antifungals.
The compounds shown in Table 5, having oxazole (38a/b) or imidazole (40) core ring structures, were evaluated in assays with H. capsulatum and C. neoformans to explore the effect of modifications to the thiazole core structure on activity. Imidazole 40 was weakly active against H. capsulatum and inactive against C. neoformans whereas 38a and 38b were inactive against both fungal species. These results are consistent with those previously reported by Khalil et al.,22 which strongly suggests that the thiazole core structure is essential for activity.
Linking tricyclic fluorene rings via methylene spacer either through the 2- (16a/b), 1- (18), or 4-position (19) (Scheme 4) to the 5-position of the thiazole core structure resulted in loss of activity both in H. capsulatum and C. neoformans (IC50s = >20 µM/MIC = >40 µM) indicating that substituents bulkier than bicyclic naphthalene are not tolerated at this position. Compounds 14, 15, and 33a/b/c (Schemes 4 and 5) were designed and synthesized with the intention of molecular target identification (vide supra). Unfortunatley, these compounds were inactive (IC50s = >20 µM/MIC = >40 µM) both against H. capsulatum and C. neoformans, which indicates that the identification of the molecular target of 41F5-derived antifungals via unbiased affinity chromatography may be difficult to accomplish.
A small libray of highly active compounds, composed of 17b, 21b, 22b, 26b, and 36b, was evaluated in a comparative assay against C. neoformans using both fluconazole-sensitive (H99 and B3501) and -resistant (TES9 and MRL862) strains (Table 6).44 Fluconazole and 41F5 were used as reference compounds in this assay. The results indicate that 17b, 21b, 22b, 26b, 36b, and 41F5 were active to approximately the same extent against both fluconazole-sensitive and -resistant strains. In contrast, fluconazole was notably less active in the resistant strains as compared to the sensitive strains. These observations suggest that 14α-demethylase is not the molecular target of 17b, 21b, 22b, 26b, 36b, and 41F5. It should be noted that clinically-used fluconazole, which has fungistatic or funcicidal dose-dependent activity in Cryptococcus,45 has approximately the same activity levels against the fluconazole-sensitive H99- and B3501 C. neoformans strains as the experimental compounds 17b, 21b, 22b, 26b, 36b, and 41F5.
Table 6.
Activity of 41F5-derived antifungals against fluconazole-resistant Cryptococcus isolates
| Cmpd | H99 (serotype A-FluS) |
B3501 (serotype D-FluS) |
TES9a (serotype A-FluR) |
MRL862a (serotype A-FluR) |
||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| IC50 (µM) [±SD]b |
MIC (µM) |
IC50 (µM) [±SD] |
MIC (µM) |
IC50 (µM) [±SD] |
MIC (µM) |
IC50 (µM) [±SD] |
MIC (µM) |
|
| 41F5 | 0.67 [±0.06] | 1.25 | 0.18 [±0.17] | 0.63 | 0.20 [±0.03] | 0.63 | 0.23 [±0.04] | 0.63 |
| Fluconazole | 0.95 [±0.63] | 4.08 | 0.13 [±0.22] | 2.04 | 27.38 [±2.17] | 65.36 | 14.61 [±0.65] | 32.68 |
| 17b | 0.34 [±0.04] | 0.63 | 0.27 [±0.02] | 0.63 | 0.23 [±0.01] | 0.63 | 0.20 [±0.02] | 0.63 |
| 21b | 0.62 [±0.12] | 2.50 | 0.32 [±0.02] | 0.63 | 0.41 [±0.02] | 0.63 | 0.15 [±0.09] | 0.63 |
| 22b | 0.21 [±0.09] | 0.63 | 0.16 [±0.03] | 0.63 | 0.19 [±0.01] | 0.31 | 0.13 [±0.01] | 0.31 |
| 26b | 0.41 [±0.07] | 1.25 | 0.42 [±0.06] | 1.25 | 0.45 [±0.07] | 1.25 | 0.37 [±0.58] | 1.25 |
| 36b | 0.89 [±0.32] | 2.50 | 0.38 [±0.06] | 1.25 | 0.47 [±0.05] | 1.25 | 0.41 [±0.05] | 1.25 |
TES9 and MRL862 are fluconazole-resistant (FluR) strains due to mutation of the azole target Erg11.
