

Abstract
Polycystic kidney disease (PKD) is a life-threatening disorder, commonly caused by defects in polycystin-1 (PC1) or polycystin-2 (PC2), in which tubular epithelia form fluid-filled cysts 1, 2. A major barrier to understanding PKD is the absence of human cellular models that accurately and efficiently recapitulate cystogenesis 3, 4. Previously, we have generated a genetic model of PKD using human pluripotent stem cells and derived kidney organoids 5, 6. Here we show that systematic substitution of physical components can dramatically increase or decrease cyst formation, unveiling a critical role for microenvironment in PKD. Removal of adherent cues increases cystogenesis 10-fold, producing cysts phenotypically resembling PKD that expand massively to 1-centimeter diameters. Removal of stroma enables outgrowth of PKD cell lines, which exhibit defects in PC1 expression and collagen compaction. Cyclic AMP, when added, induces cysts in both PKD organoids and controls. These biomaterials establish a highly efficient model of PKD cystogenesis that directly implicates the microenvironment at the earliest stages of the disease.
PKD affects one in ∼ 1000 people worldwide, with no known cure 1, 2. Animal models do not fully genocopy or phenocopy human PKD, and are too complex physiologically for a minimal reconstitution approach7, 8, 9, 10. A human cellular model is needed to complement animal models and reveal the early pathophysiology of PKD. We have generated human pluripotent stem cells (hPSCs) with targeted, biallelic mutations that lack the mature form of PC1, or any detectable PC2, using the Cas9/CRISPR (clustered regularly interspaced short palindromic repeats) gene editing system (Supplementary Fig. 1a-b) 6, 11. Under adherent culture conditions, kidney organoids derived from PKD1-/- or PKD2-/- hPSCs form fluid-filled cysts, although cystogenesis is highly inefficient (∼ 7 % of organoids), and its mechanism has not been determined6. We used this system as a starting point to investigate how cysts form and identify modulators of cystogenesis. Time-lapse imaging revealed that cyst formation involved two steps: first, rearrangement of a tubule within an organoid, resulting in deformation of linear shape to form a ‘pre-cyst’ surrounding an hollow pocket; and second, partial detachment of the pre-cyst from the underlying stratum followed immediately by rapid expansion, resulting in a buoyant cyst tethered to an adherent organoid remnant (Fig. 1a, Supplementary Fig. 1c-d, and Movie 1). Cysts therefore arose from whole tubular segments that expanded and partially detached from the adherent surface.
Fig. 1. Removal of adherent cues establishes a highly efficient model of PKD cystogenesis.
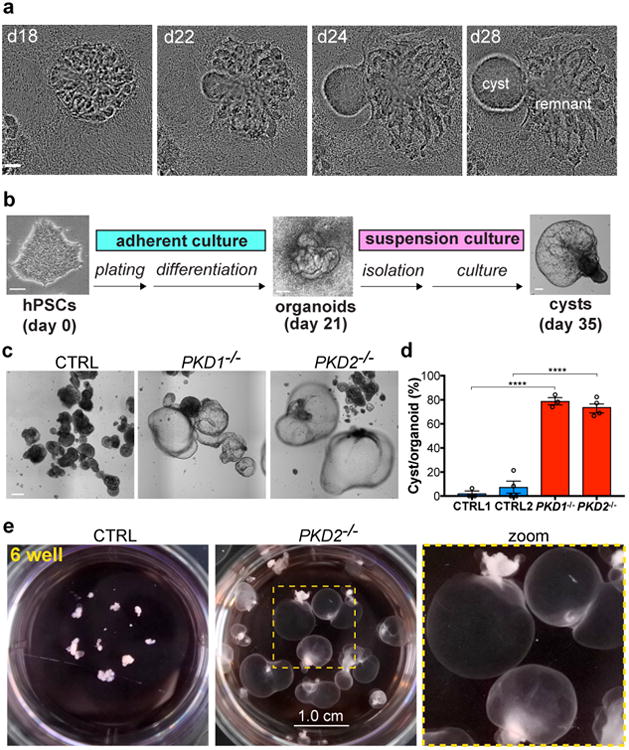
a, Still images from Movie 1 showing cyst formation from a PKD organoid in adherent culture. b, Schematic of high-efficiency organoid cystogenesis protocol. c, Representative images of kidney organoids and d, quantification of cyst formation after two weeks of suspension culture (CTRL1 vs. PKD1-/-, n=3 separate experiments, ± s.e.m., t(3.663)=21.05, p=5.8949×10-5; CTRL2 vs. PKD2-/-, n=4 separate experiments, ± s.e.m., t(5.458)=10.66, p=7.3731×10-5). e, 6-well (3.5 cm) dishes containing PKD or control organoids after 9 months of culture. Scale bars, 100 μm (a-c) and 1 cm (e).
Based on these results, we hypothesized that adherent forces played a critical role in limiting tubular deformation and subsequent cyst formation. To test this, organoids were purified on day 21 of differentiation, prior to the formation of cysts, and transferred to wells coated with an anti-adhesive hydrogel to generate suspension cultures (Fig. 1b, Supplementary Fig. 1e, and Movie 2). After two weeks in suspension, ∼75 % of PKD organoids formed large, free-floatingcysts (Fig. 1c-d), a 10-fold increase in cyst formation over adherent cultures6. Control organoids of identical genetic background formed cysts only very rarely under these conditions, indicating that cystogenesis remained a specific consequence of the PKD mutations (Fig. 1c-d). As an inherent property of this differentiation system in both PKD and control cultures, each of the organoids placed into suspension was relatively small (∼ 250 μm in diameter) and contained only ∼ 5 tubules, therefore a significant proportion (∼ 15 %) of individual tubules deformed into cysts. In long-term cultures, PKD cysts further expanded massively and reached diameters of ∼ 1 centimeter, reflecting a 4000-fold increase in size over the original organoid (Fig. 1e). Even after many months in suspension, cysts remained rare among control organoids, which formed smaller, denser aggregates (Fig. 1d-e). Thus, modification of the material environment enabled the establishment of a highly efficient minimal reconstitution system for PKD cystogenesis.
