

Abstract
Adaptive protein evolution may be facilitated by neutral amino acid mutations that confer no benefit when they first arise but which potentiate subsequent function-altering mutations via direct or indirect structural mechanisms. Theoretical and empirical results indicate that such compensatory interactions (intramolecular epistasis) can exert a strong influence on trajectories of protein evolution. For this reason, assessing the form and prevalence of intramolecular epistasis and characterizing biophysical mechanisms of compensatory interaction are important research goals at the nexus of structural biology and molecular evolution. Here I review recent insights derived from protein-engineering studies, and I describe an approach for identifying and characterizing mechanisms of epistasis that integrates experimental data on structure-function relationships with analyses of comparative sequence data.
Graphical Abstract
Introduction
A number of important questions about mechanisms of protein evolution concern the context-dependence of mutational effects (‘epistasis’). Do amino acid mutations typically produce the same functional effect regardless of sequence context (i.e., regardless of the amino acid states of other sites in the same protein)? Alternatively, do the effects of mutations often depend on the sequence context in which they occur? Epistatic interactions between mutant sites in the same protein can help explain why evolution follows some pathways rather than others [1–5,6•,7•,8••]. If the sign of a mutation’s fitness effect is conditional on genetic background (‘sign epistasis’), then pairs of mutations that are individually neutral or beneficial may be deleterious in combination. In such cases, some fraction of all possible mutational pathways connecting ancestral and descendant genotypes will be selectively inaccessible because they include incompatible mutational combinations as intermediate steps [9–15,16•,17,18]. Conversely, pairs of mutations that are individually deleterious may be neutral or beneficial in combination, thereby opening up new pathways through sequence space that previously would have been off limits.
Intramolecular epistasis has important implications for biochemical adaptation and the evolution of novel protein functions. The evolution of an advantageous change in protein function may be facilitated by neutral mutations that confer no benefit when they first arise but which lay the groundwork for subsequent function-altering mutations. For example, an amino acid mutation at site X may produce a subtle change in protein conformation or stability that – by itself – is functionally inconsequential, but the altered structural context may change the functional effect of subsequent mutations at other sites in the same protein [4,5,7•,15,19–22]. The mutation at site X is neutral when it first arises, but by facilitating the fixation of a beneficial, function-altering mutation at site Y, it then becomes deleterious to revert site X to its ancestral state. Likewise, the mutation at site Y is beneficial on a background in which the mutation at site X has already occurred, but otherwise it would be neutral or deleterious. In principle, the compensatory change at site X could precede the function-altering change at site Y (in which case it is called a ‘permissive’ substitution), or it could occur afterwards, in which case there would be a transient reduction in fitness. In principle, the two mutations could also be fixed simultaneously if they co-occurred on the same sequence haplotype [23,24]. Evidence for epistatic fitness effects prompts us to view the longstanding ‘selectionist/neutralist’ debate through a new prism since a given mutation may be neutral, beneficial, or deleterious depending on the genetic background in which it occurs.
Compensatory substitutions are central to questions about the role of historical contingency in shaping pathways and outcomes of protein evolution. If the fitness effects of amino acid mutations are conditional on genetic background, then mutations can have different effects depending on the sequential order in which they occur [2]. Consequently, the accumulated history of substitutions in the past will influence the set of allowable mutations in the future, and evolutionary outcomes will be historically contingent on ancestral starting points [7•,8••,12,14,25–27].
The role of intramolecular epistasis in protein evolution
Insights from protein-engineering experiments
The question of whether mutations have different effects in different genetic backgrounds can be decisively tested with site-directed mutagenesis experiments. Lunzer et al. [28] investigated the effects of swapping amino acid states at residue positions that differ between two highly divergent orthologs of isopropymalate dehydrogenase (IMDH). They introduced a total of 168 single mutations into wildtype IMDH of Escherichia coli that match the amino acid states at the same sites in the IMDH ortholog of Pseudomonas aeruginosa. Of these 168 swapped residues, over 1/3 of the wildtype amino acid states in P. aeruginosa compromised enzyme activity on the genetic background of E. coli. The fact that identical amino acid states produced different phenotypic effects on the two genetic backgrounds must be attributable to substitutions at other residue positions in one or both lineages that epistatically interact with the focal residues. These could be permissive substitutions that made a given amino acid state acceptable in the native P. aeruginosa background, and/or restrictive substitutions that made the same state deleterious in the E. coli background. Results of this experiment demonstrate how evolved changes in sequence context can reduce the number of site-specific amino acid states that are unconditionally acceptable in the divergent backgrounds of orthologous proteins (Fig. 1). Interestingly, mutagenesis screens revealed that the amino acid replacements that most strongly reduced activity on the E. coli background could be compensated by multiple mutations at structurally remote sites; the compensatory mechanisms did not generally involve direct physical interactions [28].
