

Abstract
Nitric oxide (NO) is a small gaseous signaling molecule that has important biological effects. It has been heavily implicated in migraine; and the NO donor, nitroglycerin, has been used extensively as a human migraine trigger. Correspondingly, a number of components of the NO signaling cascade have been shown to be upregulated in migraine patients. NO is endogenously produced in the body by NO synthase (NOS), of which there are three isoforms: neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS). Based on the accumulating evidence that endogenous NO regulation is altered in migraine pathogenesis, global and isoform-selective inhibitors of NOS have been targeted for migraine drug development. This review highlights the evidence for the role of NO in migraine and focuses on the use of NOS inhibitors for the treatment of this disorder. In addition, we discuss other molecules within the NO signaling pathway that may be promising therapeutic targets for migraine.
Electronic supplementary material
The online version of this article (10.1007/s13311-018-0614-7) contains supplementary material, which is available to authorized users.
Keywords: Migraine, Headache, Pain, Nitric oxide synthase, Guanylyl cyclase
Introduction
Nitric oxide (NO) is an endogenous gaseous signaling molecule that is involved in a number of physiological processes. The effect of NO on headache was first intimated in 1847 with the synthesis of the NO donor nitroglycerin (NTG) by Ascanio Sobrero, who reported “great precaution should be used, for a very minute quantity put upon the tongue produces a violent headache for several hours” [1]. NO is endogenously produced in the body by three isoforms of nitric oxide synthase (NOS), which are homologous but have distinct functional roles. Extensive work on the relationship between NO and many forms of primary headaches, including migraine, cluster, and tension-type headache, has revealed the importance of this signaling molecule on the induction and maintenance of headache disorders. The goal of this review will be to summarize the literature on the mechanism of action of NO and NOS specifically in migraine pathophysiology, and to examine the therapeutic potential for targeting this pathway for migraine drug development.
NO is produced in almost every mammalian cell type and regulates a host of physiological functions, including vascular tone, neurotransmission, and as an immune defense mechanism [2]. NO is produced intracellularly by the oxidation of L-arginine yielding NO and L-citrulline (Fig. 1). The formation of NO is catalyzed by three different isoforms of NOS, which share ~ 50–60% homology, with the greatest variability in the amino terminal. In addition, NOS isoforms are highly conserved between species, and homology for a given isoform can be as great as 85 to 92% [2, 4]. The production of NO requires various co-factors including tetrahydrobiopterin (BH4), flavin adenine dinucleotide, flavin mononucleotide, calmodulin, and heme (iron protoporphyrin IX) [5]. In order to be functional, the three NOS isoforms need to form dimers to then bind BH4 and the substrate L-arginine to catalyze NO production [4, 5]. The three members of the NOS family correspond to the tissue type they were discovered in, and where they are predominantly expressed: neuronal NOS (nNOS, also known as NOS1 and NOSI), endothelial NOS (eNOS, or NOS3, NOSIII), and inducible NOS (iNOS, or NOS2, NOSII) (see Fig. 2 for localization). Both nNOS and eNOS are constitutively active, and this activation is dependent on increases in intracellular Ca2+ concentrations and its subsequent binding to calmodulin [2]. nNOS is predominately expressed in neurons, and is found in both the central and peripheral nervous systems [2, 9]. Of the three isoforms, nNOS is unique in that it binds to the scaffolding protein post-synaptic density protein 95 (PSD95) which allows it to interact with the N-methyl-D-aspartate (NMDA) glutamate receptor [10]. Opening of the NMDA channel increases Ca2+ influx, which binds to calmodulin and catalytically activates nNOS [11]. Thus, manipulation of the NMDA receptor will also have significant effects on nNOS activity. eNOS was originally purified and cloned from cells in the vascular endothelium, but it has also been detected in other tissues including platelets, cardiomyocytes, and the brain [12]. NO produced by eNOS regulates vascular tone and vasodilation, and NO production by eNOS is initiated by a number of factors including shear stress, histamine, bradykinin, and acetylcholine [4, 12]. iNOS is expressed in a number of cell types including macrophages, glia, and neurons. Of the three NOS isoforms, iNOS is distinct as it is not constitutively active, but is induced by bacterial infection and pro-inflammatory cytokines, and therefore serves as part of the host immunological defense system [4]. When active, iNOS is calcium-insensitive, and can produce up to 1000× more NO than nNOS and eNOS [2].
Fig. 1.
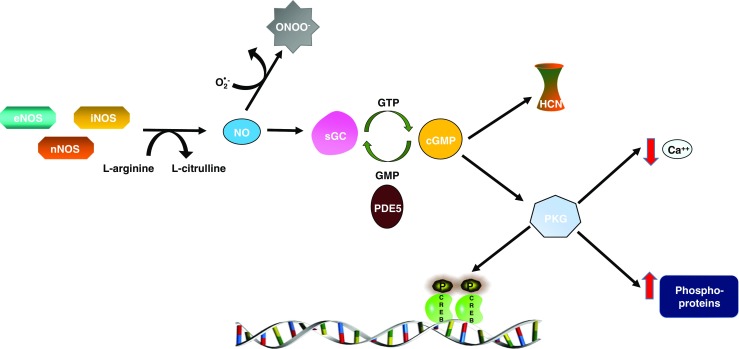
Nitric oxide synthesis and signaling. The three NO synthases: nNOS, eNOS, and iNOS produce NO through the oxidation of L-arginine. Soluble guanylyl cyclase (sGC) is the high affinity receptor for NO in the body. Upon binding of NO, sGC converts guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP), which in turn activates the cell membrane bound ion channels; hyperpolarization-activated cyclic nucleotide–gated channel (HCN), and cGMP-dependent protein kinase (PKG). PKG regulates a number of proteins through phosphorylation, and the results include altered gene expression, decreased intracellular calcium, and modified G protein coupled receptor activation [3]. Phosphodiesterase 5 (PDE5) specifically breaks down cGMP to GMP, thus acting as a negative regulator of this pathway
Fig. 2.

