

Abstract
Background
The formation of new blood vessels, known as angiogenesis, is critical for recovery from ischemic heart disease, and estrogen is considered an important factor in this process. Here, we investigated the effects of 17β-estradiol (17β-E2) on proliferation and migration of cardiac microvascular endothelial cells (CMECs) in vitro.
Material/Methods
Rat CMECs were isolated and cultured with 17β-E2 (0.001–1 μmol/l) in the absence or presence of the estrogen antagonist tamoxifen. Then, the expression level of estrogen receptor alpha was evaluated by using immunofluorescence assay, RT-PCR, and Western blot. Cell proliferation was detected by methyl thiazolyl tetrazolium analysis and the cell migration was verified by a scraping assay and quantified by a Transwell chamber assay. CMEC differentiation was examined using a tube formation assay. Vascular endothelial growth factor (VEGF) secretion was detected by enzyme-linked immunosorbent assay.
Results
CMECs exhibited homogenous, polygonal, exhibited contact inhibition, and had characteristically ovoid nuclei with 1 or 2 nucleoli, and the cytoplasm exhibited red fluorescence after staining for von Willebrand factor. 17β-E2 treatment upregulated estrogen receptor alpha expression in CMECs. 17β-E2 treatment significantly promoted the proliferation, migration, tubular structure formation, and VEGF secretion in CMECs. The maximal proliferation occurred in the presence of 0.01 μmol/l 17β-E2. Furthermore, estrogen and VEGF were found to synergistically stimulate angiogenesis.
Conclusions
Our data show that 17β-E2 promotes angiogenesis in vitro and suggests that estrogen treatment as a novel therapeutic modality in the management of arterial insufficiency.
MeSH Keywords: Angiogenesis Inducing Agents, Endothelial Cells, Estradiol
Background
Angiogenesis generates new capillaries and therefore leads to tissue vascularization [1,2]. Angiogenic activity is generally low in adults, except in the female reproductive tract, where neovascularization occurs as a normal aspect of the menstrual cycle [3]. It is considered that neovascularization in reproductive tissues is regulated by steroid hormones, particularly 17-β-estradiol [4]]. Moreover, a role for gonadal steroids in modulating endothelial cell behavior is supported by studies examining blood vessel formation in the primate endometrium [5].
Powazniak et al. demonstrated that estradiol enhanced human umbilical vein endothelial cell activity (non-endometrial endothelial cells) in vitro and in vivo [6]. These cells play an important role in neovascularization, suggesting a promoting influence of estrogen on angiogenesis. Importantly, estrogen increased vascular endothelial growth factor (VEGF) receptor-2 expression and promoted DNA synthesis in retinal microvascular endothelial cells [7]. But the role of estrogen in angiogenesis of CMECs (cardiac microvascular endothelial cells) was still unclear, so the aim of the present study was to examine the influence of exogenous estrogen in angiogenesis of CMECs.
Material and Methods
Reagents
17β-estradiol and tamoxifen, obtained from Sigma-Aldrich (St. Louis, MO, USA), were suspended in 100% ethanol and diluted to the indicated concentrations in DMEM (Dulbecco’s modified Eagle’s medium) (Gibco Laboratories, Grand Island, NY, USA). All antibodies were obtained from Abcam (Cambridge, MA, USA). All experiments conformed to the Institutional Guidelines for the Care and Use of Laboratory Animals in the Fourth Military Medical University, Xi’an, China, and conformed to the National Institutes of Health (China) Guide for Care and Use of Laboratory Animals.
Isolation and culture of rat CMECs
Primary rat CMECs were isolated and cultured as described previously [8]. Briefly, adult male Sprague-Dawley rats (170–200 g) were anesthetized and hearts were rapidly excised. The remaining left ventricular tissue was immersed in 70% ethanol for 30 s and the outer one-fourth of the left ventricular free wall and septum were removed to eliminate epicardial arteries and larger penetrating vessels. The remaining heart tissue was finely minced in 0.2% collagenase in PBS and incubated in a shaking water bath for 8 min at 37°C. Trypsin was added to a concentration of 0.02%, and the sample was incubated for another 4 min. Dissociated cells were filtered through a l00-μm mesh filter and washed with calcium-free Krebs-Henseleit bicarbonate buffer, followed by centrifugation at 1000×g for 10 min. Cells were resuspended in DMEM supplemented with 10% FBS and antibiotics (penicillin 100 IU/ml, streptomycin 100 pg/ml) at a density of 2.5×103 cells/cm2 on culture dishes pretreated with rat tail glue. After 4 h, attached cells were washed with DMEM and cultured in DMEM with 10% FBS in a 5% CO2 atmosphere at 37°C. When primary cultures were confluent, cells were diluted and transferred to new plates. The final yield of CMECs was ~4.5×105 cells/g ventricular tissue.
