

Abstract
Purpose
The purpose of this study is to summarize the effectiveness and safety of metreleptin in patients with congenital or acquired generalized lipodystrophy.
Methods
Patients (N=66) aged ≥6 months had lipodystrophy, low circulating leptin, and ≥1 metabolic abnormality (diabetes mellitus, insulin resistance, or hypertriglyceridemia). Metreleptin dose (once or twice daily) was titrated to a mean dose of 0.10 mg/kg/day with a maximum of 0.24 mg/kg/day. Means and changes from baseline to month 12 were assessed for glycated hemoglobin (HbA1c), fasting triglycerides (TGs), and fasting plasma glucose (FPG). Additional assessments included the proportions of patients achieving target decreases in HbA1c or fasting TGs at months 4, 12, and 36, medication changes, and estimates of liver size. Treatment-emergent adverse events (TEAEs) were recorded.
Results
Significant mean reductions from baseline were seen at month 12 for HbA1c (−2.2%, n=59) and FPG (−3.0 mmol/L, n=59) and mean percent change in fasting TGs (−32.1%, n=57) (all p≤0.001). Reductions from baseline over time in these parameters were also significant at month 36 (all p<0.001, n=14). At month 4, 34.8% of patients had a ≥1% reduction in HbA1c and 62.5% had a ≥30% reduction in fasting TGs; at month ≥12, 80% of patients had a 1% decrease in HbA1c or ≥30% decrease in TGs, and 66% had a decrease of ≥2% in HbA1c or ≥40% decrease in TGs. Of those on medications, 41%, 22%, and 24% discontinued insulin, oral antidiabetic medications, or lipid-lowering medications, respectively. Mean decrease in liver volume at month 12 was 33.8% (p<0.001, n=12). Most TEAEs were of mild/moderate severity.
Conclusions
In patients with generalized lipodystrophy, long-term treatment with metreleptin was well tolerated and resulted in sustained improvements in hypertriglyceridemia, glycemic control, and liver volume.
Keywords: Diabetes, Insulin resistance, Leptin, Lipodystrophy, Metreleptin
Introduction
Lipodystrophy syndromes are rare, clinically heterogeneous, inherited or acquired, and potentially life-threatening disorders [1]. The underlying pathogenesis of lipodystrophy is the irreversible widespread deficiency or dysfunction of adipose tissue. In some cases, this leads to low levels of circulating leptin, an adipokine that signals energy sufficiency [2, 3]. Lipodystrophy can be classified according to etiology and distribution of fat loss, leading to four main categories: congenital or acquired generalized lipodystrophy (GL) and familial or acquired partial lipodystrophy [1, 4]. Congenital GL has been ascribed to multiple genetic causes, whereas acquired GL may result from the autoimmune destruction of adipocytes and has been associated with several autoimmune disorders [1, 5, 6]. In some cases, genetic forms of lipodystrophy may only become clinically evident after infancy, and thus may be misdiagnosed as acquired GL (EAO and RB, unpublished observations). Regardless of the origin of lipodystrophy, the combination of adipose tissue loss and accompanying leptin deficiency (and its ensuing hyperphagia) is noted with ectopic fat deposition in the liver and muscle [7, 8]. This leads to insulin resistance, diabetes mellitus, and hypertriglyceridemia, which are often refractory to treatment with conventional lipid- and glucose-lowering agents, as these metabolic abnormalities are typically more severe than those associated with obesity and type 2 diabetes [8, 9].
To treat or prevent these severe complications of lipodystrophy requires aggressive treatment and management over the lifetime of the patient. Improvement in glycemic control is important to reduce the acute negative impact of hyperglycemia and the long-term impact of microvascular complications of diabetes. Reduction of triglyceride (TG) levels is also critical since elevated levels increase the risk of acute pancreatitis and cardiovascular disease [10]. As ectopic fat accumulation and insulin resistance are driven by the inability to store excess calories in adipocytes, one effective therapeutic approach is to reduce caloric intake; however, this is often nearly impossible in patients with GL who have very low leptin levels, causing hyperphagia.
Metreleptin, a recombinant methionyl human leptin, was approved by the Japanese Ministry of Health, Labour and Welfare in 2013 and by the US Food and Drug Administration in 2014 to treat the complications of leptin deficiency in patients with acquired or congenital GL as an adjunct to diet as replacement therapy [11, 12]. The regulatory approvals were based on data from a long-term trial conducted at the National Institutes of Health (NIH) (ClinicalTrials.gov identifier: NCT00025883), which generated several publications on interim data before the completion of the trial [13–20].
Here, we report the safety and effectiveness of metreleptin treatment in patients with GL from a final combined analysis of two open-label investigator-sponsored trials that represent the largest cohort of patients reported as one group.
Patients and methods
Study design and participants
Data collected from 2000–2014 for a pilot study (NIH Study 991265 [NCT00005905]) and its long-term extension (NIH Study 20010769 [NCT00025883]) were integrated into one final analysis. Although conducted as separate studies, these can be considered a single extended study because they employed similar protocols and almost all patients in the pilot study continued long-term treatment in the second study.
An overview of the study design, key inclusion criteria, and metreleptin dosing from both studies appears in Table 1. Briefly, metreleptin was dosed according to weight (sex- and age-dependent) in both studies; dose titrations were scheduled in the pilot study (NIH Study 991265) and were based on clinical response and clinician judgment in the long-term study (NIH Study 20010769). Full metreleptin dosing information from both studies and the protocol-defined discontinuations appear in the Online Resource.
Table 1.
Study designs of trials evaluating patients with generalized lipodystrophy
| NIH Study 991265 (NCT00005905) | NIH Study 20010769 (NCT00025883) | |
|---|---|---|
| Years conducted | 2000–2003 | 2001– 2014 |
|
| ||
| Study design | Open-label pilot study | Open-label long-term study |
|
| ||
| Key inclusion criteriaa |
|
|
|
| ||
| Metreleptin dosing |
|
|
After amendments to original study protocol.