Localization of nitric oxide synthase (NOS), and possible sites of action of NOS inhibitors, relevant to migraine and treatment. Inducible NOS (iNOS; green dots) is upregulated in dural macrophages, alongside interleukin 6, after systemic nitroglycerin (NTG). It is thought that this increase is involved in NTG mediating dural mast cell degranulation and plasma protein leakage (top left panel). Plasma protein extravasation is inhibited by an iNOS inhibitor, but this did not subsequently translate to therapeutic efficacy in migraine treatment [6]. There is expression of iNOS in the trigeminal ganglion (TG) and trigeminal nucleus caudalis, most likely on afferent terminals, in a model of post traumatic headache [7]. Endothelial NOS (eNOS; red dots) is predominantly localized in the endothelium of dural and pial blood vessels, while neuronal NOS (nNOS; blue dots) is also localized in dural and pial layers, but likely restricted to nerve fibers, and nerve endings. nNOS, and eNOS to a much lesser extent, are also localized in the TG, trigeminal neurons, the periaqueductal gray (PAG), hypothalamus, thalamus and somatosensory and cingulate cortices [8], relevant to migraine. It is likely that within these sites, nNOS inhibitors act by preventing the release of neuropeptides, such as calcitonin gene-related peptide and its subsequent downstream effects on secondary messenger systems that promote neuronal activation
NO is highly membrane permeable, and once it is synthesized, it can activate signaling cascades both within the cell in which it was produced, as well as by freely passing through membranes and activating nearby cells. Soluble guanylyl cyclase (sGC) is the only known receptor of NO in the body (See Fig. 1), although NO can also have sGC independent effects. sGC is a heme-containing heterodimer consisting of an α and β subunit. Four sGC isoforms have been identified: α1, α2, β1, and β2; although only α1/β1 and α2/β1 heterodimers appear to be activated by NO [4]. Upon entering the cell, NO binds with high affinity to the heme site of sGC resulting in a conformational change and enzyme activation. sGC converts guanosine-5′-triphosphate (GTP) to the signaling molecule cyclic guanosine monophosphate (cGMP). Phosphodiesterases (PDEs) hydrolyze cGMP to GMP, and PDE5 is the only PDE that selectively catalyzes the breakdown of cGMP exclusively [4]. cGMP signals through a number of mechanisms including the activation of ion channels, specifically the hyperpolarization-activated cyclic nucleotide-gated channel (HCN). Upon activation, HCN channels can in turn activate the ionotropic glutamate receptor (iGluR) producing excitatory effects [13]. In addition, cGMP binds to, and activates, cGMP dependent protein kinases, also known as protein kinase G (PKG) (Fig. 1, [2, 9]). PKG is a serine/threonine kinase that phosphorylates a number of proteins, and plays an important role in Ca2+ homeostasis and smooth muscle contractility [3]. Among other things, PKG also phosphorylates the transcription factor, cAMP response element-binding protein (CREB); therefore affecting expression of CREB target genes and neuronal plasticity [3]. In addition, NO can have sGC independent effects. NO is a major source of peroxynitrite, and this reactive nitrogen species allows for NO to be used as a defense against infection especially in the high concentrations seen when iNOS is activated [2]. However, this form of oxidative stress may also play a role in pathological conditions, including migraine [14].
Nitroglycerin and Migraine
NO has been implicated in pain processing [15], and is most commonly associated with migraine headache [16, 17]. In particular, the NO donor nitroglycerin (NTG) has been used extensively as a human experimental model of migraine [17]. An initial study using sublingual NTG found that migraine sufferers and their first-order relatives were more sensitive to NTG-provoked headache compared to healthy volunteers. They also observed that migraineurs developed a delayed migraine-like headache several hours following NTG [18]. The Danish Headache Center has used this provocation model extensively to characterize human migraine [17]. Intravenous administration of NTG has been shown to reliably trigger migraine-like headache in a high proportion of migraineurs, in a dose-dependent manner [19]. NTG has a biphasic effect; the rapid vasodilatory effects of NTG likely mediates the headache shortly following infusion, which is enhanced in migraineurs [20]. Furthermore, in these patients NTG also evokes headache [21] several hours after infusion, with similar characteristics to their spontaneous migraine attacks [22]. To support the role of the NO-sGC pathway in migraine, the same Copenhagen group has found that the PDE5 inhibitor, sildenafil, can also evoke headache in healthy controls [23] and delayed migraine headache in patients [24]. In addition, serum cGMP levels were also found to be increased during a migraine attack [25]. Interestingly, NTG does not provoke aura, even in migraine with aura patients [26], suggesting that aura may be regulated by different molecular mechanisms (see below).
The use of NTG has also been adapted to establish preclinical models of migraine, which have produced some compelling results. In rats and mice, NTG increases the expression of the immediate early gene, c-fos, in the trigeminal nucleus caudalis, implicating it in headache [27, 28]. In terms of pain, acute nitroglycerin was previously shown to produce thermal and mechanical allodynia in mice that was reversed by anti-migraine therapies, sumatriptan [28, 29] and a CGRP receptor antagonist [30]. In addition, in a transgenic mouse model of familial migraine, animals expressing a mutant human migraine gene (casein kinase 1 delta) showed a greater sensitivity to NTG-evoked hyperalgesia compared to wildtype controls [31]. Chronic intermittent treatment with NTG has also been found to evoke a long-lasting basal hypersensitivity, which is sensitive to migraine preventives [32, 33], thus serving as a behavioral model of chronic migraine. NTG can also evoke other migraine-related physiological effects, and has been shown to produce light-aversion [34, 35] and changes in the cranial vasculature of rodents [34, 36, 37]. Taken together, these results strengthen the notion of increased NO as a hallmark of migraine-associated symptoms, and establishes NTG as a useful translationally significant model of migraine.
Soluble Guanylyl Cyclase (sGC) Inhibitors and Migraine
NO produced endogenously or by NTG administration can act through the sGC pathway, but can also be converted to peroxynitrite to cause increased oxidative stress, and both of these pathways may contribute to migraine. Preclinical pharmacological profiling has identified the sGC signaling cascade as a critical regulator of migraine-related symptoms. Similar to NTG, the PDE5 inhibitor sildenafil was shown to evoke both an acute and chronic hyperalgesia [32], strengthening the role of cGMP as a pro-migraine signaling mechanism. Further, in a recent study, direct activation of sGC by the novel sGC stimulator, VL-102, also produced cephalic pain, which was sensitive to both acute and preventive migraine treatments. In addition, VL-102 treatment also increased expression of the pro-migraine neuropeptide CGRP in trigeminal ganglia [38]. In parallel, the sGC inhibitor, ODQ, completely blocked migraine-associated acute and chronic pain evoked by NTG, again supporting the action of NTG through the sGC-cGMP pathway. Interestingly, ODQ also effectively inhibited established chronic migraine-associated pain, in the absence of exogenous NO stimulation. These intriguing data indicate that a chronic migraine state could be maintained by an upregulation in endogenous NOS activity [38]. Thus, blocking this maladaptation could help to reset the chronic migraine brain. This work encourages the development of negative sGC modulators as targets for migraine therapy. Although sGC is an important regulator of blood pressure, the effect of negative modulation of this system has not been definitively characterized and should be explored further.
Nitric Oxide Synthase Inhibitors (NOSi) and Migraine
Based on the accumulating evidence that NO is likely a causative molecule in the pathophysiology of migraine, either independently, or by mediating a cascade that involves other neurobiochemical processes, targeting elements of this nitrergic cascade represents a potentially productive therapeutic approach in the treatment of migraine headache. Specifically, targeting the production of NO via NOS inhibitors has been explored over the last 20 years, with several targeted approaches towards NOS in the treatment of migraine headache, with varying degrees of success.
Non-Selective NOS Inhibition
The first clinical trial to determine if targeting NOS in the treatment of migraine could be an effective treatment approach used a non-selective NOS inhibitor, L-NG-methylarginine hydrochloride (546C88). In a double-blind trial, the effects of 546C88 (6 mg/kg, IV) were studied in acute attacks of spontaneous migraine without aura [39]. 546C88 was found to be better than placebo at providing headache relief, 2 h after infusion. Symptoms of sensory hypersensitivity were also improved. In a subsequent study, another non-selective NOSi, NG-monomethyl-L-arginine hydrochloride (L-NMMA), provided pain relief compared to placebo in chronic tension-type headache [40]. These studies provided the first proof-of-concept data that targeting NOS may be productive in the treatment of primary headaches, including migraine.