Histological analysis revealed that PKD organoid cysts were lined with a thin, squamous epithelial layer, approximately one single cell in thickness, with irregular edges, surrounding a hollow lumen (Fig. 2a). When compared to cysts from various stages and subtypes of clinical PKD in vivo, organoid cysts most closely resembled cysts in prenatal PKD, which extended radially from the medulla to the cortex and appeared prominent just beneath the nephrogenic zone (Fig. 2a and Supplementary Fig. 2a). Similar cysts were previously reported in patients with biallelic mutations in PKD112. In contrast, postnatal cysts from patients exhibited a more smooth-edged and multi-layered appearance, and were accompanied by interstitial nephritis and inflammatory infiltrates not observed prenatally or in organoids (Fig. 2a and Supplementary Fig. 2a).
Fig. 2. Organoid PKD cysts phenotypically resemble PKD patient cysts.
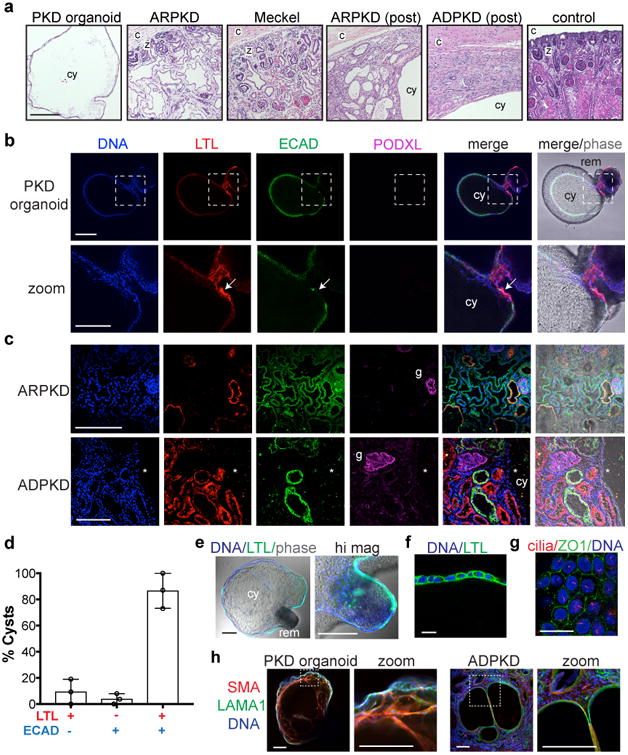
a, Paraffin sections dyed with hematoxylin and eosin from PKD organoids, or human kidney biopsies taken from patients with autosomal dominant PKD (ADPKD), autosomal recessive PKD (ARPKD), and Meckel syndrome. Identifying labels are provided for orientation and emphasis of specific histological features (c, kidney capsule; z, nephrogenic zone; i, inflammatory infiltrate; cy, large cyst; post, postnatal). b, Confocal immunofluorescence showing nephron segment markers in PKD organoid cysts or c, PKD patient kidneys. Zoom shows close-up of dotted boxed region. Arrow represents an area of specific enrichment for LTL. Glomeruli (g) do not appear cystic. Neither LTL nor ECAD is detected in a large ADPKD cyst, whose epithelium has dedifferentiated (*). d, Percentage of PKD organoid cysts labeling positive for LTL, ECAD, or both markers (n=3 separate experiments, ± s.e.m.). e, Confocal optical sections showing LTL affinity in a representative cyst in suspension. Higher magnification (hi mag) image shows LTL in the adjoining organoid remnant portion of this cyst. f, LTL in cyst-lining epithelial cells. g, Cilia (acetylated α-tubulin) and tight junctions (ZO1) in representative cyst-lining epithelial cells. h, Representative confocal images showing stromal markers in PKD organoid cyst and patient cysts. Scale bars, 200 μm or 25 μm (f-g).
We previously identified Lotus tetragonolobus lectin (LTL) and E-cadherin (ECAD), respectively, as markers of organoid proximal and distal tubules in these cultures6. In non-cystic organoids and tissues, these markers were not mutually exclusive, but rather formed a continuum, with enrichment in their respective nephron segments (Supplementary Fig. 2b). In PKD organoid cysts, LTL and ECAD largely overlapped within the cyst-lining epithelium, exhibiting patches of specific enrichment, whereas PODXL, a marker enriched in kidney podocytes, was not detected in cysts, similar to prenatal and postnatal PKD patient kidneys (Fig. 2b-c and Supplementary Fig. 2c) 12, 13, 14. Approximately 80 % of organoid cysts expressed both LTL and ECAD, which were also detected within the tubular remnants continuous with these cysts (Fig. 2b-f and Supplementary Fig. 2c). Cyst-lining cells in organoids were heavily coated with primary cilia and formed tight junctions between cells in a cobblestone pattern (Fig. 2g). Interestingly, organoid cysts in long-term suspension cultures also contained a sub-population of stromal cells that co-expressed smooth muscle α-actinin (SMA) and laminin, markers expressed in cystic stroma of PKD patient kidneys (Fig. 2h and Supplementary Fig. 2d). Collagen deposition in these cysts remained scant, similar to cysts in prenatal PKD kidneys, whereas postnatal PKD kidney cysts exhibited prominent fibrosis (Supplementary Fig. 2e). PKD organoid cysts in vitro therefore recapitulated hallmark features of PKD patient cysts, particularly the very early stages of PKD.
PKD organoid cysts in adherent cultures exhibited a two-fold increase in phosphorylated histone H3 (pH3), compared to LTL+ tubular cells from non-mutants, indicating increased proliferation (Fig. 3a-b). Similarly, in large cysts in long-term suspension cultures, dividing cells were detected within the cyst-lining epithelium and in anaphases oblique and internal to the plane of the cyst (Fig. 3c, Supplementary Fig. 3a-b, and Movie 3). When large PKD cysts were microdissected away from their remnant tubules, they immediately deflated, reflecting the loss of accumulated fluid (Fig. 3d). Cysts contained from ∼30,000 to ∼ 600,000 cells, whereas the original organoids from which they derived contained only∼ 3,000 cells, indicating extensive proliferation (Fig. 3e). Pathway-based global gene expression microarray analysis revealed significant enrichment of hallmark gene sets for cell cycle progression, mTOR signaling, and MYC activity in cysts, compared to remnant tubules (Fig. 3f). PKD cysts therefore arose from hyperproliferative kidney tubular epithelial cells (KTECs), a hallmark of mouse and human ADPKD8, 9, 14, 15.