ADA American Diabetes Association, LD lipodystrophy, NIH National Institutes of Health, TGs triglycerides.
The trial was conducted at the NIH in Bethesda, MD, with patients originating from the United States; the European Union, including Belgium, Germany, Italy, Lithuania, Spain, and the United Kingdom; the Eastern Mediterranean region, including Turkey, Albania, Israel, and Serbia; and other locations, including Argentina, Canada, India, Madagascar, Pakistan, Peru, and Saudi Arabia.
Vital signs and laboratory assessments were recorded at baseline and at scheduled postbaseline visits. The co-primary efficacy endpoints were actual change from baseline in glycated hemoglobin (HbA1c) and percent change from baseline in fasting TGs, both at month 12. Key secondary and additional endpoints included the proportion of patients achieving target decreases in HbA1c or fasting TGs from baseline at month 12 of 1) a ≥1% decrease in HbA1c or a ≥30% decrease in TGs, 2) a ≥1.5% decrease in HbA1c or a ≥35% decrease in TGs, or 3) a ≥2% decrease in HbA1c or a ≥40% decrease in TGs, as well as actual and percent changes from baseline in HbA1c, TGs, and fasting plasma glucose (FPG) over time. Early changes in individual target parameters were assessed at month 4 for HbA1c and TGs. While responder analyses were not conceived in the original study protocol, they were prespecified before the final data analysis. Changes in insulin and other antidiabetic and lipid-lowering medications were evaluated. Until 2006, patients aged ≥5 years underwent magnetic resonance imaging to estimate liver size. Serum leptin concentration was measured via a radioimmunoassay (lower limit of quantitation, 0.5 ng/mL) using a Linco Research/Millipore Assay kit (St. Charles, MO, USA), which recognizes both human leptin and metreleptin.
Assessed safety endpoints included metreleptin exposure parameters (duration and dose), incidence of treatment-emergent adverse events (TEAEs) and serious adverse events (SAEs), and changes from baseline in laboratory parameters. Adverse events (AEs) were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 19.0. Events possibly associated with hypersensitivity were evaluated and defined as standardized MedDRA queries (SMQs) of angioedema, anaphylactic reaction, anaphylactic/anaphylactoid shock conditions, and severe cutaneous reactions.
Statistical analyses
The analysis populations were defined as follows: the safety analysis set included all enrolled patients who received ≥1 dose of study drug; the full analysis set comprised all patients in the safety analysis who had either of the co-primary efficacy parameters of interest measured at baseline and ≥1 postbaseline visit; and the controlled concomitant medication full analysis set included all patients in the full analysis set who had controlled concomitant medication use, described as no change or a decrease in baseline concomitant medications (antidiabetic or lipid-lowering therapies) before month 12. For all analyses of the controlled concomitant medication full analysis set population, only patients with no increase in the relevant concomitant medication were included for each parameter (eg, only patients with no increase in antidiabetic medications in the first 12 months were analyzed for change in HbA1c).
Descriptive statistics were used to summarize demographics and baseline characteristics, metreleptin exposure, changes from baseline in efficacy endpoints, and the incidence of TEAEs. The co-primary efficacy analyses were performed using the full analysis set and reported as mean actual and percent changes. P values for changes from baseline to month 12 in efficacy endpoints were computed using paired t tests at a one-sided α-level of 0.025. The last observation carried forward (LOCF) method was used to impute missing month 12 values (in 18 patients) and only took into account results that were at least 6 months postbaseline. Thus, the analysis of primary efficacy endpoints included patients with baseline and at least day 180 measurements. The proportion achieving target decreases in HbA1c and TGs were evaluated by the number and percent of responders with two-sided exact binomial 95% confidence intervals. The original study protocol specified the sample size required to detect clinically meaningful changes with 80% power (α set at 0.05) as 10 patients to detect an actual decrease of 1.5% in HbA1c, and 12 patients to detect a 30% decrease in fasting TGs. As noted, the final sample size across the two protocols was 107 patients. As it became clear that treatment with metreleptin improved the metabolic abnormalities associated with lipodystrophy and could be safely administered to patients in the long term, the protocol was amended to expand the eligibility criteria and to increase the sample size.
A mixed-effects model for repeated measures (MMRM) analysis assessed changes over time by evaluating average levels of a parameter across all assessment time points.
Results
Patient disposition and demographics
A total of 66 patients with GL (congenital, n=45; acquired, n=21) were enrolled and treated across both studies, comprising the safety analysis population. (There were 62 patients in the full analysis and 54 in the concomitant medication full analysis populations.) Overall, 23 patients ended study participation during the 14-year duration of the study because of transfer to another metreleptin treatment program (n=8, 12.1%), noncompliance (n=5, 7.6%), death (n=3, 4.5%), ineligibility (n=2, 3.0%), other reasons (bipolar disorder, gastric bypass surgery) (n=2, 3.0%), an AE (n=1, 1.5%), lost to follow-up (n=1, 1.5%), and lack of efficacy (n=1, 1.5%).
Baseline demographics in the safety analysis population are presented in Table 2. Most patients (77.3%) were female, and the median age was 15.0 years. The majority of patients (74%) met the diagnostic criteria for diabetes with an HbA1c ≥6.5%, and 80.3% were taking antidiabetic medications. Congenital GL was present in 68.2% of patients. The baseline values for HbA1c, TGs, and FPG reflected the severity of the patients’ condition, with respective mean values of 8.6%, 14.5 mmol/L, and 10.3 mmol/L. Over the course of the study, the mean (standard deviation [SD]) overall duration of metreleptin exposure was 62.5 (45.7) months, with a mean (SD) daily dose of metreleptin of 5.0 (3.0) mg (range, 0.8–19.0 mg) and a weighted mean (SD) dose of 0.098 (0.04) mg/kg (range, 0.025–0.21 mg/kg).