Immunofluorescence
Immunofluorescence staining was performed as reported previously [9]. In brief, rat CMECs on coverslips were washed once with PBS, fixed in 4% paraformaldehyde (pH 7.2~7.6) for 20 min at room temperature, washed with PBS, and then blocked with 5% bovine serum albumin in TBST for 15 min. The cells were incubated with PBS (negative control) or mouse antibody against estrogen receptor alpha (1: 100, Santa Cruz, Santacruz, CA, US) overnight at 4°C and then for 1 h at 37°C, then washed with PBS 3 times for 5 min each. The cells were then incubated with FITC-labeled goat anti-mouse IgG (1: 200 in TBST, Santa Cruz, Santacruz, CA, US) for 45 min and washed 3 times. Coverslips were analyzed using DPController and DPManager software (Olympus, Tokyo, Japan).
RT-PCR
Total RNA was prepared using Trizol reagent (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Total RNA was reverse transcribed using Advantage RT-for-PCR kit (Clontech, Palo Alto, CA) with oligo dT primers. The forward primer for estrogen receptor alpha was 5′-CAAGTCCACTTGTGATCAAGC-3′, and the reverse primer was 5′-GCCTTCTACACATTTACCTTG-3′. The PCR products were analyzed on a 1.0% agarose gel using LumiAnalyst Image Analysis software (Roche, Mannheim, Germany).
Western blot
For Western blot analysis, 50–100 μg of proteins were separated by SDS-PAGE on polyacrylamide gels. After electrophoresis, the proteins were electro-transferred to a nitrocellulose membrane, blocked with 5% non-fat milk, and probed with estrogen receptor alpha antibody (1: 5000, Santa Cruz, Santacruz, CA, US) overnight at 4°C. The blots were then washed, incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h (1: 15000, Santa Cruz, Santacruz, CA, US), and exposed to an enhanced chemiluminescence (ECL) reagent (Amersham, Piscataway, NJ).
Cell proliferation assay
Cell proliferation was quantified by counting and by the methyl thiazolyl tetrazolium (MTT) assay. Briefly, when cells reached 70–75% confluence, they were treated with increasing concentrations of 17β-E2 (0.001, 0.01, 0.1, or 1 μmol/l) or carrier (0.1% methanol) in DMEM containing 5% FBS. After incubation for 24 h, 20 μl MTT reagent (Millipore Corp., Billerica, MA, USA) was added into each well, then the plates were incubated in a humidified atmosphere for 4 h. The supernatant was then removed, and 150 μl dimethyl sulfoxide (Sigma-Aldrich) was added per well to solubilize formazan salt crystals. The formazan product was spectrophotometrically quantified at 490 nm using an ELISA reader (Bio-Rad, Hercules, CA, USA).
Wounding migration assay and Transwell chamber assay
Rat CMECs cultured for 48 h in hormone-free medium were plated at ~90% confluent in 24-well culture plates. After 12 h, the cell monolayer was scraped with a 1-ml pipette tip to create a cell-free zone [6]. The medium and dislodged cells were then aspirated, and the plates were rinsed with PBS. Fresh hormone-free medium and the indicated concentration of 17β-E2 were added to the plates. Tamoxifen is a selective estrogen receptor (ER) modulator reported to affect cancer cell survival; therefore, tamoxifen (0.1 μmmol/l) was also added to inhibit cell proliferation, allowing observation of migration in the absence of proliferation. Images of each scratch were taken at random at 4 separate sites along the length of the scratch, starting proximally and ending distally. Images were obtained using an inverted microscope immediately after the scratch was made and then again at 24 h.