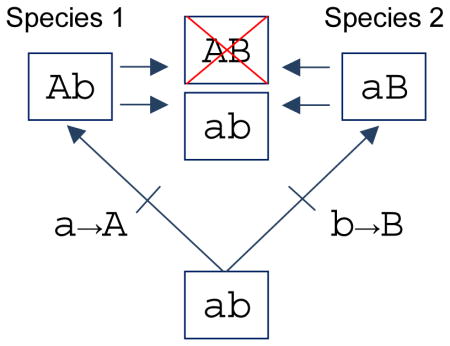
Fig. 1.

Amino acid states that are allowable in one genetic background may be deleterious in other backgrounds. (A) Two species diverge from a common ancestor with the two-site genotype, ab. The substitution a→A occurs at the first site in species 1 (yielding Ab) and the substitution b→B occurs at the second site in species 2 (yielding aB). A negative epistatic interaction (‘Dobzhanksy-Müller imcompatibility’) is revealed by moving mutation A from species 1 into the orthologous background of species 2, or by moving mutation B from species 2 into the orthologous background of species 1. Mutations A and B are individually neutral on the genetic backgrounds in which they occurred during evolution, but they are deleterious in combination. Swapping mutations a or b to form genotype ab results in a reversion to the ancestral state. (B) The same type of incompatibility can arise if both substitutions occur in one lineage, while the other species retains the ancestral states at both sites. In this case, swapping mutations a or B yields a low-fitness genotype (aB). Swapping mutations A or b yields genotype Ab, one of two possible mutational intermediates in the ancestry of species 1. Note that since mutations a and B are deleterious in combination, substitution a→A must have preceded substitution b→B in the ancestry of species 1 because Ab is the only viable single-mutant intermediate connecting the ancestral (ab) and descendant (AB) genotypes.
Similar to the ‘horizontal’ swapping of amino acids between divergent orthologs of extant taxa, Gong et al. [15] performed ‘vertical’ exchanges between ancestral and descendant genotypes of the influenza nucleoprotein that were isolated from different timepoints over the span of several decades. Capitalizing on their ability to infer the temporal order of observed substitutions in a single line of descent, Gong et al. individually introduced each of 39 observed amino acid replacements into the genetic background of an extinct viral strain that approximated the ancestral genotype. The experiments revealed that three of the 39 substitutions that occurred as intermediate steps in the pathway had strongly deleterious effects on the ancestral genetic background even though they must have been neutral, nearly neutral, or possibly even beneficial on the derived genetic background in which they actually fixed. The three mutations that had deleterious effects on the ancestral background severely compromised thermal stability of the native protein fold. These same mutations were tolerated at later steps in the pathway because they were preceded by stabilizing substitutions in the same protein. These substitutions provided a stability buffer and therefore permitted the fixation of mutations with destabilizing effects.
As in the Lunzer et al. [28] study on IMDH orthologs, the identified compensatory interactions in influenza nucleoprotein involved pairs of structurally remote residues. The permissive mutations did not directly alter the effects of destabilizing mutations; instead, they simply increased overall thermal stability so that the destabilizing mutations did not cause the fraction of folded protein to fall below the critical threshold where the assayed functional property (viral RNA transcription) was compromised. This illustrates how mutations that have additive effects on structural properties can have nonadditive effects on higher-level properties due to nonlinear relationships between structure and function or between function and fitness [18•,19,22,29–31].
In summary, neutral or nearly neutral mutations that fix in one species may have deleterious effects if they were to occur on the divergent genetic background of a different species. Likewise, within a single line of descent, substitutions may have different phenotypic effects depending on the sequential order in which they occur. Site-directed mutagenesis experiments that swap residues between orthologs of contemporary species [28,32,33] or between ancestral and descendant sequences [9,17,18•,34–37] have revealed pervasive epistasis for functional properties or fitness proxies. These experimental findings are generally consistent with results of theoretical and computational analyses [38–44,45•,46] and in silico simulations [47,48••]. By contrast, other experimental studies that focused on mutational perturbations of structural stability have suggested a less important role for epistasis during longterm protein evolution [49–51].
Genetic compensation of pathogenic mutations
Additional insights into the prevalence of epistasis and the nature of genetic compensation are provided by cases where a pathogenic amino acid mutation in a human protein appears as the wildtype residue at the same site in the orthologous protein of one or more nonhuman species [52](Fig. 2). In such cases, the pathogenic variant is invariably present at low frequency in the human gene pool, but the same amino acid is fixed (present at a frequency of 1.0) in the nonhuman species. In order for the disease-associated residue (DAR) to become fixed in the nonhuman species, its deleterious effects must have been compensated by one or more substitutions at other sites in the same protein or in an interacting protein. This permits an indirect inference of sign epistasis for fitness: the DAR produces a deleterious effect in the human protein, but is neutral in the nonhuman ortholog due to genetic compensation [52–58,59•].
Fig. 2.

