

Abstract
In acute myeloid leukemia (AML), high Galectin 3 (LGALS3) expression is associated with poor prognosis. The role of LGALS3 derived from mesenchymal stromal cells (MSC) in the AML microenvironment is unclear; however, we have recently found high LGALS3 expression in MSC derived from AML patients is associated with relapse. In this study, we used reverse phase protein analysis (RPPA) to correlate LGALS3 expression in AML MSC with 119 other proteins including variants of these proteins such as phosphorylated forms or cleaved forms to identify biologically relevant pathways. RPPA revealed that LGALS3 protein was positively correlated with expression of thirteen proteins including MYC, phosphorylated beta-Catenin (p-CTNNB1), and AKT2 and negatively correlated with expression of six proteins including integrin beta 3 (ITGB3). String analysis revealed that proteins positively correlated with LGALS3 showed strong interconnectivity. Consistent with the RPPA results, LGALS3 suppression by shRNA in MSC resulted in decreased MYC and AKT expression while ITGB3 was induced. In co-culture, the ability of AML cell to adhere to MSC LGALS3 shRNA transductants was reduced compared to AML cell adhesion to MSC control shRNA transductants. Finally, use of novel specific LGALS3 inhibitor CBP.001 in co-culture of AML cells with MSC reduced viable leukemia cell populations with induced apoptosis and augmented the chemotherapeutic effect of AraC. In summary, the current study demonstrates that MSC-derived LGALS3 may be critical for important biological pathways for MSC homeostasis and for regulating AML cell localization and survival in the leukemia microenvironmental niche.
Introduction
Acute myeloid leukemia (AML) is a highly fatal disease, so understanding the mechanisms controlling chemoresistance of leukemic cells is critical for developing more effective therapies. With growing evidence of the importance of the leukemic bone marrow (BM) microenvironmental niche (1-4), therapeutic strategies for AML and other leukemias will need to target not only the malignant cell but the other components of the tumor microenvironment. Mesenchymal stromal cells (MSC) provide critical support for leukemia cells in the BM (5-10). This is achieved by diverse mechanisms that include secretion of cytokines and chemokines, activating survival signaling in tumor cells after cell-to-cell contact, and blocking immune surveillance by suppressing NK and T cells (5-11).
Galectin 3 (LGALS3) is a member of a family of beta-galactoside-binding proteins that supports cell survival by diverse mechanisms involving BCL2, p53, RAS, and many other molecules (12-18). Consistent with its role supporting leukemia cell survival, a recent report from Cheng and colleagues demonstrated that high LGALS3 levels in AML patients was associated with poor disease prognosis (19). LGALS3 exerts effects on cells when secreted or present on the cellular surface, including promoting apoptosis of T cells, suppression of NK cell function, mediating cancer cell adhesion to many cell types in the tumor niche (e.g., MSC, vascular endothelial cells, and immune cells), and promoting angiogenesis (9, 12,13, 20-22).
MSC have been shown to be an important source of secreted LGALS3 (23, 24). In our recent study, reverse phase protein analysis (RPPA) analysis examined expression of LGALS3 and over 100 other proteins in MSC derived from AML patients (25). RPPA revealed LGALS3 levels were highest in refractory and relapse patients compared to patients at diagnosis, suggesting the MSC-derived LGALS3 is important in drug resistance (25). In the current study we used RPPA to compare expression of LGALS3 with 119 other proteins as well as phosphorylation or other modified variants to identify protein networks involving LGALS3 that may be critical for AML-MSC interactions. A distinct set of proteins were identified including MYC. LGALS3 was suppressed in healthy donor-derived MSC using lenti-viral shRNA, and the effect on MSC properties, including adhesion and cell protection, were examined.
Material and Methods
Isolation and culture of primary MSC from bone marrow
MSC were isolated from bone marrow (BM) of consented AML patients undergoing diagnostic BM aspiration and from healthy donors who were undergoing BM harvest for use in allogeneic BM transplantation. BM was subjected to centrifugation (700 g for 15 minutes at 4°C) over a Ficoll-Hypaque (Sigma-Aldrich, St. Louis, MO) gradient to separate mononuclear cells. After centrifugation, the buffy coat layer was carefully extracted and suspended in αMEM (Cellgro, Manassas, VA) supplemented with 10% pooled human platelet lysate (pHPL, kindly provided by Dr. Dirk Strunk, Department of Hematology and Stem Cell Transplantation, Medical University of Graz, Austria), supplemented with 2 mM L-glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin (Sigma-Aldrich). The BM mononuclear cell content was analyzed by automated blood count (Beckman Coulter, Indianapolis, IN), and mononuclear cells were seeded at a density of 5 × 104 cells/cm2 in tissue-culture flasks and cultured at 37°C in 5% CO2 incubator. The non-adherent cells were removed by completely changing the medium after 3 days, and the adherent cells were continuously cultured. The cultures were fed twice weekly by replacing 30% of the medium with fresh supplemented medium. The cells were harvested before reaching confluence by applying 0.25% trypsin and 1 mM EDTA (Life Technologies, Carlsbad, CA). MSC were cryopreserved and early passage (passage 2-3) samples were used for study. As reported in our previous study with these samples, isolated MSC are CD73+/CD90+/CD105+ (25).
RPPA Method
Proteomic profiling was done on MSC samples from healthy donor and patients with AML using RPPA. The method and validation of the technique are fully described in previous publications (26-30). Validation data for LGALS3 antibody (Santa Cruz Biotechnology; Dallas, TX; catalog # sc32790) is provided in Figure 1A-D. The LGALS3 antibody used detects a single band at 31 kd from protein lysates from a number of different cell lines (Figure 1A). Comparison of expression detected by LGALS3 antibody on RPPA array slide (Figure 1B) with western reveals detection levels (summarized in Figure 1C) are highly correlative by both methods according to Pearson Correlation (R = 0.8827; Figure 1D). Antibodies against 119 different proteins (114 targeting total protein with 32 paired antibodies targeting phosphoepitopes on 26 proteins, and 5 with only a phosphoepitope but not total protein epitope) were used for analysis (listed in Table 1). An IgG subtype specific secondary antibody was used to amplify the signal and finally a stable dye is precipitated. The stained slides were analyzed using the Microvigene software (Version 3.0, Vigenetech, Carlisle, MA) to produce quantified data. For RPPA, supercurve algorithms were used to generate a single value from the five serial dilutions (26, 27). Loading control and topographical normalization procedures accounted for protein concentration and background staining variations. Analysis using unbiased hierarchical clustering and perturbation bootstrap clustering, and principle component analysis was then done as fully described in a previous publication using available R packages and Qlucore software (Version 3.1, Qlucore Inc. Lund Sweden; refs 26, 27). Raw data for RPPA results is uploaded as an Excel file in Supplemental materials.
Figure 1. Validation of LGALS3 antibody for RPPA.
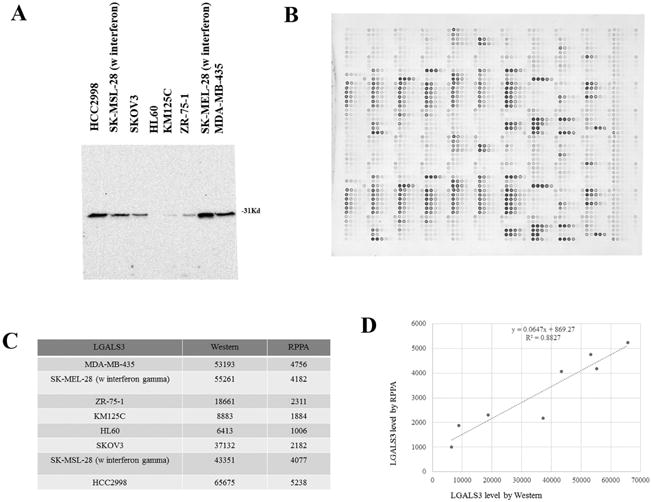
(A) Santa Cruz LGALS3 antibody sc32790 identifies a single band at 31 kd by western analysis in 8 different listed cell lines. (B) Array showing detection LGALS3 by sc32790 at 1:50 dilution. (C) Quantitation of LGAL3 expression of 8 cancer cell lines by Western and RPPA. (D) Correlation of Western with RPPA of the 8 cancer cell lines. R2 was determined by Pearson correlation.