To quantify cell invasion, a Transwell chamber housing a polycarbonate filter (8 mm pore size; Millipore) was used [10]. Filters were coated with collagen I (10 μg/ml), and the lower Transwell chamber contained reduced serum medium supplemented with VEGF (20 ng/ml) as the chemoattractant. Tamoxifen (0.1 μmmol/l) and 17β-E2 (0.001, 0.01, 0.1, or 1 μmol/l) were added to both the upper and lower chambers. Rat CMECs suspended in serum-free DMEM were added to the upper chamber at a density of 2.5×104 cells/well and allowed to migrate for 24 h at 37°C. Migrated cells attached to the underside of the filters were washed with PBS, stained with crystal violet, and counted under a microscope. The number of cells per microscopic field (the precise size) that migrated through the pores to the lower surface of each filter was determined using a Nikon-Optiphot-2 (20× magnification) (Nikon, Tokyo, Japan). Data reflect the mean number of cells per field.
Tube formation assay
For tube formation assays, matrigel (Becton Dickinson Labware, Bedford, MA, USA) at 4°C was diluted 1: 4 with pre-chilled DMEM (Gibco). The gel was allowed to form by incubation in a CO2-free incubator at 37°C for 30 to 60 min, and 300 μl of the appropriate dilution was added to each well. Rat CMECs in DMEM supplemented with 10% FBS and antibiotics were pipetted onto the gel.
After incubating under 5% CO2 at 37°C for 48 h, the medium was aspirated and the gel was fixed in 4% formaldehyde. Gels were examined using a Nikon TE300 microscope equipped with Hoffman modulation optics and a cooled CCD camera (Nikon). Images from 4 fields per gel were obtained using OpenLab (Improvision, University of Warwick Science Park, Coventry, Millburn Hill Road, UK), and the data were imported as TIFF files into the ISEE software program (Innovision Corp., Raleigh, NC, USA). The total length of each tube that formed, or the long axis of single cells or groups of adjacent cells, was measured and the mean length of tube-like formations was determined for each well.
VEGF ELISA
Supernatants from tube formation assay samples were collected, and VEGF levels in the supernatant were measured in triplicate or quadruplicate by ELISA (R&D Systems, Minneapolis, MN, USA). The optical density was measured at 490 nm.
Statistical analyses
Data are presented as means ±S.E.M. from at least 3 independent experiments. All values were processed using the statistical software GraphPad Prism 5.0, and the unpaired Student’s t-test was used for statistical analysis. ANOVA was performed for serial analyses. Differences between experimental values having p-values <0.05 were considered significant.
Results
Morphological and biological characteristics of isolated and cultured CMECs
Isolated primary rat CMECs grew as confluent monolayers in culture with the typical cobblestone morphology of normal endothelial cells. They were homogenous, polygonal, exhibited contact inhibition, and had characteristically ovoid nuclei with 1 or 2 nucleoli (Figure 1A, 1B). CD34 (an endothelial cell marker) immunostaining was observed in the nuclear perimeter (Figure 1C) and the cytoplasm exhibited red fluorescence after staining for von Willebrand factor (a constitutive and specific component of endothelial cell matrix) (Figure 1D).
Figure 1.

Culture of CMECs isolated from adult male Sprague-Dawley rat heart tissue (left ventricle). (A) Primary isolated rat CMECs (100×). (B) Rat CMECs at passage 2 (100×). (C) Immunostaining for CD34 in rat CMECs (100×). (D) Immunostaining for von Willebrand factor in rat CMECs (200×).
ER expression is detected in rat CMECs
Immunofluorescence analysis was used to examine ER expression in rat CMECs. Murine monoclonal anti-ER was used as the primary antibody, and goat-anti-mouse IgG-FITC was used as the secondary antibody. Immunofluorescent staining (green) was observed in samples treated with anti-ER, but not in control samples lacking anti-ER (Figure 2). RT-PCR and Western blot also showed that 17β-E2 treatment upregulated estrogen receptor alpha expression in CMECs.
Figure 2.