Indirect evidence for genetic compensation is provided by cases where a pathogenic amino acid mutation in a human protein appears as the wildtype residue at the same site in the orthologous protein of one or more nonhuman species. The pathogenic hemoglobin (Hb) mutant, Hb Mequon (β41Phe→Tyr), provides an illustrative example. Although the Hb Mequon mutation is associated with severe hemolytic anemia in humans, the disease-associated β41Tyr is wildtype in Mus and Rattus. In order for the Tyr variant to become fixed in the common ancestor of these rodent taxa, the deleterious effects that are manifest in human Hb must have been compensated by one or more rodent-specific substitutions at other sites in the same protein.
The pathogenicity of amino acid mutations often stems from their negative effects on protein structural stability [60], and evidence suggests that the destabilizing effects of such DARs are sometimes partly or wholly compensated by substitutions at structurally proximal residue positions [52,55,56,58, 59•,61,62]. For example, Xu and Zhang [58] demonstrated that uncompensated DARs in human proteins are associated with lower average structural stabilities than the corresponding wildtype DARs in cases where the latter are accompanied by one or more lineage-specific substitutions at residue positions within a 4 Å radius of the DAR. This finding suggests that one or more of the observed substitutions in the nonhuman background compensate for the destabilizing effect of the DAR.
In cases where compensatory substitutions have not been identified, there are other possible explanations for the existence of wild-type DARs that do not invoke sign epistasis for fitness [54]. For example, the observation that a DAR is wildtype in a given species does not rule out the possibility that it has a mildly deleterious effect, as such mutations can fix due to drift, especially in small or bottlenecked populations [63]. This is unlikely to be a general explanation, however, especially in cases where the wildtype DAR is shared by multiple species within the same clade. A number of wildtype DARs have been documented in laboratory mice (Mus domesticus)[53], but the majority of these are shared by multiple species of Mus that diverged over one million years ago, so they clearly do not represent deleterious variants that were fixed as a result of founder effects during the history of mouse breeding and domestication [54].
Rather than using the identification of wildtype DARs to indirectly infer sign epistasis for fitness, a similar approach can be used to infer sign epistasis for biochemical phenotypes without making assumptions about fitness effects. This is possible in cases where we have detailed information about structure-function relationships. For example, in the case of human hemoglobin (Hb), crystallographic and NMR studies of mutant Hbs have provided exquisitely detailed insights into the structural mechanisms responsible for observed functional effects of specific amino acid replacements. In cases where a particular mutation in human Hb is known to alter a particular functional property, it is often possible to identify Hbs from nonhuman species in which the same amino acid is wildtype and yet the property of interest is not altered in the same way. In such cases the connection between genotype and biochemical phenotype can be experimentally tested. By contrast, associations with disease states do not generally permit direct insights into the mapping functions that relate genotype to phenotype or phenotype to fitness.
Molecular basis of the Bohr effect: a case study of genetic compensation
The Hbs of jawed vertebrates are heterotetramers, composed of paired αβ dimers (α2β2) that undergo a symmetrical rotation during oxygenation-linked transitions in quaternary structure [64]. This allosteric transition is mediated by a conformational equilibrium between the low-affinity, deoxygenated ‘T state’ and the high-affinity, oxygenated ‘R state’. Hb-O2 affinity is reduced at low pH because protons preferentially bind and stabilize deoxyHb, thereby shifting the allosteric equilibrium in favor of the low-affinity T conformation [65]. Because Hb-O2 affinity changes as a positive function of pH over the physiological range (6.6 to 7.6), the metabolic acidosis of capillary blood induces Hb to release O2 to the tissues that need it most. This pH-sensitivity of Hb-O2 affinity is known as the Bohr effect.
At physiological pH and temperature, the Bohr effect of human Hb is mainly attributable to the oxygenation-linked deprotonation of surface histidines because their imidazole side chains typically have acid dissociation constants, pKa’s, in the physiological pH range [66]. The C-terminal histidine of the β-chain, β146His, makes an outsized contribution, accounting for ~60% of the Bohr effect in the presence of 0.1 M chloride [67]. In deoxy (T state) Hb, the positive charge on the imidazole sidechain of β146His is stabilized by formation of a salt bridge with the carbonyl group of β94Asp in the same β-chain subunit (Fig. 3). This ionization of the β146His side chain substantially raises its pKa in the deoxy T state. Consequently, mutational replacements of either β146His or β94Asp result in a severely diminished Bohr effect because the β94Asp-β146His salt-bridge in the T state is replaced by an unionizable hydrogen bond; thus, no protons are released in the allosteric T→R transition in quaternary structure. Surprisingly, substitutions at these highly conserved residue positions have been identified in the Hbs of several vertebrate species that do not have reduced Bohr effects [68–70]. For example, human Hb mutants such as Hb Bologna-St. Orsola (β146His→Tyr) and Hb Kodaira (β146His→Gln) exhibit increased O2-affinities (due to destabilization of the T-state) and severe reductions