Table 1. List of antibodies used for RPPA.
| MSC RPPA Staining Details | |||||
|---|---|---|---|---|---|
| Hugo name | Target | Manufacturer | Catalog# | Antibody Source | Antibody Dilution |
| AKT1 | AKT1 | Cell Signaling | 2967 | Mouse | 300 |
| AKT1/2/3 | AKT1/2/3 | Cell Signaling | 9272 | Rabbit | 150 |
| AKT1/AKT2/AKT3-phospho ser473 | AKT-p473(Ser) | Cell Signaling | 9271 | Rabbit | 50 |
| AKT1/AKT2/AKT3-phospho Thr308 | AKT-p308(Thr) | Cell Signaling | 9275 | Rabbit | 50 |
| AKT1S1 | PRAS40 | Invitrogen | AHO103 1 | Mouse | 300 |
| AKT1S1.p | p-PRAS40 (Thr246) | Cell Signaling | 2997 | Rabbit | 200 |
| AKT2 | AKT2 | Cell Signaling | 2962 | Rabbit | 500 |
| AKT3 | AKT3 | Cell Signaling | 4059 | Rabbit | 200 |
| NOL3 | ARC (NOL3) | Imgenex | IMG-171 | Rabbit | 2000 |
| ATF3 | ATF3 | Abcam | ab87213 | Rabbit | 500 |
| BAD | Bad | Cell Signaling | 9292 | Rabbit | 100 |
| BAD-phospho Ser112 | Bad-p112(Ser) | Cell Signaling | 9291 | Rabbit | 100 |
| BAD-phospho Ser136 | Bad-p136(Ser) | Cell Signaling | 9295 | Rabbit | 50 |
| BAD-phospho Ser155 | Bad-p155(Ser) | Cell Signaling | 9297 | Rabbit | 100 |
| BAK1 | Bak | Cell Signaling | 3792 | Rabbit | 50 |
| BAK1 | Bak | Abcam | ab32371 | Rabbit | 50 |
| BAX | Bax | Cell Signaling | 2772 | Rabbit | 100 |
| BCL2 | Bcl2 | DAKO | M0887 | Mouse | 200 |
| BCL2L1 | Bcl-XL | Cell Signaling | 2762 | Rabbit | 500 |
| BCL2L11 | Bim | Epitomics | 1036-1 | Rabbit | 200 |
| BECN1 | Beclin-1 | Cell Signaling | 3738 | Rabbit | 500 |
| BID | Bid | Cell Signaling | 2002 | Rabbit | 250 |
| BIRC5 | Survivin | Cell Signaling | 2802 | Rabbit | 50 |
| CAV1 | Caveolin-1 | Cell Signaling | 3238 | Rabbit | 100 |
| CCNB1 | Cyclin B1 | Santa Cruz | SC245 | Mouse | 100 |
| CCNB1 | Cyclin B1 | Epitomics | 1495-1 | Rabbit | 1000 |
| CCND1 | Cyclin D1(M-20) | Santa Cruz | sc718 | Rabbit | 500 |
| CCND3 | Cyclin D3 | Cell Signaling | 2936 | Mouse | 100 |
| CCNE1 | Cyclin E | Santa Cruz | sc-247 | Rabbit | 100 |
| CD34 | CD34 | Epitomics | 2150-1 | Rabbit | 500 |
| CDK1 | CDC2 | Calbiochem | cc01 | Mouse | 200 |
| CDK2 | CDK2 | Santa Cruz | SC6248 | Mouse | 200 |
| CDK4 | CDK4 | Cell Signaling | 2906 | Mouse | 200 |
| CDKN1A | P21/Waf | Cell Signaling | 2946 | Mouse | 250 |
| CDKN1A | P21/Waf | Santa Cruz | sc-397 | Rabbit | 200 |
| CDKN1B | P27 | Epitomic | 1591-1 | Rabbit | 100 |
| CDKN1B-pS10 | P27-phosphoSer10 | Epitomic | 2187-1 | Rabbit | 500 |
| PTEN | PTEN | Cell Signaling | 9552 | Rabbit | 400 |
| CREB1 phospho Ser133 | CREB-p(ser133) | Epitomics | 1113-1 | Rabbit | 2000 |
| CSNK2A1 | CK2α | Cell Signaling | 2656 | Rabbit | 100 |
| CTNNA1 | Catenin-alpha | Calbiochem | CA1030 | Mouse | 75 |
| CTNNB1 | Catenin-beta | Cell Signaling | 9562 | Rabbit | 50 |
| CTNNB1-phospho Ser33/37/Thr41 | catenin-beta phospho- | Cell Signaling | 9561 | Rabbit | 500 |
| DIABLO | Smac/Diablo | Cell Signaling | 2954 | Mouse | 500 |
| EGFR | EGFR | Santa Cruz | sc-03 | Rabbit | 500 |
| EGFR-phospho Tyr992 | EGFR-p tyr992 | Cell Signaling | 2235 | Rabbit | 50 |
| EGLN1 | Egln1 | Millipore | 05-1327 | Mouse | 500 |
| EIF2S1 | elF2 | Cell Signaling | 9722 | Rabbit | 3000 |
| EIF2S1-phospho Ser51 | phospho-eIF2-alpha | Cell Signaling | 9721 | Rabbit | 250 |
| EIF4E | eIF4E | Cell Signaling | 9742 | Rabbit | 200 |
| ELK1-phospho Ser383 | ElK(phospho-ser383) | Cell Signaling | 9181 | Rabbit | 100 |
| ERBB2 | HER2/Erb2 | Cell Signaling | 2242 | Rabbit | 250 |
| ERBB2 | HER2/Erb2 | Lab Vision (Fisher) | MS-325-P0(MS32 5P0) | Mouse | 1000 |
| ERBB2-phospho Tyr1248 | HER2(p-Tyr1248) | Upstate | 06-229 | Rabbit | 1500 |
| ERG | ERG1/2/3 | Santa Cruz | sc-353 | Rabbit | 1000 |
| FN1 | Fibronectin | Epitomics | 1574 | Rabbit | 30000 |
| FOXO1-phospho thr24/FOXO3-phospho thr32 | FoxO1a/3a | Cell Signaling | 9464 | Rabbit | 500 |
| FOXO3 | FoxO3a | Cell Signaling | 9467 | Rabbit | 500 |
| FOXO3 | FoxO3a | Cell signaling | 2497 | Rabbit | 50 |
| FOXO3-phospho Ser318/321 | FKHRL1/FoxO 3a (P-Ser 318/321) | Cell Signaling | 9465 | Rabbit | 10000 |
| GAB2 | Gab2 | Cell Signaling | 3239 | Rabbit | 500 |
| GAB2-phospho Tyr452 | Gab2-pTyr452 | Cell Signaling | 3882 | Rabbit | 100 |
| GAPDH | GAPDH | Ambion | AM4300 | Mouse | 2000 |
| GATA3 | Gata3 | BD bioscience | 558686 | Mouse | 500 |
| GSKA/GSKB | GSK3 | Santa Cruz | sc-7291 | Mouse | 200 |
| GSKA/GSKB-phospho Ser21/9 | GSK3a/B(p-ser21/9) | Cell Signaling | 9331 | Rabbit | 200 |
| HDAC3 | HDAC3 | Cell Signaling | 2632 | Rabbit | 100 |
| HIF1A | HIF-1α | BD Pharmingen | 610959 | Mouse | 50 |
| HNRNPK | hnRNPK | Santa Cruz | sc-28380 | Mouse | 5000 |
| HSP90AA1/HSP90AB1 | HSP90 | Cell Signaling | 4875 | Rabbit | 500 |
| HSPB1 | HSP27 | Cell Signaling | 2402 | Mouse | 100 |
| INPP5D | SHIP1 | Santa Cruz | SC-8425 | Mouse | 250 |
| IRS1-p1101 (serine) | IRS1-phospho ser1101 | Cell Signaling | 2385 | Rabbit | 250 |
| ITGA2 | CD49b | BD Transductio n Lab | 611016 | Mouse | 500 |
| ITGAL | CD11a | BD Transduction Lab | 610826 | Mouse | 500 |
| ITGB3 | Integrin-beta3 | Cell Signaling | 4702 | Rabbit | 250 |
| JMJD6 | JMJD6 | Abcam | ab50720 | Rabbit | 1000 |
| JUN-phospho Ser73 | Jun-C-phospho ser73 | Cell Signaling | 9164 | Rabbit | 100 |
| JUNB | Jun-B | Cell Signaling | 3755 | Rabbit | 100 |
| KDR | VEGFR2 | Cell Signaling | 2479 | Rabbit | 700 |
| KIT | Kit-C | Epitomics | 1522 | Rabbit | 1000 |
| LCK | Lck | Cell Signaling | 2752 | Rabbit | 50 |
| LEF1 | LEF1 | Cell Signaling | 2230 | Rabbit | 1000 |
| LGALS3 | Galectin 3 | Santa Cruz | sc-32790 | Mouse | 250 |
| LYN | Lyn | Cell Signaling | 2732 | Rabbit | 250 |
| MAP2K1 | MEK | Cell Signaling | 9122 | Rabbit | 2000 |
| MAP2K1/MAP2K2-phospho ser217/221 | MEK(p-ser217/221) | Cell Signaling | 9121 | Rabbit | 1000 |
| MAPK1 | Erk2 | Santa Cruz | Sc-154 | Rabbit | 2000 |
| MAPK1/MAPK3-phospho Thr202/Tyr204 | Erk-p42/44(Thr202/ Tyr204 | Cell Signaling | 9101 | Rabbit | 400 |
| MAPK14 | P38 | Cell Signaling | 9212 | Rabbit | 200 |
| MAPK14-phospho Thr180/tyr182 | P38-pThr180/Tyr18 2 | Cell Signaling | 9211 | Rabbit | 100 |
| MAPK8 | JNK1/3 | Santa Cruz | sc-474 | Rabbit | 1000 |
| MAPK9 | JNK2 | Cell Signaling | 4672 | Rabbit | 25 |
| MCL1 | MCL1 | BD pharmingen | 559027 | Mouse | 50 |
| MDM2 | MDM2 | Santa Cruz | sc813 | Rabbit | 5000 |
| MS4A1 | I can't find the antigen name | ||||
| MSI2 | MSI2 | Chemicon | MAB100 85 | Mouse | 500 |
| MTOR | mTor | Cell Signaling | 2983 | Rabbit | 200 |