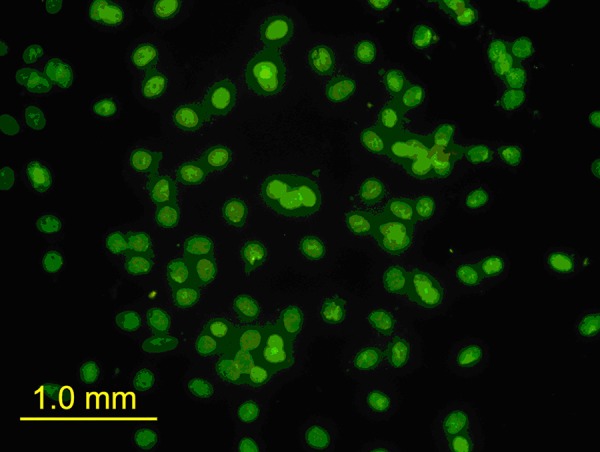
ER expression is detected in rat CMECs. Show fluorescence immunostaining of estrogen receptor alpha in rat CMECs (100×). Immunofluorescent staining in control samples lacking anti-ER alpha. Immunofluorescent staining (green) in samples treated with anti-ER alpha.
17 b-estradiol promotes rat CMEC proliferation
Because endothelial cells are important for revascularization, we examined whether 17β-E2 promotes proliferation of endothelial cells. Rat CMECs were incubated with increasing concentrations of 17β-E2 for 24 h, and proliferation was assessed by MTT analysis. Treatment with 0.001 μmol/l or 0.01 μmol/l 17β-E2 significantly promoted cell proliferation (6.71±1.55% and 26.20±3.54%, respectively, p<0.05, compared to untreated controls) (Figure 3A). There were no differences among groups treated with 0.01, 0.1, or 1 μmol/l 17β-E2. The estrogen antagonist tamoxifen reduced the 17β-E2-induced increase in proliferation by 15.90±1.51% of cells (P<0.0001, compared to 0.01 μmol/l 17β-E2) (Figure 3B).
Figure 3.

Effect of 17β-estradiol (17β-E2) on rat CMEC proliferation. (A) Cell cultures were treated with the indicated concentration of 17β-E2, and cell proliferation was quantified using the MTT assay. * P<0.05 vs. control, *** P<0.001 vs. control; n=6. (B) Tamoxifen inhibition of 17β-E2-induced cell proliferation. Cell cultures were untreated or treated with 17β-E2 (0.01 μmol/l) in the absence or presence of tamoxifen (Tamo; 0.1 μmol/l) *** P<0.001 vs. control; n=6.
Tamoxifen reduces migration of CMECs in vitro
The rat CMECs (3×104 cells/well) were seeded in 6-well plates and cultured in medium containing a reduced level of serum for 18 h. The cells were then stimulated with 17β-E2 (0.01 μmol/l) for an additional 8 h before a scratch was made across each well using a pipette tip to produce a cell-free gap of approximately 1.0 mm between 2 adjacent cell areas. 17β-E2 promoted cell migration into the scratch area at 24 h, but tamoxifen blocked this effect (Figure 4A). To quantify CMEC migration, cell cultures were grown for 24 h in Transwell chambers. Treatment with 17β-estradiol (0.01 μmol/l) resulted in migration of 59.14±6.42% of cells across the Transwell filter, and tamoxifen reduced 17β-E2-induced migration to 20.10±3.69% of cells (Figure 4B).
Figure 4.

Effect of 17β-estradiol (17β-E2) on rat CMEC migration. (A) Cell cultures were untreated (Normal) or treated with 17β-E2 (0.01 μmol/l) in the absence or presence of tamoxifen (Tamo; 0.1 μmol/l). Representative images of CMEC migration across wound edges 24 h after scraping of the culture surface are shown. (B) Cell migration was determined using a Transwell chamber assay; migration was quantified by counting cells after 24 h. Data represent the mean ±S.E.M. from 4 independent experiments. Data were compared using ANOVA. ### P<0.001 vs. control.