in the Bohr effect [71,72]. Remarkably, the same amino acid states are observed as wildtype in the adult Hbs of the dwarf caiman (Paleosuchus palpebrosus)(β146Tyr) and the golden-mantled ground squirrel (Callospermophilus lateralis)(β146Gln), and yet the Hbs of both species exhibit Bohr effects that are undiminished relative to normal human Hb [68,69](Fig. 4). In the case of both caiman and ground-squirrel Hb, the loss of a single key residue with a major effect on pH-sensitivity, β146His, appears to be compensated by the lineage-specific gain of multiple solvent-exposed, titratable histidines with individually minor effects [68,69]. In the case of β94Asp, human Hb mutants such as Hb Barcelona (β94Asp→His) and Hb Bunbury (β94Asp→Asn) also exhibit marked increases in O2-affinity and concomitant reductions in the Bohr effect due to the disruption of the β94Asp-β146His intrachain salt bridge. In two different species of high-altitude Andean waterfowl (crested duck [Lophonetta specularioides] and Puna teal [Anas puna]), β94Asp→Glu mutations have contributed to adaptive increases in Hb-O2 affinity, but the Bohr effect is not compromised relative to wildtype Hbs with the ancestral β94Asp [70]. A consideration of the crystal structure of avian Hb provides a clear explanation for this result [67], as the β94Asp-β146His salt bridge is not formed in the deoxy T conformation, so the amino acid state of β94 does not affect the pKa of β146His. This may also explain why the β146His→Tyr substitution in dwarf caiman Hb is not associated with a diminished Bohr effect relative to the Hbs of other crocodilians, and demonstrates how ‘major effect’ Bohr groups in human Hb may have minor or nonexistent effects in the Hbs of other species. These examples also illustrate how subtle changes in the three-dimensional orientation of highly conserved amino acids (caused by substitutions at other sites) can alter the functional effects of substitutions at those conserved sites [73].
Fig. 3.

The structural basis for the Bohr effect in tetrameric (α2β2) human Hb. (A, B) The C-terminal His of each β-chain subunit (β146His) participates in two electrostatic interactions in the deoxy (T) state. The positively charged imidazole side chain of β146His forms an intrasubunit salt bridge with β94Asp (which increases its pKa) and its negatively charged carboxyl group forms an intradimer salt bridge with α40Lys. (C) When Hb is oxygenated, the allosteric transition in quaternary structure shifts the triad of residues apart from one another, outside the range of electrostatic interaction. The consequent rupturing of the β146His- β94Asp salt bridge results in the deprotonation of the His side chain (two protons are released per tetramer), which makes a major contribution to the Bohr effect.
Fig. 4.

In the Hbs of amniote vertebrates, comparative sequence analysis and experimental data on structure-function relationships reveal intramolecular epistasis for the Bohr effect. In the β-chain subunit of vertebrate Hb, the highly conserved β146His generally accounts for a major fraction of the Bohr effect [66,67,71,72]. This is well-documented by experimental studies of naturally occurring human Hb mutants which demonstrate that mutational replacements of β146His (H) with Asp (D), Tyr (Y), Arg (R), Leu (L), Pro (P), or Gln (Q) invariably result in a severely diminished Bohr effect. Surprisingly, however, two of these amino acid states, Q (Hb Bologna-St. Orsola) and Y (Hb Kodaira), occur as wildtype in the Hbs of two nonhuman vertebrates, dwarf caiman (Paleosuchus palpebrosus) and golden-mantled ground squirrel (Callospermophilus lateralis), respectively, and yet the Hbs of both species exhibit Bohr effects that are undiminished relative to normal human Hb [68,69]. In both species, the aggregate effect of other lineage-specific substitutions (e.g., gains of solvent-exposed histidines at other sites in the α- and/or β-chains of the Hb tetramer) may have rendered β146His redundant with respect to oxygenation-linked proton binding, so it could therefore be replaced without unduly compromising the Bohr effect.
Conclusions and future directions
Efforts to elucidate mechanisms of epistasis represent a nexus between the fields of structural biology and molecular evolution [1,5,74]. An especially important question is whether permissive/compensatory mutational effects are typically localized and specific, or whether they typically involve generalized effects on global properties such as structural stability. If function-altering mutations can only be compensated by mutations with localized and specific effects (e.g., via direct steric or electrostatic sidechain interactions between structurally proximal residues), then accessible mutational pathways to novel functions may be fortuitously contingent on the acquisition of exceedingly rare mutations [7•,25]. By contrast, there is a much larger mutational target size for perturbations of overall structural stability, so destabilizing, function-altering mutations may be effectively compensated by stabilizing mutations at many possible residue positions in the same protein, and the compensatory effect would not require direct site-site interaction. If deleterious pleiotropic effects of adaptive, function-altering mutations can be effectively compensated by such global suppressor mutations, then the optimizing power of selection should be considerably less constrained and evolutionary outcomes will be less strongly contingent on ancestral starting points.
Highlights.
Adaptive protein evolution may be facilitated by conditionally neutral mutations.
Mutations can have different effects on different genetic backgrounds.
Contingency plays a key role in shaping trajectories of protein evolution.
Insights into epistasis are provided by compensated function-altering mutations.