| MTOR-phospho Ser2448 | mTor(p-Ser2448) | Cell Signaling | 2971 | Rabbit | 100 |
| MYC | Myc | Cell Signaling | 9402 | Rabbit | 100 |
| NOTCH1 cleaved val1744 | Notch1-cleaved (Val1744) | Cell Signaling | 4147 | Rabbit | 400 |
| NOTCH3 | Notch3 | Santa Cruz | sc5593 | Rabbit | 200 |
| NPM1 | NPM | Invitrogen | 32-5200 | Mouse | 10000 |
| NR4A1 | Nur77 | Imgenex | IMG-528 | Rabbit | 200 |
| NRP1 | NRP1(neuropili n) | Santa Cruz | SC-5307 | Mouse | 10 |
| PA2G4.pS65 | 4EBP1-phosph Ser65 | Cell Signaling | 9456 | Rabbit | 400 |
| PA2G4.pT37_46 | 4EBP1-phospho Thr37/46 | Cell Signaling | 9459 | Rabbit | 20000 |
| PA2G4.pT70 | 4EBP1-phospho Thr70 | Cell Signaling | 9455 | Rabbit | 400 |
| PARK7 | DJ-1 | Private-Andreeff Lab in house antibody | Rabbit | 500 | |
| PARP1 | PARP | Cell Signaling | 9542 | Rabbit | 200 |
| PDK1 | PDK1 | Cell Signaling | 3062 | Rabbit | 200 |
| PDK1-phospho ser241 | PDK1-p241(Ser) | Cell Signaling | 3061 | Rabbit | 500 |
| PECAM1 | CD31/PECAM | DAKO | M0823 | Mouse | 200 |
| PPARG | PPARγ | Santa Cruz | sc7273 | Mouse | 75 |
| PPP2R2A/PP2R2B/PPP2R2 C/PPP2R2D | PP2A-B55 | Santa Cruz | sc-18330 | Goat | 500 |
| PRKAA1/PRKAA2 | AMPKα | Cell Signaling | 2532 | Rabbit | 200 |
| PRKAA1/PRKAA2-phospho Thr172 | AMPKα P(Thr172) | Cell Signaling | 2535 | Rabbit | 200 |
| PTEN | PTEN | Cell Signaling | 9552 | ||
| PTGS2 | cox-2 | Epitomics | 2169-1 | Rabbit | 250 |
| PTGS2 | cox-2 | Cell Signaling | 4842 | Rabbit | 100 |
| PTK2 | Fak | Cell Signaling | 3285 | Rabbit | 500 |
| RAC1/RAC2/RAC3 | Rac1/2/3 | Cell Signaling | 2465 | Rabbit | 500 |
| RELA | NF-kB p65 | Cell Signaling | 3034 | Rabbit | 500 |
| RPS6 | S6 Ribosomal protein | Cell Signaling | 2217 | Rabbit | 250 |
| RPS6-phospho ser235/236 | S6 Ribosomal protein(phospho-ser235/236) | Cell Signaling | 2211 | Rabbit | 1500-2000 |
| RPS6-phospho ser240/244 | S6 Ribosomal protein(phospho-ser240/244) | Cell Signaling | 2215 | Rabbit | 750-1000 |
| RPS6KB1 | p70S6K | Cell Signaling | 9202 | Rabbit | 250 |
| RPS6KB1 | p70S6K | Epitomics/a bcam | 1494-1(ab3252 9) | ||
| RPS6KB1-phospho thr389 | p70S6K(p-thr389) | Cell Signaling | 9205 | Rabbit | 250 |
| SFN | X14.3.3Sigma | Upstate | 05-632 | mouse | 200 |
| SIRT1 | SIRT1 | Abcam | ab32441 | Rabbit | 1000 |
| SMAD1 | smad1 | Epitomics | 1649-1 | Rabbit | 200 |
| SMAD4 | smad4 | Santa Cruz | sc7966 | Mouse | 1000 |
| SMAD6 | Smad6 | Cell Signaling | 9519 | Rabbit | 100 |
| SPP1 | Osteopotin | Santa Cruz | sc-21742 | Mouse | 500 |
| SQSTM1 | P62 | Santa Cruz | sc-28359 | Mouse | 250 |
| SRC | Src | Upstate | 05-184 | Mouse | 600 |
| SRC-phospho tyr416 | Src(phosphotyr416) | Cell Signaling | 2101 | Rabbit | 400 |
| SRC-phospho tyr527 | Src(phosphotyr527) | Cell Signaling | 2105 | Rabbit | 100 |
| STAT1 | stat1 | Cell Signaling | 9172 | Rabbit | 250 |
| STAT1-phospho tyr701 | Stat1(phosphotyr701) | Cell Signaling | 9171 | Rabbit | 100 |
| STAT3 | Stat3 | Upstate | 06-596 | Rabbit | 50 |
| STAT3-phospho ser727 | Stat3-p727(Ser) | Cell Signaling | 9134 | Rabbit | 100 |
| STAT3-phospho tyr705 | Stat3 p705(Tyr) | Cell Signaling | 9131 | Rabbit | 500 |
| STAT5A/STAT5B | Stat5 | Cell Signaling | 9352 | Rabbit | 250 |
| STAT5A/STAT5B phospho Tyr694 | Stat5(phospho-Tyr694) | Cell Signaling | 9351 | Rabbit | 100 |
| STK11 | LKB1/STK11 | Cell Signaling | 3050 | Rabbit | 500 |
| TCF4 | TCF-4 | Santa Cruz | sc8632 | Goat | 400 |
| TGM2 | TG2 | Abcam | ab2386 | Mouse | 2000 |
| TP53 | TP53 | Cell Signaling | 9282 | Rabbit | 1000 |
| TP53-phosphoS15 | TP53-phosphoS15 | Cell Signaling | 9284 | Rabbit | 200 |
| TSC2 | TSC2 | Epitomic | 1613-1 | Rabbit | 500 |
| VHL | VHL | Novus | NB100-485 | Rabbit | 2000 |
| XIAP | XIAP | Cell Signaling | 2042 | Rabbit | 100 |
| YWHAE | X14.3.3Epsilon | Santa Cruz | sc-23957 | Mouse | 200 |
| YWHAZ | 14-3-3-zeta | Chemicon | AB9746 | Rabbit | 750 |
Pathway Analysis
String software (String 10.0; website: http://string-db.org) (31) was used to determine protein associations. Pathway analysis to identify canonical pathways, upstream regulators, and protein networks was performed using Ingenuity Pathway software (Qiagen, Germantown, MD).
MSC shRNA Transductants
LGALS3 was knocked down in a MSC derived from a presumably healthy donor by lentiviral transduction of shRNA using pLKO based transfer vectors (Open Biosystems, Huntsville, AL). As a negative control we used pLKO.1 control plasmid (Addgene, Cambridge, MA). Infected cells were selected with 1 ug/ml puromycin (Invivogen, San Diego, CA). Knockdown was verified by western blot analysis and real time PCR.
Immunoblot Analysis
Cells were sonicated in lysis buffer and protein (5 × 104 cell equivalents) was subjected to electrophoresis using 4-20% gradient acrylamide/ 0.1% SDS or 10% acrylamide/ 0.1% SDS gels. Proteins were transferred to a membrane and western blotting analysis was performed with antibodies against LGALS3, MYC, ITGB3 (Santa Cruz Biotechnology), total AKT (Cell Signaling Technology, Danvers, MA), p-AKT (S473; Cell Signaling Technology) and Tubulin (Sigma-Aldrich). Immunoblot analysis was performed at least three different times. Signals were detected by using the Odyssey Infrared Imaging System and quantitated by Odyssey software version 3.0 (both LI-COR Biosciences, Lincoln, NE). Tubulin was used as a loading control.
Gene Expression Analysis
Real-time PCR (qRT-PCR) was conducted using an ABI 7900HT Fast Real-Time PCR System (Life Technologies). TaqMan Gene Expression Assays (Life Technologies) for LGALS3 and 18s RNA were used as directed by the manufacturer. RQ Manager 1.2.1 (Life Technologies) was used to analyze the data.
Cell Treatment and Cytotoxicity and Adhesion Assessments
THP-1 cells were co-cultured with MSC at a ratio of 4 AML cells to 1 MSC cell for 18 hours. Cells were treated with vehicle (0.1% DMSO), 30 ug/ml CBP.001 (kindly provided by Kiromic Biopharma; Houston, TX) and/or 1 uM AraC (Selleck Chemicals, Houston, TX) for 48 hours. AML cells were removed by gentle trypsinization (90 seconds, RT) to examine adherent AML cell number and viability. Cells were stained with human CD90 PE antibody (Becton Dickinson, San Jose, CA) to identify AML cells (i.e CD90- cells) and stained with Annexin V to identify apoptotic cells (i.e. Annexin V +) and DAPI to identify viable cells (i.e. DAPI- cells). Counting beads were included to assess total cells.