In response, many preclinical studies have explored this biochemical mechanism to dissect the neurophysiological pathway where NO is likely released during migraine, and thus where NOS inhibitors may exert their effects. Head pain during migraine, and other primary headaches, is believed to be mediated by activation, and sensitization, of the trigeminal innervation to the nociceptive-specific dural vasculature, and its central projection to the medullary dorsal horn (trigeminocervical complex; TCC) [41, 42]. These mechanisms can be modeled in animals using dural-nociceptive activation, either via electrical stimulation of the dura mater [43], or using chemical mediators [44, 45] applied locally to the dura mater or systemically. These methods have proved to be reliable preclinical approaches against which to assess potential therapeutic efficacy. At the peripheral level, observations of changes in dural meningeal blood flow or caliber are thought to be a marker of activation of its dural-trigeminovascular innervation. NG-Nitro-L-arginine methyl ester hydrochloride (L-NAME, non-selective NOSi) lowers basal blood flow and caliber, and inhibits evoked meningeal blood flow increases and vasodilation, mediated by dural electrical stimulation [46, 47]. Indeed, CGRP-induced vasodilation is also attenuated by L-NAME [46]. At the central projection to the trigeminocervical complex, neuronal responses to dural manipulation are commonly measured using electrophysiological techniques or measurement of the immediate early gene c-fos, or its protein product Fos [48]. L-NAME inhibited both ongoing neuronal firing [49], and dural-evoked expression of Fos immunoreactivity, in the TCC [50]. It also inhibited dural-mediated cutaneous facial allodynia (believed to be the behavioral correlate of sensitization of central trigeminovascular neurons) after placement of icilin (a TRPM8 agonist) on the dura mater [51]. These data suggest that NO production is at least partly responsible for maintaining basal neuronal and vascular tone within the dural-trigeminovascular system, and is involved in the dural meningeal vascular and TCC neuronal changes mediated by activation of the dural trigeminal innervation. These studies also validate the efficacy of non-selective NOS inhibition in the treatment of migraine. Targeting NOS clearly has potential as a therapeutic approach in the treatment of migraine headache; however, a caveat to this conclusion is that in each study where blood pressure was measured, both clinical and preclinical studies, the non-selective NOS inhibitor did produce profound cardiovascular changes, with increased blood pressure. Moving forward, this is not a desirable adverse event that can be worked with clinically, particularly in patients with cardiovascular contraindications. Therefore, from a drug development perspective, more selective targeting of either inducible, endothelial, or neuronal NOS enzymes may be a better long-term strategy for safe and effective migraine treatment.
Selective NOS Inhibition–Inducible NOS Inhibition
Initially, data from several preclinical studies suggested that targeting iNOS would be the most effective non-vascular path forward for the development of selective NOSi in migraine treatment. In rodents after NTG treatment, there is a delayed upregulation of iNOS mRNA in the dural meningeal macrophage after 2 h, with a corresponding increase in protein expression after 4–10 h (Fig. 2). These findings are consistent with the hypothesis that dural inflammatory mechanisms contribute to trigeminovascular activation in migraine. Further, this was accompanied by increased levels of other inflammatory mediators in dural macrophage and CSF, as well as dural plasma protein leakage. The plasma protein leakage and NO signaling were suppressed by selective iNOS inhibition [6, 52]. Studies in patients during spontaneous migraine attacks also revealed upregulation of iNOS expression in blood samples taken from the jugular vein that reduced at the end of the attack [53]. Most recently, several studies have suggested a genetic link with an iNOS G2087A gene polymorphism and migraine susceptibility. G2087A polymorphism alone affects susceptibility for aura in migraine, while interactions with an iNOS C(−1026) A or eNOS rs743506 gene polymorphism, affects susceptibility to migraine aura, or migraine in general, respectively [54, 55]. Combined these data suggest selective iNOS inhibition as a treatment in migraine could represent an attractive, non-vascular approach that is supported by the dural inflammatory hypothesis of migraine.
Counter to this are two preclinical studies that used acute nociceptive activation of the dural-trigeminovascular pathway, without an obvious inflammatory component. Here, specific iNOS inhibition did not prevent dural-evoked meningeal blood flow increase [49] or vasodilation [46], even when mediated by systemic CGRP [46]. This suggests iNOS may not be involved in this acute trigeminovascular nociceptive activation. Subsequently, there have been two controlled clinical trials using a selective iNOS inhibitor, GW274150, in the abortive [56] or preventive [57] treatment of migraine. In each case, GW274150 was ineffective at providing pain relief or reducing headache frequency. It leads one to conclude that despite studies demonstrating the production and upregulation of iNOS in various animal approaches, it is unlikely a key mediator in migraine pathophysiology, and therefore not an ideal target. As a consequence, projects to develop selective iNOS inhibitors to treat migraine are no longer being pursued. However, as a footnote to its role in headache, iNOS may play a larger role in post-traumatic headache (PTH) where inflammation is induced by traumatic brain injury (TBI). PTH after mild TBI very closely resembles the phenotype of migraine, according to ICHD-III. In a controlled cortical impact model of PTH, iNOS protein levels were upregulated in the trigeminal ganglia and trigeminal nucleus caudalis (Fig. 2), an effect that was reversed by treatment with sumatriptan or the CGRP receptor antagonist MK8825. Further, iNOS knockout mice showed less light aversion and augmentation of CGRP in response to injury [7]. Therefore, iNOS inhibition may still be a relevant approach for the specific treatment of PTH.
Endothelial NOS Inhibition
From the outset, targeting eNOS in the treatment of migraine does not appear to be a safe option. It is found predominantly in the vascular endothelium and is thus fundamental for healthy cardiovascular function [58] . Therefore, interference by targeting eNOS with an inhibitor is very likely to produce unwanted side-effects. Despite this, there is evidence that it may have a role in migraine pathophysiology. eNOS mRNA is highly expressed in pial arteries, with detectable but low levels of expression in the dura mater, trigeminal ganglia, or trigeminal nucleus caudalis of rats [8] (Fig. 2). Levels of eNOS mRNA in the dura and pia mater are also increased in rats that underwent air-stress [59], with stress commonly associated with triggering migraine [60, 61]. In addition, in preclinical models of acute dural-trigeminovascular nociception, several selective eNOS inhibitors suppressed CGRP-evoked dural meningeal vessel dilation, but had no effect on neurogenically medicated vasodilation [46]. As a mechanism in migraine, the data suggest that production of NO, mediated by eNOS, in endothelial cells on blood vessels is likely to produce vasodilation. Although, it is currently thought that vasodilation is an epiphenomenon to neuronal responses in migraine [62]. The presence of eNOS along the central migraine pathway may also suggest a non-vascular mechanism, but at the moment it is not clear if it has a similar role to that of nNOS (see below). There have also been many studies trying to link gene polymorphisms that encode eNOS with migraine susceptibility, with several associations described, including GLU298ASP [63], rs743506 in interaction with iNOS G2087A [55], rs3918226 [64], and -786T/C and 894G/T [65]. Together, these data suggest eNOS may play some role in migraine pathophysiology, but it has never been explored for drug discovery because of the high likelihood of interference with cardiovascular regulation.