Table 2.
Baseline characteristics in the safety analysis set
| Patients with GL (N=66) | |
|---|---|
| Sex, n (%) | |
| Female | 51 (77.3) |
| Male | 15 (22.7) |
| Race, n (%) | |
| Caucasian | 31 (47.0) |
| Black | 16 (24.2) |
| Asian | 3 (4.5) |
| Native American | 2 (3.0) |
| Hispanic | 11 (16.7) |
| Other | 3 (4.5) |
| Age (years), median (min–max) | 15.0 (1.0–68.0) |
| GL subtype, n (%)a | |
| Acquired | 21 (31.8) |
| Congenital/familial | 45 (68.2) |
| Medical history, n (%) | |
| Cardiac disorders | 15 (22.7) |
| Diabetes mellitus | 32 (48.5) |
| Hepatomegaly | 36 (54.5) |
| Hepatosplenomegaly | 7 (10.6) |
| Hypertriglyceridemia | 46 (69.7) |
| Hepatic steatosis | 16 (24.2) |
| Liver cirrhosis | 6 (9.1) |
| Nonalcoholic steatohepatitis | 23 (34.8) |
| Pancreatitisb | 18 (27.3) |
| Steatohepatitis | 11 (16.7) |
| Fasting leptin (ng/mL), mean (SD) | 1.3 (1.0) |
| HbA1c (%), mean (SD) | 8.6 (2.3) |
| Fasting TGs (mmol/L), mean (SD) | 14.5 (25.3) |
| FPG (mmol/L), mean (SD) | 10.3 (5.04) |
| Liver volume (mL), mean (SD) | 3357.7 (1121.7) |
| ALT (U/L), mean (SD) | 112.5 (111.1) |
| ALT above ULN, n (%) | 49 (74.2) |
| AST (U/L), mean (SD) | 75.3 (70.0) |
| AST above ULN, n (%) | 36 (54.5) |
| On antidiabetic medications, n (%) | 53 (80.3) |
| Any insulin | 39 (59.1) |
| Insulin alone | 19 (28.8) |
| Insulin plus oral agent | 20 (30.3) |
| Oral agent only | 12 (18.2) |
| On lipid-lowering medications, n (%) | 34 (51.5) |
| Fibrates | 25 (37.9) |
| Statins | 11 (16.7) |
| Other lipid-lowering agents | 10 (15.2) |
Three patients originally classified as acquired at study entry were subsequently found to have a genetic mutation.
Includes relapsing pancreatitis and acute pancreatitis.
ALT alanine aminotransferase, AST aspartate aminotransferase, FPG fasting plasma glucose, GL generalized lipodystrophy, HbA1c glycated hemoglobin, SD standard deviation, TGs triglycerides, ULN upper limit of normal.
Efficacy
Treatment with metreleptin resulted in substantial improvements from baseline in glycemic control and hypertriglyceridemia over 12 months, with significant reductions from baseline in mean HbA1c (from 8.6% to 6.4%; −2.2%, p<0.001) and fasting TG concentrations (from 14.7 to 4.5 mmol/L; mean patient level change −32.1%, p=0.001) in the full analysis set population (Fig. 1a, c). Similar and significant decreases were observed in the controlled concomitant medication full analysis set population (from 8.5% to 6.6% [p<0.001] for HbA1c; from 8.6 to 3.2 mmol/L [p=0.026] for TGs). Treatment with metreleptin also led to clinically meaningful and statistically significant reductions in FPG levels in both the full analysis (from 10.2 to 7.0 mmol/L; p<0.001) and controlled concomitant medication full analysis (from 9.9 to 7.4 mmol/L; p=0.001) set populations (Fig. 1b). Notably, female patients in the full analysis set exhibited a greater significant reduction from baseline (−3.4; p<0.001) in mean FPG levels than did males (−1.6; p=0.191). This may be due to their higher baseline levels (10.5 mmol/L in females vs 9.0 mmol/L in males) since levels at month 12 were similar (7.0 and 7.1 mmol/L, respectively). This difference between the sexes was not observed in the controlled concomitant medication full analysis set population. When classified by lipodystrophy subtype, mean baseline HbA1c was 9.2% and 8.3% and baseline TGs were 22.9 and 10.9 mmol/L in patients with acquired and congenital GL, respectively. In the full analysis set, reductions in mean HbA1c and fasting TGs, respectively, from baseline to month 12/LOCF were numerically greater in patients with acquired GL (−2.9% and −53.5%) than in patients with congenital GL (−1.8% and −22.2%).
Fig. 1.

Mean (standard deviation) levels of a) HbA1c, b) FPG, c) fasting TGs, and d) liver volume at baseline and month 12/LOCF. a) Mean values for HbA1c 8.6% at baseline and 6.4% at month 12 in the FAS and 8.5% at baseline and 6.6% at month 12 in the CFAS. b) Mean values for FPG were 10.2 mmol/L at baseline and 7.0 mmol/L at month 12 in the FAS and 9.9 mmol/L at baseline and 7.4 mmol/L at month 12 in the CFAS. c) Mean (median) values for fasting TGs were 14.7 mmol/L (4.6 mmol/L) at baseline and 4.5 mmol/L (2.3 mmol/L) at month 12 in the FAS and 8.6 mmol/L (3.8 mmol/L) at baseline and 3.2 mmol/L (2.1 mmol/L) at month 12 in the CFAS. Percent change reflects mean of individual differences. d) Mean (median) liver volume was 3357.7 mL (3318.0 mL) at baseline and 2193.0 mL (2110.0 mL) at month 12. *p≤0.001; **p=0.026. an=30 in the CFAS group includes only patients whose antidiabetes medications were unchanged or decreased from baseline. Δ, mean change from baseline; BL, baseline; CFAS, controlled concomitant medication full analysis set; FAS, full analysis set; FPG, fasting plasma glucose; HbA1c, glycated hemoglobin; LOCF, last observation carried forward; TGs, triglycerides
Significant mean changes from baseline in HbA1c, FPG, and TG levels are reported for months 6, 12, 24, 36, and 48 with no loss of efficacy over time (p<0.001 for all parameters) (Fig. 2).
