

Abstract
Tumourigenic tissue uses modified metabolic signalling pathways in order to support hyperproliferation and survival. Cancer-associated aerobic glycolysis resulting in lactic acid production was described nearly 100 years ago. Furthermore, increased reactive oxygen species (ROS) and lactate quantities increase metabolic, survival and proliferation signalling, resulting in increased tumourigenesis. In order to maintain redox balance, the cell possesses innate antioxidant defence systems such as superoxide dismutase, catalase and glutathione. Several stimuli including cells deprived of nutrients or failure of antioxidant systems result in oxidative stress and cell death induction. Among the cell death machinery is autophagy, a compensatory mechanism whereby energy is produced from damaged and/or redundant organelles and proteins, which prevents the accumulation of waste products, thereby maintaining homeostasis. Furthermore, autophagy is maintained by several pathways including phosphoinositol 3 kinases, the mitogen-activated protein kinase family, hypoxia-inducible factor, avian myelocytomatosis viral oncogene homolog and protein kinase receptor-like endoplasmic reticulum kinase. The persistent potential of cancer metabolism, redox regulation and the crosstalk with autophagy in scientific investigation pertains to its ability to uncover essential aspects of tumourigenic transformation. This may result in clinical translational possibilities to exploit tumourigenic oxidative status and autophagy to advance our capabilities to diagnose, monitor and treat cancer.
Keywords: Warburg effect, Autophagy, Oxidative stress, Cancer
Background
In non-tumourigenic cells, glycolysis is a highly regulated and conserved metabolic process in the cytoplasm in which glucose is converted to pyruvate by a series of enzymatic steps. Pyruvate is converted to acetyl coenzyme A, which produces adenosine triphosphate (ATP) via the tricarboxylic acid cycle in the mitochondria involving oxidative phosphorylation (OXPHOS) [1]. Glutaminolysis is a process whereby glutamine is converted to intermediates of the tricarboxylic acid cycle through a series of enzymatic steps. Initially, glutamine is oxidized, forming glutamate that is subsequently converted into α-ketoglutarate, which enters the tricarboxylic acid cycle to provide the metabolic intermediates [2]. However, tumourigenic cells possess aberrant metabolic programming that supports hyperproliferation, survival and long-term maintenance that is characterised by the Warburg effect and increased glycolysis and glutaminolysis (Fig. 1) [3, 4].
Fig. 1.
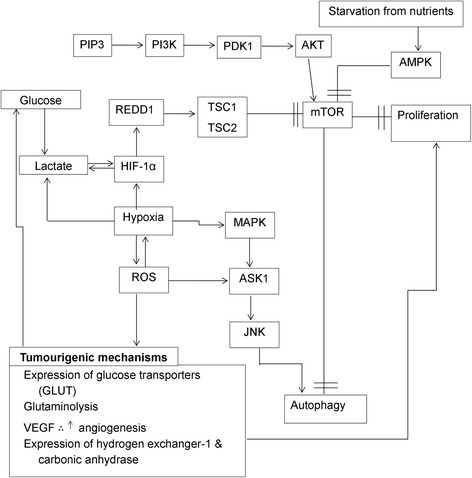
Benefits of tumourigenic metabolism. Tumourigenic metabolism is characterised by the Warburg effect with high ATP and lactate quantities produced in aerobic glycolysis. Furthermore, increased glycolysis and glutaminolysis are present in tumourigenic cells by means of increased expression of glucose and amino acid transporters resulting in increased production of ATP. Lactate production generates an acidic and hypoxic microenvironment which promotes tumourigenesis, invasion and metastasis. The hypoxic microenvironment results in autophagy induction via increased ROS production and subsequent JNK activation. Furthermore, hypoxia also induces expression of HIF-1α, which leads to inhibition of mTOR. In addition, PI3K/Akt signaling also results in inhibition of mTOR. The inhibition of mTOR results in subsequent autophagy induction. Images were created using Microsoft Word 2010 software
OXPHOS in the mitochondria generates 36 ATP molecules from 1 molecule of glucose compared to tumourigenic aberrant glycolysis that only produces 2 ATP molecules from 1 molecule of glucose. In order to compensate for the low ATP output per glucose molecule, tumourigenic cells increase the uptake of glucose by several ways including upregulated glucose transporter 1 (GLUT1) expression (Fig. 1) [5]. GLUT1 overexpression is found in several types of cancer and is associated with poor clinical outcomes in lung cancer, breast cancer, oesophageal cancer, hepatocellular carcinoma, gallbladder carcinoma, colorectal cancer, ovarian cancer and bladder cancer [6–8]. Lee et al. [9] reported that phosphorylation of GLUT1 on Ser226 by protein kinase C regulates glucose transport and GLUT1 deficiency syndrome also demonstrated impaired Ser226 phosphorylation. In addition, a known glucose uptake inducer, 12-O-tetradecanoyl-phorbol-13-acetate (TPA), increases GLUT1 cell surface localization and GLUT1 phosphorylation on Ser226 [9].
The increased requirement for energy production and synthesis of macromolecules results in increased transport of the required nutrients from the environment. Two key nutrients amongst these are glucose and glutamine. Tumourigenic cells consume glucose and glutamine at a higher rate when compared to non-tumourigenic or differentiated tissue and are therefore described as being addicted to glucose and glutamine [4, 10]. The increased consumption of glucose and glutamine yields upregulated carbon sources for anabolic processes and proliferation. The excess carbon is directed towards multiple pathways including glycolysis, glutaminolysis, fatty acid synthesis, nucleic acid production and the pentose phosphate pathway (PPP), resulting in synthesis of macromolecules including nucleotides, lipids and proteins [11].
The Warburg effect describes a hallmark of cancer where glycolysis results in lactate production even in aerobic conditions [1, 3, 4]. Furthermore, Warburg reported that tumourigenic cells exhibit defective mitochondrial OXPHOS that results in switching metabolic energy production to glycolysis. However, decades later we now have research indicating that the mitochondria in tumourigenic cells are not damaged or defective and OXPHOS still takes place in tumourigenic cells proportionally to oxygen supply. However, the rate of glycolysis is drastically upregulated, resulting in lactate production irrespective of oxygen availability [12]. Tumourigenic cells also metabolise glucose by means of the PPP, resulting in nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) production. NADPH also promotes anabolism and favours tumourigenesis by enhancing the antioxidant defence system against hostile environments, radiation and chemotherapeutic compounds [13]. Furthermore, PPP increases generation of ribose-5-phosphate, which is required for the production of nucleic acids. In addition, NADPH also promotes fatty acid synthesis [14].
Increased glucose metabolism by means of tumourigenic glycolysis generates lactic acid and subsequently generates an acidic and hypoxic microenvironment which promotes tumourigenesis, invasion, metastasis and survival and is correlated with clinical prognosis and outcome. Hypoxia increases glucose consumption and subsequent glycolysis, re-enforcing the acidic and hypoxic microenvironment that is beneficial for tumourigenesis [15]. Acidic microenvironments are toxic to non-tumourigenic cells; however, in tumourigenic acidic microenvironments this promotes degradation of the extracellular matrix by proteinases, increases angiogenesis through vascular endothelial growth factor (VEGF) and inhibits the immune response to tumour antigens. As the extracellular matrix is degraded by the proteinases, the open space is invaded by tumourigenic cells. Further adaptations to promote acidic and hypoxic conditions include upregulation of hydrogen exchanger-1 and carbonic anhydrase [16].
Tumourigenesis is characterised by several genotypic and phenotypic metabolic alterations that allow for the increased metabolic activity and proliferation [17]. Cell metabolism is a complicated network of interconnected pathways and impacts overall tumour survival. Research is still ongoing in aberrant cancer metabolic processing and novel therapeutics may selectively target tumourigenic cells by obstructing the metabolic evolution. This is of great importance since the altered tumourigenic metabolism has been reported to be essential in limitless proliferation and resistance to apoptosis induction [18].
Tumourigenic redox regulation
A hallmark of cancer is increased production of reactive oxygen species (ROS) due to upregulated proliferation and glycolytic activity [19]. ROS are a group of molecules produced during metabolism and are fundamental signalling molecules at low concentrations. Production of mitochondrial ROS primarily occurs at the electron transport chain situated on the inner mitochondrial membrane during OXPHOS. ROS include the hydroxyl radical (OH), hydrogen peroxide (H2O2) and superoxide (O2−) [20]. Oxygen reduction by one electron produces superoxide, which is known as the precursor of most intracellular ROS. Superoxide is catalysed into hydrogen peroxide via an antioxidant defence mechanism in an effort to reduce danger elicited by ROS (Fig. 2) [21]. Subsequently, hydrogen peroxide is reduced to water and oxygen or partially reduced via transition elements to the hydroxyl radical [20, 21].
Fig. 2.