Flow cytometry was performed using a Becton Dickinson LSR II flow cytometer (Becton Dickinson, San Jose, CA). To assess effect of LGALS3 knockdown on cell adhesion, AML OCI-AML3 cells were co-cultured with MSC transduced with control lentivial vector (LKO) or MSC transduced with LGALS3 shRNA lentiviral plasmid at a ratio of 4 AML cells to 1 MSC cell. After 72 hours, media was removed, cell co-culture washed with 1X PBS, and then cells were visualized in the flask using a light microscope (Nikon TMS inverted microscope; Melville, NY). Representative pictures of cells were taken with a Nikon Coolpix 930 camera that was attached to the microscope. Next, cells were trypsinized (3 minutes, 37° C) and cells were stained with human CD45 PE antibody (Becton Dickinson) and subject to flow cytometry. Counting beads were included to assess total cells. Flow cytometry was performed using a Becton Dickinson LSR II flow cytometer (Becton Dickinson).
Statistical Analyses
All means ± SD for triplicate samples were calculated with Microsoft Excel 2003 SP2 software (Microsoft Corporation, Seattle, WA). In all statistical analyses, the results were considered statistically significant when p < 0.05 using a two tailed Student t-test. Comparison of the protein levels between paired samples was done by performing paired t-test. Association between protein expression levels and categorical clinical variables were assessed in R using standard t tests, linear regression, or mixed effects linear models. Unbiased hierarchical clustering was performed using the weighted average method and the associated figures show expression normalized to median = 0, variance = 1. The P-value and the associated Q-value (a measure of the false discovery rate) are shown for each clustering analysis. Association between continuous variable and protein levels were assessed by using the Pearson and Spearman correlation and linear regression. Bonferroni corrections were done to account for multiple statistical parameters for calculating statistical significance.
Results
LGALS3 protein levels are elevated in MSC from AML patients
RPPA is used in our laboratory to analyze protein expression and modifications in cells from clinical samples representing many hematologic malignancies (25-30). RPPA depends on the availability of a validated antibody panel. Validation for the LGALS3 antibody used for RPPA is presented in Figure 1A-D and described in “Materials and Methods”. We used RPPA to study LGALS3 protein expression in MSC from presumed healthy donors (N = 71) and MSC from newly diagnosed AML patients (N = 106). As shown in Figure 2A, levels of LGALS3 protein were statistically significantly (p = 0.0001) higher in the MSC from the AML patients compared to normal donor MSC. To determine if increased protein expression in the AML derived MSC could be attributed to a transcriptional mechanism, gene expression in AML MSC (N = 10) and presumed healthy donor derived MSC (N =9) was performed using qRT-PCR. As shown in Figure 2B, though LGALS3 expression relative to 18S was slightly higher in AML MSC compared to healthy donor MSC, this difference was not significant (p = 0.284). These findings suggest that a post-transcriptional or post-translational mechanism was involved in LGALS3 expression regulation.
Figure 2. LGALS3 protein but not mRNA is elevated in MSC derived from AML patients.
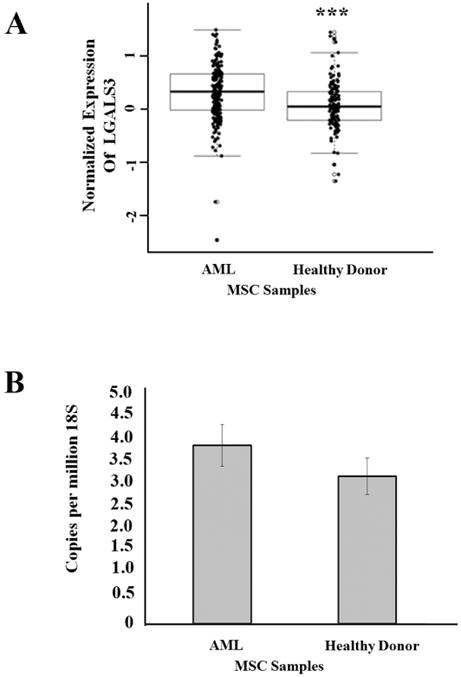
(A) RPPA was used to determine LGALS3 expression in AML derived MSC (N = 54) and MSC derived from heathy donor (N =71). Student t-test comparison of the two groups determined a significant difference (***, p = 0.0001). (B) Gene expression LGALS3 and 18S rRNA expression was determined by qRT-PCR in AML derived MSC (N = 10) and MSC derived from presumed heathy donor (N = 9). Student t-test comparison of the two groups determined no significant difference (p > 0.05).
LGALS3 levels correlate with expression of a distinct set of proteins including MYC
RPPA was used to examine correlations of LGALS3 with other proteins in the MSC from AML patients. In addition to LGALS3 antibody, the RPPA was probed with 150 other antibodies targeting 119 different proteins (114 targeting total protein with 32 paired antibodies targeting phosphoepitopes on 26 proteins, and 5 with only a phosphoepitope but not total protein epitope) (Table 1). Raw data for RPPA results is uploaded as an Excel file in Supplemental materials. The targeted proteins regulate a wide variety of cellular functions. Thirteen proteins were positively correlated with LGALS3 in AML-derived MSC, while six proteins are negatively correlated (Figure 3A). The two proteins exhibiting highest positive correlation with LGALS3 were phosphorylated beta catenin (p -CTNNB1; S33/S37/T41) and MYC. Though phosphorylated forms of AKT were not correlated with LGALS3 expression, expression of AKT2 was positively correlated with expression of the galectin. Proteins negatively correlated with LGALS3 included STMN1, LYN, SIRT1, and ITGB3.
Figure 3. LGALS3 protein expression correlates with a distinct set of other proteins in MSC derived from AML patients.
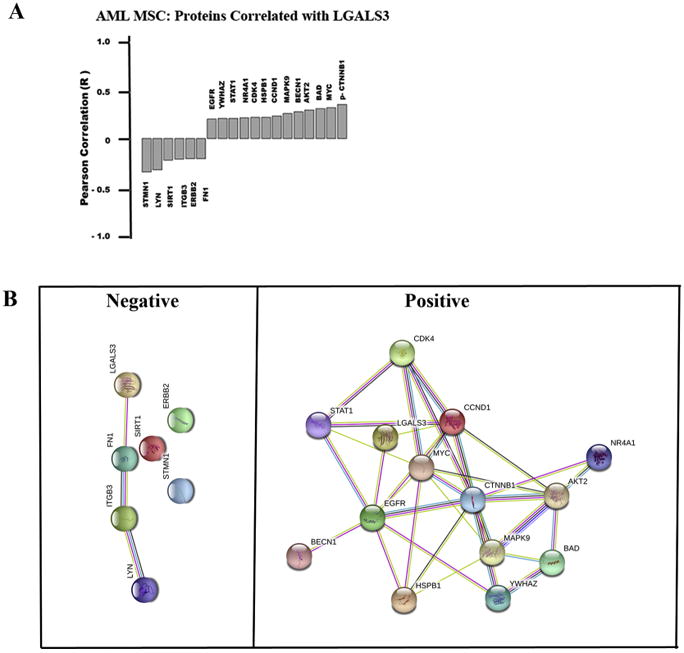
(A) RPPA was used to determine expression of 151 proteins in AML derived MSC (N = 54). Pearson correlation was performed to identify proteins that correlate with LGALS3 expression based on R ≥ 0.25. (B) String analysis was performed on proteins identified in (A). Sets were grouped based on those with negative correlation and those with positive correlation.
String 10.0 (http://string-db.org) was used to examine associations among the groups of proteins that were negatively or positively correlated with LGALS3 (listed in Figure 3A). LGALS3 was included as a member in each set. As shown in Figure 3B, there were few connections among the proteins negatively correlated with LGALS3, but the proteins that were positively correlated with LGALS3 showed many interconnections. Proteins negatively correlated with LGALS3 appeared to have no apparent association as determined by String 10.0. Protein:Protein Interaction (PPI) enrichment, and the p-value for this group of proteins was 0.139 (http://string-db.org).
However, for the proteins positively correlated with LGALS3, String 10.0 determined the PPI enrichment p-value for this group of proteins is 7.63e-10, suggesting that the proteins were, at least in part, biologically connected (http://string-db.org). To determine possible pathways associated with this group of proteins, Ingenuity Pathway Analysis (IPA) was used. The top canonical pathways identified involved networks of proteins involved in metastasis in colorectal cancer (p = 4.45e-11; data not shown). IPA identifies upstream molecules that are associated with the set of proteins listed. A total of 1877 upstream molecules were identified by IPA (Excel file available in Supplemental data). Among the top 10 upstream molecules associated with the protein network was transforming growth factor beta (TGF beta; p = 8.2 e-11). The identification of TGF beta as a possible regulator of this set of LGALS3 associated proteins was intriguing since this molecule has emerged as a key MSC-derived cytokine (reviewed in 32, 33). TGF beta and LGALS3 also cross-regulate with the AKT and CTNNB1 pathways as common targets in the TGF beta/LGALS3 signaling axis (34, 35).
Suppression of LGALS3 alters protein expression in MSC including a reduction of MYC expression
MSC from a healthy donor were transduced with control lentivirus (LKO) or lentivirus containing LGALS3 shRNA. We achieved ∼ 90% reduction of LGALS3 in MSC according to immunoblot analysis (Figure 4A). To validate correlation of LGALS3 with MYC, immunoblot analysis was performed using a single filter transferred from a 4-20% SDS polyacrylamide gel. As shown in Figure 4A, MYC levels were reduced by roughly 50% in MSC with LGALS3 shRNA. This reduction of MYC is consistent with expression levels found in MSC by RPPA (Figure 2A).