Fig. 2.

Mean (standard error) change from baseline in a) HbA1c, b) FPG, and c) fasting TGs over time in the full analysis set. Least-squares mean, standard error, and p values for the change from baseline parameters are computed using a mixed-effects model for repeated measures. p<0.001 for the change from baseline for all time points. FPG, fasting plasma glucose; HbA1c, glycated hemoglobin; TGs, triglycerides
Long-term treatment with metreleptin led to clinically meaningful and statistically significant reductions from baseline in metabolic parameters. Although the number of patients in the study decreased over time, the MMRM analyses showed significant least-squares mean actual decreases from baseline in HbA1c (−1.4%) and FPG (−2.1 mmol/L) and percent change from baseline in fasting TGs (−22.4%) over all patients and analysis visits (p<0.001 for all).
At month 4, 62.5% of patients in the full analysis set population achieved a ≥30% reduction in fasting TGs and 34.5% of patients had a ≥1% reduction in HbA1c (Fig. 3a). High proportions of patients achieved target reductions in HbA1c or TGs at month 12/LOCF, with 79.7% of patients in the full analysis set achieving the target of a ≥1% decrease in HbA1c or a ≥30% decrease in TGs, and 66.1% achieving the highest target decreases of ≥2% in HbA1c or a ≥40% decrease in TGs. These trends were consistent in the month12/LOCF data for the controlled concomitant medication full analysis set (Fig. 3b). High proportions of patients with abnormal baseline levels of HbA1c (≥7%) or TGs (≥2.26 or ≥5.65 mmol/L) also met their target reductions (Fig. 3c). The composite responder analysis revealed that 55% of patients achieved both an actual decrease in HbA1c of ≥1% and a ≥30% reduction in TGs at month 12/LOCF, with more than one-third of patients achieving the highest target reductions of both a 2% actual decrease in HbA1c and a ≥40% reduction in TGs (Fig. 4). Consistent with the trend for increased efficacy with worse baseline metabolic characteristics as evidenced in the primary efficacy outcome (ie, change from baseline to month 12 in HbA1c and TGs), higher proportions of responders were observed for patients with higher baseline HbA1c or TG levels (Table 3).
Fig. 3.
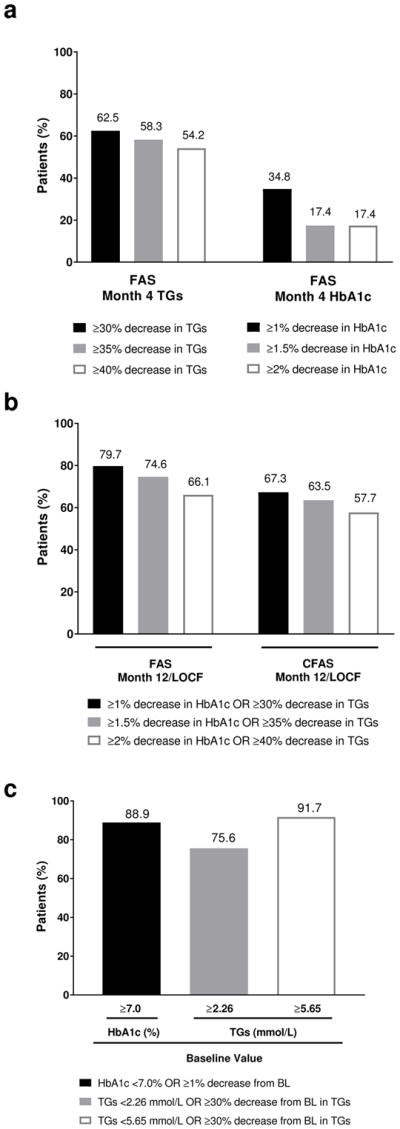
Proportion achieving a) target actual decreases in HbA1c and percent decreases in fasting TGs in the FAS at month 4, b) HbA1c or percent decreases in fasting TGs at month 12/LOCF in the FAS and the CFAS, and c) by abnormal BL levels of HbA1c or fasting TGs at month 12/LOCF. BL, baseline; CFAS, controlled concomitant medication full analysis set; FAS, full analysis set; HbA1c, glycated hemoglobin; LOCF, last observation carried forward; TGs, triglycerides
Fig. 4.

Proportion achieving target decreases in HbA1c and fasting TGs in the FAS at month 12/LOCF. FAS, full analysis set; HbA1c, glycated hemoglobin; LOCF, last observation carried forward; TGs, triglycerides
Table 3.
Baseline characteristics by responder status in the full analysis set
| Respondera (n=47) | Nonresponder (n=12) | |
|---|---|---|
| GL subtype, n (%) | ||
| Acquired | 18 (38.3) | 1 (8.3) |
| Congenital | 29 (61.7) | 11 (91.7) |
| Fasting leptin (ng/mL), mean (SD) | 1.5 (1.06) | 1.0 (0.52) |
| HbA1c (%), mean (SD) | 9.2 (2.02) | 6.2 (1.98) |
| HbA1c category, n (%) | ||
| <6.5% | 5 (10.6) | 9 (75.0) |
| ≥6.5% | 42 (89.4) | 3 (25.0) |
| ≥ 8.0% | 37 (78.7) | 2 (16.7) |
| Fasting TGs (mmol/L), mean (SD) | 17.9 (28.57) | 4.8 (8.62) |
| Fasting TG category, n (%) | ||
| <2.26 mmol/L | 7 (14.9) | 6 (50.0) |
| ≥2.26 mmol/L | 39 (83.0) | 6 (50.0) |
| ≥5.65 mmol/L | 23 (48.9) | 1 (8.3) |
| FPG (mmol/L), mean (SD) | 11.0 (4.84) | 6.1 (2.54) |
| ALT (U/L), mean (SD) | 104.7 (75.26) | 147.8 (211.11) |
| ALT above ULN, n (%) | 37 (78.7) | 7 (58.3) |
| AST (U/L), mean (SD) | 74.9 (58.66) | 83.9 (114.73) |
| AST above ULN, n (%) | 30 (63.8) | 3 (25.0) |
| Insulin use, n (%) | 31 (66.0) | 3 (25.0) |
| Antidiabetic medications, n (%) | 40 (85.1) | 7 (58.3) |
| Lipid-lowering medications, n (%) | 24 (51.1) | 5 (41.7) |
Responders are those patients with a ≥1% actual decrease in HbA1c or ≥30% decrease in fasting TGs at month 12 (or last observation carried forward).