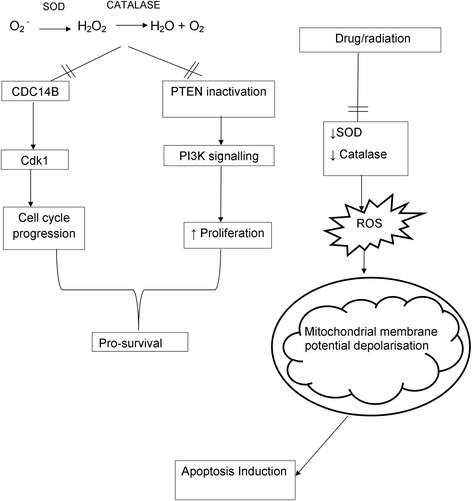
Tumourigenic ROS signalling. ROS inhibits PTEN activity that results in increased augmentation of PI3K/ Akt signalling and subsequently promotes proliferation. Furthermore, ROS promotes cell cycle progression by means of inhibiting phosphatase Cdc14B resulting in the activation of cyclin dependent kinase 1 (Cdk1). Furthermore, anti-tumourigenic compounds induce ROS production by means of inhibiting FOXM1 activity. This results in inhibition of enzymatic activity of antioxidant enzymes including SOD and catalase. The resulting increase ion ROS leads to the depolarisation of the mitochondrial membrane potential and apoptosis induction. Images were created using Microsoft Word 2010 software
In addition to superoxide produced in mitochondrial metabolism, superoxide is also produced by NADPH oxidases that are located in the cellular membrane of various cell types including endothelial cells, neutrophils, eosinophils, monocytes and macrophages. Another source of superoxide generation is from the electron transport chain. The electron transport complexes include complex I (nicotinamide adenine dinucleotide hydrogen (NADH) dehydrogenase), complex II (succinate dehydrogenase), complex III (cytochrome c reductase), complex IV (cytochrome c oxidase) and complex V (mitochondrial F1F0 ATP synthase) [21–23]. Research indicates that the superoxide radical anion is produced by transfer of an unpaired electron from ubisemiquinone of complex I or complex III to an oxygen molecule [24].
The higher quantities of ROS produced in tumourigenic cells are beneficial for tumourigenesis and promote signalling pathways responsible for proliferation, aberrant metabolic activities and angiogenesis [25]. Hydrogen peroxide is well known for being membrane-permeable and is often utilised for intracellular signalling pertaining to proliferation and survival by means of reversible cysteine oxidation within proteins [25, 26]. Hydrogen peroxide oxidises cysteine residues on phosphatase and tensin homolog (PTEN), resulting in PTEN inactivation and subsequent enhancement of phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signalling and promotes proliferation [19].
Upregulated ROS quantities in tumourigenic cells result in increased susceptibility to oxidative stress which causes damage to protein, lipids and deoxyribonucleic acid (DNA). The DNA damage is a double-edged sword since the DNA mutations potentially promote tumourigenesis and genomic instability or the cell undergoes cell death. Survival of tumourigenic cells is highly dependent on their capacity to regulate gene expression that controls innate antioxidant activity [27, 28]. Antioxidant defences include a superoxide dismutase (SOD) with manganese (Mn), which reduces superoxide that is created in the matrix. Superoxide quantities in the intermembrane space are regulated by copper and zinc SOD isozyme which is found in the cytoplasm. The intermembrane also contains cytochrome c, which is reduced by superoxide to produce oxygen. Subsequently, cytochrome c is still capable of transferring electrons to the terminal oxidase during the electron transport chain process [21]. Hydrogen peroxide can be detoxified by glutathione peroxidase, which is mainly found in the liver. Phospholipid-hydroperoxide glutathione is associated with detoxifying peroxides that are found in the membrane. Catalase, which is found in the peroxisomes, is known to detoxify hydrogen peroxide without cofactors, to water and oxygen. All these mechanisms are involved in maintaining a steady state between ROS and the antioxidant activity [29].
A cancer-specific isoform of pyruvate kinase, pyruvate kinase M2 (PKM2), confers an additional advantage to tumourigenic cells by providing them the advantage to withstand oxidative stress [30]. PKM2 is activated by fructose 1,6-bisphosphate and induces an active tetrameric confirmation. Phosphotyrosine-marked proteins activated by extracellular growth signals bind to PKM2 and convert PKM2 to a low activity confirmation by inducing expression of fructose 1,6-bisphosphate [31]. Furthermore, PKM2 promotes glycolysis and subsequent production of lactate in tumourigenic cells [32]. Modulation of PKM catalytic activity also regulates DNA and lipid synthesis required for cell proliferation and NADPH required for redox homeostasis [32].. Exposure to hydrogen peroxide or a thiol-oxidizing compound (diamide) induced oxidation of PKM2 at Cys358 in lung cancer (A549) cells. Furthermore, treatment with a reducing agent (dithiothreitol) restored PKM2 activity. Also, diamide induced impaired coimmunoprecipitation of endogenous PKM2 with flagged-tagged PKM2, and this was totally inhibited by exposure to a reducing agent (dithiothreitol) prior to immunoprecipitation. This suggests that oxidative stress induces PKM2 subunits to dissociate and results in less stable subunit association. Thus, PKM2 provides tumourigenic cells with an additional advantage to withstand oxidative stress. However, increasing PKM2 will compromise its pro-anabolic and antioxidant functions. Activation of PKM2 by small molecule compounds may be a potential future anti-cancer therapy to interfere with tumourigenic metabolism and antioxidant advantage [30]. These PKM2 activators bind to a pocket at the PKM2 subunit interface distinct from the binding site for fructose 1,6-bisphosphate and promote the association of PKM2 subunits into stable tetramers [33, 34]. Small molecule PKM2 activators mimic PKM1 in PKM2 expressing cells and interfere with anabolic metabolic processes including compromised production of ribose-phosphate and serine, which are essential precursors for generation of nucleotides, lipids and amino acids. Potential anticancer activity by small molecule PKM2 activators was confirmed when small molecule PKM2 activators abrogated the ability of tumourigenic cells to form cancer tumours in mice [33].
Nuclear factor kappa B (NF-kB), a redox-sensitive transcription factor, regulates the expression of several genes (cyclin D1, cyclin B1, interleukin (IL)-1, IL-2, granulocyte-macrophage colony stimulating factor (GM-CSF) and baculoviral IAP repeat-containing 3) that regulate inflammation, stress responses due to oxidative stress and apoptosis induction. Moreover, it has been suggested that NF-kB is a sensor for oxidative stress [35–37]. Low hydrogen peroxide quantities activate NF-kB and antioxidants inhibit NF-kB activation [26]. Reactive oxygen species activate NF-kB directly and additionally are involved in NF-kB activation by various other stimuli including tumour necrosis factor alpha (TNFα), phorbol ester, and IL-1 [37]. Oxidative stress resulting in TNFα activation leads to upregulation of antioxidant activity including SOD and catalase. However, NF-kB is essential for tumour progression since genes associated with NF-kB are frequently found in cancer. An NF-kB family member, v-Rel oncogene, has been shown to transform cells in vitro and in vitro [35].
Anticancer drugs that increase ROS production in cancer include bortezomib (Velcade), siomycin, thiostrepton and MG132, since these compounds inhibit the transcription activity of Forkhead Box M1 (FOXM1) [38]. FOXM1 is overexpressed in several types of cancer (prostate, lung, bladder, ovarian, colon, liver, breast stomach and pancreatic cancer) and reduces ROS by means of inducing expression of antioxidant enzymes including catalase, SOD2 and thioredoxin-dependent peroxidase reductase (PRDX3). Furthermore, exposure to FOXM1 inhibitors results in decreased tumour growth in breast and liver xenografts and sensitisation to cell death induced by DNA-damaging agents including doxorubicin and radiation [27, 39].
Metabolic symbiosis between oxidative/aerobic tumour cells and hypoxic/glycolytic cells
Monocarboxylate transporters (MCTs) of the SLC16A gene family determine substrate availability, the metabolic path of lactate and acidity balance within the tumour and the microenvironment [40]. MCT-1, MCT-2, MCT-3 and MCT-4 are essential for proton-linked transportation, and previous studies have shown that MCT-1, MCT-2 and MCT-4 are upregulated in several types of cancer including breast, ovary, colon, liver and lung cancer [41]. MCT-4 is upregulated by hypoxia and contributes to acidification of the microenvironment by means of glycolysis that produces lactate. MCT-1 is upregulated by oxygen and MCT-1 expression is repressed by hypoxia. Furthermore, MCT-1 promotes the transport of lactate into the cell and subsequent utilization of lactate to produce energy [42]. Research further demonstrated that inhibition of MCT-1 did not affect cell survival in cells propagated in growth medium containing glucose; however, cell death induction was prominent when MCT-1 was silenced in cells propagated in growth medium not containing glucose. Inhibition of MCT-1 in mouse models injected with Lewis lung carcinoma cells or colon colorectal tumourigenic cells (WiDr) resulted in decreased tumour growth [43]. Cells expressing MCT-4 thus promote uptake of glucose and subsequent utilization via glycolysis to produce lactate. MCT-1-expressing cells import lactate and utilize it as a substrate in the tricarboxylic acid and as a consequence exhibit stem cell-like characteristics. Cells expressing MCT-1 are rich in mitochondria and produce high quantities of metabolic intermediates and ATP [44, 45]. Inhibition of MCT-1 can thus be a potential therapeutic option since silencing of MCT-1 abrogates the metabolic symbiosis between oxidative/aerobic tumour cells and hypoxic/glycolytic cells [44].
Autophagy
Autophagy is another crucial defence mechanism utilised by the cells in response to oxidative stress [39]. Autophagy (derived from Greek and meaning “self-eating”) is a compensatory mechanism that produces energy from cytoplasmic contents including damaged and/or redundant organelles and proteins which subsequently prevent the accumulation of waste products, thereby maintaining homeostasis [20].