Acknowledgments
My work on protein evolution is funded by grants from the National Institutes of Health (HL087216) and the National Science Foundation (MCB-1517636). I thank anonymous reviewers for constructive comments, and C. Natarajan and H. Moriyama for help with figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.DePristo MA, Weinreich DM, Hartl DL. Missense meanderings in sequence space: A biophysical view of protein evolution. Nature Reviews Genetics. 2005;6:678–687. doi: 10.1038/nrg1672. [DOI] [PubMed] [Google Scholar]
- 2.Weinreich DM, Watson RA, Chao L. Sign epistasis and genetic constraint on evolutionary trajectories. Evolution. 2005;59:1165–1174. [PubMed] [Google Scholar]
- 3.Poelwijk FJ, Kiviet DJ, Weinreich DM, Tans SJ. Empirical fitness landscapes reveal accessible evolutionary paths. Nature. 2007;445:383–386. doi: 10.1038/nature05451. [DOI] [PubMed] [Google Scholar]
- 4.Bloom JD, Arnold FH. In the light of directed evolution: Pathways of adaptive protein evolution. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9995–10000. doi: 10.1073/pnas.0901522106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harms MJ, Thornton JW. Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nature Reviews Genetics. 2013;14:559–571. doi: 10.1038/nrg3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6•.Kondrashov DA, Kondrashov FA. Topological features of rugged fitness landscapes in sequence space. Trends in Genetics. 2015;31:24–33. doi: 10.1016/j.tig.2014.09.009. A review of movement rules and accessibility relations that govern the evolution of proteins through the multidimensional space of all possible genotypes. [DOI] [PubMed] [Google Scholar]
- 7•.Starr TN, Thornton JW. Epistasis in protein evolution. Protein Science. 2016;25:1204–1218. doi: 10.1002/pro.2897. A thoughtful synthesis that discusses different modes of compensatory interaction, and the the role of epistasis in shaping trajectories of protein evolution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8••.Starr TN, Picton LK, Thornton JW. Alternative evolutionary histories in the sequence space of an ancient protein. Nature. 2017;549:409–413. doi: 10.1038/nature23902. A creative study that combined ancestral protein resurrection with deep mutational scanning to evaluate a multitude of might-have-been histories in the evolution of a transcription factor’s switch from one DNA specificity to another. The derived specificity could have evolved from many possible starting points within the network of genotypes that encode the ancestral specificity, but the biophysical basis of the switch was highly contingent on the starting point. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weinreich DM, Delaney NF, DePristo MA, Hartl DL. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312:111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- 10.Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW. Crystal structure of an ancient protein: Evolution by conformational epistasis. Science. 2007;317:1544–1548. doi: 10.1126/science.1142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bridgham JT, Ortlund EA, Thornton JW. An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature. 2009;461:515–519. doi: 10.1038/nature08249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salverda MLM, Dellus E, Gorter FA, Debets AJM, van der Oost J, Hoekstra RF, Tawfik DS, de Visser JAGM. Initial mutations direct alternative pathways of protein evolution. PLoS Genetics. 2011;7:e1001321. doi: 10.1371/journal.pgen.1001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.da Silva J, Coetzer M, Nedellec R, Pastore C, Mosier DE. Fitness epistasis and constraints on adaptation in a human immunodeficiency virus type 1 protein region. Genetics. 2010;185:293–303. doi: 10.1534/genetics.109.112458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dickinson BC, Leconte AM, Allen B, Esvelt KM, Liu DR. Experimental interrogation of the path dependence and stochasticity of protein evolution using phage-assisted continuous evolution. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:9007–9012. doi: 10.1073/pnas.1220670110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong LI, Suchard MA, Bloom JD. Stability-mediated epistasis constrains the evolution of an influenza protein. eLife. 2013;2:e00631. doi: 10.7554/eLife.00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16•.Kaltenbach M, Jackson CJ, Campbell EC, Hollfelder F, Tokuriki N. Reverse evolution leads to genotypic incompatibility despite functional and active site convergence. eLife. 2015;4:e06492. doi: 10.7554/eLife.06492. The authors conducted a directed evolution experiment which revealed that a PTE enzyme that had evolved a novel function (arylesterase activity) could be reverted to its ancestral function (phosphotriesterase activity). Interestingly, however, the ancestral catalytic activity was restored via the fixation of new mutations, not by reverting mutations that had fixed during evolution of the derived arylesterase activity. This finding demonstrates how the ancestral state of a given site can become incompatible with derived states at other sites in the same protein, thereby inhibiting mutational reversions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tufts DM, Natarajan C, Revsbech IG, Projecto-Garcia J, Hoffman FG, Weber RE, Fago A, Moriyama H, Storz JF. Epistasis constrains mutational pathways of hemoglobin adaptation in high-altitude pikas. Molecular Biology and Evolution. 