ALT alanine aminotransferase, AST aspartate aminotransferase, FPG fasting plasma glucose, GL generalized lipodystrophy, HbA1c glycated hemoglobin, TGs triglycerides, ULN upper limit of normal.
Among the 39 patients receiving insulin at baseline, 91.2% achieved a ≥1% decrease in HbA1c or ≥30% decrease in TGs by month 12/LOCF; of these patients, 16 (41.0%) were able to discontinue insulin during the study and 11 did so within the first year of metreleptin use. Among patients taking oral antidiabetic and lipid-lowering medications at baseline, 7 of 32 (21.9%) and 8 of 34 (23.5%), respectively, were able to discontinue use of those drugs with metreleptin treatment.
Most patients had elevated hepatic transaminase levels at baseline (Table 2). Reduction in alanine aminotransferase levels occurred in the full analysis set population during treatment with metreleptin at month 12, with a mean (SD) change of −53.1 (126.6) U/L (p=0.010) from a baseline of 111.9 (112.6) U/L, but no change was observed in aspartate aminotransferase levels (−23.8 [142.4] U/L [p=0.291] from a baseline of 75.0 [71.1] U/L). Mean liver volume decreased by 33.8% at month 12 (p<0.001) (Fig. 1d).
Safety
Overall, 59 of 66 patients (89.4%) experienced ≥1 TEAE, most of which were mild to moderate in severity, and 32 patients (48.5%) experienced a drug-related TEAE (Table 4). Nearly 35% of patients had an SAE, with 4.5% experiencing a drug-related SAE. The overall incidence of TEAEs and the incidence of drug-related TEAEs were similar among patients with acquired GL and those with congenital GL. Patients with acquired GL were more likely to report events of severe intensity (67%) and SAEs (52%) compared to patients with congenital GL (33% and 27%, respectively). Over the duration of this 14-year study, three deaths (none assessed as drug-related) were reported (renal failure [n=1], cardiac arrest due to pancreatitis and shock [n=1], and progressive end-stage liver disease [n=1]). In addition to the three deaths, two events led to discontinuation of study treatment (peripheral T-cell lymphoma [n=1] and increased blood TGs and inadequate control of diabetes [n=1]).
Table 4.
Summary of adverse events in the safety analysis set
| Patients with GL (N=66)
|
||
|---|---|---|
| Adverse event, n (%) | Acquired (n=21) | Congenital (n=45) |
| ≥1 TEAE | 21 (100.0) | 38 (84.4) |
| Drug-related TEAE | 11 (52.4) | 21 (46.7) |
| Severe TEAE | 14 (66.7) | 15 (33.3) |
| Drug-related severe TEAE | 2 (9.5) | 5 (11.1) |
| Treatment-emergent SAE | 11 (52.4) | 12 (26.7) |
| Drug-related treatment-emergent SAE | 1 (4.8) | 2 (4.4) |
| TEAE leading to study drug discontinuation | 2 (9.5) | 3 (6.7) |
| Pancreatitis and shock, subsequent cardiac arresta | 0 (0.0) | 1 (2.2) |
| Peripheral T-cell lymphoma | 1 (4.8) | 0 (0.0) |
| Progressive end-stage liver disease | 1 (4.8) | 0 (0.0) |
| Renal failure | 1 (2.2) | |
| Worsening of TGs (increased)/worsening glycemic control | 0 (0.0) | 1 (2.2) |
| On-study deaths | 1 (4.8) | 2 (4.4) |
| Pancreatitis and shock, subsequent cardiac arresta | 0 (0.0) | 1 (2.2) |
| Progressive end-stage liver disease | 1 (4.8) | 0 (0.0) |
| Renal failure | 0 (0.0) | 1 (2.2) |
| TEAE by preferred term (≥10% incidence)b | ||
| Decreased weight | 7 (33.3) | 10 (22.2) |
| Abdominal pain | 4 (19.0) | 7 (15.6) |
| Hypoglycemia | 4 (19.0) | 6 (13.3) |
| Decreased appetite | 2 (9.5) | 6 (13.3) |
| Headache | 3 (14.3) | 5 (11.1) |
Patient was hospitalized with a diagnosis of pancreatitis, did not receive metreleptin during hospitalization, and died after cardiac arrest on day 3 of hospitalization.
Medical Dictionary for Regulatory Activities, version 19.0.
GL generalized lipodystrophy, SAE serious adverse event, TEAE treatment-emergent adverse event, TGs triglycerides.
The most common TEAEs were consistent with the expected pharmacological effects of metreleptin, including weight decrease, hypoglycemia, and decreased appetite. Mean (SD) and median changes in weight were −2.8 (3.24) kg and −2.0 kg, respectively, from baseline to month 6 (n=26 patients) and −2.7 (5.21) kg and −2.2 kg, respectively, from baseline to month 12 (n=39 patients). Of the 10 patients who experienced hypoglycemia, eight events were judged to be treatment-related. All hypoglycemia events were mild to moderate in severity, and none of the events led to treatment discontinuation. Patients who experienced hypoglycemic events were on insulin therapy with or without oral agents.