Neuronal NOS Inhibition
Targeting neuronal NOS seems to be the next best opportunity for selective NOS inhibition in migraine treatment. It is strongly associated with migraine pathophysiology. It is expressed all along the migraine pain pathway, including the dura and pia mater, where its expression is likely restricted to nerve fibers and nerve endings of both trigeminal ganglionic and post-ganglionic parasympathetic projections [8, 66, 67] (Fig. 2). It is also expressed along the central migraine pain pathway, including the periaqueductal gray (PAG), hypothalamus, thalamus, and somatosensory and cingulate cortices [8]. It is therefore ideally placed to modulate mechanisms in migraine. Many studies have demonstrated an association with activation of the trigeminovascular system and the production or upregulation of nNOS. NTG provokes increased nNOS immunoreactivity in the trigeminal ganglion and trigeminal nucleus caudalis [67–69]. It is also increased in the trigeminal dural afferents in a preclinical model of triptan-overuse headache [70]. At the peripheral level, a selective nNOS inhibitor decreased basal meningeal blood vessel diameter and inhibited neurogenically-mediated dural meningeal blood vessel vasodilation, but it had no effect on CGRP-induced vasodilation [46]. Although conflicting data have been demonstrated when meningeal blood flow is measured rather than vasodilation, using a different nNOS inhibitor [47]. In an in vitro CGRP release assay, using a dissected hemi-skull with dura intact, NXN-413, a selective nNOS inhibitor, developed as part of a nNOS drug development program for pain disorders, inhibited KCl-induced CGRP release from the dura mater [71]. NXN-413 also attenuated capsaicin-induced dural meningeal vasodilation in an in vivo cranial preparation [71].
At the central projection, direct iontophoresis of a selective nNOS inhibitor, Nω-propyl-l-arginine (p-ARG), onto trigeminocervical neurons suppressed both dural- and l-glutamate-evoked neuronal action potentials [72]. Using the preclinical model of triptan-overuse headache and central trigeminovascular sensitization, another selective nNOS inhibitor, NXN-323, reversed established periorbital and hind-paw allodynia [70]. It also prevented the development of allodynic symptoms in this model, whereas selective iNOS and eNOS inhibitors had no effects [70]. NXN-323 also reversed and prevented stress-induced allodynia in triptan-overuse rats, mediated by bright-light stimulus. Although, it has no effects on allodynia mediated by a NO donor. Combined, these data suggest selectively targeting nNOS may be beneficial therapeutically, either as an acute or preventive treatment option. Currently, there are no completed clinical trials for any selective nNOS inhibitors, although the novel compound NXN-462 (NeurAxon Inc.) is listed as in phase 2 development for the treatment of chronic migraine.
Another approach to targeting nNOS in the treatment of migraine has come in the form of combining it with a 5-HT1B/1D receptor agonist (triptan), using NXN-188 (developed by NeurAxon Inc.). NXN-188 has been studied in both in vitro and in vivo preclinical assays. In vitro, NXN-188 had no effects on CGRP levels per se, but it dose-dependently blocks KCl-induced CGRP release from the hemi-skull dura mater preparation, trigeminal ganglia, and trigeminal nucleus caudalis. Using the in vivo preparation studying dural meningeal responses, NXN-188 dose-dependently attenuated capsaicin and electrically-mediated dural meningeal vasodilation [71]. Overall, this preclinical profile would suggest a potentially effective novel treatment approach for migraine. Two clinical trial studies have been completed with NXN-188 for the treatment of migraine with (NCT00920686) and without aura (NCT00959751). Preliminary studies suggest that this molecule may be effective in migraine treatment [73, 74], As a therapeutic target nNOS alone, or in combination with a 5-HT1B/1D agonist, may prove to be a novel and effective target for the treatment of migraine.
nNOSi Mechanism of Action
As a mechanism of action in migraine, it is likely that the production of NO, via an increase in expression of nNOS, mediates the production and release of migraine-relevant neuropeptides, specifically CGRP, but also very likely pituitary adenylyl-cyclase activating polypeptide (PACAP). It is still debated quite how these peptides mediate activation and sensitization of trigeminovascular pathways, whether it is by directly causing neuronal firing, or by mediating other messenger systems. Similarly, it is possible that NO also acts directly to excite and activate trigeminal neurons. However, given the delay in mediating migraine, and also the delay in mediating meningeal nociceptor hypersensitivity [75], indirect mechanisms seem far more likely. Also, given that nNOS is expressed along the entire migraine pain pathway, these mechanisms could be mediated in both the periphery and centrally. One study has suggested that dural vascular signaling mechanisms may be responsible for mediating changes to primary afferent trigeminal ganglion (TG) cells [75]. Several studies also demonstrate the production of CGRP in the TG, co-localized with nNOS, which might suggest a neuronal mechanism. Finally, given that NO is highly volatile, and readily crosses the blood-brain barrier, coupled to the expression of nNOS in many brain nuclei related to migraine mechanisms, it suggests that central mechanisms may also play a part. Indeed, in the TG recording study, only dural mechanical hypersensitivity was demonstrated after NTG [75], but not an increase in ongoing firing. Whereas, previous studies have demonstrated an increase in ongoing discharge firing in medullary trigeminal neurons, in response to NO donors [76, 77]. One explanation is that this medullary activation is mediated by central facilitation of the neuronal response, potentially directly on medullary dural-trigeminovascular neurons, or higher up the pain pathway, to produce central sensitization. Thus, both peripheral and central mechanisms may be involved, and therefore the activity of potential nNOS inhibitors may be more efficacious in the treatment of migraine if they gain access to the brain, but may not be a prerequisite.
Non-Selective NOS Inhibitors (NOSi) and Migraine Aura
Cortical spreading depression (CSD) is believed to be the experimental correlate of migraine aura. It is a transient wave of neuronal and glial depolarization that is associated with a loss of ion homeostasis, involving vascular and neuronal synaptic changes, and a subsequent depression in electrical activity (spreading depression). Production of NO is thought to be involved in mechanisms of CSD, particularly the vascular component. Electrical changes associated with cortical hyperaemia and oligaemia are accompanied with NO changes, which are significantly attenuated with NOS inhibition [78]. Further, in NTG primed cats, CSD induction significantly enhanced NO release compared to controls [79], and causes increased expression of nNOS mRNA and protein in the cerebral cortex [80]. Also, as reported above, a polymorphism in the G2087A gene affects susceptibility for aura in migraine [54]. However, several studies demonstrate that while NOS inhibition might reduce NO release mediated by CSD, it has little effects on the overall cerebrovascular changes [81, 82], suggesting NO release does not directly regulate the regional cerebral vascular changes during CSD. A further study looked at the effect of a selective nNOS inhibitor, 7-nitroindazole (7-NINA), on local electrical DC changes mediated by KCl-induced CSD. 7-NINA was able to inhibit the number and DC negativity shift of CSD; however, the effective dose was 100-fold higher than its Ki for nNOS [83]. This effect was also not reversed by the NO precursor, L-arginine. This adds further support to the suggestion NO formation is not involved in eliciting neuronal and vascular changes in CSD, despite its release. These data are perhaps in line with the clinical findings that NTG is not able to trigger migraine aura in susceptible patients, only migraine without aura; and NTG does not affect CSD in mice [28]. Furthermore, preliminary data from a single study, using a NXN-188, the nNOS/5-HT1B/1D receptor agonist, for the treatment of migraine aura only suggested that it may alleviate migraine aura symptoms [73, 74]. Although, it is impossible to determine whether either, or both, the nNOS or 5-HT components were involved in symptom alleviation. Together, these data suggest that NO and the action of nNOS, may not be involved in mechanisms of migraine aura, and that the positive preclinical data represent neural findings that do not translate to the clinical condition observed as haemodynamic changes.