Autophagy of non-selective cytosolic contents by lysosomes is classified into 3 categories, namely macroautophagy, microautophagy and chaperone-mediated autophagy. Microautophagy involves cytosolic contents being taken up by lysosomes directly through an invagination of the lysosomal membrane. In chaperone-mediated autophagy, proteins are translocated across the lysosomal membrane accompanied with chaperone proteins (including heat shock protein 70) that are recognised by the lysosomal membrane receptor, lysosomal-associated membrane protein 2A (LAMP-2A), leading to their unfolding and degradation [46]. In macroautophagy (hereafter referred to as autophagy), a part of the cytoplasm is taken up by a membrane sac called the isolation membrane (also known as the phagophore). The isolation membrane continues to expand and engulfs the aggregates by creating double membrane vesicles called autophagosomes. The latter matures and fuses with lysosomes, creating autolysosomes [47, 48]. Autophagy is generally a non-selective degradation process. However, in rare cases, autophagy is selective of damaged or excess organelles. Selective autophagy is characterised depending on the contents of the degraded cargo: mitophagy (mitochondria), pexophagy (peroxisomes), reticulophagy (endoplasmic reticulum), ribophagy (ribosomes), lipophagy (lipid droplets), aggrephagy (protein aggregates), nucleophagy (nuclear contents) and xenophagy (invasive microbes) [48].
Autophagy serves in an adaptive capacity, promoting cell survival during periods of cellular stress including nutrient starvation, hypoxia, ATP/adenosine monophosphate (AMP) ratio alteration and intracellular ROS. However, certain conditions may result in pro-death autophagy induction [20, 49]. The mechanisms in which autophagy switches from pro-survival to pro-death is not well understood yet, but the involvement of Beclin 1 is indicated [50]. Beclin 1 is a protein associated with autophagy and is essential for embryonic development and autophagy initiation in non-tumourigenic cells and, if present, will distinguish whether the fate of the cell is pro-death or pro-survival [51]. Autophagy is frequently used as a pro-survival mechanism in stressful conditions and contributes to the turnover of cellular contents at a basal level, whereas starvation-induced autophagy provides amino acids for the gluconeogenesis and protein synthesis that is essential in nutrient-deprived conditions [48, 50, 52]. In other pro-survival adaption roles, autophagy contributes to chemotherapy and radiotherapy resistance. Radiation therapy, chemotherapy (doxorubicin, temozolomide and etoposide), histone deacetylase inhibitors, rapamycin, imatinib and anti-hormonal therapy (tamoxifen) induce pro-survival autophagy in human cancer cell lines. If autophagy is inhibited, the efficacy of these therapies increases [49, 50].
Molecular pathways involved in autophagy
Autophagy is a process that requires multiple signalling aspects creating a network of regulatory complexes and protein-protein interactions which aid in a fully functional process [53] (Fig. 1). The process of autophagy can be broken down into several steps, namely induction, vesicle nucleation, expansion and maturation, breakdown and recycling [54]. Autophagy is induced by the unc-51 like autophagy activating kinase (ULK1) complex that consists of the ULK1/ULK2 protein, focal adhesion kinase family interacting protein 200 kDa (FIP 200), scaffold and the Hop/Rev7/Mad2 (HORMA) domain-containing proteins autophagy-related protein (ATG) 13 and ATG101 [55]. Subsequently, the ULK1 complex regulates the formation of the vacuolar protein sorting 34 (VPS34) complex consisting of VPS34 protein, VPS15 protein, Beclin 1 and ATG14 [55, 56]. The phagophore formation in response to stress is dependent on VSP 34 ultraviolet irradiation resistance-associated gene (UVRAG), B-cell lymphoma-2 associated X protein-interacting factor 1 (BIF 1), ATG14-like protein (ATG14L) and RUN domain and cysteine-rich domain containing Beclin 1-interacting protein (Rubicon) [46, 57].
Mammalian target of rapamycin
Autophagy induction is regulated by a serine/threonine protein kinase, mammalian target of rapamycin (mTOR) (Fig. 1). The mTOR signalling pathway is integrated by mammalian target of rapamycin complex 1&2 (mTORC1/mTORC2) [50]. mTORC1 consists of mTOR, regulatory-associated protein mTOR complex 1 (Raptor), and mammalian lethal with SEC13 protein 8 (mLST8) and regulates protein synthesis, proliferation, autophagy, metabolism and the stress response [58, 59]. Raptor facilitates recruitment of the substrate to mTORC1 by means of binding to the TOR signalling (TOS) motif. mTORC1 also contains proline-rich AKT substrate 40 kDa (PRAS40) and DEP domain-containing mTOR-interacting protein (Deptor) [45]. mTORC2 consists of mTOR, mLST8, rapamycin-insensitive companion of mTOR complex 2 (Rictor), mammalian stress-activated map kinase interacting protein (mSIN1), and protein observed with Rictor (Protor1/2) and controls cell survival and polarity [58–60].
Both mTORC complexes are stimulated by growth complexes; however, only mTORC1 is activated by amino acids. Glutaminolysis generates α-ketoglutarate, which induces the translocation of mTORC1 to the lysosome and also activates mTORC1, as demonstrated by increased ribosomal protein S6 kinase (S6K) and S6 phosphorylation [61]. This subsequently leads to inhibition of ULK1, which results in autophagy inhibition. However, a decrease in nutrients or oxygen results in mTORC1 inhibition by means of ULK1 phosphorylation and subsequently autophagy is induced and an autophagosome is formed [11]. The elongation of this structure is dependent on the ATG 12&5 aided by ATG 7&10. ATG 12&5 interact with ATG 16, which then forms a complex and is subsequently added onto the phospholipids in the membrane, in a process that is similar to ubiquitination and SUMOylation. ATG 4 protease aids the previously mentioned reaction and continues to cleave microtubule-associated protein 1A/1B-light chain 3 (LC3), which is further incorporated into the membrane subsequently closing the double membrane structure. The latter fuses with the lysosome and is further degraded, leading to autolysosome formation [25, 57, 62].
Amino acids are the major stimuli for mTORC activation, but the specific mechanistic profile remains elusive; for example, the specific amino acids that regulate mTORC1 are still unclear. Studies have indicated that mTOR senses amino acids (leucine, glutamine and arginine) indirectly within the lysosomal lumen, which requires Rag guanine triphosphatases (GTPases). Activated Rag GTPases recruit mTORC to the lysosome, where Rheb GTPase activates mTORC1 [63]. There are four Rag proteins of interest, namely RagA, RagB, RagC and RagD. RagA and RagB form a heterodimer with RagC and RagD, respectively. RagA/RagB complexes bind directly to mTORC1, and overexpression of constitutively active RagA/RagB protein renders mTORC1 insensitive to amino acid deprivation [64]. However, mTORC is active in fibroblasts lacking RagA and RagB (RagA/B knockout (KO)). Leucine and arginine stimulate mTORC1 activation. However, arginine and leucine failed to induce activation of mTORC in RagA/B KO fibroblasts. Glutamine stimulated mTORC1 irrespective of Rag /B expression (control and RagA/B KO fibroblasts) at the lysosome, indicating that glutamine stimulation of mTORC1 is independent of Rag GTPases [63].
Class I phosphoinositol 3 kinase pathway
The phosphoinositol 3 kinases (PI3K), a family of lipid enzymes, are crucial in autophagy, proliferation, differentiation, motility, intracellular trafficking and cell survival. PI3K phosphorylates the 3′-hydroxyl position of the inositol ring in phosphatidylinositides including phosphatidylinositol (PI) and phosphatidylinositol-4,5-bisphosphate (PIP2) [65]. PIP2 phosphorylation results in the formation of phosphatidylinositol-3,4,5-triphosphate, which activates protein kinase B (Akt) [50]. PIP3 binds to Akt and subsequently Akt moves to the cell membrane, where Akt is activated by PDK1 and mTORC2 phosphorylation by Thr308 and Ser473, respectively [66]. Akt inhibits tuberous sclerosis complex (TSC) by phosphorylating TSC2 at Ser939 and binding to 14–3-3 protein (Fig. 1). Subsequently TSC promotes activation of RAS homolog enriched in brain (Rheb) and mTOR stimulation inhibition and inhibits autophagy induction [66]. Furthermore, activation of autophagy by means of rapamycin increases phosphorylation of Akt, which provides negative feedback and inhibits autophagy induction [67].
Deregulation of PI3K/Akt/mTOR signalling is associated with high-grade tumours (grade 3–grade 4) and tumourigenesis [68]. Thus, modulation of the PI3K/Akt pathway would modify autophagy inhibition or induction and could be a target for chemotherapy [69]. Compounds that target PI3K are currently being investigated for anticancer activity by means of autophagy induction include NVP-BEZ235 (dactolisib), XL-765, PX-866, NVP-BKM120 (buparlisib), GDC-0941 (pictilisib), XL-147, NVP-BYL719, GSK-2636771 and GDC-0032 (taselisib) [43]. NVP-BEZ235 (dactolisib), a dual inhibitor of PI3K and mTOR, exerts dose-dependent antiproliferative activity, inhibits Akt phosphorylation (Ser473) and induces apoptosis in human glioblastoma cells (U87). However, in vivo studies utilizing nude rats and NOD/SCID mice in orthotopic xenograft models of glioblastoma demonstrated that NVP-BEZ235 (dactolisib) exerted no survival benefit or inhibition of tumour growth [70]. NVP-BKM120 (buparlisib) targets PI3K by inhibiting the catalytic subunit p110α of PI3K by competitive binding of the lipid kinase domain on the ATP binding site [71]. NVP-BKM120 (buparlisib) possesses antiproliferative and pro-apoptotic activity in several types of tumourigenic cell lines including breast, glioblastoma, osteosarcoma, ovary and prostate and is to date enlisted in more than 80 clinical trials. Thus, targeting the PI3K/Akt/mTOR pathway for increased effective chemotherapy remains promising, but requires more research [71, 72].