Fig. 3. PKD organoid cysts arise from hyperproliferative KTECs.
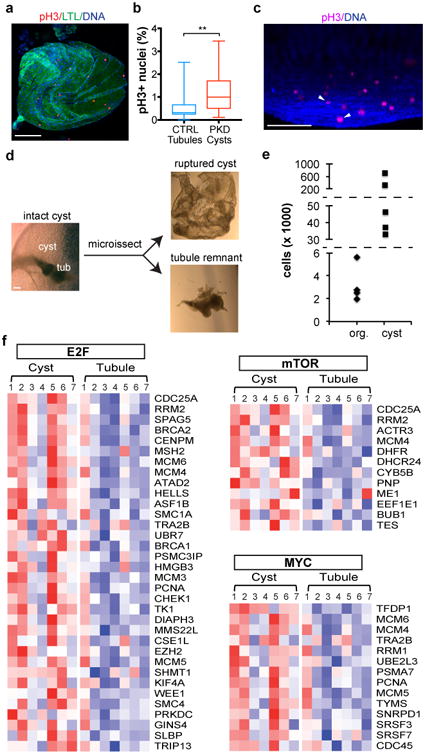
a, Representative images and b, quantification of pH3 in adherent PKD organoid cysts under adherent conditions. Boxes show 25th and 75th percentiles, whiskers indicate min and max values (n = 115 tubules pooled from 7 separate experiments and 26 cysts pooled from 6 separate experiments, ± s.e.m., t(37.16)=3.491, p=0.0013). c, Three-dimensional confocal reconstruction of a large cyst in suspension. Arrowhead indicates anaphases. See also Movie 3. d, Representative images showing microdissection of large cysts in suspension. e, Cell counts in organoids immediately after placement in suspension (org.) or in microdissected cysts grown for several months (cyst). Dashed lines represent non-linear breaks in the y-axis. f, Heat maps from microarray analysis of cysts and tubule remnants from cultured organoids, showing differentially expressed genes (p-value ≤ 0.05) contributing to activation of E2F targets, mTORC1 signaling, and Myc. Columns represent samples and row represent gene; red indicates greater than the mean (white) and blue, less than the mean values. Scale bars, 100 μm.
The process of purifying organoids and transferring them into suspension might induce cyst formation by provoking an injury response16, 17. To test this, organoids were purified and immediately replated onto wells coated with a thin layer of extracellular matrix (ECM) but lacking stroma. The replated organoids re-adhered but did not form cysts, indicating that injury alone was not sufficient to promote cystogenesis (Fig. 4a). Under these conditions, we observed that both control and PKD organoids formed expanding cell outgrowths very quickly, which could be further expanded as monolayers up to three passages (Fig. 4a and Supplementary Fig. 3c). Cells derived from organoid outgrowths exhibited a cobblestone-like epithelial morphology and predominantly expressed markers specific to KTECs, including LTL and kidney injury molecule-1 (KIM-1), similar to flow-sorted LTL+ cells from organoid cultures (Fig. 4b-d and Supplementary Fig. 3d-e). Removal of stroma thus stimulated proliferation and migration of organoid cells.
Fig. 4.
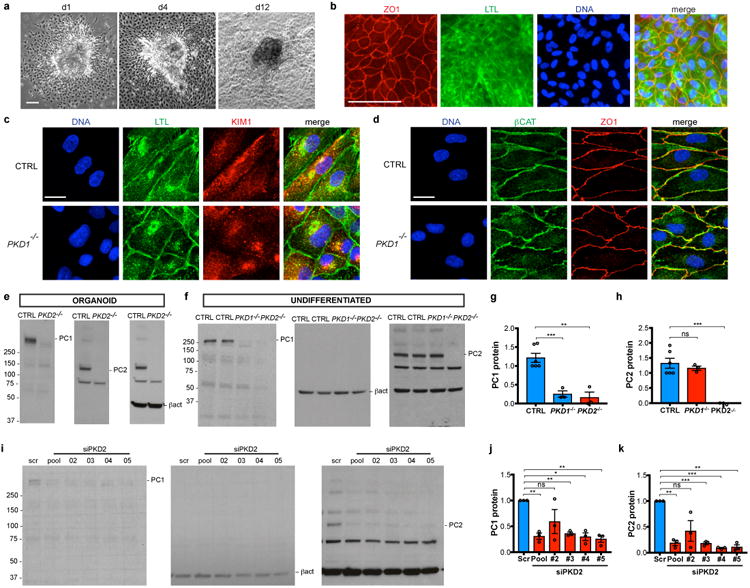
Outgrowth of PKD cell lines reveals a critical deficiency in PC1 expression. a, Phase contrast image of organoid explants on days 1, 4, and 12 after replating. b, Wide-field fluorescence and c-d, confocal sections showing epithelial and kidney-specific marker expression in representative kidney organoid cell monolayers. e, Representative immunoblots of PC1 and PC2 in kidney organoids and f, undifferentiated hPSCs. g, PC1 protein levels in undifferentiated hPSCs, normalized to b-actin loading control (ctrl, n = 6; PKD1-/- and PKD2-/-, n=3, ± s.e.m., ctrl vs. PKD1-/-, t(6.936)=6.603, p=0.00031 (***); ctrl vs. PKD2-/-, t(4.837)=5.669, p=0.0026 (**). h, PC2 protein levels in undifferentiated hPSCs, normalized to b-actin loading control (ctrl, n=6; PKD1-/- and PKD2-/-, n=3, ± s.e.m., ctrl vs. PKD1-/-, t(6.451)=0.9247, p=0.3884 (ns); ctrl vs. PKD2-/-, t(5)=8.006, p=0.0005 (***)). i, Representative immunoblot and j-k, quantification of PC1 and PC2 levels in hPSCs treated with four different PKD2 siRNAs (pooled or individually) or a scrambled (scr) siRNA control (n=3). j, Unpaired t test with Welch's correction, scr vs. pool, t(2)=11, p=0.0075; #2 vs. scr, t(2)=1.747, p=0.2227; #3 vs. scr, t(2)=22.66, p=0.0019; #4 vs. scr, t(2)=9.467, p=0.0110; #5 vs. scr, t(2)=11.56, p=0.0074. k, Unpaired t test with Welch's correction, scr vs. pool, t(2)= 16.92, p=0.0035; #2 vs. scr, t(2)=2.912, p=0.1005 (ns, not significant); #3 vs. scr, t(2)=31.93, p=0.0010; #4 vs. scr, t(2)=77.64, p=0.0002; #5 vs. scr, t(2)=20.28, p=0.0024. Scale bars, 100 μm (a-b) or 10 μm (c-d). ns, not significant.