SD: Standard deviation
The activities of compounds 41F5, 17b, 22b and 36b were also evaluated against H. capsulatum yeasts phagocytozed into murine P388D1 macrophages. Compound 36b displayed slightly improved IC50/MIC values as compared to those of 41F5 (0.71±0.15 µM/1.25 µM v.s. 1.26±0.17 µM/2.5 µM) whereas 17b (1.38±0.10 µM/2.5 µM) and 22b (1.21±0.0.7 µM/2.5 µM) had similar inhibitory activities. As already discussed (vide supra), compounds 17b, 22b and 36b showed ~2-fold increase in activity against H. capsulatum yeast in vitro as compared to 41F5. However, the same trend was only observed in the case of 36b for intramacrophage H. capsulatum. In addition, 17b, 22b, 36b, and 41F5 showed moderately reduced activity against intramacrophage H. capsulatum as compared to H. capsulatum yeast in vitro (Tables), which might be due to the fact that the compounds have to permeate into the phagosome to inhibit the intracellular yeasts.
3. Summary and Conclusions
The highest activities against both C. neoformans and H. capsulatum were found for compounds containing [5,6]- and [6,5]-bicyclic ring systems, such as benzothiophene or indane groups, at the 5-position (17b, 22a, 22b, 23a, 26a, 26b) and a [6,6]-bicyclic ring system linked via carbonyl spacer (36b) to the same position of the thiazole core structure. These compounds also showed low toxicities against murine P388D1 macrophages resulting in excellent selectivity indices (SIs). Highly active compounds, such as 17b, 21b, 22b, 26b, and 36b, remained active against fluconazole-resistent C. neoformans TES9 and MRL862 strains and H. capsulatum yeasts phagocytozed into murine P388D1 macrophages. Compounds with reduced activity had ester functions in cyclic systems either at the 2- or the 5-position (14, 15, 33), a tricyclic ring system at the 5-position (16, 18, 19), an ionizable nitrogen atom in aromatic systems at the 5-position (28, 29, 31, 32), a small ethyl substituent at the 5-position (30), or a modified core structure (38, 40). Several other modifications did also not lead to improved activity (17a, 20, 21, 23b, 24, 25, 27, 36a). Based on the obtained results we were able to extend our understaning of the H. capsulatum specific SAR for 41F5-derived antifungals, which is illustrated in Figure 1. As discussed (vide supra), the C. neoformans specific SAR deviates from this pattern to some degree.
Figure 1.

Updated Histoplasma SAR for 41F5-derived antifungal compounds. Areas circled with dotted lines indicate findings described in this paper. All other findings were reported previously by Khalil et al.
The results obtained for the 36a/b tandem (carbonyl linker at the 5-position) indicate that the exploration of e.g. ether-, thioether-, thiocarbonyl-, thionyl- and sulfonamide linkers at the 2- and 5 position have the potential to further improve activity of 41F5-derived antifungals. Overall, however, the chemical space specific for this compound class has been explored fairly exhaustively by our group in the present and a previous study.22 Although compounds such as 17b, 21b, 22b, 26b, and 36b appeared to have a ~2 fold increased activity both against H. capsulatum and C. neoformans as compared to 41F5, compounds with markedly improved potency could not be indentified. Therefore, it is doubtful if additional SAR studies using a phenotypic screening approach with a further extended compound library can produce any dramatic advancement.
As discussed (vide supra), it is conceivable that structural information for the molecular target(s) of 41F5-derived antifungal compounds could potentially facilitate a more rational structure-based drug design approach. However, low antifungal activities obtained for ester-modified compounds make it questionable if an unbiased affinity chromatography approach for molecular target indentification is a viable strategy.
Supplementary Material
Acknowledgments
We thank June Kwon-Chung at the NIH for providing fluconazole-resistant Cryptococcus strains for testing. The project described was supported by Award Number Grant 8UL1TR000090-05 from the National Center For Advancing Translational Sciences and, in part, NIH (NIAID) grant R21AI109437. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Advancing Translational Sciences or the National Institutes of Health. This work was initiated with funds provided by the OSU Public Health Preparedness for Infectious Diseases (PHPID) program (phpid.osu.edu) and by the OSU College of Pharmacy.