Figure 4. Suppression of LGALS3 by shRNA in MSC reduces MYC expression and suppresses activation of AKT.
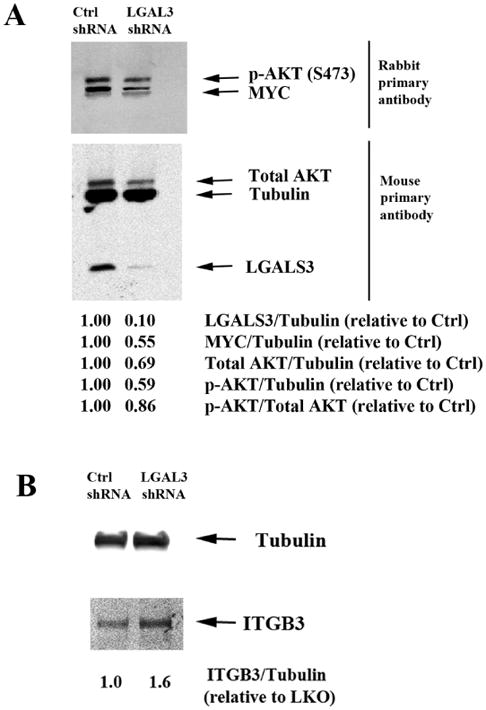
Protein lysates from control MSC (LKO) or MSC expressing LGALS3 shRNA were subject to electrophoresis and immunblot analysis performed using 2 separate filters. (A) Antibodies against Tubulin, LGALS3, MYC, p-AKT (S473), and total AKT were used. (B) Tubulin and ITGB3 antibodies were used. Densitometry using LiCor software was performed and ratio of protein relative to Tubulin assessed relative to LKO MSC are listed. Results for both (A) and (B) are representative of a single experiment that was performed three different times.
A role for LGALS3 as a positive regulator of AKT is well established (36, 37), and LGALS3's regulation of AKT appears to be important for migration in pig MSC (38). We examined AKT expression and AKT phosphorylation in MSC transduced with control lentivirus and MSC transduced with LGALS3 shRNA and found phosphorylation of AKT at serine 473 was reduced by roughly half (Figure 4A). Consistent with the RPPA finding that LGALS3 positively correlated with AKT2 expression (Figure 3A), LGALS3 suppression resulted in a 30% reduction of total AKT (Figure 4A). This result suggests that the decreased levels of phosphorylated AKT may be due to reduced levels of total AKT. These findings suggest that LGALS3 plays a role in supporting MYC expression and positively regulating AKT expression and activity. RPPA determined that ITGB3 was negatively correlated with LGALS3 (Figure 3A). A filter from a separate 4-20% SDS polyacrylamide gel was probed with an antibody against ITGB3. The level of detection of ITGB3 with the antibody used was much weaker than the bands for proteins surveyed on Figure 4A so a separate western was required. As shown in Figure 4B, suppression of LGALS3 in MSC by shRNA promoted elevated expression of ITGB3. This result suggests that LGALS3 may act as an upstream negative regulator of ITGB3.
Suppression of LGALS3 in MSC inhibits adhesion of OCI-AML3 cells
CBP.001 (Carbohydrate Binding Protein version 001; also known as Gal3C) is a truncated recombinant version of LGALS3 that acts as a dominant negative inhibitor (39-42). CBP.001 is generated by expression of a truncated protein in E. coli and His-tag purification and was extensively validated as reported in reference 41. Protein vehicle was used as a negative control. Endotoxin concentration was evaluated by Limulus amebocyte lysate (LAL) method and revealed less than 1 unit/mL of purified protein. Prior to use for the experiments shown here, CBP.001 was validated by Gal3 laminin-binding inhibition assay (data not shown). The molecule has shown anticancer efficacy in models of multiple myeloma and ovarian cancer (39, 40). The ability of CBP.001 to affect AML cells in the microenvironment was tested in the MSC co-culture system using THP-1 cells. THP-1 AML cells were allowed to adhere to MSC for 18 h and then vehicle (PBS) or 30 ug/ml CBP.001 or 1 uM AraC, or a combination of both agents at the single agent dose was added to the co-culture mix. After 48 h, AML cells were isolated by gentle trypsinization and flow cytometry performed to determine total viable cell number and percentage of apoptotic cells. As shown in Figure 5A, CBP.001 or CBP.001/AraC combination reduced viable cell number by approximately 85% while AraC reduced viable cells by roughly 75%. AraC was more potent inducing apoptosis compared to CBP.001 (22.0% versus 7.8%, respectively; Figure 5B). As shown in Figure 5B, combination of both agents did exhibit slightly higher but statistically significant induction apoptosis (29.9%; p = 0.05 compared to AraC alone). These results suggested that CBP.001 could augment AML cell killing by AraC.
Figure 5. CBP.001 alone or with AraC reduces viable AML cells in the presence of MSC.
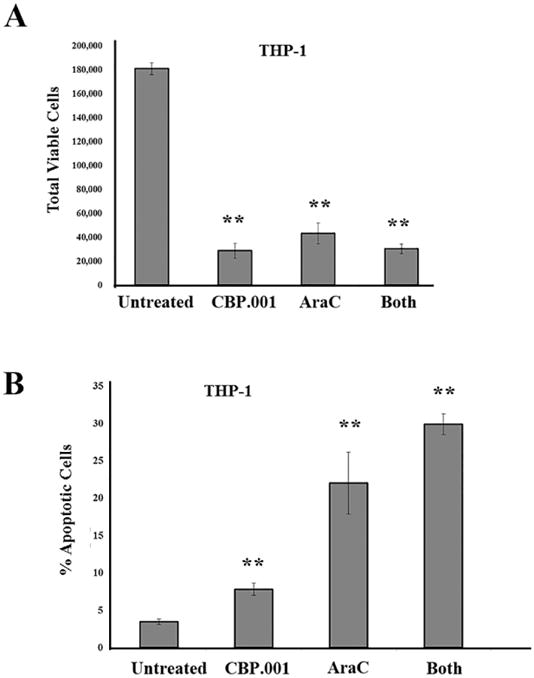
(A) THP-1 cells were co-cultured with MSC for 24 hours and then were treated with vehicle, 30 ug/ml CBP.001, 1 uM AraC, or combination of both agents at single agent dose for 48 hours and then adherent AML cells were isolated. Flow cytometry was performed to determine (A) total viable THP1 cells (CD90-/Annexin V-/DAPi-) and (B) % apoptotic cells (CD90-/Annexin V +). For A & B, data represents average of three different replicates. Student t-test comparison of groups compared to control (untreated) determined a significant difference in all treated groups (**, p < 0.01).
To examine the effect of LGALS3 knock down in MSC on the adhesion of leukemia cells, OCI-AML3 cells were co-cultured with MSC expressing control lentivirus or with MSC expressing LGALS3 shRNA. After 72 h, the medium was removed and replaced with fresh medium. Adherent cells were imaged using a light microscope. As shown in Figure 6A, a greater number of AML cells are visualized in co-culture with control MSC compared to MSC expressing LGALS3 shRNA. To quantify total adherent cells in each co-culture, cells were trypsinized and AML cells counted by flow cytometry using CD45 antibody, which binds AML cells and not MSC. As shown in Figure 6B, there was approximately 50% reduction in AML cells adhering to control MSC compared to MSC expressing LGALS3 shRNA.
Figure 6. OCI-AML3 cells adhere less efficiently to MSC with reduced LGALS3.
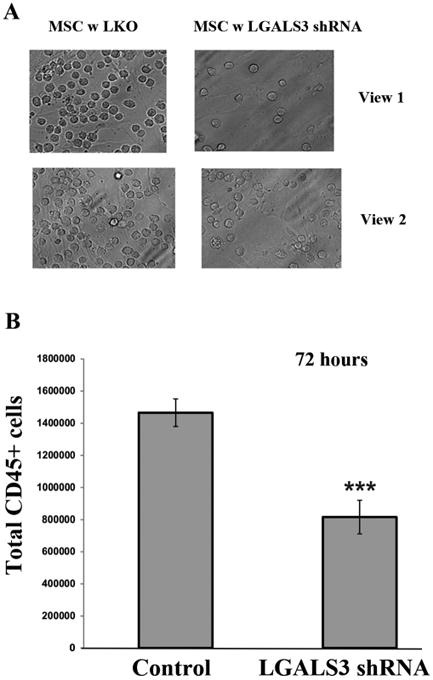
OCI-AML3 cells were co-cultured with control MSC or MSC expressing LGALS3 shRNA for 72 hours. (A) Cell co-culture was washed and cells visualized in a representative flask using an inverted light microscope. Two views from the same representative flask are shown. The plane of focus is on the AML cells. (B) Cells were trypsinized, stained with CD45 antibody, and flow cytometry was performed with counting beads included. Data represents average of three different replicates. Student t-test comparison of the two groups determined a significant difference (***, p < 0.001).