We further utilized these organoid KTEC cell lines to investigate the expression of PC1, whose structure suggests a possible role in cell adhesion18, 19. In contrast to many cell types, KTECs derived from organoid outgrowths expressed sufficient quantities of endogenous PC1 to detect in lysates by immunoblot (Fig. 4e) 20, 21. Surprisingly, we found that PC1 protein was nearly undetectable in KTECs derived from PKD2-/- organoids, using an antibody against the amino terminal fragment (Fig. 4e) 22. Undifferentiated PKD2-/- hPSCs exhibited a similarly strong decrease in PC1 expression levels (Fig. 4f-g). PKD1 transcripts were expressed at normal levels in PKD2-/- hPSCs, suggesting that PC1 loss occurs through a post-transcriptional mechanism (Supplementary Fig. 4a-b). Conversely, in PKD1-/- cells, PC2 expression levels were unchanged from isogenic controls, although its localization to primary cilia was strikingly decreased, consistent with previous reports (Fig. 4f,h and Supplementary Fig. 4c-d) 5, 20, 21. Furthermore, treatment of control hPSCs with three different siRNAs, which knocked down PC2 protein to 12.6 ± 0.02 % of normal levels (avg. ± s.e.m.), induced a corresponding decrease in PC1 protein to 30.5 ± 0.03 % of normal levels (Fig. 4i-k). These studies revealed, unexpectedly, that PC2 was required for PC1 amino-terminus expression in human cells, in contrast to reports in mouse Pkd2-/- cells 20, 21. Differences between species, cell types, or exogenous versus endogenous expression levels may account for this discrepancy, as PC1 is a low abundance protein in humans, who appear to be highly sensitive to reductions in its expression, compared to mice 6, 7, 23, 24. A unifying hypothesis that emerges from these studies is that the ECM microenvironment functions to maintain tubular shape and adhesion through interactions involving PC1'slong, extracellular domain18, 19, 25. To directly test the effect of PKD mutations on the matrix microenvironment, we embedded individual organoids (∼ 250 μm diameter) into larger collagen droplets (∼ 4 mm diameter), and placed these in suspension. Droplets implanted with organoids did not form cysts, but rather contracted dramatically over a period of approximately 1-2 weeks (Fig. 5a). PKD1-/- organoids were quantitatively impaired in their ability to compact collagen droplets, compared to isogenic controls (Fig. 5b). Contracted droplets comprised an inner core of solid collagen encompassed by a thin, continuous epithelium of LTL+ECAD+KTECs (Fig. 5c-d). During the formation of these structures, KTECs could be observed migrating out of the implanted organoid to coat the surface of the droplet (Fig. 5e). Collagen staining appeared more intense after contraction, and collagen fibers appeared denser ultrastructurally, indicating that the changes in droplet size involved physical compression (Fig. 5f-g). Collectively, these findings revealed that kidney organoid epithelia were capable of dramatically remodeling their ECM microenvironment through migratory forces, and that this property was partially dependent on PC1 (Fig. 5h).
Fig. 5. Organoids remodel their matrix microenvironment in a PKD-dependent manner.
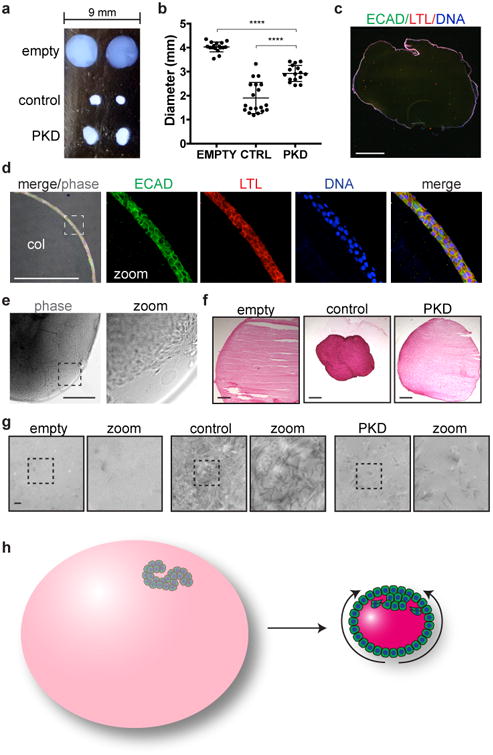
a, Photograph of organoids implanted into collagen balls and cultured in suspension for two weeks. b, Diameters of empty (n=17) and organoid-implanted (ctrl, n=19; PKD, n=16) collagen droplets, pooled from three experiments (empty vs. ctrl, t(21.96)=13.53, p=3.9334×10-12; ctrl vs. PKD, t(24.74)=11.33, p=1.7989×10-6). Each droplet is indicated by a single data point. c, Wide-field immunofluorescence image of whole droplet compacted by a PKD organoid, stained for tubule segment markers. Zoom of dashed boxed region is shown for fluorescent channels. d, Confocal immunofluorescence at the edge of a representative droplet after contraction, adjacent to the collagen interior (col). The dashed boxed region is shown for each individual channel at higher magnification. e, Phase contrast images showing the edge of a representative droplet at an early stage of compaction. f, Sirius red staining of collagen droplets. g, TEM 25,000X images of collagen filament structure in the interior of droplets. Zoom of dashed boxed region is shown for each image. h, Schematic of collagen droplet compaction by organoids. KTECs migrate out and surround the scaffold, which contracts towards the outgrowth (curved arrows). Scale bars, 500 μm (b-f) or 500 nm (g).