Overall, 18 of 66 patients (27.3%) experienced TEAEs, potentially associated with a potential hypersensitivity event (evaluated based on the broad SMQs). Events in this category were of moderate intensity, nonserious, and did not lead to treatment withdrawal. Within this broad SMQ category, the most commonly reported events were cough (n=4, 6%), edema, pruritus, and dyspnea (n=3 each, 5%), and asthma (n=2, 3%). Injection-site reactions, including erythema and urticaria, were reported in four patients (6.1%).
Three patients with events in this category that were reported as SAEs included one patient with severe respiratory distress associated with chest pain and dyspepsia and another patient with a history of asthma with severe chest discomfort, flushing, and dyspnea with related panic attack; in both of these patients, metreleptin dosing was resumed without recurrence of the adverse reaction. A third patient experienced cardiac arrest as a likely consequence of pancreatitis and septic shock, deemed unrelated to study treatment. None of the other events possibly associated with hypersensitivity led to death or treatment withdrawal.
Ten patients (15.2%) developed a neoplasm during the study: two cases of benign breast neoplasm; two cases of peripheral T-cell lymphoma; and one case each of anaplastic large-cell lymphoma, basal cell carcinoma, breast cancer (in one of the patients with peripheral T-cell lymphoma), intraductal proliferative breast lesion, uterine leiomyoma, ovarian neoplasm, papillary thyroid cancer, and granular cell tumor. Only the case of anaplastic large-cell lymphoma, which developed after approximately 1 year and 10 months on metreleptin treatment, was assessed as treatment-related.
Ten patients (15.2%) experienced a hepatic disorder, the most common of which were autoimmune hepatitis (n=3) and liver disorder (n=2; worsening of preexisting nonalcoholic steatohepatitis). These events were reported as SAEs in five patients (three hospitalizations and two important medical events that may have jeopardized the patient), and all were considered to be unrelated to study treatment. Eight of the 10 patients who experienced a hepatic disorder during the study had a past medical history of liver disease (hepatomegaly or hepatosplenomegaly [n=6], nonalcoholic steatohepatitis or steatohepatitis [n=4], autoimmune hepatitis [n=3], hepatic cirrhosis [n=3], hepatic fibrosis [n=2], hepatitis [n=2], and steatosis [n=2]).
Seven patients (10.6%) experienced possible autoimmune disorders, which were nonserious and assessed as unrelated to study drug. Of these, three were autoimmune hepatitis, two membranoproliferative glomerulonephritis, two focal segmental glomerulosclerosis, and one autoimmune thyroiditis (in a patient who also had focal segmental glomerulosclerosis).
Serious infections occurred in seven patients (10.6%), and none were assessed as related to study treatment.
Three patients (4.5%) experienced pancreatitis or relapsing pancreatitis, all of which were judged to be severe in intensity and unrelated to study drug. All three of these patients had a history of pancreatitis and hypertriglyceridemia.
Discussion
This combined analysis of two open-label trials examined the largest published cohort of patients with GL over an extended time period (mean overall duration on metreleptin of 5 years). Metreleptin administration and dose adjustments have been previously described [8]. Here, clinically meaningful and significant improvements were observed from baseline to month 12 for HbA1c, fasting TGs, and FPG. High percentages of patients achieved target decreases in fasting TGs as early as month 4, with even greater proportions achieving target reductions in HbA1c or fasting TGs at month 12. Metreleptin likely shows beneficial effects even earlier than 4 months, since a Japanese study of seven patients treated with metreleptin showed improvements in FPG and TGs within 1 week and a French study of five patients found significant decreases in HbA1c and TGs after 1 month of metreleptin treatment, which were maintained at 12 months [21, 22].
Many patients lacked 36-month data because they did not have a laboratory value within the 36-month window, were enrolled in the final 3 years of the study, or transferred to another site before 36 months of metreleptin treatment. However, meaningful mean changes from baseline in HbA1c, fasting TGs, and FPG were sustained at month 36 in this trial in 17 patients who had assessments at month 36. Moreover, the MMRM analysis showed significant decreases in HbA1c, fasting TGs, and FPG over all patients and visits. Other trials of metreleptin with small numbers of patients with GL (n≤8) have also shown consistent long-term reductions in HbA1c, TGs, and/or FPG, indicating that, at least for some patients, metreleptin shows continued effectiveness [21, 23, 24].
Another benefit of treatment with metreleptin is that many patients were able to discontinue their use of insulin, antidiabetic medications, or lipid-lowering medications. The similarity of the efficacy results between the full analysis and controlled concomitant medication full analysis set populations indicated that the results were minimally influenced by any increases in, or additions to, background medication, but rather were due to treatment with metreleptin.
Ectopic fat deposition is an important characteristic of lipodystrophy, and significant reductions in alanine aminotransferase levels and liver volume in the study are consistent with an effect of metreleptin treatment to reduce hepatic steatosis, as reported in previous studies [21, 25].
Over the 14-year study duration, treatment with metreleptin was generally well tolerated and most TEAEs were mild or moderate in severity. The most common TEAEs were consistent with the expected mechanism of action of metreleptin. Weight loss occurred by 6 months of therapy and was stable thereafter. Although 26% of patients experienced AEs that were coded as potential hypersensitivity reactions per broad MedDRA terminology, almost none of these were assessed as true drug hypersensitivity. The three deaths that occurred during the study were consistent with the underlying morbidity of lipodystrophy, and none were considered to be related to treatment.
The US label for metreleptin carries a boxed warning that T-cell lymphoma has been reported in patients with acquired GL, in those both treated and untreated with metreleptin. Three patients developed lymphoma during this trial. An analysis of these three metreleptin-treated patients, as well as four patients with acquired GL who developed lymphoma without metreleptin treatment, indicated that treatment with metreleptin did not appear to directly cause the development of lymphoma [26]. Rather, patients with acquired GL appear to be at a higher risk for lymphoma than the general population, likely due to underlying autoimmunity. Overall, there was a relatively high incidence of diagnosis of new malignant and nonmalignant neoplasms. Greater reporting of neoplasms in this young population is likely due to the careful surveillance of the population in a clinical trial and may potentially reflect the presence of insulin resistance, which is a risk factor for development of multiple neoplastic conditions [27–29].