Discussion
In AML cells, LGALS3 supports survival and promotes resistance to chemotherapy including BH3 mimetics (43). At present, little is known about the role of LGALS3 in the AML niche but it has been implicated in the microenvironment of other hematologic malignancies including ALL and CML (reviewed in ref. 44). The Heisterkamp group has demonstrated that LGALS3 is important in the ALL microenvironment (45, 46). A role for LGALS3 biology in MSC is emerging. LGALS3 contributes to the immunosuppressive ability of MSC. Suppression of LGALS3 in MSC reduces MSC abilit y to inhibit T cell function (24, 44). Recent studies have suggested LGALS3 plays an important role in MSC migration (38, 47). A study of LGALS3 in murine MSC revealed that the galectin is elevated in MSC as mice age (48). The ageing mice also exhibited greater oxidative stress and senescent phenotype. Interestingly, senescence appears to promote LGALS3 expression and secretion in a colorectal cancer model (49). In our previous study, AML derived MSC were found to exhibit a greater degree of senescence compared to MSC from healthy donor (25). The senescent phenotype of MSC appears to be critical in myelodysplastic syndrome (50). In that study, senescent MDS derived MSC were found to express and secrete higher levels of interleukin-6 (IL6) and TGF beta compared to MSC derived from healthy donor (50). Blockade of TGF beta in AML cell co-culture with MSC reverses the protective effect of MSC suggesting the cytokine has an important role in MSC mediated survival function (51). TGF beta is one of the top 10 upstream regulatory molecules identified by IPA that is connected with the LGALS3 associated proteins. It is tempting to speculate that a LGALS3/TGF beta axis may be at work in the AML derived MSC.
In a previous study we determined that MSC from salvage samples (i.e., relapse/refractory) expressed higher levels of LGALS3 (25). In that study, LGALS3 was one of only nine proteins that were identified as differentially expressed in MSC from AML salvage samples compared to AML MSC samples taken at first diagnosis. Interestingly, LGALS3 and p-CTNNB1 were two of three proteins found elevated in salvage MSC, while LYN and ITGB3 were two of six proteins found reduced in salvage MSC (25). Our current finding that LGALS3 expression is positively correlated with p-CTNNB1 and negatively correlated with LYN and ITGB3 expression suggests that these proteins may be part of an axis that may influence MSC mediated therapy resistance. Activating mutation of CTNNB1 in osteoblasts (which are derived from MSC) is associated with niche-induced myeloid leukemogensis (52). Canonical WNT signaling has been shown to be important in many cancers including lymphoid and myeloid leukemias (53, 54). Stromal mediated protection of leukemia cells is supported by active WNT/CTNNB1 signaling (52-59). It is not clear yet how LGALS3 might regulate CTNNB1 in MSC populating the AML niche, however, a role for LGALS3 as a positive regulator of CTNNB1 is well established (60-62).
Recent studies have determined that MSC in AML patients have unique characteristics and that the stromal cells can contribute to the disease state (25, 63-66). Of over 119 proteins surveyed in AML MSC, LGALS3 was only one of three that were elevated in MSC derived from patients during relapse or who were refractory to therapy (25). MYC emerged as one of the important proteins that positively correlated with expression of LGALS3. As LGALS3 supports ERK and AKT survival signaling and these proteins are crucial for MYC stability, the galectin would be expected to support MYC expression. That suppression of LGALS3 results in reduced MYC supports a role for LGALS3 as an upstream regulator of MYC. LGALS3 positive regulation of MYC in MSC could impact the biology of the stromal cells. Sato and colleagues have established that MYC is important in MSC proliferation particularly during hypoxia (67). MYC overexpression when combined with inactivation of p16 (INK4a) in MSC results in conversion to osteosarcoma with loss of adipocytic potential (68). Though AML MSC are not transformed, they appear to be defective in the ability to differentiate to adipocytes (65). In addition, MYC suppresses p21 in MSC and promotes cell proliferation (69). Perhaps LGALS3 induction of MYC contributes to cell proliferation of AML MSC while preventing their potential for adipocytic differentiation. The Raz group has shown that LGALS3 prevents osteoblast differentiation via a Notch mediated mechanism (70).
LGALS3 regulates endocytosis and function of a number of different molecules including integrins. LGALS3's ability to pentamerize and interact with glycoproteins and other glycosylated molecules such as glycosphingoipids enables the lectin to organize dynamic lattices in cell membranes (71,72). The LGALS3 lattice regulates the mobility and endocytosis of key regulatory molecules such as CD44 and integrins. Integrins are heterodimeric transmembrane glycoproteins comprised of an alpha subunit and beta subunit (73). These proteins are critical for communicating signals from cell to cell or cell to extracellular matrix. Integrin-mediated signaling is bi-directional; that is integrins can direct external signals into the cell or they can direct signaling initiated in the cell to extracellular targets (73).
LGALS3 and other Galectins are critical regulators of integrin function (71-77). Recombinant LGALS3 induces production of inflammatory cytokines in pancreatic stellate cells via an integrin beta 1 (ITGB1) mediated mechanism (77). Yang and colleagues demonstrated that recombinant LGALS3 promotes lateral mobility of ITGB1 on the cell surface of HeLa cells resulting in increased cell migration though there were no effects on cell viability (72). A recent study implicates LGALS3/ITGB3 mediated signaling as important for KRAS signaling in lung cancer (76). Sensitivity of lung cancer cells to modified pectin GCS-100 was linked to LGALS3 and ITGB3 expression. In this study we find suppression of LGALS3 resulted in induction of ITGB3. The result would appear paradoxical as the Seguin study suggests LGALS3/ITGB3 axis in important in K-RAS signaling, at least in lung cancer cells (76). The results from the Seguin study would suggest that ITGB3 regulates LGALS3 as knock down of ITGB3 in H1792 cells results in decreased LGALS3 expression and knock down of LGALS3 appears to have little effect on ITGB3 expression in those cells (76). However, knock down of LGALS3 in murine embryonic fibroblasts (MEFs) did promote an increase in ITGB3 in MEFs especially in MEFs containing mutant KRAS (i.e. G12D; ref. 76). These findings suggest potential regulation of ITGB3 by LGALS3 is cell type specific. Perhaps induction of ITGB3 with suppression of LGALS3 in MSC represents a feedback mechanism. Identification of such a feedback loop is under investigation.
In summary, our data support a role for LGALS3 in regulating MSC chemoprotection of leukemia cells likely involving a mechanis m of cell adhesion. A model displaying LGALS3 role in MSC and effect on AML cells in the microenvironment is depicted in Figure 7. The identification of MYC as a LGALS3 target in MSC suggests that LGALS3 may have key roles in supporting survival and proliferation. AKT phosphorylation was increased in the cells though the effect in part involves increased expression of total AKT kinase. Interestingly ITGB3 was found to be induced when expression of LGALS3 was suppressed though whether this represents a feedback loop remains to be determined. These results suggest that LGALS3 is important in MSC to support leukemia cell survival in the tumor microenvironment. Therefore, targeting LGALS3 could be an effective microenvironment based strategy for AML therapy.
Figure 7. Model of role of LGALS3 in MSC in the leukemia niche.
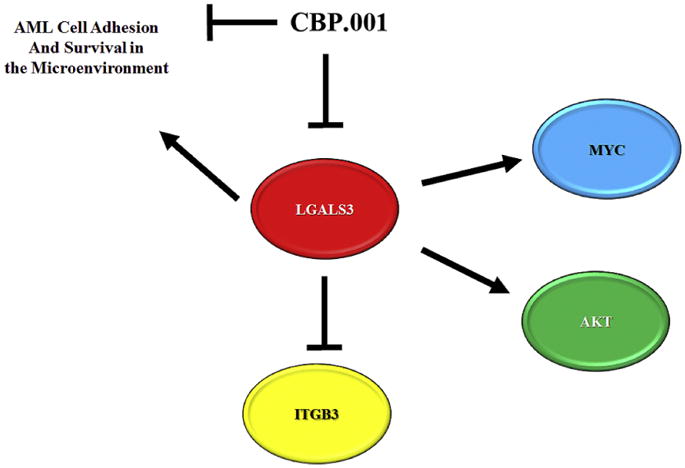
LGALS3 positively regulates MYC and AKT while negatively regulating ITGB3. LGALS3 supports AML cell adhesion and survival in the leukemia microenvironment. LGALS3 inhibitor CBP.001 blocks LGALS3 ability to support MSC mediated survival and adhesion of AML cells.
Supplementary Material
Highlights.
In mesenchymal stromal cells (MSC) from AML patients, Galectin 3 (LGALS3) protein but not RNA expression is elevated compared to MSC from presumably healthy donor and LGALS3 protein expression in AML derived MSC is correlated with a distinct set of biologically interconnected proteins that include MYC and AKT.
Suppression of LGALS3 in MSC reduces AKT and MYC and renders the stromal cell less able to adhere to AML cells in co-culture.
Kiromic Biopharma's novel LGALS3 inhibitor CBP.001 reduces AML cell viability in the presence of MSC and sensitizes AML cells to AraC in co-culture suggesting targeting LGALS3 may be an effective microenvironment based strategy for AML therapy.
Acknowledgments
We would like to thank Kiromic Biopharma (Houston, TX) for generous research support (PPR). The work was supported in part by research funding from the National Institutes of Health (P01CA49639; MA, SMK) and the Paul and Mary Haas Chair in Genetics (MA). Research was performed in the Flow Cytometry & Cellular Imaging Core Facility, which is supported in part by the National Institutes of Health through M. D. Anderson's Cancer Center Support Grant CA016672 (MA, JKB).