Another approach to dissect the potential of NOS inhibition to treat migraine aura is to explore the potential impact of other molecules thought to modulate NO levels during CSD. The only other molecule developed for its association with NO production, CSD and treatment for migraine aura is tonabersat (SB-220453), a benzopyran derivative, gap junction modulator. In preclinical studies, SB-220453 was shown to inhibit NO release and CSD following KCl induction [84, 85]. Several clinical trials have been conducted for acute and preventive use. For acute use, the data are inconsistent but generally negative in aborting headache [86, 87]. However, as tonabersat has slow absorption, it was thought daily administration as a preventive may prove more beneficial. As a preventive tonabersat did show some efficacy in reducing headache days, but studies had a high placebo rate, complicating conclusions derived from the study [88]. In another study, the effects on migraine with and without aura were compared. While tonabersat did not reduce the number of migraine headache days with and without aura, compared to placebo, it did reduce the number of aura attacks [89]. This is consistent with its ability to inhibit CSD. While preclinical studies with tonabersat demonstrate that its effects on CSD occurrence and NO release coincide, overwhelmingly it appears that the release of NO may be independent, or unnecessary, to induce the neuronal and vascular mechanisms as a consequences of CSD induction, which might be related to migraine aura mechanisms. Therefore, these data cannot confirm the potential efficacy of targeting NO and its production in the prevention of migraine aura, only that targeting CSD may be effective.
Conclusions and Future Directions
Multiple lines of evidence indicate that endogenous NO production is a key mediator in migraine initiation and maintenance. However, because NO also plays an important role in a number of biological processes, including cardiovascular tone, it has been difficult to develop therapies that lack significant adverse events. The development of isoform selective NOSi appears to be the most promising strategy for migraine, especially nNOS inhibitors. NO was only identified as a biological signaling molecule in the late 1970s and 1980s, and therefore compared to other signaling mechanisms, it is still relatively poorly characterized. A better understanding of the downstream signal regulation and targets of NO may provide more selective and effective therapeutic options. For example, very little is known about HCN channel regulation in migraine [90], and this may be one promising avenue. In addition, NO has been shown to act as an epigenetic regulator, that can affect histone acetylation, DNA methylation, and miRNA expression [91]. Identification of migraine specific genes that are epigenetically regulated in a NO-dependent manner could also yield novel therapeutic targets. Overall, NO and NOS are clearly important regulators of migraine, and a better understanding of this pathway will have significant biological and therapeutic implications.
Electronic supplementary material
(PDF 498 kb)
(PDF 506 kb)
Acknowledgements
ZB is a member of the Graduate Program in Neuroscience at UIC.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding Information
AAP and ZB are supported by NIH-NIDA DA040688.
Compliance with Ethical Standards
Conflict of Interest
AAP and ZB report no conflicts.
SA reports an unrestricted grant, honoraria and travel reimbursements from electroCore LLC unrelated to the submitted work.
Contributor Information
Amynah A. Pradhan, Phone: +1 312 355 1557, Email: pradhan4@uic.edu
Simon Akerman, Phone: +1 410 706 2172, Email: sakerman@umaryland.edu.
References
- 1.Hughes RDJ. Cyclopedia of drug pathogenesy. New York: Boerike and Tafel; 1888. [Google Scholar]
- 2.Moncada S, Higgs EA. The discovery of nitric oxide and its role in vascular biology. British journal of pharmacology 2006 Jan;147 Suppl 1:S193–201. PubMed Pubmed Central PMCID: PMC1760731. Epub 2006/01/13. eng. [DOI] [PMC free article] [PubMed]
- 3.Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacological reviews. 2010 Sep; 62(3):525–63. PubMed Pubmed Central PMCID: PMC2964902. Epub 2010/08/19. eng. [DOI] [PMC free article] [PubMed]
- 4.Bryan NS, Bian K, Murad F. Discovery of the nitric oxide signaling pathway and targets for drug development. Frontiers in bioscience (Landmark edition). 2009 Jan 01;14:1–18. PubMed Epub 2009/03/11. eng. [DOI] [PubMed]
- 5.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. The Biochemical journal 2001 Aug 01;357(Pt 3):593–615. PubMed Pubmed Central PMCID: PMC1221991. Epub 2001/07/21. eng. [DOI] [PMC free article] [PubMed]
- 6.Reuter U, Bolay H, Jansen-Olesen I, Chiarugi A, Sanchez del Rio M, Letourneau R, et al. Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain 2001 Dec;124(Pt 12):2490–502. PubMed [DOI] [PubMed]
- 7.Daiutolo BV, Tyburski A, Clark SW, Elliott MB. Trigeminal pain molecules, allodynia, and photosensitivity are pharmacologically and genetically modulated in a model of traumatic brain injury. J Neurotrauma 2016 Apr 15; 33(8):748–60. PubMed Pubmed Central PMCID: PMC4840831. [DOI] [PMC free article] [PubMed]
- 8.Ramachandran R, Ploug KB, Hay-Schmidt A, Olesen J, Jansen-Olesen I, Gupta S. Nitric oxide synthase (NOS) in the trigeminal vascular system and other brain structures related to pain in rats. Neurosci Lett. 2010 Nov 05; 484(3):192–6. PubMed [DOI] [PubMed]
- 9.Chachlaki K, Garthwaite J, Prevot V. The gentle art of saying NO: how nitric oxide gets things done in the hypothalamus. Nature reviews Endocrinology. 2017 Sep;13(9):521–35. PubMed Epub 2017/06/18. eng. [DOI] [PubMed]
- 10.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996 Mar 08;84(5):757–67. PubMed Epub 1996/03/08. eng. [DOI] [PubMed]
- 11.Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988 Nov 24;336(6197):385–8. PubMed Epub 1988/11/24. eng. [DOI] [PubMed]
- 12.Michel T, Feron O. Nitric oxide synthases: which, where, how, and why? The Journal of clinical investigation. 1997 Nov 01;100(9):2146–52. PubMed Pubmed Central PMCID: PMC508408. Epub 1997/12/31. eng. [DOI] [PMC free article] [PubMed]
- 13.He C, Chen F, Li B, Hu Z. Neurophysiology of HCN channels: from cellular functions to multiple regulations. Progress in neurobiology. 2014 Jan; 112:1–23. PubMed Epub 2013/11/05. eng. [DOI] [PubMed]
- 14.Taffi R, Vignini A, Lanciotti C, Luconi R, Nanetti L, Mazzanti L, et al. Platelet membrane fluidity and peroxynitrite levels in migraine patients during headache-free periods. Cephalalgia: an international journal of headache. 2005 May; 25(5):353–8. PubMed Epub 2005/04/21. eng. [DOI] [PubMed]
- 15.Schmidtko A, Tegeder I, Geisslinger G. No NO, no pain? The role of nitric oxide and cGMP in spinal pain processing. Trends Neurosci. 2009 Jun; 32(6):339–46. PubMed Epub 2009/05/06. eng. [DOI] [PubMed]
- 16.Olesen J. Nitric oxide-related drug targets in headache. Neurotherapeutics: the journal of the American Society for Experimental Neuro Therapeutics. 2010 Apr; 7(2):183–90. PubMed Epub 2010/05/01. eng. [DOI] [PMC free article] [PubMed]