Mitogen-activated protein kinase family
Mitogen-activated protein kinase (MAPK) signalling is essential in proliferation, migration, differentiation, apoptosis induction, autophagy induction and sensitivity to chemotherapy. The MAPK family consists of c-Jun N-terminal kinase or stress-activated protein kinase (JNK or SAPK), extracellular signal-regulated kinase (ERK1/2), big MAP kinase 1 (Erk1/2) and p38 and is involved in cell proliferation and survival processes [73–75]. The ERK pathway is activated by growth factors, cytokines, ultraviolet irradiation, genotoxic agents, oxidative stress and hormones. This results in the phosphorylation of cytoplasmic signalling proteins including G alpha interacting protein, which abolishes the inhibitory protein (Gi3) of autophagy. ERK also targets nuclear components such as ternary complex factor (TCF) transcription factors which are essential for inducing expression of c-Fos and myelocytomatosis oncogene (c-Myc), subsequently promoting cell survival proliferation and motility [73, 74].
Various stressful conditions including oxidative stress activate JNK and p38 pathway signalling by means of apoptosis signal-regulated kinase 1 (ASK1) in several types of cancer including human colon cancer cells (HCT116) (Fig. 1) [76]. This is due to ROS oxidizing two cysteine residues in the redox centre located on thioredoxin. This induces formation of a disulphide bond between Cys32 and Cys35, leading to thioredoxin dissociating from ASK1 and oligomerisation of ASK1. Furthermore, ROS activates JNK by inhibiting JNK-inactivating phosphatases by means of oxidation of a catalytic site, cysteine to sulfenic acid. This results in promotion of JNK activation [75]. Thus, activation and inhibition of phosphatases by ROS are essential for cell signalling pertaining to JNK and p38 [73–75].
JNK, another member of the MAPK signalling family, comprises 3 isoforms of which the third isoform influences autophagy by phosphorylating B-cell lymphoma 2 (Bcl-2), which has proapoptotic functions. This results in a disturbance of the bond between Beclin 1 and Bcl-2, thereby inducing autophagy [62, 77]. Beclin 1 is regulated by JNK by phosphorylating c-Jun (transcription factor), which regulates Beclin 1 expression. Furthermore, JNK stimulate ATG5 to induce autophagy in which ATG expression is influenced by transcriptional factors including Forkhead Box 3 (FOXO3). AMPK aids in the accumulation of nuclear FOXO3; thus activating it which will eventually lead to the induction of autophagy [50].
The RAS family is the regulator of the MAPK/ERK pathway, and mutations in the RAS family are associated with cancer even under sufficient nutrient supply [74]. Human cancer cell lines presenting with H-ras- or K-ras-activating mutations demonstrate higher levels of autophagy induction and increased ROS levels. Inhibition of autophagy correlates with decreased cell growth, suggesting that autophagy is essential for tumour cell survival, and that suppression of autophagy in Ras-driven cancers may be an effective treatment [47].
5′-adenosine monophosphate-activated protein kinase
5′-Adenosine monophosphate-activated protein kinase (AMPK), a heterotrimeric protein complex, is an essential stress and energy sensor [78]. AMPK consists of alpha (catalytic), beta and gamma (both regulatory) subunits and is activated by numerous kinases through phosphorylation and the increase of calcium in the cytosol through calmodulin-dependent protein kinase kinase beta (CAMKK-ß). AMPK senses energy by binding directly to AMP, ATP or ADP, resulting in conformational changes of the enzyme and activation, thus promoting phosphorylation and subsequently preventing further dephosphorylation of the energy molecules [79].
Oxidative stress and starvation lead to AMPK activation, resulting in mTOR inhibition and subsequently autophagy induction accompanied characterised by autophagosome formation [80]. Isoquinoline alkaloids including liensinine, isoliensinine, dauricine, cepharanthine and hernandezine directly activate AMPK and induce autophagy in several cancer cell lines including a cervical cancer cell line (HeLa), alveolar adenocarcinoma cell line (A549), breast adenocarcinoma cell line (MCF-7), prostate cancer cell line (PC3), liver cancer cell lines (HepG2 and Hep3B) and human non-small cell lung carcinoma cell line (H1299) [81].
Studies have suggested that low DNA damage results in pro-survival autophagy induction. However, high levels of DNA damage result in pro-death autophagy induction via activation of ATM, activation of AMPK and repression of mTORC1 [82]. The DNA damage response (DDR) is associated with increased ROS generation and initiates ATM signalling that activates AMPK and subsequent autophagy induction. Furthermore, ionizing radiation activates ATM, which in turn activates tumour suppressor tuberin (TSC2) by means of serine/threonine-protein kinase 11 (STK11) and the AMPK metabolic pathway, resulting in inhibition of mTORC1, and subsequently activates ATG1 [83].
Tumourigenic cells are autophagy-addicted since tumourigenic cells tend to induce autophagy constitutively by means of metabolic reprogramming and AMPK activation. AMPK regulates the autophagy-dependent amino acid recycling system with FIP200 and ULK1 [84–86]. Even under nutrient-sufficient conditions, increased mTOR activity inhibits ULK1 activation by ULK1 phosphorylation on Ser757, thereby abrogating the interaction between ULK1 and AMPK [87].
AMPK is also responsible for the turnover of mitochondria via mitophagy. ULK 1 and ULK2 form stable associations with AMPK in which AMPK phosphorylates and activates ULK1, triggering mitophagy [88]. In cells where the endogenous ULK was replaced with AMPK (and where the phosphorylation sites were defective and replaced by alanine) damaged mitochondrial morphology and reduced mitochondrial membrane potential after nutrient deprivation were present [89].
Hypoxia-inducible factor
Tumourigenic cells depend on autophagy induction in order to survive hypoxia and lower ROS production [90]. Hypoxia-inducible factor (HIF) is a transcriptional regulator during hypoxia that constitutively expresses the beta subunit and an oxygen regulated alpha subunit, which is degraded under normal conditions (oxygen-rich) [91]. Hypoxia results in decreased ubiquitination of the alpha subunit, thereby providing stability. The alpha subunit binds to hypoxia responsive element sequences, thus facilitating the metabolic shift from OXPHOS to glycolysis. In oxidative stressful environments or nutrient-deprived conditions, HIF-1 is capable of stimulating AMPK and subsequently induces autophagy. Furthermore, HIF-1 stimulates Regulated in development and DNA damage response 1 (REDD1) in hypoxia. REDD1 activates the TSC1/2 complex, thereby halting mTOR and activating autophagy [57].
HIF-1 also activates transcription of the gene encoding the BH3 domain protein BNIP3, which induces mitochondrial autophagy (mitophagy) by means of competing with Beclin-1 for binding privileges with Bcl-2. Therefore Beclin-1 is free to induce autophagy. BNIP3-induced autophagy was originally suggested to be hypoxia-specific, but studies conducted in HIF-1α-null mouse embryo fibroblasts demonstrated that mitochondrial autophagy was essential for cell survival in prolonged hypoxic conditions [92].
Cancer stem cells
The significant intra-tumour heterogeneity in tumour tissue plays an essential role in resistance to conventional anticancer treatments including chemotherapy and radiation [93]. Tumour heterogeneity refers to heterogeneous cellular populations with a hierarchical organization governed by cancer stem cells also known as progenitor-like cells or stem cell-like cells [94, 95]. Furthermore, cancer recurrence and subsequent metastasis are largely due to disseminated tumour cells which are cancer stem cells that survived the initial treatment, generally referred to as minimal residual disease. The presence of these disseminated tumour cells in the bone marrow and lymph nodes is associated with poor prognosis and high mortality rates [96]. In addition, cancer stem cells are the cause of all tumourigenic cells contained within the tumour and are capable of forming genetic clones of tumourigenic cells contained within the malignant tissue [97, 98]. Cancer stem cells possess high tumourigenic activity and stem cell-like abilities including capabilities of self-renewal, multi-potent differentiation and multiple DNA repair mechanisms, making them cell death resistant [99]. Cancer stem cells renew themselves by remaining in an undifferentiated state. Cancer stem cells also express specific surface markers, including CD44, CD133 and enzyme aldehyde dehydrogenase (ALDH) [100].
Presently, there is no consensus on the metabolic characteristics of cancer stem cells; however, numerous studies suggest glycolytic dependency instead of mitochondrial respiration. Research confirms that cancer stem cells are more glycolytic than other differentiated cancer cells in vitro and in vivo [101]. However, there are also studies that support cancer stem cell proliferation that utilizes mitochondrial respiration. Moreover, cancer stem cells’ mitochondria possess increased mass and mitochondrial membrane potential, which is a reflection of mitochondrial function, higher mitochondrial ROS production and higher oxygen consumption compared to differentiated cancer cells that generate their energy mainly via glycolysis [102–105]. Cancer stem cells adapt metabolic- and signalling pathways contingent to the microenvironmental changes by shifting energy production from one pathway to the other [106]. Differentiation or a hypoxic microenvironment results in cancer stem cells from several tumour types switching from oxidative to glycolytic metabolism in order to compensate for deficient mitochondrial machinery [107].
Studies indicate that intracellular ROS levels are significantly lower when compared to non-tumourigenic stem cells, which may be due to upregulated expression of members of the free radical scavengers system [108]. ROS have been implicated in metastasis via cell migration, invasion and angiogenesis. One of the proposed signalling mechanisms involves CD13 negatively regulating ROS with a resultant increase of stem cell-like abilities and increased ROS scavenger capacity in human liver cancer stem cells [109]. Furthermore, hydrogen peroxide activates canonical wingless-type mouse mammary tumour virus (MMTV) integration site family (Wnt) signalling, which is vital for malignancy regulation and tumourigenesis [110, 111].