Footnotes
Conflict of Interest Statement for Ruvolo et al: Drs. Mirandola and Chiriva-Internati are employees of Kiromic Biopharma in Houston, TX. While the company provided drug, technical advice on use of the drug, and research funds to Dr. Ruvolo, all experiments were conducted and data interpreted at MD Anderson Cancer Center.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ho AD, Wagner W. Bone marrow niche and leukemia. Ernst Schering Found Symp Proc. 2006:125–139. doi: 10.1007/2789_2007_048. [DOI] [PubMed] [Google Scholar]
- 2.Lane SW, Scadden DT, Gilliland DG. The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood. 2009;114:1150–1157. doi: 10.1182/blood-2009-01-202606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konopleva M, Tabe Y, Zeng Z, et al. Therapeutic targeting of microenvironmental interactions in leukemia: mechanisms and approaches. Drug Resist Updat. 2009;12:103–113. doi: 10.1016/j.drup.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tabe Y, Konopleva M. Advances in understanding the leukaemia microenvironment. Br J Haematol. 2014;164:767–778. doi: 10.1111/bjh.12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng Z, Shi YX, Samudio IJ, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113:6215–6224. doi: 10.1182/blood-2008-05-158311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kojima K, McQueen T, Chen Y, et al. p53 activation of mesenchymal stromal cells partially abrogates microenvironment-mediated resistance to FLT3 inhibition in AML through HIF-1α-mediated down-regulation of CXCL12. Blood. 2011;118:4431–4439. doi: 10.1182/blood-2011-02-334136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barcellos-de-Souza P, Gori V, Bambi F, et al. Tumor microenvironment: bone marrow-mesenchymal stem cells as key players. Biochim Biophys Acta. 2013;1836:321–335. doi: 10.1016/j.bbcan.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Jacamo R, Chen Y, Wang Z, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood. 2014;123:2691–2702. doi: 10.1182/blood-2013-06-511527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fortuna-Costa A, Gomes AM, Kozlowski EO, et al. Extracellular galectin-3 in tumor progression and metastasis. Front Oncol. 2014;4:138. doi: 10.3389/fonc.2014.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guilloton F, Caron G, Ménard C, et al. Mesenchymal stromal cells orchestrate follicular lymphoma cell niche through the CCL2-dependent recruitment and polarization of monocytes. Blood. 2012;119:2556–2567. doi: 10.1182/blood-2011-08-370908. [DOI] [PubMed] [Google Scholar]
- 11.Poggi A, Musso A, Dapino I, Zocchi MR. Mechanisms of tumor escape from immune system: role of mesenchymal stromal cells. Immunol Lett. 2014;159:55–72. doi: 10.1016/j.imlet.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Fukumori T, Kanayama HO, Raz A. The role of galectin-3 in cancer drug resistance. Drug Resist Updat. 2007;10:101–108. doi: 10.1016/j.drup.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newlaczyl AU, Yu LG. Galectin-3--a jack-of-all-trades in cancer. Cancer Lett. 2011;313:123–128. doi: 10.1016/j.canlet.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Cecchinelli B, Lavra L, Rinaldo C, et al. Repression of the antiapoptotic molecule galectin-3 by homeodomain-interacting protein kinase 2-activated p53 is required for p53-induced apoptosis. Mol Cell Biol. 2006;26:4746–4757. doi: 10.1128/MCB.00959-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elad-Sfadia G, Haklai R, Balan E, Kloog Y. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J Biol Chem. 2004;279:34922–34930. doi: 10.1074/jbc.M312697200. [DOI] [PubMed] [Google Scholar]
- 16.Song S, Ji B, Ramachandran V, et al. Overexpressed galectin-3 in pancreatic cancer induces cell proliferation and invasion by binding Ras and activating Ras signaling. PLoS One. 2012;7:e42699. doi: 10.1371/journal.pone.0042699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshii T, Fukumori T, Honjo Y, et al. Galectin-3 phosphorylation is required for its anti-apoptotic function and cell cycle arrest. J Biol Chem. 2002;277:6852–6857. doi: 10.1074/jbc.M107668200. [DOI] [PubMed] [Google Scholar]
- 18.Levy R, Biran A, Poirier F, et al. Galectin-3 mediates cross-talk between K-Ras and Let-7c tumor suppressor microRNA. PLoS One. 2011;6:e27490. doi: 10.1371/journal.pone.0027490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng CL, Hou HA, Lee MC, et al. Higher bone marrow LGALS3 expression is an independent unfavorable prognostic factor for overall survival in patients with acute myeloid leukemia. Blood. 2013;121:3172–3180. doi: 10.1182/blood-2012-07-443762. [DOI] [PubMed] [Google Scholar]
- 20.Nakahara S, Oka N, Raz A. On the role of galectin-3 in cancer apoptosis. Apoptosis. 2005;10:267–275. doi: 10.1007/s10495-005-0801-y. [DOI] [PubMed] [Google Scholar]
- 21.Dumic J, Dabelic S, Flögel M. Galectin-3: an open-ended story. Biochim Biophys Acta. 2006;1760:616–635. doi: 10.1016/j.bbagen.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 22.Giordano M, Croci DO, Rabinovich GA. Galectins in hematological malignancies. Curr Opin Hematol. 2013;20:327–335. doi: 10.1097/MOH.0b013e328362370f. [DOI] [PubMed] [Google Scholar]
- 23.Kasper G, Mao L, Geissler S, et al. Insights into mesenchymal stem cell aging: involvement of antioxidant defense and actin cytoskeleton. Stem Cells. 2009;27:1288–1297. doi: 10.1002/stem.49. [DOI] [PubMed] [Google Scholar]
- 24.Sioud M. New insights into mesenchymal stromal cell-mediated T-cell suppression through galectins. Scand J Immunol. 2011;73:79–84. doi: 10.1111/j.1365-3083.2010.02491.x. [DOI] [PubMed] [Google Scholar]
- 25.Kornblau SM, Ruvolo P, Wang R, et al. Distinct protein signatures of acute myeloid leukemia bone marrow-derived stromal cells are prognostic for patient survival. Haematologica. 2018 doi: 10.3324/haematol.2017.172429. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kornblau SM, Womble M, Qiu YH, et al. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006;108:2358–2365. doi: 10.1182/blood-2006-02-003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tibes R, Qiu Y, Lu Y, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512–2521. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 28.Kornblau SM, Singh N, Qiu Y, Chen W, Zhang N, Coombes KR. Highly phosphorylated FOXO3A is an adverse prognostic factor in acute myeloid leukemia. Clin Cancer Res. 2010;16:1865–1874. doi: 10.1158/1078-0432.CCR-09-2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruvolo PP, Qui YH, Coombes KR, et al. Low expression of PP2A regulatory subunit B55alpha is associated with T308 phosphorylation of AKT and shorter complete remission duration in acute myeloid leukemia patients. Leukemia. 2011;25:1711–1717. doi: 10.1038/leu.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruvolo PP, Qiu Y, Coombes KR, et al. Phosphorylation of GSK3α/β correlates with activation of AKT and is prognostic for poor overall survival in acute myeloid leukemia patients. BBA Clin. 2015;4:59–68. doi: 10.1016/j.bbacli.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crane JL, Cao X. Bone marrow mesenchymal stem cells and TGF-β signaling in bone remodeling. J Clin Invest. 2014;124:466–472. doi: 10.1172/JCI70050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grafe I, Alexander S, Peterson JR, et al. TGF-β Family Signaling in Mesenchymal Differentiation. Cold Spring Harb Perspect Biol. 2017 doi: 10.1101/cshperspect.a022202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mackinnon AC, Gibbons MA, Farnworth SL, et al. Regulation of transforming growth factor-β1-driven lung fibrosis by galectin-3. Am J Respir Crit Care Med. 2012;185:537–546. doi: 10.1164/rccm.201106-0965OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gong D, Shi W, Yi SJ, et al. TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012;13:31. doi: 10.1186/1471-2172-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saegusa J, Hsu DK, Liu W, et al. Galectin-3 protects keratinocytes from UVB-induced apoptosis by enhancing AKT activation and suppressing ERK activation. J Invest Dermatol. 2008;128:2403–2411. doi: 10.1038/jid.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song S, Mazurek N, Liu C, et al. Galectin-3 mediates nuclear beta-catenin accumulation and Wnt signaling in human colon cancer cells by regulation of glycogen synthase kinase-3beta activity. Cancer Res. 2009;69:1343–1349. doi: 10.1158/0008-5472.CAN-08-4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao Q, Xia Y, Liu L, et al. Galectin-3 Enhances Migration of Minature Pig Bone Marrow Mesenchymal Stem Cells Through Inhibition of RhoA-GTP Activity. Sci Rep. 2016;6:26577. doi: 10.1038/srep26577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mirandola L, Yu Y, Chui K, et al. Galectin-3C inhibits tumor growth and increases the anticancer activity of bortezomib in a murine model of human multiple myeloma. PLoS One. 2011;6:e21811. doi: 10.1371/journal.pone.0021811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mirandola L, Nguyen DD, Rahman RL, et al. Anti-galectin-3 therapy: a new chance for multiple myeloma and ovarian cancer? Int Rev Immunol. 2014;33:417–427. doi: 10.3109/08830185.2014.911855. [DOI] [PubMed] [Google Scholar]