- 17.Ashina M, Hansen JM, BO AD, Olesen J. Human models of migraine - short-term pain for long-term gain. Nature reviews Neurology. 2017 Oct 06. PubMed Epub 2017/10/07. eng. [DOI] [PubMed]
- 18.Sicuteri F, Bene ED, Poggioni M, Bonazzi A. Unmasking latent dysnociception in healthy subjects. Headache. 1987;27:180–5. doi: 10.1111/j.1526-4610.1987.hed2704180.x. [DOI] [PubMed] [Google Scholar]
- 19.Iversen HK, Olesen J, Tfelt-Hansen P. Intravenous nitroglycerin as an experimental model of vascular headache. Basic characteristics. Pain. 1989;38:17–24. doi: 10.1016/0304-3959(89)90067-5. [DOI] [PubMed] [Google Scholar]
- 20.Olesen J, Iversen HK, Thomsen L. Nitric oxide supersensitivity: a possible molecular mechanism of migraine pain. Neuro Report. 1993;4(8):1027–30. doi: 10.1097/00001756-199308000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Headache Classification Committee of the International Headache Society. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013 Jul;33(9):629–808. PubMed [DOI] [PubMed]
- 22.Thomsen LL, Kruuse C, Iversen HK, Olesen J. A nitric oxide donor (nitroglycerin) triggers genuine migraine attacks. European journal of neurology. 1994 Sep;1(1):73–80. PubMed Epub 1994/09/01. eng. [DOI] [PubMed]
- 23.Kruuse C, Thomsen LL, Jacobsen TB, Olesen J. The phosphodiesterase 5 inhibitor sildenafil has no effect on cerebral blood flow or blood velocity, but nevertheless induces headache in healthy subjects. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2002 Sep; 22(9):1124–31. PubMed Epub 2002/09/10. eng. [DOI] [PubMed]
- 24.Kruuse C, Thomsen LL, Birk S, Olesen J. Migraine can be induced by sildenafil without changes in middle cerebral artery diameter. Brain: a journal of neurology. 2003 Jan; 126 (Pt 1):241–7. PubMed Epub 2002/12/13. eng. [DOI] [PubMed]
- 25.Stepien A, Chalimoniuk M. Level of nitric oxide-dependent cGMP in patients with migraine. Cephalalgia: an international journal of headache. 1998 Nov; 18(9):631–4. PubMed Epub 1999/01/07. eng. [DOI] [PubMed]
- 26.Christiansen I, Thomsen L, Daugaard V, Ulrich V, Olesen J. Glyceryl trinitrate induces attacks of migraine without aura in sufferers of migraine with aura. Cephalalgia. 1999;19:660–07. doi: 10.1046/j.1468-2982.1999.019007660.x. [DOI] [PubMed] [Google Scholar]
- 27.Ramachandran R, Bhatt DK, Ploug KB, Olesen J, Jansen-Olesen I, Hay-Schmidt A, et al. A naturalistic glyceryl trinitrate infusion migraine model in the rat. Cephalalgia: an international journal of headache. 2012 Jan; 32(1):73–84. PubMed Epub 2011/12/17. eng. [DOI] [PubMed]
- 28.Bates EA, Nikai T, Brennan KC, Fu YH, Charles AC, Basbaum AI, et al. Sumatriptan alleviates nitroglycerin-induced mechanical and thermal allodynia in mice. Cephalalgia: an international journal of headache. 2010 2/2010; 30(2):170–8. [DOI] [PMC free article] [PubMed]
- 29.Pradhan AA, Smith ML, Zyuzin J, Charles A. delta-Opioid receptor agonists inhibit migraine-related hyperalgesia, aversive state and cortical spreading depression in mice. British journal of pharmacology. 2014 May; 171(9):2375–84. PubMed Pubmed Central PMCID: PMC3997277. Epub 2014/01/29. eng. [DOI] [PMC free article] [PubMed]
- 30.Capuano A, Greco MC, Navarra P, Tringali G. Correlation between algogenic effects of calcitonin-gene-related peptide (CGRP) and activation of trigeminal vascular system, in an in vivo experimental model of nitroglycerin-induced sensitization. European journal of pharmacology. 2014 Oct 5; 740:97–102. PubMed Epub 2014/07/08. eng. [DOI] [PubMed]
- 31.Brennan KC, Bates EA, Shapiro RE, Zyuzin J, Hallows WC, Huang Y, et al. Casein kinase idelta mutations in familial migraine and advanced sleep phase. Sci Transl Med. 2013 May 1; 5(183):183ra56, 1–11. PubMed [DOI] [PMC free article] [PubMed]
- 32.Pradhan AA, Smith ML, McGuire B, Tarash I, Evans CJ, Charles A. Characterization of a novel model of chronic migraine. Pain. 2014 Feb; 155(2):269–74. PubMed Pubmed Central PMCID: PMC3920577. Epub 2013/10/15. eng. [DOI] [PMC free article] [PubMed]
- 33.Tipton AF, Tarash I, McGuire B, Charles A, Pradhan AA. The effects of acute and preventive migraine therapies in a mouse model of chronic migraine. Cephalalgia: an international journal of headache. 2015 Dec 17. PubMed Epub 2015/12/20. Eng. [DOI] [PMC free article] [PubMed]
- 34.Markovics A, Kormos V, Gaszner B, Lashgarara A, Szoke E, Sandor K, et al. Pituitary adenylate cyclase-activating polypeptide plays a key role in nitroglycerol-induced trigeminovascular activation in mice. Neurobiol Dis. 2012 1/2012; 45(1):633–44. [DOI] [PubMed]
- 35.Farajdokht F, Babri S, Karimi P, Mohaddes G. Ghrelin attenuates hyperalgesia and light aversion-induced by nitroglycerin in male rats. Neuroscience letters. 2016 Sep 6; 630:30–7. PubMed Epub 2016/07/21. Eng. [DOI] [PubMed]
- 36.Greco R, Meazza C, Mangione AS, Allena M, Bolla M, Amantea D, et al. Temporal profile of vascular changes induced by systemic nitroglycerin in the meningeal and cortical districts. Cephalalgia : an international journal of headache. 2011 Jan;31(2):190–8. PubMed Epub 2010/08/10. eng. [DOI] [PubMed]
- 37.Pryazhnikov E, Kislin M, Tibeykina M, Toptunov D, Ptukha A, Shatillo A, et al. Opposite reactivity of meningeal versus cortical microvessels to the nitric oxide donor glyceryl trinitrate evaluated in vivo with two-photon imaging. PloS one. 2014;9(2):e89699. PubMed Pubmed Central PMCID: PMC3938546. Epub 2014/03/04. eng. [DOI] [PMC free article] [PubMed]
- 38.Ben Aissa M, Tipton AF, Bertels Z, Gandhi R, Moye LS, Novack M, et al. Soluble guanylyl cyclase is a critical regulator of migraine-associated pain. Cephalalgia : an international journal of headache. 2017 Jan 01:333102417737778. PubMed Epub 2017/10/13. eng. [DOI] [PMC free article] [PubMed]
- 39.Lassen LH, Ashina M, Christiansen I, Ulrich V, Grover R, Donaldson J, et al. Nitric oxide synthase inhibition: a new principle in the treatment of migraine attacks. Cephalalgia : an international journal of headache. 1998 Jan;18(1):27–32. PubMed Epub 1998/05/28. eng. [DOI] [PubMed]
- 40.Ashina M, Lassen LH, Bendtsen L, Jensen R, Olesen J. Effect of inhibition of nitric oxide synthase on chronic tension-type headache: a randomised crossover trial. Lancet. 1999 Jan 23;353(9149):287–9. PubMed [DOI] [PubMed]
- 41.Goadsby PJ, Holland PR, Martins-Oliveira M, Hoffmann J, Schankin C, Akerman S. Pathophysiology of migraine: a disorder of sensory processing. Physiol Rev. 2017 Apr;97(2):553–622. PubMed [DOI] [PMC free article] [PubMed]
- 42.Noseda R, Burstein R. Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. Pain. 2013 Dec;154 Suppl 1:S44–53. PubMed Epub 2013/07/31. eng. [DOI] [PubMed]
- 43.Goadsby PJ, Zagami AS. Stimulation of the superior sagittal sinus increases metabolic activity and blood flow in certain regions of the brainstem and upper cervical spinal cord of the cat. Brain. 1991;114(Pt 2):1001–11. doi: 10.1093/brain/114.2.1001. [DOI] [PubMed] [Google Scholar]
