

Abstract
Aberrant expression of proteins often underlies many diseases, including cancer. A recently developed approach in drug development is small molecule-mediated, selective degradation of dysregulated proteins. We have devised a protein-knockdown system that utilizes chimeric molecules termed specific and nongenetic IAP-dependent protein erasers (SNIPERs) to induce ubiquitylation and proteasomal degradation of various target proteins. SNIPER(ER)-87 consists of an inhibitor of apoptosis protein (IAP) ligand LCL161 derivative that is conjugated to the estrogen receptor α (ERα) ligand 4-hydroxytamoxifen by a PEG linker, and we have previously reported that this SNIPER efficiently degrades the ERα protein. Here, we report that derivatization of the IAP ligand module yields SNIPER(ER)s with superior protein-knockdown activity. These improved SNIPER(ER)s exhibited higher binding affinities to IAPs and induced more potent degradation of ERα than does SNIPER(ER)-87. Further, they induced simultaneous degradation of cellular inhibitor of apoptosis protein 1 (cIAP1) and delayed degradation of X-linked IAP (XIAP). Notably, these reengineered SNIPER(ER)s efficiently induced apoptosis in MCF-7 human breast cancer cells that require IAPs for continued cellular survival. We found that one of these molecules, SNIPER(ER)-110, inhibits the growth of MCF-7 tumor xenografts in mice more potently than the previously characterized SNIPER(ER)-87. Mechanistic analysis revealed that our novel SNIPER(ER)s preferentially recruit XIAP, rather than cIAP1, to degrade ERα. Our results suggest that derivatized IAP ligands could facilitate further development of SNIPERs with potent protein-knockdown and cytocidal activities against cancer cells requiring IAPs for survival.
Keywords: ubiquitin, proteasome, X-linked inhibitor of apoptosis protein (XIAP), estrogen receptor, tumor therapy, SNIPER, protein knockdown, ERalpha
Introduction
Aberrant or increased expression of pathogenic proteins often results in the initiation of many diseases, including a variety of cancers. However, a limited number of pathogenic proteins are currently targeted by pharmacological inhibitors, primarily because many proteins do not have suitable binding pockets where a pharmacophore may interact to regulate protein activity (1, 2). An alternative approach has been to down-regulate expression of the pathogenic proteins; this is usually achieved in vitro through use of genetic methods involving antisense oligonucleotides, dsRNAs, and CRISPR-Cas9 technology. However, clinical application of these technologies remains challenging because delivery of oligonucleotides to the target tissues is not easily accomplished (3, 4).
As a novel strategy to down-regulate pathogenic proteins in a nongenetic manner, we and others have devised a protein-knockdown system that uses small molecules with sufficient membrane permeability to induce selective degradation of target proteins. These small-molecule compounds, designated as proteolysis-targeting chimeras (PROTACs)7 and specific and non-genetic IAP-dependent protein erasers (SNIPERs), are chimeric molecules that contain two different ligands connected by a linker; one ligand is specific for an E3 ubiquitin ligase, and the other is specific for a target protein (5–7). The PROTACs and SNIPERs are designed to cross-link the E3 ubiquitin ligase and the target protein to induce polyubiquitylation and proteasomal degradation of the target protein within cells. To recruit the von Hippel–Lindau (VHL) E3 ligase complex and the cereblon (CRBN) E3 ligase complex, a VHL inhibitor (based on the HIF-1α peptide) and a phthalimide moiety have been respectively integrated into PROTAC constructs (8–11). In a similar manner, an IAP antagonist has been incorporated into SNIPERs to recruit either cellular inhibitor of apoptosis protein 1 (cIAP1) or X-linked inhibitor of apoptosis protein (XIAP) E3 ligase (12–18). To date, a range of PROTAC and SNIPER compounds have been developed, allowing degradation of a variety of proteins, such as estrogen receptor α (ERα), oncogenic kinase BCR-ABL, and epigenetic regulator bromodomain-containing protein 4 (BRD4) (19–30). Some PROTACs and SNIPERs also have demonstrated the ability to degrade target proteins in vivo, suggesting that this technology is feasible for use in novel drug development. These include VHL-based and CRBN-based PROTACs directed against BRD4, which are capable of inducing BRD4 protein degradation in mouse xenograft models (10, 31). By incorporating an LCL161 derivative, a high-affinity IAP ligand, we developed SNIPER(ER)-87, which induces in vivo degradation of ERα and growth inhibition of an ERα-positive human breast tumor in a xenograft model (13).