Cancer stem cells have a superior antioxidant defence system that includes a CD44 variant isoform containing variable exon 8–10 (CD44v8–10) and cysteine/glutamate antiporter (xCT). Both have been implicated to be significant when considering resistance to anticancer treatment [112, 113]. CD44v8–10 interacts with cysteine transporter xCT (SLC7A11), resulting in stabilization of cysteine transporter xCT, thereby possibly providing protection against redox stress due to ROS generation. The cysteine transporter xCT acts as a sodium-independent transporter that regulates the exchange of extracellular cysteine for intracellular glutamate and supports reduced glutathione (GSH) synthesis, which acts as an intracellular buffer. CD44v-xCT-GSH is associated with resistance to anticancer agents. Cysteine availability is rate-limiting to GSH synthesis, and the activity of xCT is therefore crucial to resistance to oxidant-dependent anticancer agents [114]. Ectopic expression of CD44v8–10 was induced after ROS-enhancing chemotherapy in a patient with a hereditary cancer predisposition syndrome commonly associated with p53 mutations (Li-Fraumeni syndrome) [115]. Thus, the CD44v-xCT-GSH axis is of importance when considering resistance to a microenvironment rich in oxidative stress and subsequent anticancer treatments dependent on ROS production [110, 113, 115].
Oxidative stress activates the Wnt pathway and promotes expression of CD44 and c-Myc. Furthermore, high quantities of hydrogen peroxide activate the canonical beta-catenin/Wnt signalling pathway by means of the thioredoxin-like protein nucleoredoxin (NRX) [116, 117]. Furthermore, CD44v8–10 and c-Myc have an inversed expression pattern in vitro and in vivo. The latter negative correlation is especially prominent at the invasive front of the tumour enriched with cancer stem cells [110]. Another study verified the existence of crosstalk between ROS and the Wnt pathway. Wnt signalling induced ROS production mediated by NADPH oxidase 1 (Nox1). This occurred by increased levels of the Ras-related C3 botulinum toxin substrate 1 (Rac1)-GTP through activation of Rac1, guanine nucleotide exchange factors (GEFs) and vav guanine nucleotide exchange factor 2 (Vav2) by proto-oncogene tyrosine-protein kinase Src-dependent tyrosine phosphorylation [118]. Moreover, Nox1-generated ROS inactivate NRX through oxidation and disrupt the NRX-dishevelled (Dvl) complexes, thereby activating the Wnt-β-catenin pathway involved in proliferation of both healthy and malignant cells [118].
A γ-secretase-mediated intracellular domain of CD44 promotes aggressive glioma growth and a stem cell-like phenotype by means of CREB-binding protein (CBP)/p300-dependent enhancement of HIF-2α activity. Furthermore, expression of CD44 correlates with hypoxia-induced gene signatures and poor survival rates in human glioblastoma multiforme patients [119]. A hypoxic bone marrow microenvironment is a stem- and progenitor cell niche that acts as a sanctuary for cancer stem cells. HIF-1α expression is upregulated in the hypoxic bone marrow niche. HIF-1α induces expression of stromal cell-derived factor 1 (also known as C-X-C motif chemokine 12 (CXCL12)), resulting in an increase in adhesion, migration and homing of circulating C-X-C chemokine receptor type 4 (CXCR4)-positive progenitor cells into the ischemic tissue [120]. This suggests that a hypoxic bone marrow microenvironment represents a conditional stem and progenitor cell niche in which HIF-1α-induced stabilization and activation of CXCL12 and CXCR4 facilitate recruitment and retention of progenitor cells. In this context, HIF-1α may represent an important molecular target within the tumour microenvironment [121].
Recent research indicates that autophagy promotes the stem cell-like characteristics present in cancer stem cells in several types of cancer including breast cancer, pancreatic cancer, colon cancer, hepatocarcinoma, osteosarcoma and bladder cancer [122, 123]. Inhibition of autophagy resulted in a decrease in stem cell surface markers (POU Class 5 Homeobox 1 Transcript Variant OCT4B4, SRY-Box 2, Homeobox Transcription Factor Nanog and CD44) in breast cancer cell lines (MCF-7 and MDA-MB-468) [123, 124]. Furthermore, autophagy inhibition in osteosarcoma cancer stem cells decreased cancer cell surface markers and enhanced sensitivity to hypoxic conditions and exposure to cisplatin, resulting in increased cell death induction [122–124]. Thus, identification of mechanisms underlying cancer stem cell characteristics and the development of novel approaches to target them holds promise for the therapeutic elimination of cancer stem cells and the complete eradication of tumours [120–124].
Conclusion
Research has shown that autophagy mediates cancer cell metabolism, allowing for hyperproliferation by providing amino acids and other building blocks when the macromolecules are unavailable from other sources [125]. Autophagy has also been shown to be required for tumourigenic cell survival during hypoxic stress in order to combat oxidative stress [90]. Hypoxia, which is a characteristic of cancerous cells, produces moderately high levels of ROS leading to the induction of autophagy [126]. ROS enhances the PI3K pathway, which is already hyperactivated in cancerous cells. It is stated in the literature that activation of the PI3K pathway, which is hyperactivated by cell growth producing growth factor signalling, leads to increased proliferation and cellular mobility and promotes cell survival. ROS also activate HIF, which is a pathway activated upon hypoxic conditions and as is known to be a prominent state in tumour cells [77]. Tumourigenic cells activate HIF-1, resulting in transcription activation, in order to adapt to the hypoxic condition. The transcription targets of the HIFs promote cell survival in hypoxic conditions, shifting metabolism in order to increase aerobic glycolysis and angiogenesis [127, 128].
Hypoxia and nutrient deprivation increase the AMP/ATP ratio, which subsequently leads to the activation of AMPK, thereby inhibiting mTORC1 and activating autophagy [62]. Furthermore, ROS (hydrogen peroxide) is also known to induce autophagy in cancer by means of oxidising ATG4, followed by the removal of lipids and protein maturation of ATG4, resulting in LC3-associated autophagosomes [125]. Increased H2O2 production causes oxidative stress and AMPK phosphorylation by an upstream kinase (AMPKK), resulting in autophagy induction [62].
Autophagy and oxidative stress are both involved in a complex signalling network in tumourigenesis pertaining to major processes including proliferation, metabolism, angiogenesis, oxidative stress and resistance to cell death (autophagy and apoptosis) and oxidative stress [129]. In addition, tumourigenic cells possess increased metabolic activities which produce high ROS quantities contributing to hyperproliferation and increased cell survival, in turn contributing to chemo- and radiosensitivity [130, 131]. The interactions of the pathways and processes are evidently tightly linked, but are not well documented in the literature as one complete or linked process. The manner in which metabolism autophagy as well as ROS interacts along with the pathways involved will contribute to novel and more effective methods to target tumourigenic cells in order to reduce the high cancer prevalence. The bimodal nature of ROS and its influence on autophagy by means of pro-survival and pro-death signalling requires elucidation in order to identify novel biochemical targets for chemotherapy and increased efficacy in current treatment options.
Funding
This study was supported by grants from the Cancer Association of South Africa, the Medical Research Council, the National Research Foundation, Struwig Germeshuysen Trust and the School of Medicine Research Committee of the Faculty of Health Sciences, University of Pretoria.
Abbreviations
- ALDH
Aldehyde dehydrogenase
- AMP
Adenosine monophosphate
- AMPK
5′-Adenosine monophosphate-activated protein kinase
- ASK1
Apoptosis signal-regulated kinase 1
- ATG
Autophagy-related protein
- ATG14L
ATG14-like protein
- ATP
Adenosine triphosphate
- Bcl-2
B-cell lymphoma 2
- BIF 1
B-cell lymphoma-2 associated X protein-interacting factor 1
- CAMKK-ß
Calmodulin-dependent protein kinase kinase beta
- CBP
CREB-binding protein
- CD44v8-10
CD44 variant isoform containing variable exon 8-10
- Cdk1
Cyclin dependent kinase 1
- CXCL12
C-X-C motif chemokine 12
- CXCR4
C-X-C chemokine receptor type 4
- DDR
DNA damage response
- Deptor
DEP domain-containing mTOR-interacting protein
- DNA
Deoxyribonucleic acid
- Dvl
Dishevelled
- ERK1/2
Extracellular signal-regulated kinase
- FIP 200
Focal adhesion kinase family interacting protein 200 kDa
- FOXM1
Forkhead Box M1
- FOXO3
Forkhead box 3
- GLUT1
Glucose transporter 1
- JNK
C-Jun N-terminal kinase
- TPA
12-O-Tetradecanoyl-phorbol-13-acetate
- xCT
Cysteine/glutamate antiporter
- GSH
Glutathione
- GM-CSF
Granulocyte-macrophage colony stimulating factor
- GTPases
Guanine triphosphatases
- HIF
Hypoxia-inducible factor
- H2O2
Hydrogen peroxide
- OH
Hydroxyl radical
- LAMP-2A
Lysosomal-associated membrane protein 2A
- Mn
Manganese
- mLST8
Mammalian lethal with SEC13 protein 8
- mSIN1
Mammalian stress-activated map kinase interacting protein
- mTOR
Mammalian target of rapamycin
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- MAPK
Mitogen-activated protein kinase
- MCTs
Monocarboxylate transporters
- MMTV
Mouse mammary tumour virus
- c-Myc
Myelocytomatosis oncogene
- Nox1
NADPH oxidase 1
- NADH
Nicotinamide adenine dinucleotide hydrogen
- NADPH
Nicotinamide adenine dinucleotide phosphate hydrogen
- NF-kB
Nuclear factor kappa B
- OXPHOS
Oxidative phosphorylation
- PPP
Pentose phosphate pathway
- PTEN
Phosphatase and tensin homolog
- PI
Phosphatidylinositol
- PIP2
Phosphatidylinositol-4,5-bisphosphate
- PI3K
Phosphoinositide 3-kinase
- PRAS40
Proline-rich AKT substrate 40 kDa
- Protor
Protein observed with Rictor
- Akt
Protein kinase B
- PKM2
Pyruvate kinase M2
- Rac1
Ras-related C3 botulinum toxin substrate 1
- S6K
Ribosomal protein S6 kinase
- Rictor
Rapamycin-insensitive companion of mTOR complex 2
- Raptor
Regulatory-associated protein mTOR complex 1
- REDD1
Regulated in development and DNA damage response 1
- Rheb
RAS homolog enriched in brain
- ROS
Reactive oxygen species
- Rubicon
RUN domain protein as Beclin 1 interacting and cysteine-rich containing
- HORMA
Scaffold and the Hop/Rev7/Mad2
- STK11
Serine/threonine-protein kinase 11
- SAPK
Stress-activated protein kinase
- O2
Superoxide
- SOD
Superoxide dismutase
- PRDX3
Thioredoxin-dependent peroxidase reductase
- NRX
Thioredoxin-like protein nucleoredoxin
- TCF
Ternary complex factor
- TOS
TOR signalling
- TSC
Tuberous sclerosis complex
- TNFα
Tumour necrosis factor alpha
- TSC2
Tumour suppressor tuberin
- UVRAG
Ultraviolet irradiation resistance-associated gene
- ULK1
Unc-51 like autophagy activating kinase
- VPS34
Vacuolar protein sorting 34
- VEGF
Vascular endothelial growth factor
- Vav2
Vav guanine nucleotide exchange factor 2
- Wnt
Wingless-type MMTV integration site family
Authors’ contributions
Initial compilation of manuscript was done by MVG and MHV. Editing and final compilation of manuscript was performed by MVG, AMJ and MHV. MHV and AMJ were responsible for supervision and acquisition of funds. All authors have read and approve of the final manuscript.