Cyclic AMP (cAMP) signaling is hypothesized to contribute significantly to PKD, but can also promote fluid accumulation in non-PKD epithelia 3, 26. Forskolin, a powerful agonist of adenylyl cyclase, induced rapid and dose-dependent swelling of adherent organoids into round, cyst-like structures that retained the shape of the original tubules (Supplementary Fig. 5a-b and Movie 4). The non-degradable cAMP analog 8-Br-cAMP also induced swelling, although the effect was much less pronounced (Supplementary Fig. 5b-c). Upon withdrawal of these agents, the swollen structures deflated and the organoids returned towards their original size (Supplementary Fig. 5a-c). Both PKD and non-PKD organoids swelled and deflated to a similar degree after cAMP stimulation, indicating a modifier effect (Supplementary Fig. 5a-d and Movie 4). One limitation of this system is that we were unable to examine collecting ducts, which are the primary target of cAMP-mediated candidate therapeutics10, 27, because these structures do not mature in kidney organoids 6, 28, 29, 30.
In addition to gene-edited mutants, we also investigated the potential of using induced pluripotent stem cells (iPSCs) derived from PKD patients to model disease 5. We found that iPSCs from human patients exhibited dramatic line-to-line variability in their abilities to form organoids, regardless of PKD genotype and organoid differentiation protocol (Supplementary Fig. 6a-c). The morphology of tubular structures also varied noticeably between different lines (Supplementary Fig. 6c). As such differences reflected a degree of heterogeneity that would confound analysis of PKD-specific effects, we focused our studies on the CRISPR-mutant hPSCs. Although patient-derived organoids presented much variability, they could eventually represent valuable tools to develop personalized medicine approaches.
In conclusion, by combining PKD organoids with modular physical environments, we have established a human cellular system that models PKD with high efficiency and specificity. Comparison of PKD and non-PKD organoids suggests a specific, primary role for microenvironment and adhesion in early stages of the disease. Interventions that strengthen stromal or scaffolding components can provide a critical cue favoring migratory repair over cystogenesis. Our biochemical studies indicate a central requirement for PC1, which we propose functions as an adhesion regulator that maintains tubular architecture through interactions with the microenvironment. The efficiency, specificity, and modularity of organoid cultures provide critical insight into the biomaterial basis of human disease, with great potential for mechanistic studies and therapeutics development.
Methods
Kidney Organoid Differentiation
WA09 (H9) hPSCs with CRISPR-targeted PKD1-/- or PKD2-/- mutations, or passage- and procedure-matched non-mutant isogenic controls, were maintained feeder-free on 1 % Reduced Growth Factor GelTrex (Life Technologies) in mTeSR1 (Stem Cell Technologies) and dissociated with Accutase (Stem Cell Technologies), as previously described 6. hPSC lines were derived from either the WA09 hESC line (WiCell) or the WTC11 iPSC line (Gladstone Institute). 60,000 cells from each cell line were plated per well of a 24-well plate pre-coated with GelTrex in mTeSR1 supplemented with 10 μM Rho-kinase inhibitor Y27632 (StemGent). The media was replaced with 500 μL mTeSR1 + 1.5 % GelTrex at 16 hours, 500 μL mTeSR1 at 36 hours, Advanced RPMI + Glutamax (from Life Technologies) + 12 μM CHIR99021 (Stemgent) at 50 hours, and RB (Advanced RPMI + Glutamax + B27 Supplement, from Life Technologies) at 86 hours. RB was changed two days later and every three days thereafter. Alternatively (Protocol B, Extended Fig. 5b), undifferentiated hPSCs were plated overnight and then treated with 8 μM CHIR99021 in APEL media (Stem Cell Technologies) for 48-72 hours, 30 ng/ml FGF9 or FGF2 (Peprotech) + 1 μg/ml heparin (Stem Cell Technologies) in APEL for 96 hours, and subsequently cultured in APEL which was replaced every three days. Organoids typically became visible by light microscopy 12-18 days after plating.
Cyst Formation
In adherent cultures (untreated), ‘cysts’ were identified as large, balloon-like, translucent structures that swayed in response to agitation. Flat rings and dilated tubules were not counted as cysts and occasionally appeared even in non-PKD controls. Forskolin and 8-Br-cAMP (Sigma) were added to adherent cultures on the 21st day of differentiation, resulting in rapid formation of cysts that typically did not sway in response to agitation. Wells were imaged using a Nikon TiE inverted widefield microscope and cysts were quantified using ImageJ cell counter. To generate large cysts in suspension, adherent organoids were microdissected with a 23-gauge syringe needle from 24-well plates on an inverted phase-contrast microscope, and carefully transferred using a transfer pipette into a low-adhesion 6-well plate (Corning) containing 2 mL RB. The organoids were isolated on the 21st day of differentiation, before cysts formed. RB media was changed by gravity every three days, and cystogenesis was assessed at two weeks after replating.
Generation of KTEC lines from organoids
To prepare monolayers of kidney cells for analysis, freshly isolated organoids were immediately plated onto tissue culture dishes pre-coated with 1 % GelTrex (Thermo Scientific) and cultured for one week in RB, and the resulting epithelial outgrowths were processed for immunoblot and immunofluorescence. Alternatively, to isolate KTECs using flow cytometry, entire organoid cultures were incubated with fluorescein-conjugated LTL (FL-1321, Vector Labs) diluted 1:500 into RB for four hours, dissociated with Accutase, pelleted, resuspended in flow sorting buffer: 1 % FBS, 10 mM HEPES buffer in PBS (Thermo Scientific). LTL+ cells were isolated on a FACSAria Cell Sorter (BD Biosciences) and replated onto GelTrex-coated tissue plates in RB.