Limitations of this work include the relatively small sample size and open-label design. However, GL is a rare condition, and this study represents the largest cohort in the world, with a wide range of regions of origin and races/ethnicities. Furthermore, after the pilot study demonstrated improvements with metreleptin treatment, and given the risk of this patient population for life-threatening metabolic complications, it was considered unethical to assign patients to a placebo group in the long-term study.
A strength of this study is that it allowed for an assessment of TEAEs over a long period of time. Metreleptin was well tolerated, which is important for a chronic disease such as GL that requires long-term treatment. The long-term tolerability of metreleptin will be further monitored in a safety registry (NCT02325674).
In summary, this trial showed that HbA1c, fasting TGs, and FPG levels decreased, along with liver volume and enzymes, during treatment with metreleptin for GL. Results from this trial further support the recommendation of metreleptin as first-line treatment (with diet) for patients with GL, including children [1].
Supplementary Material
Acknowledgments
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript and take responsibility for the integrity of the work as a whole. Aegerion Pharmaceuticals provided funding for medical writing and/or editing support in the development of this manuscript; Jennifer L. Giel, PhD, of inScience Communications, Springer Healthcare (Philadelphia, PA, USA), based on input from authors, wrote the first draft and revised subsequent drafts of the manuscript, and Adrienne M. Schreiber of inScience Communications, Springer Healthcare (Philadelphia, PA, USA) copyedited and styled the manuscript per journal requirements. Aegerion Pharmaceuticals reviewed and provided feedback to the authors. The authors had full editorial control of the manuscript and provided their final approval of all content. Jean-Karl Sirois provided statistical analyses on behalf of Veristat LLC, Montreal, Canada, which was funded by Aegerion Pharmaceuticals, Inc. The authors acknowledge the services of the Clinical Core Laboratory of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health for measurement of leptin levels.
Funding This work was supported by the intramural research program of the National Institute of Diabetes and Digestive and Kidney Diseases. EAO is supported by NIH grant RO-1 DK-088114 as well as UM Lipodystrophy Fund donated by the Sopha Family and White Point Foundation of Turkey.
Footnotes
Compliance with ethical standards
Conflict of interest
R.J.B., E.C., and P.G. report no conflicts of interest and have nothing to disclose. E.A.O. has served as a consultant to Aegerion, Akcea, AstraZeneca, and Regeneron, received grants from Aegerion, Akcea, AstraZeneca, Gemphire, GIDynamics, and received nonmaterial support from Aegerion and Boehringer Ingelheim. D.A.V. and D.B.S. have served as consultants to Aegerion Pharmaceuticals. A.L. and G.F. are employees of Aegerion Pharmaceuticals. T.S. is a former employee of Aegerion Pharmaceuticals.
Ethical approval For this type of study formal consent is not required.
References
- 1.Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, Mungai L, Oral EA, Patni N, Rother KI, von Schnurbein J, Sorkina E, Stanley T, Vigouroux C, Wabitsch M, Williams R, Yorifuji T. The diagnosis and management of lipodystrophy syndromes: a multi-society practice guideline. J Clin Endocrinol Metab. 2016;101:4500–4511. doi: 10.1210/jc.2016-2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haque WA, Shimomura I, Matsuzawa Y, Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab. 2002;87:2395. doi: 10.1210/jcem.87.5.8624. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 4.Handelsman Y, Oral EA, Bloomgarden ZT, Brown RJ, Chan JL, Einhorn D, Garber AJ, Garg A, Garvey WT, Grunberger G, Henry RR, Lavin N, Tapiador CD, Weyer C. The clinical approach to the detection of lipodystrophy–an AACE consensus statement. Endocr Pract. 2013;19:107–116. doi: 10.4158/endp.19.1.v767575m65p5mr06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bingham A, Mamyrova G, Rother KI, Oral E, Cochran E, Premkumar A, Kleiner D, James-Newton L, Targoff IN, Pandey JP, Carrick DM, Sebring N, O'Hanlon TP, Ruiz-Hidalgo M, Turner M, Gordon LB, Laborda J, Bauer SR, Blackshear PJ, Imundo L, Miller FW, Rider LG. Predictors of acquired lipodystrophy in juvenile-onset dermatomyositis and a gradient of severity. Medicine (Baltimore) 2008;87:70–86. doi: 10.1097/MD.0b013e31816bc604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oral EA. Lipoatrophic diabetes and other related syndromes. Rev Endocr Metab Disord. 2003;4:61–77. doi: 10.1023/a:1021827520301. [DOI] [PubMed] [Google Scholar]
- 7.Garg A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350:1220–1234. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- 8.Meehan CA, Cochran E, Kassai A, Brown RJ, Gorden P. Metreleptin for injection to treat the complications of leptin deficiency in patients with congenital or acquired generalized lipodystrophy. Expert Rev Clin Pharmacol. 2016;9:59–68. doi: 10.1586/17512433.2016.1096772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patni N, Garg A. Congenital generalized lipodystrophies—new insights into metabolic dysfunction. Nat Rev Endocrinol. 2015;11:522–534. doi: 10.1038/nrendo.2015.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN, Goldberg AC, Howard WJ, Jacobson MS, Kris-Etherton PM, Lennie TA, Levi M, Mazzone T, Pennathur S. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123:2292–2333. doi: 10.1161/CIR.0b013e3182160726. [DOI] [PubMed] [Google Scholar]