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mokgadi Violet Gwangwa, Email: mokgadigwangwa@gmail.com.
Anna Margaretha Joubert, Email: annie.joubert@up.ac.za.
Michelle Helen Visagie, Phone: +27 12 3192245, Email: michelle.visagie@up.ac.za.
References
- 1.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parsklow D, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetics function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 2.Fu X, Hu X, Li N, Zheng F, Dong X, Duan J, et al. Glutamine and glutaminolysis are required for efficient replication of infectious spleen and kidney necrosis virus in Chinese perch brain cells. Oncotarget. 2017;8(2):2400–2412. doi: 10.18632/oncotarget.13681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christofk HR, van der Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform and pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–234. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 4.Zhu C, Martinez AF, Martin HL, Li M, Crouch BT, Carlson DA, et al. Near-simultaneous intravital microscopy of glucose uptake and mitochondrial membrane potential, key endpoints that reflect major metabolic axes in cancer. Sci Rep. 2017;7:13722. doi: 10.1038/s41598-017-14226-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 6.Chung FY, Huang MY, Yeh CS, Chang HJ, Cheng TL, Yen LC, et al. GLUT1 gene is a potential hypoxic marker in colorectal cancer patients. BMC Cancer. 2009;9:241. doi: 10.1186/1471-2407-9-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J, Ye C, Chen C, Xiong H, Xie B, Zhou J. Glucose transporter GLUT1 expression and clinical outcome in solid tumours: a systematic review and meta-analysis. Oncotarget. 2017;8(10):16875–16886. doi: 10.18632/oncotarget.15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bravata V, Stefano A, Cammarata FP, Minafra L, Russo G, Nicolosi S, et al. Genotyping analysis and (1)(8)FDG uptake in breast cancer patients: a preliminary research. J Exp Clin Cancer Res. 2013;32:23. doi: 10.1186/1756-9966-32-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee EE, Ma J, Sacharidou A, Mi W, Salato VK, Nguyen N, et al. A protein kinase C phosphorylation motif in Glut1 affects glucose transport and is mutated in glut 1 deficiency syndrome. Mol Cell. 2015;58(5):875–853. doi: 10.1016/j.molcel.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Belestrieri C, et al. Oncogenic K-ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keibler MA, Wasylenko TM, Kelleher JK, Iliopoulos O, van der Heiden MG, Stephanopoulos G. Metabolic requirements for cancer cell proliferation. Cancer Metab. 2016;4:16. doi: 10.1186/s40170-016-0156-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schulz TJ, Thierbach R, Voigt A, Drewes G, Mietzner B, Steinberg P, et al. Induction of oxidative metabolism by mitochondrial frataxin inhibits cancer growth Otto Warburg revisited. J Biol Chem. 2005;281:977–981. doi: 10.1074/jbc.M511064200. [DOI] [PubMed] [Google Scholar]
- 13.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz L, Seyfried T, Alfarouk KO, Da Veiga MJ, Fais S. Out of Warburg effect: an affective cancer treatment targeting the tumour specific metabolism and dysregulated pH. Semin Cancer Biol. 2017;43:134–138. doi: 10.1016/j.semcancer.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 15.Höckel M, Vaupel P. Tumour hypoxia: definitions and current clinical, biological, and molecular aspects. J Natl Cancer Inst. 2001;93:266–276. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- 16.Estrella V, Chen T, Lloyd M, Wojkowiak J, Cornwell HH, Ibrahim-Hashim A, et al. Acidity generated by tumor microenvironment drives local invasion. Cancer Res. 2012;73(5):1524–1535. doi: 10.1158/0008-5472.CAN-12-2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robertson-Tessi M, Gillies RJ, Gatenby RA, Anderson ARA. Impact of metabolic heterogeneity on tumor growth, invasion, and treatment outcomes. Cancer Res. 2015;75(8):1567–1579. doi: 10.1158/0008-5472.CAN-14-1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodríguez-Lirio A, Pérez-Yarza G, Fernández-Suárez MR, Alonso-Tejerina E, Boyano MD, Asumendi A. Metformin induces cell cycle arrest and apoptosis in drug-resistant leukemia cells. Leuk Res 2015;2015. 516460. [DOI] [PMC free article] [PubMed]
- 19.Chandel NS, Diebold L. Mitochondrial ROS regulation of proliferating cells. Free Radic Biol Med. 2016;100:86–93. doi: 10.1016/j.freeradbiomed.2016.04.198. [DOI] [PubMed] [Google Scholar]
- 20.Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2014;4:184–192. doi: 10.1016/j.redox.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pérez-Carreras M, Del Hoyo P, Martin MA, Rubio JC, Martin A, Castellano G, et al. Defective heptaic mitochondrial respiratory chain in patients with nonalchoholic steatohepatitis. Hepatology. 2003;38(4):999–1007. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 23.Schuett J, Schuett H, Oberoi R, Koch A-K, Pretzer S. Luchtefeld, et al. NADPH oxidase NOX2 mediates TLR2/6-dependent release of GM-CSF from endothelial cells. FASEB J. 2017;31(6):2612–2624. doi: 10.1096/fj.201600729R. [DOI] [PubMed] [Google Scholar]
- 24.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron acceptor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237(2):408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 25.Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014;2:17. doi: 10.1186/2049-3002-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mantzaris MD, Bellou S, Skiada V, Kitsati V, Fotsis T, Galaris D. Intracellular labile iron determines H2O2-induced apoptotic signaling via sustained activation of ASK1/JNK-p38 axis. Free Radic Biol Med. 2016;97:454–465. doi: 10.1016/j.freeradbiomed.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 27.Nogueira V, Hay N. Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin Cancer Res. 2013;19:4309–4314. doi: 10.1158/1078-0432.CCR-12-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aybastier Õ, Dawbaa S, Demir C, Akgün O, Ulukaya E, Ari F. Quantification of DNA damage products by gas chromatography tandem mass spectrophotometry in lung cell lines and prevention effect of thyme antioxidants on oxidative induced DNA damage. Mutat Res. 2018;808:1–9. doi: 10.1016/j.mrfmmm.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 29.Szymonik-Lesiuk S, Czechowska G, Stryjecka-Zimmer M, Słomka M, Madro A, Celiński K, et al. Catalase, superoxide dismutase, and glutathione peroxidase activities in various rat tissues after carbon tetrachloride intoxication. J Hepatobiliary Pancreat Sci. 2003; 10.1007/s00534-002-0824-5. [DOI] [PubMed]
- 30.Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J-J, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contribute to cellular antioxidant responses. Science. 2011;334(6060):1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kung C, Hixon J, Choe S, Marks K, Gross S, Murphy E, et al. Small molecule activation of PKM2 in cancer cells induces serine auxotrophy. Chem Biol. 2012;19(9):1187–1198. doi: 10.1016/j.chembiol.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su B-Q, Han Y-Q, Fan S-S, Ming S-L, Wan B, Lu W-F, et al. PKM2 knockdown influences SREBP activation and lipid synthesis in bovine mammary-gland epithelial MAC-T cells. Biotechnol Lett. 2018:1–8. [DOI] [PubMed]
- 33.Pamell KM, Foulks JM, Nix RN, Clifford A, Bullough J, Luo B, et al. Pharmacologic activation of PKM2 slows lung tumor xenograft growth. Mol Cancer Ther. 2013;12(8):1453–1460. doi: 10.1158/1535-7163.MCT-13-0026. [DOI] [PubMed] [Google Scholar]
- 34.Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8:839–847. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stortz P. Reactive oxygen species in tumor progression. Front Biosci. 2005;10:1881–1896. doi: 10.2741/1667. [DOI] [PubMed] [Google Scholar]
- 36.Wang T, Zhang X, Li JJ. The role of NF-kB in the regulation of cell stress responses. Int Immunopharmacol. 2002;2(1):1509–1520. doi: 10.1016/s1567-5769(02)00058-9. [DOI] [PubMed] [Google Scholar]
- 37.Chen AC-H, Arany PR, Huang Y-Y, Tomkinson EM, Sharma SK, Kharkwal GB, et al. Low-level laser therapy activates NF-kB via generation of reactive oxygen species in mouse embryonic fibroblasts. PLoS One. 2011;6(7):e22453. doi: 10.1371/journal.pone.0022453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halasi M, Pandit B, Wang M, Nogueira V, Hay H, Gartel A. Combination of oxidative stress and FOXM1 inhibitors induces apoptosis in cancer cells and inhibits xenograft tumor growth. Am J Pathol. 2013;183(1):257–265. doi: 10.1016/j.ajpath.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin J, Duan J, Cui Z, Ren W, Li T, Yin Y. Hydrogen peroxide-induced oxidative stress activates NF-kB and NRF2/Keap1 signals and triggers autophagy in piglets. R Soc Chem. 2015;5:15479–15486. [Google Scholar]