2015;32:287–298. doi: 10.1093/molbev/msu311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18•.Kumar A, Natarajan C, Moriyama H, Witt CC, Weber RE, Fago A, Storz JF. Stability-mediated epistasis restricts accessible mutational pathways in the functional evolution of avian hemoglobin. Molecular Biology and Evolution. 2017;34:1240–1251. doi: 10.1093/molbev/msx085. This study dissected the molecular basis of an evolved change in hemoglobin function that was caused by the combined effect of four amino acid substitutions, two of which occurred in highly conserved intradimeric interfaces of the tetrameric protein. The experiments revealed that half of all possible forward pathways connecting functionally distinct ancestral and descendant genotypes included functionally aberrant intermediates because particular combinations of mutations promoted tetramer-dimer dissociation. The results demonstrate how epistasis for particular functional properties of proteins may be mediated indirectly by mutational effects on quaternary structural stability. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Protein stability promotes evolvability. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bloom JD, Raval A, Wilke CO. Thermodynamics of neutral protein evolution. Genetics. 2007;175:255–266. doi: 10.1534/genetics.106.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bloom JD, Romero PA, Lu Z, Arnold FH. Neutral genetic drift can alter promiscuous protein functions, potentially aiding functional evolution. Biology Direct. 2007;2:17. doi: 10.1186/1745-6150-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bloom JD, Gong LI, Baltimore D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science. 2010;328:1272–1275. doi: 10.1126/science.1187816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura M. The role of compensatory neutral mutations in molecular evolution. Journal of Genetics. 1985;64:7–19. [Google Scholar]
- 24.Meer MV, Kondrashov AS, Artzy-Randrup Y, Kondrashov FA. Compensatory evolution in mitochondrial tRNAs navigates valleys of low fitness. Nature. 2010;464:279–282. doi: 10.1038/nature08691. [DOI] [PubMed] [Google Scholar]
- 25.Harms MJ, Thornton JW. Historical contingency and its biophysical basis in glucocorticoid receptor evolution. Nature. 2014;512:203–207. doi: 10.1038/nature13410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Natarajan C, Hoffmann FG, Weber RE, Fago A, Witt CC, Storz JF. Predictable convergence in hemoglobin function has unpredictable molecular underpinnings. Science. 2016;354:336–340. doi: 10.1126/science.aaf9070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Storz JF. Causes of molecular convergence and parallelism in protein evolution. Nature Reviews Genetics. 2016;17:239–250. doi: 10.1038/nrg.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lunzer M, Golding GB, Dean AM. Pervasive cryptic epistasis in molecular evolution. PloS Genetics. 2010;6:e1001162. doi: 10.1371/journal.pgen.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lunzer M, Milter SP, Felsheim R, Dean AM. The biochemical architecture of an ancient adaptive landscape. Science. 2005;310:499–501. doi: 10.1126/science.1115649. [DOI] [PubMed] [Google Scholar]
- 30.Bloom JD, Silberg JJ, Wilke CO, Drummond DA, Adami C, Arnold FH. Thermodynamic prediction of protein neutrality. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:606–611. doi: 10.1073/pnas.0406744102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKeown AN, Bridgham JT, Anderson DW, Murphy MN, Ortlund EA, Thornton JW. Evolution of DNA specificity in a transcription factor family produced a new gene regulatory module. Cell. 2014;159:58–68. doi: 10.1016/j.cell.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Maille PE, Malone A, Dellas N, Hess BA, Jr, Smentek L, Sheehan I, Greenhagen BT, Chappell J, Manning G, Noel JP. Quantitative exploration of the catalytic landscape separating divergent plant sesquiterpene synthases. Nature Chemical Biology. 2008;4:617–623. doi: 10.1038/nchembio.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melero C, Ollikainen N, Harwood I, Karpiak J, Kortemme T. Quantification of the transferability of a designed protein specificity switch reveals extensive epistasis in molecular recognition. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:15426–15431. doi: 10.1073/pnas.1410624111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lozovsky ER, Chookajorn T, Brown KM, Imwong M, Shaw PJ, Kamchonwongpaisan S, Neafsey DE, Weinreich DM, Hartl DL. Stepwise acquisition of pyrimethamine resistance in the malaria parasite. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12025–12030. doi: 10.1073/pnas.0905922106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Natarajan C, Inoguchi N, Weber RE, Fago A, Moriyama H, Storz JF. Epistasis among adaptive mutations in deer mouse hemoglobin. Science. 2013;340:1324–1327. doi: 10.1126/science.1236862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacquier H, Birgy A, Le Nagard H, Mechulam Y, Schmitt E, Glodt J, Bercot B, Petit E, Poulain J, Barnaud G, et al. Capturing the mutational landscape of the beta-lactamase TEM-1. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:13067–13072. doi: 10.1073/pnas.1215206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schenk MF, Szendro IG, Salverda MLM, Krug J, de Visser JAGM. Patterns of epistasis between beneficial mutations in an antibiotic resistance gene. Molecular Biology and Evolution. 2013;30:1779–1787. doi: 10.1093/molbev/mst096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Povolotskaya IS, Kondrashov FA. Sequence space and the ongoing expansion of the protein universe. Nature. 2010;465:922–926. doi: 10.1038/nature09105. [DOI] [PubMed] [Google Scholar]