Organoid embedding in collagen droplets
To embed organoids in collagen droplets, a sheet of Parafilm was soaked in 70 % ethanol, air dried, and pressed against the holes of a box of gel-loading pipette tips (1-200 μl, Fisher Scientific 02-707-138) to create a dimpled mold31. One organoid was placed in each dimple and 30 μL of 7 mg/mL collagen I (Corning) was added. The droplets were incubated 25 minutes at 37°C and carefully resuspended in 3 ml of RB media in an untreated 6-well plate. The media was changed weekly and the droplets were imaged after 2 weeks of incubation using a Nikon Ti Inverted Widefield microscope and a Nikon 1 J1 Camera. Droplet diameters were measured using NIS Elements imaging software (Nikon) and normalized to the diameter of empty droplets from the same set, for a total of three sets. Droplets that failed to undergo compaction (∼ 20 % in control and ∼ 50 % in PKD samples) were excluded. Droplets were fixed with 4% paraformaldehyde for 20 minutes at room temperature, incubated 16 hours in 30 % sucrose (Sigma) in water, mounted in Tissue-Tek (Sakura), flash frozen, and cryosectioned onto SuperfrostPlus slides (Fisher). Sections were stained in Picro-sirius red solution (Sky-Tek laboratories) for one hour, rinsed in two changes of 0.5% acetic acid solution, and dehydrated in two changes of absolute ethanol before mounting. Immunofluorescence was performed as described below.
siRNA and Immunoblotting
16 hours after plating, hPSCs were transfected with Dharmacon Smartpool siRNAs (Fisher Scientific) directed against PKD2 or scrambled control in mTeSR1 without antibiotics. Media was changed 24 hours later. 72 hours after siRNA treatment, cells were lysed in RIPA buffer (Thermo Scientific) containing 1× Complete mini EDTA-free protease inhibitor, PhosSTOP, and benzonase nuclease (all Sigma). Protein lysates were quantified using a BCA protein assay kit (Thermo Fisher). To prepare the samples, 50 μg of protein were mixed with Pierce Lane Marker Reducing Sample (Thermo Fisher) and incubated at 40°C for 5 minutes. Samples were separated in a 4-20% protein gel (Bio-Rad) and transferred into a PVDF membrane using standard protocols and 0.01% SDS in the transfer buffer (0.25M Tris Base, 1.92M Glycine, 0.1% SDS). Gels included pre-stained molecular weight markers (Precision Plus Protein Kaleidoscope Standards, Bio-Rad), which were annotated manually by overlay of the film onto the nitrocellulose membrane. The antibodies used for the immunoblots were anti-PC1 (sc-130554, Santa Cruz), anti-PC2 (sc-10376, Santa Cruz) and anti-β-actin (4970S, Cell signaling). The intensity ratio of the experimental band to the loading control was normalized to 1 in the negative control (unmodified or untreated hPSCs). The remaining bands were normalized to the control and averaged for at least three independent experiments. Examples of unprocessed immunoblots with the original standards hand-marked are provided in Supplementary Fig. 7.
Global Gene Expression and Bioinformatics Analysis
For systems biology analysis of cysts, cystic epithelium or tubule remnants were manually separated from PKD1-/- organoids (71-87 days of culture from three experiments) using a 22-gauge needle under a dissecting microscope and flash frozen separately in liquid nitrogen. Total RNA containing small RNA was extracted from seven paired samples (cysts and tubule remnants from the same organoid) using RNeasy Micro Kit (Qiagen) with an on-column DNA digestion step to minimize genomic DNA contamination. The sample integrity of the RNA was assessed using the RNA 6000 Nano Assay on 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA) to ensure that RNA integrity number (RIN) was greater than 7. Microarray experiments were performed at The Centre for Applied Genomics (TCAG) at the Hospital for Sick Children (Toronto, Ontario, Canada). Following the manufacturer's protocol, 10 ng of total RNA was labeled using the GeneChip WT Pico Reagent Kit (Affymetrix). Fragmented and biotin-labeled ss-cDNAs were then hybridized to GeneChip Human Gene 2.0 ST Arrays (Affymetrix) for 16 hours at 45°C. Hybridized arrays were stained and washed in the Affymetrix Fluidics Station 450. Thereafter, the arrays were scanned on an Affymetrix GeneChip Scanner 3000 and the image (.DAT) files were preprocessed using the Affymetrix GeneChip Command Console (AGCC) software v.4.3 to generate cell intensity (.CEL) files. After image processing, the array data were uploaded to the Affymetrix Expression Console software v1.4.1 for further processing and quality control. All quality assessment metrics, including spike-in controls during target preparation and hydrization were found within the boundaries. The probe set signal intensities were then extracted and normalized using the robust multi-array average (RMA) algorithm embedded in the Expression Console software, which consists of convolution background correction, quantile normalization, and median polish summarization. Downstream paired sample t-test was carried out via Partek Genomics Suite 6.6 (Partek Inc., Chesterfield, MO, USA) to determine differentially expressed genes between cysts and tubules. Gene set enrichment analysis (GSEA, http://software.broadinstitute.org/gsea/index.jsp) was used as the primary tool to identify potential gene pathways or gene sets that may modulate cystic kidney organoid growth 32. Before running GSEA, Affymetrix probe sets were collapsed to one gene level, paired sample t-test statistics scores were used to create a ranked list of genes of the entire data set (in total, 29406 unique genes with gene symbols). GSEA was performed using the Hallmark gene sets from Molecular Signatures Database (MSigDB, http://software.broadinstitute.org/gsea/msigdb/collections.jsp#H) 33. The description of each gene set can be found on MSigDB website. We defined overrepresented gene sets with a FDR ≤ 0.25. For RNA-Seq analysis of hPSCs, RNA was prepared from isogenic sets of cell lines using the RNEasy Mini Kit (Qiagen), checked for high integrity on an Agilent Bioanalyzer, prepared using the TruSeq stranded mRNA library kit (Illumina), and sequenced on an Illumina NextSeq500 75×75 paired end high output run. Samples were aligned to hg19 reference sequence.