17β-estradiol induces tube-like structure formation in vitro
Compared with untreated controls, the number of tube-like structures formed in the 17β-E2 (0.01 μmol/l) group increased; tubule spacing also decreased and was interlaced with an extracellular matrix latticework (Figure 5). The average number of tubular branching points was analyzed and determined to be 12 per field in the control group and 23 per field in the 17β-E2-treated group (P<0.01). However, the average number of tubular branching points in CMECs treated with 17β-E2 and tamoxifen was 12 per field – the same as for the control group (P>0.05). These data showed that 17β-E2 induced tubule-like structures in CMEC cultures in vitro.
Figure 5.

17β-estradiol (17β-E2) induces the formation of tube-like structures in CMEC cultures. Cell cultures were untreated (Control) or treated with 17β-E2 (0.01 μmol/l) in the absence or presence of tamoxifen (Tamo; 0.1 μmol/l) for 48 h. Representative images are shown.
17β-estradiol promotes VEGF secretion
After stimulation of tube-like structure formation, culture supernatants from control and 17β-E2-treated CMECs were collected and the concentration of VEGF was determined by ELISA. 17β-E2 (0.01 μmol/l) markedly promoted the secretion of VEGF in rat CMECs (P<0.01 vs. control; n=4). In addition, tamoxifen blocked the secretion of VEGF induced by 17β-E2 (Figure 6).
Figure 6.
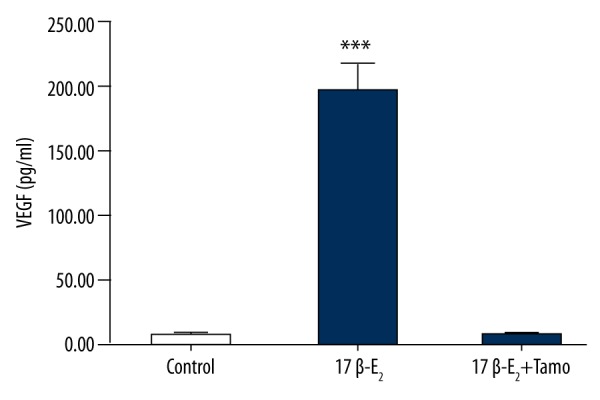
17 β-estradiol (17β-E2) induces VEGF expression in CMECs. Cell cultures were untreated or treated with 17β-E2 (0.01 μmol/l) in the absence or presence of tamoxifen (Tamo; 0.1 μmol/l) for 48 h, and VEGF expression in culture supernatants was analyzed by ELISA. Data represent the means ±S.E.M. from 4 independent experiments. Data were compared by ANOVA. *** P<0.001 vs. control.
Discussion
Coronary heart disease is one of the major risks for human health. Although using some techniques to stimulate coronary revascularization improves clinical symptoms and lifespan for some patients, many problems still persist, such as a high rate of restenosis, the occurrence of the “no-reflow” phenomenon after coronary artery intervention therapy, atherosclerotic occlusion of vein vessels after vascular bypass surgery, and the considerable number of heart disease patients for whom revascularization therapy would be inappropriate and/or ineffective [11].
Angiogenesis is a biological process whereby new blood vessels grow from the existing vascular structure. VEGF, fibroblast growth factor, and many other factors stimulate neovascularization [12]. A new strategy for treatment of ischemic cardiovascular disease via stimulation of angiogenesis has been reported and may be particularly attractive for patients with diffuse coronary artery disease and who are not candidates for traditional revascularization procedures [13]. Unfortunately, the angiogenesis-promoting drugs currently available for clinical application are inadequate, partly because angiogenesis is an extremely complex process and partly because these drugs have limited efficacy and cause adverse effects. Elucidation of angiogenic mechanisms will therefore be required to improve the treatment and prevention of cardiovascular disease and to develop more effective angiogenesis drugs.