- 39.Breen MS, Kemena C, Vlasov PK, Notredame C, Kondrashov FA. Epistasis as the primary factor in molecular evolution. Nature. 2012;490:535–538. doi: 10.1038/nature11510. [DOI] [PubMed] [Google Scholar]
- 40.Naumenko SA, Kondrashov AS, Bazykin GA. Fitness conferred by replaced amino acids declines with time. Biology Letters. 2012;8:825–828. doi: 10.1098/rsbl.2012.0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Draghi JA, Plotkin JB. Selection biases the prevalence and type of epistasis along adaptive trajectories. Evolution. 2013;67:3120–3131. doi: 10.1111/evo.12192. [DOI] [PubMed] [Google Scholar]
- 42•.Bazykin GA. Changing preferences: deformation of single position amino acid fitness landscapes and evolution of proteins. Biology Letters. 2015;11:20150315. doi: 10.1098/rsbl.2015.0315. Since a given site in a protein can be occupied by up to 20 amino acids, the set of fitness values conferred by each possible amino acid defines a vector of site-specific amino acid propensities for a given genetic background. This single-position fitness landscape can change through time due to substitutions at other sites in the same protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou Z, Zhang J. Are convergent and parallel amino acid substitutions in protein evolution more prevalent than neutral expectations? Molecular Biology and Evolution. 2015;32:2085–2096. doi: 10.1093/molbev/msv091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldstein RA, Pollard ST, Shah SD, Pollock DD. Nonadaptive amino acid convergence rates decrease over time. Molecular Biology and Evolution. 2015;32:1373–1381. doi: 10.1093/molbev/msv041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45•.Usmanova DR, Ferretti L, Povolotskaya IS, Vlasov PK, Kondrashov FA. A model of substitution trajectories in sequence space and long-term protein evolution. Molecular Biology and Evolution. 2015;32:542–554. doi: 10.1093/molbev/msu318. This paper describes an amino acid substitution model that incorporates epistatic interactions between sites. The authors conclude that observed patterns of convergence and divergence in longterm protein evolution cannot be explained without invoking changes in site-specific amino acid propensities through time. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCandlish DM, Shah P, Plotkin JB. Epistasis and the dynamics of reversion in molecular evolution. Genetics. 2016;203:1335–1351. doi: 10.1534/genetics.116.188961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pollock DD, Thiltgen G, Goldstein RA. Amino acid coevolution induces an evolutionary Stokes shift. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E1352–E1359. doi: 10.1073/pnas.1120084109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48••.Shah P, McCandlish DM, Plotkin JB. Contingency and entrenchment in protein evolution under purifying selection. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:7627–7627. doi: 10.1073/pnas.1412933112. This simulation study demonstrated that the set of acceptable amino acid mutations at a given site is contingent on the amino acid states of other sites in the same protein; likewise, once a given substitution has occurred, mutational reversions to the ancestral amino acid become increasingly deleterious due to changes in structural context caused by substitutions at other sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ashenberg O, Gong LI, Bloom JD. Mutational effects on stability are largely conserved during protein evolution. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:21071–21076. doi: 10.1073/pnas.1314781111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doud MB, Ashenberg O, Bloom JD. Site-specific amino acid preferences are mostly conserved in two closely related protein homologs. Molecular Biology and Evolution. 2015;32:2944–2960. doi: 10.1093/molbev/msv167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Risso VA, Manssour-Triedo F, Delgado-Delgado A, Arco R, Barroso-delJesus A, Ingles-Prieto A, Godoy-Ruiz R, Gavira JA, Gaucher EA, Ibarra-Molero B, et al. Mutational studies on resurrected ancestral proteins reveal conservation of site-specific amino acid preferences throughout evolutionary history. Molecular Biology and Evolution. 2015;32:440–455. doi: 10.1093/molbev/msu312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kondrashov AS, Sunyaev S, Kondrashov FA. Dobzhansky-Muller incompatibilities in protein evolution. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14878–14883. doi: 10.1073/pnas.232565499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 54.Gao LZ, Zhang JZ. Why are some human disease-associated mutations fixed in mice? Trends in Genetics. 2003;19:678–681. doi: 10.1016/j.tig.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 55.Baresic A, Hopcroft LEM, Rogers HH, Hurst JM, Martin ACR. Compensated pathogenic deviations: analysis of structural effects. Journal of Molecular Biology. 2010;396:19–30. doi: 10.1016/j.jmb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 56.Ferrer-Costa C, Orozco M, de la Cruz X. Characterization of compensated mutations in terms of structural and physico-chemical properties. Journal of Molecular Biology. 2007;365:249–256. doi: 10.1016/j.jmb.2006.09.053. [DOI] [PubMed] [Google Scholar]