- 44.Burstein R, Yamamura H, Malick A, Strassman AM. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J-Neurophysiol. 1998;79(2):964–82. doi: 10.1152/jn.1998.79.2.964. [DOI] [PubMed] [Google Scholar]
- 45.Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996 Dec 12; 384 (6609):560–4. PubMed [DOI] [PubMed]
- 46.Akerman S, Williamson DJ, Kaube H, Goadsby PJ. Nitric oxide synthase inhibitors can antagonize neurogenic and calcitonin gene-related peptide induced dilation of dural meningeal vessels. Br J Pharmacol. 2002 Sep; 137(1):62–8. PubMed [DOI] [PMC free article] [PubMed]
- 47.Messlinger K, Suzuki A, Pawlak M, Zehnter A, Schmidt RF. Involvement of nitric oxide in the modulation of dural arterial blood flow in the rat. Br J Pharmacol. 2000 Apr; 129(7):1397–404. PubMed [DOI] [PMC free article] [PubMed]
- 48.Akerman S, Holland PR, Hoffmann J. Pearls and pitfalls in experimental in vivo models of migraine: Dural trigeminovascular nociception. Cephalalgia. 2013 Jun; 33(8):577–92. PubMed [DOI] [PubMed]
- 49.De Col R, Koulchitsky SV, Messlinger KB. Nitric oxide synthase inhibition lowers activity of neurons with meningeal input in the rat spinal trigeminal nucleus. Neuroreport. 2003 Feb 10; 14(2):229–32. PubMed [DOI] [PubMed]
- 50.Hoskin KL, Bulmer DCE, Goadsby PJ. Fos expression in the trigeminocervical complex of the cat after stimulation of the superior sagittal sinus is reduced by L-NAME. Neuroscience Letters. 1999;266(3):173–6. doi: 10.1016/S0304-3940(99)00281-5. [DOI] [PubMed] [Google Scholar]
- 51.Burgos-Vega CC, Ahn DD, Bischoff C, Wang W, Horne D, Wang J, et al. Meningeal transient receptor potential channel M8 activation causes cutaneous facial and hindpaw allodynia in a preclinical rodent model of headache. Cephalalgia : an international journal of headache. 2016 Feb;36(2):185–93. PubMed Pubmed Central PMCID: PMC4635063. Epub 2015/05/07. eng. [DOI] [PMC free article] [PubMed]
- 52.Reuter U, Chiarugi A, Bolay H, Moskowitz MA. Nuclear factor-kappaB as a molecular target for migraine therapy. Ann Neurol. 2002 Apr;51(4):507–16. PubMed [DOI] [PubMed]
- 53.Sarchielli P, Floridi A, Mancini ML, Rossi C, Coppola F, Baldi A, et al. NF-kappaB activity and iNOS expression in monocytes from internal jugular blood of migraine without aura patients during attacks. Cephalalgia. 2006 Sep; 26(9):1071–9. PubMed [DOI] [PubMed]
- 54.de OSMT, Goncalves FM, Martins-Oliveira A, Speciali JG, Dach F, Lacchini R, et al. Inducible nitric oxide synthase haplotype associated with migraine and aura. Mol Cell Biochem. 2012 May; 364 (1–2):303–8. PubMed [DOI] [PubMed]
- 55.Goncalves FM, Luizon MR, Speciali JG, Martins-Oliveira A, Dach F, Tanus-Santos JE. Interaction among nitric oxide (NO)-related genes in migraine susceptibility. Mol Cell Biochem. 2012 Nov; 370 (1–2):183–9. PubMed [DOI] [PubMed]
- 56.Palmer JE, Guillard FL, Laurijssens BE, Wentz AL, Dixon RM, Williams PMA. randomised, single-blind, placebo-controlled, adaptive clinical trial of GW274150, a selective iNOS inhibitor, in the treatment of acute migraine. Cephalalgia. 2009;29:124. [Google Scholar]
- 57.Hoivik HO, Laurijssens BE, Harnisch LO, Twomey CK, Dixon RM, Kirkham AJ, et al. Lack of efficacy of the selective iNOS inhibitor GW274150 in prophylaxis of migraine headache. Cephalalgia: an international journal of headache. 2010 Dec; 30 (12):1458–67. PubMed Epub 2010/10/27. eng. [DOI] [PubMed]
- 58.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43(2):109–42. [PubMed] [Google Scholar]
- 59.Zinck T, Illum R, Jansen-Olesen I. Increased expression of endothelial and neuronal nitric oxide synthase in dura and pia mater after air stress. Cephalalgia. 2006 Jan; 26(1):14–25. PubMed [DOI] [PubMed]
- 60.Hauge AW, Kirchmann M, Olesen J. Trigger factors in migraine with aura. Cephalalgia. 2010 Mar; 30(3):346–53. PubMed [DOI] [PubMed]
- 61.Andress-Rothrock D, King W, Rothrock J. An analysis of migraine triggers in a clinic-based population. Headache. 2010 Sep; 50(8):1366–70. PubMed [DOI] [PubMed]
- 62.Amin FM, Asghar MS, Hougaard A, Hansen AE, Larsen VA, de Koning PJ, et al. Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: a cross-sectional study. Lancet Neurol. 2013 May; 12(5):454–61. PubMed [DOI] [PubMed]
- 63.Borroni B, Rao R, Liberini P, Venturelli E, Cossandi M, Archetti S, et al. Endothelial nitric oxide synthase (Glu298Asp) polymorphism is an independent risk factor for migraine with aura. Headache. 2006 Nov-Dec; 46 (10):1575–9. PubMed [DOI] [PubMed]
- 64.Schurks M, Kurth T, Buring JE, Zee RY. A candidate gene association study of 77 polymorphisms in migraine. J Pain. 2009 Jul; 10(7):759–66. PubMed Pubmed Central PMCID: PMC2704575. [DOI] [PMC free article] [PubMed]
- 65.Eroz R, Bahadir A, Dikici S, Tasdemir S. Association of endothelial nitric oxide synthase gene polymorphisms (894G/T, −786T/C, G10T) and clinical findings in patients with migraine. Neuromolecular Med. 2014 Sep;16(3):587–93. PubMed [DOI] [PubMed]
- 66.Csati A, Tajti J, Kuris A, Tuka B, Edvinsson L, Warfvinge K. Distribution of vasoactive intestinal peptide, pituitary adenylate cyclase-activating peptide, nitric oxide synthase, and their receptors in human and rat sphenopalatine ganglion. Neuroscience. 2012 Jan 27;202:158–68. PubMed [DOI] [PubMed]
- 67.Dieterle A, Fischer MJ, Link AS, Neuhuber WL, Messlinger K. Increase in CGRP- and nNOS-immunoreactive neurons in the rat trigeminal ganglion after infusion of an NO donor. Cephalalgia. 2011 Jan;31(1):31–42. PubMed [DOI] [PubMed]
- 68.Srikiatkhachorn A, Suwattanasophon C, Ruangpattanatawee U, Phansuwan-Pujito P. 2002 Wolff Award. 5 -HT2A receptor activation and nitric oxide synthesis: a possible mechanism determining migraine attacks. Headache. 2002 Jul-Aug;42(7):566–74. PubMed [DOI] [PubMed]
- 69.Pardutz A, Krizbai I, Multon S, Vecsei L, Schoenen J. Systemic nitroglycerin increases nNOS levels in rat trigeminal nucleus caudalis. Neuroreport. 2000 Sep 28;11(14):3071–5. PubMed [DOI] [PubMed]
- 70.De Felice M, Ossipov MH, Wang R, Dussor G, Lai J, Meng ID, et al. Triptan-induced enhancement of neuronal nitric oxide synthase in trigeminal ganglion dural afferents underlies increased responsiveness to potential migraine triggers. Brain : a journal of neurology. 2010 Aug;133(Pt 8):2475–88. PubMed Pubmed Central PMCID: PMC3139937. Epub 2010/07/16. eng. [DOI] [PMC free article] [PubMed]
- 71.Bhatt DK, Gupta S, Jansen-Olesen I, Andrews JS, Olesen J. NXN-188, a selective nNOS inhibitor and a 5-HT1B/1D receptor agonist, inhibits CGRP release in preclinical migraine models. Cephalalgia. 2013 Jan;33(2):87–100. PubMed [DOI] [PubMed]
- 72.Lambert GA, Hoskin KL, Zagami AS. Nitrergic and glutamatergic neuronal mechanisms at the trigeminovascular first-order synapse. Neuropharmacology. 2004 Jul; 47(1):92–105. PubMed Epub 2004/05/29. [DOI] [PubMed]
- 73.Medve RA, Lategan TW. A phase 2 multicenter, randomized, double-blind, parallel-group, placebo-controlled study of NXN-188 dihydrochloride in acute migraine without aura. Journal of Headache and Pain. 2010 Oct; 11:S38-S. PubMed PMID: WOS:000208791900107. English.