Abbreviations
- Boc
tert-butyloxycarbonyl
- DMAP
4-dimethylaminopyridine
- DCM
dichloromethane
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- EDCI
1-ethyl-3-(-3-dimethylaminopropyl) carbodiimide
- HIV
human immunodeficiency virus
- HOBt
hydroxybenzotriazole
- HR-ESI
high resolution–electrospray ionization
- LDA
lithium diisopropyl amide
- MIC
minimal inhibitory concentration
- rxn
reaction
- SAR
structure-activity-relationship
- SD
standard deviation
- SM
supplementary material
- SI
selectivity index
- TFA
trifluoroacedic acid
- THF
tetrahydrofurane
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary material associated with this article can be found, in the online version, at…… This material includes data on antifungal activities and calculated aqueous solubilities of 41F5-derived antifungal compounds at pH5 and pH7, calculated ionization energy levels of 41F5-derived antifungal compounds, a full experimental section, and NMR/MS spectra for target compound
References
- 1.Woods JP. Curr Opin Microbiol. 2003;6:327–331. doi: 10.1016/s1369-5274(03)00080-8. [DOI] [PubMed] [Google Scholar]
- 2.Hammerman KJ, Powell KE, Tosh FE. Sabouraudia. 1974;12:33–45. doi: 10.1080/00362177485380061. [DOI] [PubMed] [Google Scholar]
- 3.Pfaller MA, Diekema DJ. Crit Rev Microbiol. 2010;36:1–53. doi: 10.3109/10408410903241444. [DOI] [PubMed] [Google Scholar]
- 4.Chu JH, Feudtner C, Heydon K, Walsh TJ, Zaoutis TE. Clin Infect Dis. 2006;42:822–825. doi: 10.1086/500405. [DOI] [PubMed] [Google Scholar]
- 5.Vallabhaneni S, Mody RK, Walker T, Chiller T. Infect Dis Clin North Am. 2016;30:1–11. doi: 10.1016/j.idc.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Sabiiti W, May RC. Future Microbiol. 2012;7:1297–1313. doi: 10.2217/fmb.12.102. [DOI] [PubMed] [Google Scholar]
- 7.Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. AIDS. 2009;23:525–530. doi: 10.1097/QAD.0b013e328322ffac. [DOI] [PubMed] [Google Scholar]
- 8.Brown GD, Denning DW, Gow NAR, Levitz SM, Netea MG, White TC. Sci Transl Med. 2012;4:1–9. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 9.Denning DW, Hope WW. Trends Microbiol. 2010;18:195–204. doi: 10.1016/j.tim.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Jinna S, Finch J. Drug Des Devel Ther. 2015;9:6185–6190. doi: 10.2147/DDDT.S81944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dodds Ashley ES, Lewis R, Lewis JS, Martin C, Andes D. Clin Infect Dis. 2006;43:S28–S39. [Google Scholar]
- 12.Hage CA, Connolly P, Horan D, Durkin M, Smedema M, Zarnowski R, Smith P, Wheat LJ. Antimicrob Agents Chemother. 2011;55:4447–4450. doi: 10.1128/AAC.01681-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maligie MA, Selitrennikoff CP. Antimicrob Agents Chemother. 2005;49:2851–2856. doi: 10.1128/AAC.49.7.2851-2856.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Messer SA, Jones RN, Moet GJ, Kirby JT, Castanheira M. J. Clin. Microbiol. 2010;48:2984–2987. doi: 10.1128/JCM.00328-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Darkes MJM, Scott LJ, Goa KL. Am. J. Clin. Dermatol. 2003;4:39–65. doi: 10.2165/00128071-200304010-00005. [DOI] [PubMed] [Google Scholar]
- 16.Lewis RE. Mayo Clin Proc. 2011;86:805–817. doi: 10.4065/mcp.2011.0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramana KV, Kandi S, P Bharatkumar V, Sharada CV, Rao R, Mani R, Rao SD. Am J Infect Dis Microbiol. 2013;1:64–69. [Google Scholar]