Estrogen protects against cardiovascular disease by preventing atherosclerosis and improving coronary insufficiency. Protection is both direct, such as improvement of endothelial cell functions and inhibition of proliferation and migration of vascular smooth muscle cells, and indirect, such as regulation of lipid metabolism, improvement of hemodynamic parameters, coagulation of fibrinolysis systems, and antioxidation. Animal and human experiments have confirmed that the protective effect of estrogen on the cardiovascular system mainly depends on specific ERs [14]. ERs are expressed in the majority of arterial wall cells, and in smooth muscle cells, endothelial cells, and cardiac myocytes. The present study also identified ER expression in rat CMECs. Protection of endothelial cells by estrogen occurs mainly by inhibition of endothelial cell apoptosis via increasing tyrosine phosphorylation of the pp125 focal adhesion kinase, promotion of vascular endothelial cell DNA synthesis through reactive oxygen species signaling, and promotion of vascular endothelial cell NO synthesis [15–19]. Morales et al. found that estrogen can accelerate the proliferation and migration rates of human umbilical vein endothelial cells and inhibit their apoptosis [6]. Schnlze et al. cultivated umbilical vein endothelial cells and coronary artery endothelial cells, and found that estrogen stimulated endothelial cell proliferation and that proliferation was blocked by a specific ER antagonist [20]. We therefore examined rat CMECs and confirmed that estrogen promotes CMEC proliferation, migration, and the formation of tube-like structures. These effects were blocked by tamoxifen, a specific ER antagonist. In addition, we found that VEGF secretion was upregulated in estrogen-treated CMEC cultures. VEGF and basic fibroblast growth factor are highly specific vascular endothelial cell growth stimulating factors that specifically stimulate vascular endothelial cell proliferation and promote the formation of collateral vessels [21].
Estrogen promotes the occurrence and development of neovascularization in ischemic myocardium tissues, which is conducive to the formation of coronary collateral circulation and the decrease of myocardial ischemia and hypoxia. This suggests that estrogen may be efficacious for treatment of cardiovascular and cerebrovascular infarctions, but further experimental evidence will be required to confirm these preliminary findings.
Conclusions
Taken together, the findings of this study demonstrated that treatment with 17β-E2 promoted angiogenesis in vitro, indicating that estrogen or its signaling pathway might represent a new therapeutic modality in the management of arterial insufficiency.
Acknowledgement
The authors of this manuscript have certified that they comply with the Principles of Ethical Publishing in the International Journal of Cardiology [22].
Footnotes
Source of support: Departmental sources
Conflict of interest
None.
References
- 1.Paku S, Dezso K, Bugyik E, et al. A new mechanism for pillar formation during tumor-induced intussusceptive angiogenesis: inverse sprouting. Am J Pathol. 2011;179:1573–85. doi: 10.1016/j.ajpath.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ambrose CT. The role of capillaries in the lesser ailments of old age and in Alzheimer’s disease and vascular dementia: The potential of pro-therapeutic angiogenesis. J Alzheimers Dis. 2016;54:31–43. doi: 10.3233/JAD-160303. [DOI] [PubMed] [Google Scholar]
- 3.Elkilani OA, Soliman MA. Angiogenesis mediators in women with idiopathic heavy menstrual bleeding. Int J Gynaecol Obstet. 2017;136:280–84. doi: 10.1002/ijgo.12068. [DOI] [PubMed] [Google Scholar]
- 4.Zhang L, Xiong W, Xiong Y, et al. 17 beta-Estradiol promotes vascular endothelial growth factor expression via the Wnt/beta-catenin pathway during the pathogenesis of endometriosis. Mol Hum Reprod. 2016;22:526–35. doi: 10.1093/molehr/gaw025. [DOI] [PubMed] [Google Scholar]
- 5.Joseph K, Tholanikunnel BG, Kaplan AP. Cytokine and estrogen stimulation of endothelial cells augments activation of the prekallikrein-high molecular weight kininogen complex: Implications for hereditary angioedema. J Allergy Clin Immunol. 2017;140(1):170–76. doi: 10.1016/j.jaci.2016.09.032. [DOI] [PubMed] [Google Scholar]