IAPs are a family of antiapoptotic proteins containing one or three baculoviral IAP repeat (BIR) domains (32–34). Some family members, such as cIAP1, cIAP2, and XIAP, directly interact with and regulate caspases via the BIR domain, thus inhibiting apoptosis (35–38). These IAPs are attractive targets for tumor therapy because of their frequent overexpression in multiple human malignancies and their implications in tumor progression, treatment failure, and poor prognosis (39–45). Based on the IAP-binding tetrapeptides of second mitochondria-derived activator of caspases/direct IAP-binding protein with low pI (SMAC/DIABLO), many potent and cell-permeable peptidomimetic IAP antagonists (also known as SMAC mimetics) have been developed; some of these are under evaluation in clinical phase studies as antitumor drugs (32, 46, 47). These IAP antagonists interact with BIR domains in IAP proteins to directly inhibit XIAP or to induce autoubiquitylation and proteasomal degradation of cIAP1 and cIAP2 (48–51). Because SNIPERs utilize IAP antagonists as IAP-ligand modules, SNIPERs are able to down-regulate IAPs beyond the initial target proteins (12–18); this is likely to be advantageous when attempting to kill cancer cells that require IAPs for survival.
In this paper, we demonstrate that, by derivatizing the IAP-ligand module, we have developed novel SNIPER(ER)s whose protein knockdown and antitumor activities are more potent than those of SNIPER(ER)-87. These SNIPER(ER)s have higher affinity for IAPs and exhibit more consistent abilities to degrade ERα and IAPs. In addition, we discuss the significance of IAP down-regulation in pharmacological efforts to induce cancer cells to undergo apoptosis.
Results
Structure-activity relationship of SNIPER(ER)s with novel IAP ligands
To discover IAP ligands that are useful for the development of SNIPERs with potent protein-knockdown activity, we substituted the IAP ligand moiety of SNIPER(ER)-87 with several IAP antagonists that have been reported elsewhere (52–55) (Fig. 1 and Table S1) and examined their abilities to reduce ERα expression by MCF-7 human breast tumor cells (Fig. 2). In a series of SNIPER(ER)s with different IAP ligand modules, we found that five compounds (SNIPER(ER)-105 (4), -110 (31a), -113 (31c), -119 (31d), and -126 (50)) reduced ERα expression comparably with, and more potently than, SNIPER(ER)-87 for durations of 4 and 48 h, respectively (Fig. 2a). SNIPER(ER)-130 (69) and -131 (71) exhibited attenuated knockdown activities, whereas the others (SNIPER(ER)-104 (9), -118 (55a), -121 (55b), -134 (31b), and -136 (40)) did not induce effective knockdown at 1–100 nm (Fig. 1b). The DC50 (the concentration required for 50% reduction of expression) for each SNIPER(ER) compound is summarized in Table 1.
Figure 1.

Chemical structures of novel SNIPER(ER)s. The IAP ligand moiety of SNIPER(ER)-87 was substituted by various IAP antagonists. Chemical structures shown in red indicate parts changed from the original SNIPER(ER)-87.
Figure 2.

Novel SNIPER(ER)s with potent protein-knockdown activity. a and b, MCF-7 cells were treated with the indicated concentrations of SNIPER(ER)s for the indicated periods. Whole-cell lysates were analyzed by Western blotting with the indicated antibodies. Numbers below the ERα, cIAP1, or XIAP panels represent ERα/actin, cIAP1/actin, or XIAP/actin ratios, normalized by designating the expression from the vehicle control condition as 100%. a, data in the bar graph are the mean ± S.D. (error bars) of three independent experiments.
Table 1.