- 41.Mirandola L, Yu Y, Cannon MJ, et al. Galectin-3 inhibition suppresses drug resistance, motility, invasion and angiogenic potential in ovarian cancer. Gynecol Oncol. 2014;135:573–579. doi: 10.1016/j.ygyno.2014.09.021. [DOI] [PubMed] [Google Scholar]
- 42.Ebrahim AH, Alalawi Z, Mirandola L, et al. Galectins in cancer: carcinogenesis, diagnosis and therapy. Ann Transl Med. 2014;2:88. doi: 10.3978/j.issn.2305-5839.2014.09.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruvolo PP, Ruvolo VR, Benton CB, et al. Combination of galectin inhibitor GCS-100 and BH3 mimetics eliminates both p53 wild type and p53 null AML cells. Biochim Biophys Acta. 2016;1863:562–571. doi: 10.1016/j.bbamcr.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruvolo PP. Galectin 3 as a guardian of the tumor microenvironment. Biochim Biophys Acta. 2016;1863:427–437. doi: 10.1016/j.bbamcr.2015.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Fei F, Abdel-Azim H, Lim M, et al. Galectin-3 in pre-B acute lymphoblastic leukemia. Leukemia. 2013;27:2385–2388. doi: 10.1038/leu.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fei F, Joo EJ, Tarighat SS, et al. B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3. Oncotarget. 2015;6:11378–11394. doi: 10.18632/oncotarget.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Souza BSF, da Silva KN, Silva DN, et al. Galectin-3 Knockdown Impairs Survival, Migration, and Immunomodulatory Actions of Mesenchymal Stromal Cells in a Mouse Model of Chagas Disease Cardiomyopathy. Stem Cells Int. 2017;2017:3282656. doi: 10.1155/2017/3282656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kasper G, Mao L, Geissler S, et al. Insights into mesenchymal stem cell aging: involvement of antioxidant defense and actin cytoskeleton. Stem Cells. 2009;27:1288–1297. doi: 10.1002/stem.49. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Xu X, Wang L, et al. Senescent mesenchymal stem cells promote colorectal cancer cells growth via galectin-3 expression. Cell Biosci. 2015;5:21. doi: 10.1186/s13578-015-0012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Y, Wu D, Fei C, et al. Down-regulation of Dicer 1 promotes cellular senescence and decreases the differentiation and stem cell-supporting capacities of mesenchymal stromal cells in patients with myelodysplastic syndrome. Haematologica. 2015;100:194–204. doi: 10.3324/haematol.2014.109769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Y, Tabe Y, Jin L, et al. TGF-beta receptor kinase inhibitor LY2109761 reverses the anti-apoptotic effects of TGF-beta 1 in myelo-monocytic leukaemic cells co-cultured with stromal cells. Br J Haematol. 2008 Jun;142(2):192–201. doi: 10.1111/j.1365-2141.2008.07130.x. [DOI] [PubMed] [Google Scholar]
- 52.Kode A, Manavalan JS, Mosialou I, et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature. 2014;506:240–244. doi: 10.1038/nature12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou HS, Carter BZ, Andreeff M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol Med. 2016;13:248–259. doi: 10.20892/j.issn.2095-3941.2016.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming targeted therapy and tumor plasticity (Review) Int J Oncol. 2017;51:1357–1369. doi: 10.3892/ijo.2017.4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, O'Leary H, Fortney J, et al. Ph+/VE-cadherin+ identifies a stem cell like population of acute lymphoblastic leukemia sustained by bone marrow niche cells. Blood. 2007;110:3334–3344. doi: 10.1182/blood-2007-01-068122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Y, Mallampati S, Sun B, Zhang J, Kim SB, Lee JS, Gong Y, Cai Z, Sun X. Wnt pathway contributes to the protection by bone marrow stromal cells of acute lymphoblastic leukemia cells and is a potential therapeutic target. Cancer Lett. 2013;333:9–17. doi: 10.1016/j.canlet.2012.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang B, Li M, McDonald T, et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-β-catenin signaling. Blood. 2013;121:1824–1838. doi: 10.1182/blood-2012-02-412890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yi H, Zeng D, Shen Z, et al. Integrin alphavbeta3 enhances β-catenin signaling in acute myeloid leukemia harboring Fms-like tyrosine kinase-3 internal tandem duplication mutations: implications for microenvironment influence on sorafenib sensitivity. Oncotarget. 2016;7:40387–40397. doi: 10.18632/oncotarget.9617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang X, Mak PY, Mu H, et al. Disruption of Wnt/β-catenin exerts anti-leukemia activity and synergizes with FLT3 inhibition in FLT3-mutant acute myeloid leukemia. Clin Cancer Res. 2018 Feb 20; doi: 10.1158/1078-0432.CCR-17-1556. ePub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shimura T, Takenaka Y, Tsutsumi S, et al. Galectin-3, a novel binding partner of beta-catenin. Cancer Res. 2004;64:6363–6367. doi: 10.1158/0008-5472.CAN-04-1816. [DOI] [PubMed] [Google Scholar]
- 61.Shimura T, Takenaka Y, Fukumori T, et al. Implication of galectin-3 in Wnt signaling. Cancer Res. 2005;65:3535–3537. doi: 10.1158/0008-5472.CAN-05-0104. [DOI] [PubMed] [Google Scholar]
- 62.Song S, Mazurek N, Liu C, et al. Galectin-3 mediates nuclear beta-catenin accumulation and Wnt signaling in human colon cancer cells by regulation of glycogen synthase kinase-3beta activity. Cancer Res. 2009;69:1343–1349. doi: 10.1158/0008-5472.CAN-08-4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raaijmakers MH, Mukherjee S, Guo S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–857. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Battula VL, Chen Y, Cabreira Mda G, et al. Connective tissue growth factor regulates adipocyte differentiation of mesenchymal stromal cells and facilitates leukemia bone marrow engraftment. Blood. 2013;122:357–366. doi: 10.1182/blood-2012-06-437988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Battula VL, Le PM, Sun JC, et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight. 2017;2 doi: 10.1172/jci.insight.90036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Diaz de la Guardia R, Lopez-Millan B, Lavoie JR, et al. Detailed Characterization of Mesenchymal Stem/Stromal Cells from a Large Cohort of AML Patients Demonstrates a Definitive Link to Treatment Outcomes. Stem Cell Reports. 2017;8:1573–1586. doi: 10.1016/j.stemcr.2017.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sato Y, Mabuchi Y, Miyamoto K, et al. Notch2 Signaling Regulates the Proliferation of Murine Bone Marrow-Derived Mesenchymal Stem/Stromal Cells via c-Myc Expression. PLoS One. 2016;11:e0165946. doi: 10.1371/journal.pone.0165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shimizu T, Ishikawa T, Sugihara E, et al. c-MYC overexpression with loss of Ink4a/Arf transforms bone marrow stromal cells into osteosarcoma accompanied by loss of adipogenesis. Oncogene. 2010;29:5687–99. doi: 10.1038/onc.2010.312. [DOI] [PubMed] [Google Scholar]
- 69.Dombrowski C, Helledie T, Ling L, et al. FGFR1 signaling stimulates proliferation of human mesenchymal stem cells by inhibiting the cyclin-dependent kinase inhibitors p21(Waf1) and p27(Kip1) Stem Cells. 2013;31:2724–36. doi: 10.1002/stem.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakajima K, Kho DH, Yanagawa T, et al. Galectin-3 inhibits osteoblast differentiation through notch signaling. Neoplasia. 2014;16:939–49. doi: 10.1016/j.neo.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nabi IR, Shankar J, Dennis JW. The galectin lattice at a glance. J Cell Sci. 2015;128:2213–9. doi: 10.1242/jcs.151159. [DOI] [PubMed] [Google Scholar]
- 72.Yang EH, Rode J, Howlader MA, et al. Galectin-3 alters the lateral mobility and clustering of β1-integrin receptors. PLoS One. 2017;12:e0184378. doi: 10.1371/journal.pone.0184378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nieberler M, Reuning U, Reichart F, et al. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers (Basel) 2017;9 doi: 10.3390/cancers9090116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sasaki T, Brakebusch C, Engel J, et al. Mac-2 binding protein is a cell-adhesive protein of the extracellular matrix which self-assembles into ring-like structures and binds beta1 integrins, collagens and fibronectin. EMBO J. 1998;17:1606–1613. doi: 10.1093/emboj/17.6.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cardoso AC, Andrade LN, Bustos SO, et al. Galectin-3 Determines Tumor Cell Adaptive Strategies in Stressed Tumor Microenvironments. Front Oncol. 2016;6:127. doi: 10.3389/fonc.2016.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seguin L, Camargo MF, Wettersten HI, et al. Galectin-3, a Druggable Vulnerability for KRAS-Addicted Cancers. Cancer Discov. 2017;7:1464–1479. doi: 10.1158/2159-8290.CD-17-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao W, Ajani JA, Sushovan G, et al. Galectin-3 Mediates Tumor Cell-Stroma Interactions by Activating Pancreatic Stellate Cells to Produce Cytokines via Integrin Signaling. Gastroenterology. 2017 Dec 21;:36719–36717. doi: 10.1053/j.gastro.2017.12.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.