- 74.Medve RA, Andrews JS. Effects of fixed dose combination of nNOS inhibition and 5HT agonism on progrression of migraine with and without aura. Cephalalgia. 2009;29(1):126. [Google Scholar]
- 75.Zhang X, Kainz V, Zhao J, Strassman AM, Levy D. Vascular extracellular signal-regulated kinase mediates migraine-related sensitization of meningeal nociceptors. Ann Neurol. 2013 Jun; 73(6):741–50. PubMed Pubmed Central PMCID: PMC3688635. [DOI] [PMC free article] [PubMed]
- 76.Koulchitsky S, Fischer MJ, De Col R, Schlechtweg PM, Messlinger K. Biphasic response to nitric oxide of spinal trigeminal neurons with meningeal input in rat—possible implications for the pathophysiology of headaches. J Neurophysiol. 2004 Sep; 92(3):1320–8. PubMed [DOI] [PubMed]
- 77.Koulchitsky S, Fischer MJ, Messlinger K. Calcitonin gene-related peptide receptor inhibition reduces neuronal activity induced by prolonged increase in nitric oxide in the rat spinal trigeminal nucleus. Cephalalgia. 2009 Apr; 29(4):408–17. PubMed [DOI] [PubMed]
- 78.Read SJ, Smith MI, Hunter AJ, Parsons AA. The dynamics of nitric oxide release measured directly and in real time following repeated waves of cortical spreading depression in the anaesthetised cat. Neurosci Lett. 1997;232(3):127–30. doi: 10.1016/S0304-3940(97)00604-6. [DOI] [PubMed] [Google Scholar]
- 79.Read SJ, Smith MI, Hunter AJ, Parsons AA. Enhanced nitric oxide release during cortical spreading depression following infusion of glyceryl trinitrate in the anaesthetized cat. Cephalalgia. 1997;17(3):159–65. doi: 10.1046/j.1468-2982.1997.1703159.x. [DOI] [PubMed] [Google Scholar]
- 80.Shen PJ, Gundlach AL. Prolonged induction of neuronal NOS expression and activity following cortical spreading depression (SD): implications for SD- and NO-mediated neuroprotection. Exp Neurol. 1999 Dec; 160(2):317–32. PubMed [DOI] [PubMed]
- 81.Read SJ, Parsons AA. Nitric oxide does not mediate cerebral blood flow changes during cortical spreading depression in the anaesthetised rat. Neurosci Lett. 1998 Jul 03;250(2):115–8. PubMed [DOI] [PubMed]
- 82.Zhang ZG, Chopp M, Maynard KI, Moskowitz MA. Cerebral blood flow changes during cortical spreading depression are not altered by inhibition of nitric oxide synthesis. J Cereb Blood Flow Metab. 1994 Nov;14(6):939–43. PubMed [DOI] [PubMed]
- 83.Urenjak J, Obrenovitch TP. Pharmacological investigation into the involvement of nitric oxide in K+−induced cortical spreading depression. Naunyn Schmiedebergs Arch Pharmacol. 2000 Aug; 362(2):137–44. PubMed [DOI] [PubMed]
- 84.Read SJ, Smith MI, Hunter AJ, Upton N, Parsons AA. SB-220453, a potential novel antimigraine agent, inhibits nitric oxide release following induction of cortical spreading depression in the anaesthetized cat. Cephalalgia. 2000 Mar; 20(2):92–9. PubMed [DOI] [PubMed]
- 85.Smith MI, Read SJ, Chan WN, Thompson M, Hunter AJ, Upton N, et al. Repetitive cortical spreading depression in a gyrencephalic feline brain: inhibition by the novel benzoylamino-benzopyran SB-220453. Cephalalgia. 2000 Jul; 20(6):546–53. PubMed [DOI] [PubMed]
- 86.Dahlof CG, Hauge AW, Olesen J. Efficacy and safety of tonabersat, a gap-junction modulator, in the acute treatment of migraine: a double-blind, parallel-group, randomized study. Cephalalgia. 2009 Nov; 29 Suppl 2:7–16. PubMed [DOI] [PubMed]
- 87.Silberstein SD, Schoenen J, Gobel H, Diener HC, Elkind AH, Klapper JA, et al. Tonabersat, a gap-junction modulator: efficacy and safety in two randomized, placebo-controlled, dose-ranging studies of acute migraine. Cephalalgia. 2009 Nov; 29 Suppl 2:17–27. PubMed [DOI] [PubMed]
- 88.Goadsby PJ, Ferrari MD, Csanyi A, Olesen J, Mills JG. Randomized, double-blind, placebo-controlled, proof-of-concept study of the cortical spreading depression inhibiting agent tonabersat in migraine prophylaxis. Cephalalgia. 2009 Jul; 29(7):742–50. PubMed [DOI] [PubMed]
- 89.Hauge AW, Asghar MS, Schytz HW, Christensen K, Olesen J. Effects of tonabersat on migraine with aura: a randomised, double-blind, placebo-controlled crossover study. Lancet Neurol. 2009 Aug; 8(8):718–23. PubMed [DOI] [PubMed]
- 90.Eroli F, Vilotti S, van den Maagdenberg A, Nistri A. Hyperpolarization-activated current Ih in mouse trigeminal sensory neurons in a transgenic mouse model of familial hemiplegic migraine type-1. Neuroscience. 2017 May 20; 351:47–64. PubMed Epub 2017/04/02. eng. [DOI] [PubMed]
- 91.Socco S, Bovee RC, Palczewski MB, Hickok JR, Thomas DD. Epigenetics: the third pillar of nitric oxide signaling. Pharmacological research. 2017 Jul; 121:52–8. PubMed Epub 2017/04/22. eng. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 498 kb)
(PDF 506 kb)