- 57.Soylemez O, Kondrashov FA. Estimating the rate of irreversibility in protein evolution. Genome Biology and Evolution. 2012;4:1213–1222. doi: 10.1093/gbe/evs096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu J, Zhang J. Why human disease-associated residues appear as the wild-type in other species: genome-scale structural evidence for the compensation hypothesis. Molecular Biology and Evolution. 2014;31:1787–1792. doi: 10.1093/molbev/msu130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59•.Jordan DM, Frangakis SG, Golzio C, Cassa CA, Kurtzberg J, Davis EE, Sunyaev SR, Katsanis N Task Force Neonatal G. Identification of cis-suppression of human disease mutations by comparative genomics. Nature. 2015;524:225–229. doi: 10.1038/nature14497. Results of this genome-scale analysis indicate that conditionally deleterious amino acid mutations are often compensated by second-site mutations that are in close spatial proximity in protein tertiary structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yue P, Li Z, Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. Journal of Molecular Evolution. 2005;50:56–68. doi: 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 61.Poon AFY, Chao L. Functional origins of fitness effect-sizes of compensatory mutations in the DNA bacteriophage phi X174. Evolution. 2006;60:2032–2043. [PubMed] [Google Scholar]
- 62.Davis BH, Poon AFY, Whitlock MC. Compensatory mutations are repeatable and clustered within proteins. Proceedings of the Royal Society B-Biological Sciences. 2009;276:1823–1827. doi: 10.1098/rspb.2008.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ohta T. The nearly neutral theory of molecular evolution. Annual Review of Ecology and Systematics. 1992;23:263–286. [Google Scholar]
- 64.Storz JF. Gene duplication and evolutionary innovations in hemoglobin-oxygen transport. Physiology. 2016;31:223–232. doi: 10.1152/physiol.00060.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perutz MF. Mechanisms regulating the reactions of human hemoglobin with oxygen and carbon monoxide. Annual Review of Physiology. 1990;52:1–25. doi: 10.1146/annurev.ph.52.030190.000245. [DOI] [PubMed] [Google Scholar]
- 66.Lukin JA, Ho C. The structure-function relationship of hemoglobin in solution at atomic resolution. Chemical Reviews. 2004;104:1219–1230. doi: 10.1021/cr940325w. [DOI] [PubMed] [Google Scholar]
- 67.Berenbrink M. Evolution of vertebrate haemoglobins: Histidine side chains, specific buffer value and Bohr effect. Respiratory Physiology & Neurobiology. 2006;154:165–184. doi: 10.1016/j.resp.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 68.Revsbech IG, Tufts DM, Projecto-Garcia J, Moriyama H, Weber RE, Storz JF, Fago A. Hemoglobin function and allosteric regulation in semi-fossorial rodents (family Sciuridae) with different altitudinal ranges. Journal of Experimental Biology. 2013;216:4264–4271. doi: 10.1242/jeb.091397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weber RE, Fago A, Malte H, Storz JF, Gorr TA. Lack of conventional oxygen-linked proton and anion binding sites does not impair allosteric regulation of oxygen binding in dwarf caiman hemoglobin. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2013;305:R300–R312. doi: 10.1152/ajpregu.00014.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Natarajan C, Projecto-Garcia J, Moriyama H, Weber RE, Munoz-Fuentes V, Green AJ, Kopuchian C, Tubaro PL, Alza L, Bulgarella M, et al. Convergent evolution of hemoglobin function in high-altitude Andean waterfowl involves limited parallelism at the molecular sequence level. PLoS Genetics. 2015;11:e1005681. doi: 10.1371/journal.pgen.1005681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shih DTB, Luisi BF, Miyazaki G, Perutz MF, Nagai K. A mutagenic study of the allosteric linkage of His(HC3)146β in hemoglobin. Journal of Molecular Biology. 1993;230:1291–1296. doi: 10.1006/jmbi.1993.1242. [DOI] [PubMed] [Google Scholar]
- 72.Ivaldi G, David O, Paradossi V, Baffico M, Degani VS, Leone D, Baldi M, Parodi MI, Bernardi P, Ricco G. Hb Bologna-St. Orsola beta 146(HC3)His→Tyr : a new high oxygen affinity variant with halved Bohr effect and highly reduced reactivity towards 2,3-diphosphoglycerate Hemoglobin. 1999;23:353–359. doi: 10.3109/03630269909090751. [DOI] [PubMed] [Google Scholar]
- 73.Naoi Y, Chong KT, Yoshimatsu K, Miyazaki G, Tame JRH, Park S-Y, Adachi S, Morimoto H. The functional similarity and structural diversity of human and cartilaginous fish hemoglobins. Journal of Molecular Biology. 2001;307:259–270. doi: 10.1006/jmbi.2000.4446. [DOI] [PubMed] [Google Scholar]
- 74.Ivankov DN, Finkelstein AV, Kondrashov FA. A structural perspective of compensatory evolution. Current Opinion in Structural Biology. 2014;26:104–112. doi: 10.1016/j.sbi.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]