- 40.Izumi H, Takahashi M, Uramoto H, Nakayama Y, Oyama T, Wang K-Y, et al. Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells. Cancer Sci. 2011;102(5):1007–1013. doi: 10.1111/j.1349-7006.2011.01908.x. [DOI] [PubMed] [Google Scholar]
- 41.Xu K, Mao X, Mehta M, Cui J, Zhang C, Mao F, et al. Elucidation of how cancer cells avoid acidosis through comparative transcriptomic data analysis. PLoS One. 2013;8(8):e71177. doi: 10.1371/journal.pone.0071177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Semenza GL. Tumor metabolism: cancer cells give and take lactate. J Clin Invest. 2008;118(12):3835–3837. doi: 10.1172/JCI37373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118(12):3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015; 10.1186/s13046-015-0221-y. [DOI] [PMC free article] [PubMed]
- 45.Pertega-Gomes N, Vizcaino JR, Attig J, Jurmeister S, Lopes C, Baltazar F. A lactate shuttle system between tumour and stromal cells is associated with poor prognosis in prostate cancer. BMC Cancer. 2014;14:352. doi: 10.1186/1471-2407-14-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ozpolat B, Benbrook DM. Targeting autophagy in cancer management – strategies and developments. Cancer Manag Res. 2015;7:291–299. doi: 10.2147/CMAR.S34859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo M, Zhao X, Song Y, Cheng H, Zhou R. Nuclear autophagy: an evolutionary conserved mechanism of nuclear degradation in the cytoplasm. Autophagy. 2016;12:1973–1983. doi: 10.1080/15548627.2016.1217381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang R, Zeh HJ, Lotze MT, Tang D. The beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 51.Qin JZ, Xin H, Nickoloff BJ. Targeting glutamine metabolism sensitizes melanoma cells to TRAIL-induced death. Biochem Biophys Res Commun. 2010;398:146–152. doi: 10.1016/j.bbrc.2010.06.057. [DOI] [PubMed] [Google Scholar]
- 52.Ezaki J, Matsumoto N, Takeda-Ezaki M, Komatsu M, Takahashi K, Hiraoka Y. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy. 2011;7(7):727–736. doi: 10.4161/auto.7.7.15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Popelka H, Uversky VN, Klionsky DJ. Identification of Atg3 as an intrinsically disordered polypeptide yields insights into the molecular dynamics of autophagy-related proteins in yeast. Autophagy. 2014;10:103–114. doi: 10.4161/auto.28616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic death in Caenorhabditis elegnas. Cell Death Differ. 2008;15:105–112. doi: 10.1038/sj.cdd.4402231. [DOI] [PubMed] [Google Scholar]
- 55.Wallot-Hieke N, Verma N, Schlütermann D, Derleth N, Deitersen J, Böhler P. Systematic analysis of ATGF13 domain requirements for autophagy induction. Autophagy. 2017; 10.1080/15548627.2017.1387342. [DOI] [PMC free article] [PubMed]
- 56.Liu C-C, Lin Y-C, Chen Y-H, Chen C-M, Pang L-Y, Chen H-A. Cul3-KLHL20 ubiquitin ligase governs the turnover of ULK1 and VS34 complexes to control autophagy termination. Mol Cell. 2016;61:84–97. doi: 10.1016/j.molcel.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 57.Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaur A, Sharmna S. Mammalian target of rapamycin (mTOR) as a potential therapeutic target in various diseases. Immunopharmacology. 2017;25:293–312. doi: 10.1007/s10787-017-0336-1. [DOI] [PubMed] [Google Scholar]
- 59.Zou Z, Chen J, Yang J, Bai X. Targeted inhibition of rictor/mTORC2 in cancer treatment: a new era after rapamycin. Curr Cancer Drug Targets. 2016;16:288–304. doi: 10.2174/1568009616666151113120830. [DOI] [PubMed] [Google Scholar]
- 60.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Durán RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell. 2012;47(3):649–358. doi: 10.1016/j.molcel.2012.05.043. [DOI] [PubMed] [Google Scholar]
- 62.Nagelkerke A, Sweep FCGJ, Geurts-Moespot A, Bussink J, Span PN. Therapeutic targeting of autophagy in cancer. Part I: molecular pathways controlling autophagy. Semin Cancer Biol. 2014;16:26–36. doi: 10.1016/j.semcancer.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 63.Jewell JL, Guan K-L. Differential regulation of mTORC1 by leucine and glutamine. Science. 2015;347(6218):194–198. doi: 10.1126/science.1259472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shen K, Choe A, Sabatini DM. Intersubunit crosstalk in the rag GTPase heterodimer enables mTORC1 to respond rapidly to amino acid availability. Mol Cell. 2017;38(3):552–565. doi: 10.1016/j.molcel.2017.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shanware NP, Bray K, Abraham RT. The PI3K, metabolic, and autophagy networks: interactive partners in cellular health and disease. Ann Rev Pharmacol Toxicol. 2013;53:89–106. doi: 10.1146/annurev-pharmtox-010611-134717. [DOI] [PubMed] [Google Scholar]
- 66.Comb WC, Hutti JE, Cogswell P, Cantley LC, Baldwin AS. P85α SH2 domain phosphorylation by IKK promotes feedback inhibition of PI3K and Akt in response to cellular starvation. Mol Cell. 2012;45:719–730. doi: 10.1016/j.molcel.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65(8):3336–3346. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 68.Scrima M, De Marco C, Fabiana F, Franco R, Pirrozzi G, Rocco G, et al. Signaling networks associated with AKT activation in non-small cell lung cancer (NSCLC): new insights on the role of phosphatydil-inositol-3 kinase. PLoS One. 2012;7(2):e30427. doi: 10.1371/journal.pone.0030427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saiki S, Sasazawa Y, Imamichi Y, Kawajiri S, Fujimaki T, Tanida I, et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 2011;7:176–187. doi: 10.4161/auto.7.2.14074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Netland IA, Forde HE, Sleire L, Leiss L, Rahman MA, Skeie BS. Dactolisib (NVP-BEZ235) toxicity in murine brain tumour models. BMC Cancer. 2016;16:657. doi: 10.1186/s12885-016-2712-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lu Y, Wang Q, Fan S, Hu B, Sun L, Xue H, et al. Effective use of PI3K inhibitor BKM120 to treat human osteosarcoma. Int J Clin Exp Med. 2017;10:6378–6386. [Google Scholar]
- 72.Bohnacker T, Prota AE, Beaufils F, Burke JE, Melone A, Inglis AJ. Deconvolution of Buparlisib’s mechanism of action defines specific PI3K and tubulin inhibitors for therapeutic intervention. Nat Commun. 2017;8:14683. doi: 10.1038/ncomms14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, et al. P38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344:174–179. doi: 10.1016/j.canlet.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 74.Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways : crosstalk and compensation. Trends Biochem Sci. 2011;36:320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ray PD, Huang B-W, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baregamian N, Song J, Bailey CE, Papaconstantinou J, Evers BM, Chung DH. Tumor necrosis factor-α and apoptosis signal-regulating kinase 1 control reactive oxygen species release, mitochondrial autophagy and c-Jun N-terminal kinase p38 phosphorylation during necrotizing enterocolitis. Oxidative Med Cell Longev. 2009;2:297–306. doi: 10.4161/oxim.2.5.9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davies C, Tournier C. Exploring the function of the JNK ( c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem Soc Trans. 2012;40:85–96. doi: 10.1042/BST20110641. [DOI] [PubMed] [Google Scholar]
- 78.Houde VP, Donzelli S, Sacconi A, Galic S, Hammill JA, Bramson JL, et al. AMPK β1 reduces tumour progression and improves survival in p53-null mice. Mol Oncol. 2017;11(9):1143–1155. doi: 10.1002/1878-0261.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang D, Wang L, Yan L, Miao X, Gong C, Xiao M, et al. Vacuolar protein sorting 4B regulates apoptosis of intestinal epithelial cells via p38 MAPK in Crohn’s disease. Exp Mol Pathol. 2015;98:55–64. doi: 10.1016/j.yexmp.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 80.Li L, Chen Y, Gibson SB. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal. 2013;25:50–65. doi: 10.1016/j.cellsig.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 81.Law BYK, Mok SWF, Chan WK, Xu SW, Wu AG, Yao XJ, et al. Hernandezine, a novel AMPK activator induces autophagic cell death in drug resistant cancers. Oncotarget. 2016;7:8090–8104. doi: 10.18632/oncotarget.6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Orlotti NI, Cimino-Reale G, Borghini E, Pennati M, Sissi C, Perronne F, et al. Autophagy acts as a safeguard mechanism against G-quadruplex ligand-mediated DNA damage. Autophagy. 2012;8(8):1185–1196. doi: 10.4161/auto.20519. [DOI] [PubMed] [Google Scholar]
- 83.Alexander A, Chai S-L, Kim J, Nanaez A, Sahin M, MacLean KH, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci. 2010;107:4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. J Hematol Oncol. 2017;10(1):67. doi: 10.1186/s13045-017-0436-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Phan LM, Yeung SC, Lee MH. Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol Med 2014;11(1):1–19. [DOI] [PMC free article] [PubMed]