- 6.Powazniak Y, Kempfer AC, de la Paz Dominguez M, et al. Effect of estradiol, progesterone and testosterone on apoptosis- and proliferation-induced MAPK signaling in human umbilical vein endothelial cells. Mol Med Rep. 2009;2:441–47. doi: 10.3892/mmr_00000119. [DOI] [PubMed] [Google Scholar]
- 7.Suzuma I, Mandai M, Takagi H, et al. 17 Beta-estradiol increases VEGF receptor-2 and promotes DNA synthesis in retinal microvascular endothelial cells. Invest Ophthalmol Vis Sci. 1999;40:2122–29. [PubMed] [Google Scholar]
- 8.Yang F, Liu W, Yan X, et al. Effects of mir-21 on cardiac microvascular endothelial cells after acute myocardial infarction in rats: role of phosphatase and tensin homolog (PTEN)/vascular endothelial growth factor (VEGF) signal pathway. Med Sci Monit. 2016;22:3562–75. doi: 10.12659/MSM.897773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei Y, Yang Q, Zhang Y, et al. Plumbagin restrains hepatocellular carcinoma angiogenesis by suppressing the migration and invasion of tumor-derived vascular endothelial cells. Oncotarget. 2017;8(9):15230–41. doi: 10.18632/oncotarget.14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miao L, Xin X, Xin H, et al. Hydrogen sulfide recruits macrophage migration by integrin beta1-Src-FAK/Pyk2-rac pathway in myocardial infarction. Sci Rep. 2016;6:2236. doi: 10.1038/srep22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu H, Li G, Zhao W, Hu Y. Inhibition of MiR-92a may protect endothelial cells after acute myocardial infarction in rats: Role of KLF2/4. Med Sci Monit. 2016;22:2451–62. doi: 10.12659/MSM.897266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin W, Xie W, Xia N, et al. Silencing of transient receptor potential channel 4 alleviates oxLDL-induced angiogenesis in human coronary artery endothelial cells by inhibition of VEGF and NF-kappaB. Med Sci Monit. 2016;22:930–36. doi: 10.12659/MSM.897634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nessa A, Latif SA, Siddiqui NI, et al. Angiogenesis-a novel therapeutic approach for ischemic heart disease. Mymensingh Med J. 2009;18:264–72. [PubMed] [Google Scholar]
- 14.Wang M, Ji Y, Cai S, Ding W. MiR-206 suppresses the progression of coronary artery disease by modulating vascular endothelial growth factor (VEGF) expression. Med Sci Monit. 2016;22:5011–20. doi: 10.12659/MSM.898883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sienkiewicz P, Surazynski A, Palka J, Miltyk W. Nutritional concentration of genistein protects human dermal fibroblasts from oxidative stress-induced collagen biosynthesis inhibition through IGF-I receptor-mediated signaling. Acta Pol Pharm. 2008;65:203–11. [PubMed] [Google Scholar]
- 16.Flebus L, Lombart F, Sevrin C, et al. Low molecular weight poly (2-dimethylamino ethylmethacrylate) polymers with controlled positioned fluorescent labeling: Synthesis, characterization and in vitro interaction with human endothelial cells. Int J Pharm. 2015;478:278–87. doi: 10.1016/j.ijpharm.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 17.Bailey J, Shaw A, Fischer R, et al. A novel role for endothelial tetrahydrobiopterin in mitochondrial redox balance. Free Radic Biol Med. 2017;104:214–25. doi: 10.1016/j.freeradbiomed.2017.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chuaiphichai S, Crabtree MJ, McNeill E, et al. A key role for tetrahydrobiopterin-dependent endothelial NOS regulation in resistance arteries: Studies in endothelial cell tetrahydrobiopterin-deficient mice. Br J Pharmacol. 2017;174(8):657–71. doi: 10.1111/bph.13728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rochette L, Lorin J, Zeller M, et al. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol Ther. 2013;140:239–57. doi: 10.1016/j.pharmthera.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 20.Masuda H, Kalka C, Takahashi T, et al. Estrogen-mediated endothelial progenitor cell biology and kinetics for physiological postnatal vasculogenesis. Circ Res. 2007;101:598–606. doi: 10.1161/CIRCRESAHA.106.144006. [DOI] [PubMed] [Google Scholar]
- 21.Cartland SP, Genner SW, Zahoor A, Kavurma MM. Comparative evaluation of TRAIL, FGF-2 and VEGF-A-induced angiogenesis in vitro and in vivo. Int J Mol Sci. 2016;17(12) doi: 10.3390/ijms17122025. pii: E2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coats AJ. Ethical authorship and publishing. Int J Cardiol. 2009;131:149–50. doi: 10.1016/j.ijcard.2008.11.048. [DOI] [PubMed] [Google Scholar]