Immunofluorescence
To fix organoids, an equal volume of 8 % paraformaldehyde (Electron Microscopy Sciences) in PBS was added to the culture media (4 % final concentration) for 15 minutes at room temperature. After fixing, samples were washed in PBS, blocked in 5 % donkey serum (Millipore)/0.3 % Triton-X-100/PBS, and incubated overnight in 3 % bovine serum albumin/PBS with primary antibodies. The next day, samples were washed in PBS and incubated overnight with Alexa-Fluor secondary antibodies and DAPI (Thermo Scientific), followed by PBS washes. For frozen tissue sections, fresh tissues were incubated in 4 % paraformaldehyde for one hour at 4°C, incubated 16 hours in 30 % sucrose (Sigma) in water, mounted in Tissue-Tek (Sakura), flash frozen, and cryosectioned onto SuperfrostPlus slides (Fisher) before blocking. Paraffin sections were prepared by fixing overnight in methacarn (60 % absolute methanol, 30 % chloroform, 10 % glacial acetic acid, Sigma) or in 4 % PFA, followed by paraffin embedding, sectioning, deparaffinization in 100 % xylene (3 washes 5 minutes each), dehydration in graded 70 % - 100 % ethanol (5 minutes each), and heating in citrate buffer pH 6.0 (Sigma) in a pressure cooker (Instant Pot IPDUO60) for three minutes prior to immunostaining. Kidney organoid cysts were embedded in 2 % agarose prior to paraffin embedding. Histology stains (Hematoxylin and eosin, or Masson's trichrome) were applied to paraffin sections by UW Pathology. Primary antibodies included acetylated alpha-tubulin (051M4770; Sigma), ZO-1(339100; Invitrogen), LTL (FL-1321, Vector Labs), PC2 (sc-25749, Santa Cruz), NPHS1 (R&DAF4269, 1:500), KIM-1 (MAB1750, R&D), PODXL (R&DAF1658, 1:500), ECAD (Abcam ab11512), SMA (Sigma A2547, 1:500), LAMA1 (Sigma L9393a, 1:500), and pH3 (sc-8656, Santa Cruz). Fluorescence images were captured using a Nikon epifluorescence 90-I (upright), Nikon Ti Inverted Widefield microscope, or Nikon A1R and C1 confocal microscopes. pH3+ cells were scored manually in cysts or tubular organoids of similar sizes.
Electron microscopy
Droplets were gently transferred with a cut-off transfer pipette into EM fix: 0.15 M sodium cacodylate trihydrate (Sigma) dissolved in water (pH 7.3) containing 4% formaldehyde and 2% glutaraldehyde (Electron Microscopy Sciences), and stored overnight. Samples were post-fixed with osmium tetroxide solution (Sigma), dehydrated in serial ethanol dilutions (Sigma), and embedded in epoxy resin. Ultrathin sections (80 nm) were mounted on 200 mesh copper grids and stained with uranyl acetate and lead citrate (Electron Microscopy Sciences). Imaging was performed with a JEOL JEM-1010 TEM and a FEI Tecnai G2 Spirit TEM.
PKD patients
All human samples were obtained with informed consent and in compliance with all ethical regulations under the auspices of protocols approved by the UW Institutional Review Board. These included an ADPKD kidney donated by a 46 yr. female (generous gift of Virginia Mason Hospital, Seattle, WA), an anonymized biopsy of a 15 wk control kidney (UW Laboratory of Developmental Biology), and anonymized biopsies of kidneys with clinically-diagnosed Meckel syndrome (20 wk.), ARPKD (29 wk., 6 mo., or 6 yr.), and orofaciodigital syndrome (18 yr.) from Seattle Children's Hospital Histopathology. Patients with kidney disease were enrolled in our study for the purposes of collecting cells and tissues as positive controls. These samples were collected from patients of all ages without any discrimination with respect to gender, age, race, family history, or other co-variates. PKD samples were chosen from this collection at random and based on availability to represent a range of disease severities, ages, and genotypes.
Statistical Analysis
Experiments were performed using a cohort of PKD hPSCs, generated as described previously 6, including three PKD1-/-and two PKD2-/- hPSC lines, and a total of six isogenic control lines that were subjected to CRISPR mutagenesis but were found to be unmodified at the targeted locus. Quantification was performed on experiments performed side-by-side on at least three different occasions. Error bars are mean ± standard error (s.e.m.). Statistical analyses were performed using GraphPad Prism Software. To test significance, p values were calculated using two-tailed, unpaired t test with Welch's correction (unequal variances). Statistical significance was defined as p< 0.05. Exact p values, t values and degrees of freedom are provided in the figure legends.
Supplementary Material
Acknowledgments
The authors thank Stuart Shankland, Ed Kelly, David Beier, Hannele Ruohola-Baker, and Charles Murry (UW), and Joseph Bonventre, Jing Zhou, and Ted Steinman (Harvard Medical School) for helpful discussions. We thank Dale Hailey (ISCRM Garvey Imaging Core) and Ed Parker (UW Vision Core Lab) for microscopy support. The Laboratory of Developmental Biology was supported by NIH Award Number 5R24HD000836 (NICHD). Studies were supported by an NIH Career Development Award DK102826 (NIDDK), PKD Foundation Research Award, National Kidney Foundation Young Investigator Grant, and American Society of Nephrology Career Development Award (BSF). The work was also supported by start-up funds from the University of Washington, NIH UH2/UH3 Award TR000504 (JH) and an unrestricted gift from the Northwest Kidney Centers to the Kidney Research Institute.
Footnotes
Author Contributions: N.M.C., S.M.C., R.E.G., A.J.C., Y.K.K., K.W., L.M.T., M.A.D., and B.S.F. performed experiments and analysis of hPSCs and kidney organoids. X.S. and Y.P. performed global gene analysis of kidney organoid cysts. L.S.F., M.D., and B.S.F. analyzed tissues from PKD patients. N.M.C., X.S., S.M.C., H.F., L.S.F., Y.P., J.H., and B.S.F. contributed to experimental design and analysis. B.S.F. and N.M.C. wrote the first draft of the manuscript. All authors revised the manuscript.
Competing Financial Interests: None of the authors have any competing financial interests to declare. B.S.F. is an inventor on patent applications for kidney differentiation and disease modeling from human pluripotent stem cells (PCT/US14/34031, PCT/US16/50271).
Data Availability Statement: Microarray data are MIAME compliant and publicly available in Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) (ID: GSE101308). All remaining datasets are available from the corresponding author upon reasonable request.