- 86.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8(10):774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 87.Joungmok K, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2010;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rouschop KMA, Ramaekers CHMA, Schaaf MBE, Keulers TGH, Savelkouls GM, Lambin P, et al. Autophagy is required during hypoxia to lower production of reactive oxygen species. Radiother Oncol. 2009;92:411–416. doi: 10.1016/j.radonc.2009.06.029. [DOI] [PubMed] [Google Scholar]
- 91.Saito S, Lin Y-C, Tsai M-H, Lin C-S, Murayama Y, Sato R, et al. Emerging roles of hypoxia-inducible factors and reactive oxygen species in cancer and pluripotent stem cells. Kaohsiung J Med Sci. 2015;31:1–8. doi: 10.1016/j.kjms.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 92.Semenza GL. Hif-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yoshida GJ, Saya H. Therapeutic strategies targeting cancer stem cells. Cancer Sci. 2016;107(1):5–11. doi: 10.1111/cas.12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yoshida GJ, Saya H. EpCAM expression in the prostate cancer makes the difference in the response to growth factors. Biochem Biophys Res Commun. 2014;443(1):239–245. doi: 10.1016/j.bbrc.2013.11.093. [DOI] [PubMed] [Google Scholar]
- 96.Klein CA, Blankenstein TJF, Schmidt-Kittler O, Petronio M, Polzer B, Stoecklein NH, et al. Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet. 2002;360(9334):683–689. doi: 10.1016/S0140-6736(02)09838-0. [DOI] [PubMed] [Google Scholar]
- 97.Bischof J, Westhoff MA, Wagner JE, Halatsch M-E, Trentmann S, Knippschild U, et al. Cancer stem cells: the potential role of autophagy, proteolysis, and cathepsins in glioblastoma stem cells. Tumour Biol. 2017:1–13. [DOI] [PubMed]
- 98.Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133–143. doi: 10.1038/nrc3184. [DOI] [PubMed] [Google Scholar]
- 99.Jiang X, Gwye Y, Russell D, Cao C, Douglas D, Hung L, et al. CD133 expression in chemo-resistant Ewing sarcoma cells. BMC Cancer. 2010;10:116. doi: 10.1186/1471-2407-10-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fan F, Bellister S, Lu J, Ye X, Boulbes DR, Tozzi F, et al. The requirement for freshly isolated colorectal cancer (CRC) cells in isolating CRC stem cells. Br J Cancer. 2015;112:539–546. doi: 10.1038/bjc.2014.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Peiris-Pagè M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res. 2016;18:55. doi: 10.1186/s13058-016-0712-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.De Luca A, Fiorillo M, Peiris-Pages M, Ozsvari B, Smith DL, Sanchez-Alvarez R, et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget. 2015;6(17):14777–14795. doi: 10.18632/oncotarget.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lamb R, Bonuccelli G, Ozsvari B, Peiris-Pages M, Fiorillo M, Smith DL, et al. Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: understanding WNT/FGF-driven anabolic signaling. Oncotarget. 2015;6(31):30453–30471. doi: 10.18632/oncotarget.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vlashi E, Lagadec C, Vergnes L, Reue K, Frohnen P, Chan M, et al. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res Treat. 2014;146(3):525–534. doi: 10.1007/s10549-014-3051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Farnie G, Sotgia F, Lisanti MP. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget. 2015;6(31):30472–30486. doi: 10.18632/oncotarget.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu P-P, Liao J, Tang Z-J, Wu W-J, Yang J, Zeng ZL. Metabolic regulation of cancer cell side population by glucose through activation of the AKT pathway. Cell Death Differ. 2014;21:124–135. doi: 10.1038/cdd.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cipolleschi MG, Marzi I, Santini R, Fredducci D, Vinci MC, D’Amico M, et al. Hypoxia-resistant profile implies vulnerability of cancer stem cells to physiological agents, which suggests new therapeutic targets. Cell Cycle. 2014;13(2):268–278. doi: 10.4161/cc.27031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mizuno T, Suzuki N, Makino H, Furui T, Morii E, Aoki H, et al. Cancer stem-like cells of ovarian clear cell carcinoma are enriched in the ALDH-high population associated with an accelerated scavenging system in reactive oxygen species. Gynecol Oncol. 2015;137:299–305. doi: 10.1016/j.ygyno.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 109.Kim HM, Haraguchi N, Ishii H, Ohkuma M, Okano M, Mimori K, et al. Increased CD13 expression reduces reactive oxygen species, promoting survival of liver cancer stem cells via an epithelial–mesenchymal transition-like phenomenon. Ann Surg Oncol. 2012;19:539–548. doi: 10.1245/s10434-011-2040-5. [DOI] [PubMed] [Google Scholar]
- 110.Yoshida GJ, Saya H. Inversed relationship between CD44 variant and c-Myc due to oxidative stress-induced canonical Wnt activation. Biochem Biophys Res Commun. 2014;443:622–627. doi: 10.1016/j.bbrc.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 111.Funato Y, Michiue T, Asashima M, Miki H. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling through dishevelled. Nat Cell Biol. 2006;8:501–508. doi: 10.1038/ncb1405. [DOI] [PubMed] [Google Scholar]
- 112.Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(−) and thereby promotes tumor growth. Cancer Cell. 2011;19:387–400. doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 113.Lei Y, Zhang D, Yu J, Dong H, Zhang J, Yang S. Targeting autophagy in cancer stem cells as an anticancer therapy. Cancer Lett. 2017;393:33–39. doi: 10.1016/j.canlet.2017.02.012. [DOI] [PubMed] [Google Scholar]
- 114.Yoshida GJ, Saya H, Zouboulis CC. Three-dimensional culture of sebaceous gland cells revealing the role of prostaglandin E2-induced activation of canonical Wnt signaling. Biochem Biophys Res Commun. 2013;438:640–646. doi: 10.1016/j.bbrc.2013.07.129. [DOI] [PubMed] [Google Scholar]
- 115.Yae T, Tsuchihashi K, Ishimoto T, Motohara T, Yoshikawa M, Yoshida GJ, et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun. 2012;3:883. doi: 10.1038/ncomms1892. [DOI] [PubMed] [Google Scholar]
- 116.Yoshida GJ, Fuchimoto Y, Osumi T, Shimada H, Hosaka S, Morioka H, et al. Li-Fraumeni syndrome with simultaneous osteosarcoma and liver cancer: increased expression of a CD44 variant isoform after chemotherapy. BMC Cancer. 2012;12:444. doi: 10.1186/1471-2407-12-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Korswagen HC. Regulation of the Wnt/beta-catenin pathway by redox signaling. Dev Cell. 2006;10(6):687–688. doi: 10.1016/j.devcel.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 118.Yoshida GJ. The heterogeneity of cancer stem-like cells at the invasive front. Cancer Cell Int. 2017;17:23. doi: 10.1186/s12935-017-0393-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kajla S, Mondol AS, Nagasawa A, Zhang Y, Kato M, Matsuno K, et al. A crucial role for nox1 in redox-dependent regulation of Wnt-β-catenin signaling. FASEB J. 2012;26(5):2049–2059. doi: 10.1096/fj.11-196360. [DOI] [PubMed] [Google Scholar]
- 120.Pietras A, Katz AM, Ekström EJ, Wee B, Halliday JJ, Pitter KL, et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014;14:357–369. doi: 10.1016/j.stem.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Konopleva M, Tabe Y, Zeng Z, Andreeff M. Therapeutic targeting of microenvironmental interactions in leukemia: mechanisms and approaches. Drug Resist Updat. 2009;12(4–5):103–113. doi: 10.1016/j.drup.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lee K, Qian DZ, Rey R, Wei H, Liu JO, Semenza GL. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc Natl Acad Sci U S A. 2009;106:2353–2358. doi: 10.1073/pnas.0812801106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 123.Lei Y, Zhang D, Yu J, Dong H, Zang J, Yang S. Targeting autophagy in cancer stem cells as an anticancer therapy. Cancer Lett. 2017;393:33–39. doi: 10.1016/j.canlet.2017.02.012. [DOI] [PubMed] [Google Scholar]
- 124.Zhang D, Zhao Q, Sun H, Yin L, Wu J, Xu J, et al. Defective autophagy leads to the suppression of stem-like features of CD271+ osteosarcoma cells. J Biomed Sci. 2016;23:82. doi: 10.1186/s12929-016-0297-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang L, Xu L, Zhang F, Vlashi E. Doxycycline inhibits the cancer stem cell phenotype and epithelial-to-mesenchymal transition in breast cancer. Cell Cycle. 2016;16(8):737–745. doi: 10.1080/15384101.2016.1241929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gozuacik D, Kimchi A. Autophagy as a cell death and tumour suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 127.Kongara S, Karantza V. The interplay between autophagy and ROS in tumorigenesis. Front Oncol. 2012;21:1–13. doi: 10.3389/fonc.2012.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.DeBerardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 131.Tong L, Chuang C-C, Wu S. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015;367:18–25. doi: 10.1016/j.canlet.2015.07.008. [DOI] [PubMed] [Google Scholar]