- 18.Sharma N, Sharma D. J Pharmacol Pharmacother. 2015;6:236–239. doi: 10.4103/0976-500X.171870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roemer T, Krysan DJ. Cold Spring Harbor Perspect Med. 2014;4:a019703/1–a019703/14. doi: 10.1101/cshperspect.a019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loyse A, Dromer F, Day J, Lortholary O, Harrison T. J Antimicrobial Chemother. 2013;68:2435–2444. doi: 10.1093/jac/dkt221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards JA, Kemski MM, Rappleye CA. Antimicrob Agents Chemother. 2013;57:4349–4359. doi: 10.1128/AAC.00459-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khalil A, Edwards JA, Rappleye CA, Tjarks W. Bioorg Med Chem. 2015;23:532–547. doi: 10.1016/j.bmc.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siddiqui N, Arshad MF, Ahsan W, Alam MS. Int J Pharm Sci Drug Res. 2009;1:136–143. [Google Scholar]
- 24.Borelli C, Schaller M, Niewerth M, Nocker K, Baasner B, Berg D, Tiemann R, Tietjen K, Fugmann B, Lang-Fugmann S, Korting HC. Chemotherapy. 2008;54:245–259. doi: 10.1159/000142334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostrosky-Zeichner L, Casadevall A, Galgiani JN, Odds FC, Rex JH. Nat Rev Drug Discovery. 2010;9:719–727. doi: 10.1038/nrd3074. [DOI] [PubMed] [Google Scholar]
- 26.Ayati A, Emami S, Asadipour A, Shafiee A, Foroumadi A. Eur J Med Chem. 2015;97:699–718. doi: 10.1016/j.ejmech.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 27.Nes WD, Zhou W, Ganapathy K, Liu J, Vatsyayan R, Chamala S, Hernandez K, Miranda M. Arch Biochem Biophys. 2009;481:210–218. doi: 10.1016/j.abb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 28.Meanwell NA. J Med Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 29.Lima LM, Barreiro EJ. Curr Med Chem. 2005;12:23–49. doi: 10.2174/0929867053363540. [DOI] [PubMed] [Google Scholar]
- 30.Patani GA, LaVoie EJ. Chem Rev. 1996;96:3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- 31.Daniel J, Mastrodonato C, Sourdon A, Clermont G, Vabre J-M, Goudeau B, Voldoire H, Arbault S, Mongin O, Blanchard-Desce M. Chem Commun. 2015;51:15245–15248. doi: 10.1039/c5cc04573h. [DOI] [PubMed] [Google Scholar]
- 32.Joshi SD, Jangade NM, Dixit SR, Joshi AS, Kulkarni VH. Indo Am J Pharm Res. 2016;6:5033–5044. [Google Scholar]
- 33.Cravatt BF, Wright AT, Kozarich JW. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 34.Matuszak CA, Matuszak AJ. J Chem Educ. 1976;53:280–284. [Google Scholar]
- 35.Kanao M, Watanabe Y, Kimura Y, Kanno H, Kubo H, Ashida S. Chem Pharm Bull. 1988;36:2968–2976. doi: 10.1248/cpb.36.2968. [DOI] [PubMed] [Google Scholar]
- 36.Schiavi B, Ahond A, Al-Mourabit A, Poupat C, Chiaroni A, Gaspard C, Potier P. Tetrahedron. 2002;58:4201–4215. [Google Scholar]
- 37.Shi B-F, Maugel N, Zhang Y-H, Yu J-Q. Angew Chem Int Ed Engl. 2008;47:4882–4886. doi: 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]
- 38.Larson GL, Fry JL. Org. React. 2008;71:1–737. [Google Scholar]
- 39.Parnes ZN, Lyakhovetskii YI, Kalinkin MI, Kursanov DN, Belen'kii LI. Tetrahedron. 1978;34:1703–1705. [Google Scholar]
- 40.Nitz TJ, Salzwedel K, Finnegan C, Brunton S, Flanagan S, Montalbetti C, Coulter TS. WO 2009085256 A1. PCT Int. Appl. 2009
- 41.Kitbunnadaj R, Zuiderveld OP, Christophe B, Hulscher S, Menge WMPB, Gelens E, Snip E, Bakker RA, Celanire S, Gillard M, Talaga P, Timmerman H, Leurs R. J Med Chem. 2004;47:2414–2417. doi: 10.1021/jm049932u. [DOI] [PubMed] [Google Scholar]
- 42.Uto Y, Ogata T, Kiyotsuka Y, Miyazawa Y, Ueno Y, Kurata H, Deguchi T, Yamada M, Watanabe N, Takagi T, Wakimoto S, Okuyama R, Konishi M, Kurikawa N, Kono K, Osumi J. Bioorg Med Chem Lett. 2009;19:4159–4166. doi: 10.1016/j.bmcl.2009.05.123. [DOI] [PubMed] [Google Scholar]
- 43.Baumann K, Goetschi E, Jolidon S, Limberg A, Luebbers TF. WO 2010052199 A1. PCT Int. Appl. 2010
- 44.Sionov E, Chang YC, Garraffo HM, Dolan MA, Ghannoum MA, Kwon-Chung KJ. Antimicrob Agents Chemother. 2012;56:1162–1169. doi: 10.1128/AAC.05502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Longley N, Muzoora C, Taseera K, Mwesigye J, Rwebembera J, Chakera A, Wall E, Andia I, Jaffar S, Harrison TS. Clin Infect Dis. 2008;47:1556–1561. doi: 10.1086/593194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.