References
- 1.The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. The European Polycystic Kidney Disease Consortium. Cell. 1994;78(4):725. [PubMed] [Google Scholar]
- 2.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272(5266):1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 3.Neufeld TK, Douglass D, Grant M, Ye M, Silva F, Nadasdy T, et al. In vitro formation and expansion of cysts derived from human renal cortex epithelial cells. Kidney Int. 1992;41(5):1222–1236. doi: 10.1038/ki.1992.184. [DOI] [PubMed] [Google Scholar]
- 4.Boletta A, Qian F, Onuchic LF, Bhunia AK, Phakdeekitcharoen B, Hanaoka K, et al. Polycystin-1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells. Mol Cell. 2000;6(5):1267–1273. doi: 10.1016/s1097-2765(00)00123-4. [DOI] [PubMed] [Google Scholar]
- 5.Freedman BS, Lam AQ, Sundsbak JL, Iatrino R, Su X, Koon SJ, et al. Reduced ciliary polycystin-2 in induced pluripotent stem cells from polycystic kidney disease patients with PKD1 mutations. J Am Soc Nephrol. 2013;24(10):1571–1586. doi: 10.1681/ASN.2012111089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freedman BS, Brooks CR, Lam AQ, Fu H, Morizane R, Agrawal V, et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun. 2015;6:8715. doi: 10.1038/ncomms9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet. 2004;13(24):3069–3077. doi: 10.1093/hmg/ddh336. [DOI] [PubMed] [Google Scholar]
- 8.Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. 2006;103(14):5466–5471. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trudel M, Lanoix J, Barisoni L, Blouin MJ, Desforges M, L'Italien C, et al. C-myc-induced apoptosis in polycystic kidney disease is Bcl-2 and p53 independent. J Exp Med. 1997;186(11):1873–1884. doi: 10.1084/jem.186.11.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gattone VH, 2nd, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. 2003;9(10):1323–1326. doi: 10.1038/nm935. [DOI] [PubMed] [Google Scholar]
- 11.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vujic M, Heyer CM, Ars E, Hopp K, Markoff A, Orndal C, et al. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J Am Soc Nephrol. 2010;21(7):1097–1102. doi: 10.1681/ASN.2009101070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakanishi K, Sweeney WE, Jr, Zerres K, Guay-Woodford LM, Avner ED. Proximal tubular cysts in fetal human autosomal recessive polycystic kidney disease. J Am Soc Nephrol. 2000;11(4):760–763. doi: 10.1681/ASN.V114760. [DOI] [PubMed] [Google Scholar]
- 14.Grantham JJ, Geiser JL, Evan AP. Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int. 1987;31(5):1145–1152. doi: 10.1038/ki.1987.121. [DOI] [PubMed] [Google Scholar]
- 15.Song X, Di Giovanni V, He N, Wang K, Ingram A, Rosenblum ND, et al. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet. 2009;18(13):2328–2343. doi: 10.1093/hmg/ddp165. [DOI] [PubMed] [Google Scholar]
- 16.Takakura A, Contrino L, Zhou X, Bonventre JV, Sun Y, Humphreys BD, et al. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet. 2009;18(14):2523–2531. doi: 10.1093/hmg/ddp147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, et al. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet. 2008;17(11):1578–1590. doi: 10.1093/hmg/ddn045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. The International Polycystic Kidney Disease Consortium. Cell. 1995;81(2):289–298. doi: 10.1016/0092-8674(95)90339-9. [DOI] [PubMed] [Google Scholar]
- 19.Ibraghimov-Beskrovnaya O, Bukanov NO, Donohue LC, Dackowski WR, Klinger KW, Landes GM. Strong homophilic interactions of the Ig-like domains of polycystin-1, the protein product of an autosomal dominant polycystic kidney disease gene, PKD1. Hum Mol Genet. 2000;9(11):1641–1649. doi: 10.1093/hmg/9.11.1641. [DOI] [PubMed] [Google Scholar]
- 20.Cai Y, Fedeles SV, Dong K, Anyatonwu G, Onoe T, Mitobe M, et al. Altered trafficking and stability of polycystins underlie polycystic kidney disease. J Clin Invest. 2014;124(12):5129–5144. doi: 10.1172/JCI67273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gainullin VG, Hopp K, Ward CJ, Hommerding CJ, Harris PC. Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J Clin Invest. 2015 doi: 10.1172/JCI76972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ong AC, Harris PC, Davies DR, Pritchard L, Rossetti S, Biddolph S, et al. Polycystin-1 expression in PKD1, early-onset PKD1, and TSC2/PKD1 cystic tissue. Kidney Int. 1999;56(4):1324–1333. doi: 10.1046/j.1523-1755.1999.00659.x. [DOI] [PubMed] [Google Scholar]
- 23.Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87(6):979–987. doi: 10.1016/s0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]
- 24.Chauvet V, Qian F, Boute N, Cai Y, Phakdeekitacharoen B, Onuchic LF, et al. Expression of PKD1 and PKD2 transcripts and proteins in human embryo and during normal kidney development. Am J Pathol. 2002;160(3):973–983. doi: 10.1016/S0002-9440(10)64919-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangos S, Lam PY, Zhao A, Liu Y, Mudumana S, Vasilyev A, et al. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis Model Mech. 2010;3(5-6):354–365. doi: 10.1242/dmm.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magenheimer BS, St John PL, Isom KS, Abrahamson DR, De Lisle RC, Wallace DP, et al. Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+),K(+),2Cl(-) Co-transporter-dependent cystic dilation. J Am Soc Nephrol. 2006;17(12):3424–3437. doi: 10.1681/ASN.2006030295. [DOI] [PubMed] [Google Scholar]
- 27.Reif GA, Yamaguchi T, Nivens E, Fujiki H, Pinto CS, Wallace DP. Tolvaptan inhibits ERK-dependent cell proliferation, Cl(-) secretion, and in vitro cyst growth of human ADPKD cells stimulated by vasopressin. Am J Physiol Renal Physiol. 2011;301(5):F1005–1013. doi: 10.1152/ajprenal.00243.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taguchi A, Kaku Y, Ohmori T, Sharmin S, Ogawa M, Sasaki H, et al. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell. 2014;14(1):53–67. doi: 10.1016/j.stem.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 29.Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 2015;526(7574):564–568. doi: 10.1038/nature15695. [DOI] [PubMed] [Google Scholar]
- 30.Morizane R, Lam AQ, Freedman BS, Kishi S, Valerius MT, Bonventre JV. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol. 2015;33(11):1193–1200. doi: 10.1038/nbt.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell systems. 2015;1(6):417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.