- 11.Chou K, Perry CM. Metreleptin: first global approval. Drugs. 2013;73:989–997. doi: 10.1007/s40265-013-0074-7. [DOI] [PubMed] [Google Scholar]
- 12.Myalept [package insert] Cambridge, MA: Aegerion Pharmaceuticals, Inc; 2015. [Google Scholar]
- 13.Chan JL, Lutz K, Cochran E, Huang W, Peters Y, Weyer C, Gorden P. Clinical effects of long-term metreleptin treatment in patients with lipodystrophy. Endocr Pract. 2011;17:922–932. doi: 10.4158/EP11229.OR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chong AY, Lupsa BC, Cochran EK, Gorden P. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia. 2010;53:27–35. doi: 10.1007/s00125-009-1502-9. [DOI] [PubMed] [Google Scholar]
- 15.Christensen JD, Lungu AO, Cochran E, Collins MT, Gafni RI, Reynolds JC, Rother KI, Gorden P, Brown RJ. Bone mineral content in patients with congenital generalized lipodystrophy is unaffected by metreleptin replacement therapy. J Clin Endocrinol Metab. 2014;99:E1493–E1500. doi: 10.1210/jc.2014-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diker-Cohen T, Cochran E, Gorden P, Brown RJ. Partial and generalized lipodystrophy: comparison of baseline characteristics and response to metreleptin. J Clin Endocrinol Metab. 2015;100:1802–1810. doi: 10.1210/jc.2014-4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joseph J, Shamburek RD, Cochran EK, Gorden P, Brown RJ. Lipid regulation in lipodystrophy versus the obesity-associated metabolic syndrome: the dissociation of HDL-C and triglycerides. J Clin Endocrinol Metab. 2014;99:E1676–E1680. doi: 10.1210/jc.2014-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamran F, Rother KI, Cochran E, Safar Zadeh E, Gorden P, Brown RJ. Consequences of stopping and restarting leptin in an adolescent with lipodystrophy. Horm Res Paediatr. 2012;78:320–325. doi: 10.1159/000341398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muniyappa R, Abel BS, Asthana A, Walter MF, Cochran EK, Remaley AT, Skarulis MC, Gorden P, Brown RJ. Metreleptin therapy lowers plasma angiopoietin-like protein 3 in patients with generalized lipodystrophy. J Clin Lipidol. 2017;11:543–550. doi: 10.1016/j.jacl.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, DePaoli AM, Reitman ML, Taylor SI, Gorden P, Garg A. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346:570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 21.Ebihara K, Kusakabe T, Hirata M, Masuzaki H, Miyanaga F, Kobayashi N, Tanaka T, Chusho H, Miyazawa T, Hayashi T, Hosoda K, Ogawa Y, DePaoli AM, Fukushima M, Nakao K. Efficacy and safety of leptin-replacement therapy and possible mechanisms of leptin actions in patients with generalized lipodystrophy. J Clin Endocrinol Metab. 2007;92:532–541. doi: 10.1210/jc.2006-1546. [DOI] [PubMed] [Google Scholar]
- 22.Vatier C, Fetita S, Boudou P, Tchankou C, Deville L, Riveline J, Young J, Mathivon L, Travert F, Morin D, Cahen J, Lascols O, Andreelli F, Reznik Y, Mongeois E, Madelaine I, Vantyghem M, Gautier J, Vigouroux C. One-year metreleptin improves insulin secretion in patients with diabetes linked to genetic lipodystrophic syndromes. Diabetes Obes Metab. 2016;18:693–697. doi: 10.1111/dom.12606. [DOI] [PubMed] [Google Scholar]
- 23.Araujo-Vilar D, Sanchez-Iglesias S, Guillin-Amarelle C, Castro A, Lage M, Pazos M, Rial JM, Blasco J, Guillen-Navarro E, Domingo-Jimenez R, del Campo MR, Gonzalez-Mendez B, Casanueva FF. Recombinant human leptin treatment in genetic lipodystrophic syndromes: the long-term Spanish experience. Endocrine. 2015;49:139–147. doi: 10.1007/s12020-014-0450-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Musso C, Major ML, Andres E, Simha V. Metreleptin treatment in three patients with generalized lipodystrophy. Clin Med Insights Case Rep. 2016;9:123–127. doi: 10.4137/CCRep.S40196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beltrand J, Beregszaszi M, Chevenne D, Sebag G, De Kerdanet M, Huet F, Polak M, Tubiana-Rufi N, Lacombe D, De Paoli AM, Levy-Marchal C. Metabolic correction induced by leptin replacement treatment in young children with Berardinelli-Seip congenital lipoatrophy. Pediatrics. 2007;120:e291–e296. doi: 10.1542/peds.2006-3165. [DOI] [PubMed] [Google Scholar]
- 26.Brown RJ, Chan JL, Jaffe ES, Cochran E, DePaoli AM, Gautier JF, Goujard C, Vigouroux C, Gorden P. Lymphoma in acquired generalized lipodystrophy. Leuk Lymphoma. 2016;57:45–50. doi: 10.3109/10428194.2015.1040015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hernandez AV, Pasupuleti V, Benites-Zapata VA, Thota P, Deshpande A, Perez-Lopez FR. Insulin resistance and endometrial cancer risk: a systematic review and meta-analysis. Eur J Cancer. 2015;51:2747–2758. doi: 10.1016/j.ejca.2015.08.031. [DOI] [PubMed] [Google Scholar]
- 28.Arcidiacono B, Iiritano S, Nocera A, Possidente K, Nevolo MT, Ventura V, Foti D, Chiefari E, Brunetti A. Insulin resistance and cancer risk: an overview of the pathogenetic mechanisms. Exp Diabetes Res. 2012;2012:789174. doi: 10.1155/2012/789174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Haring MF, Rathjen T, Lockhart SM, Sorensen D, Ussar S, Rasmussen LM, Bertagnolli MM, Kahn CR, Rask-Madsen C. Insulin resistance in vascular endothelial cells promotes intestinal tumour formation. Oncogene. 2017;36:4987–4996. doi: 10.1038/onc.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.