Protein-knockdown activities and binding affinities of SNIPER(ER)s
| Compound no. | ERα DC50 |
IC50 (95% CI) |
IAP ligand |
|||||
|---|---|---|---|---|---|---|---|---|
| 4 h | 48 h | ERα | cIAP1 | cIAP2 | XIAP | Originator | Reference | |
| nm | nm | |||||||
| SNIPER(ER)-87 | <3 | 83.0 | 110 (58–200) | 450 (360–560) | 960 (600–1500) | 700 (510–960) | Novartis | WO2008016893 |
| SNIPER(ER)-104 | >100 | NDa | 61 (26–140) | >1000 | >1000 | >1000 | Novartis | WO2012080260 |
| SNIPER(ER)-105 | <3 | <3 | 69 (7–670) | 5.3 (4.6–6.1) | 7.2 (4.9–11) | 55 (38–80) | Novartis | WO2012080271 |
| SNIPER(ER)-110 | <3 | 7.7 | 120 (68–200) | 74 (62–89) | 73 (55–97) | 330 (250–430) | Abbott | Ref. 52; WO2016169989 |
| SNIPER(ER)-113 | <3 | 13.3 | 150 (82–290) | 85 (75–96) | 99 (58–200) | 810 (58–200) | Abbott | Ref. 52; WO2016169989 |
| SNIPER(ER)-118 | >100 | ND | 230 (73–540) | 140 (58–200) | 900 (58–200) | >1000 | AstraZeneca | Ref. 53 |
| SNIPER(ER)-119 | 4 | 15.7 | 200 (73–540) | 80 (62–100) | 48 (28–82) | 700 (490–1000) | Abbott | Ref. 52; WO2016169989 |
| SNIPER(ER)-121 | >100 | ND | ND | 650 (540–780) | >1000 | >1000 | AstraZeneca | Ref. 53 |
| SNIPER(ER)-126 | <3 | 3.7 | 83 (2–3800) | 68 (66–77) | 200 (150–280) | 490 (310–760) | Novartis | WO2008016893 |
| SNIPER(ER)-130 | 36.9 | ND | 41 (25–65) | 68 (60–77) | 28 (21–39) | 25 (17–37) | Genentech | Ref. 54 |
| SNIPER(ER)-131 | 33.8 | ND | 80 (39–160) | 25 (22–28) | 24 (18–32) | 140 (85–220) | Genentech | Ref. 55 |
| SNIPER(ER)-134 | >100 | ND | 47 (15–140) | 550 (450–670) | 360 (240–540) | >1000 | Abbott | Ref. 52 |
| SNIPER(ER)-136 | >100 | ND | 47 (25–89) | 890 (790–1000) | >1000 | >1000 | Genentech | Ref. 54; WO2006069063 |
a ND, not determined.
Table 1 also reports the binding affinities (IC50) of SNIPER (ER)s to ERα and IAPs in vitro. Because SNIPER(ER)-87 preferentially recruits XIAP to degrade ERα (13), we compared the binding affinity to XIAP with the DC50, for all SNIPER(ER)s. SNIPER(ER)s with effective knockdown activities (SNIPER(ER)-105, -110, -113, -119, and -126) exhibited affinities to XIAP that were greater than, or comparable with, that of SNIPER(ER)-87; in contrast, those with little or no ERα-knockdown activity (SNIPER(ER)-104, -118, -121, -134, and -136) exhibited lower binding affinities to XIAP. However, SNIPER(ER)-130 and -131 exhibited attenuated ERα-knockdown activities, despite their higher affinities for XIAP binding. These results suggest that an increased binding affinity of SNIPER(ER) for XIAP seems to be important for effective ERα knockdown but is not the sole determinant for degradation activity. Among the arene substitution isomers (SNIPER(ER)-110, -113, and -119), SNIPER(ER)-110 exhibited slightly better ability to degrade ERα; therefore, we chose SNIPER(ER)-110 as a representative. SNIPER(ER)-105, -110, and -126 also showed more potent ability to degrade ERα, compared with SNIPER(ER)-87, in assays involving other human ERα-positive breast tumor T47D and ZR-75-1 cells (Fig. 3).
Figure 3.
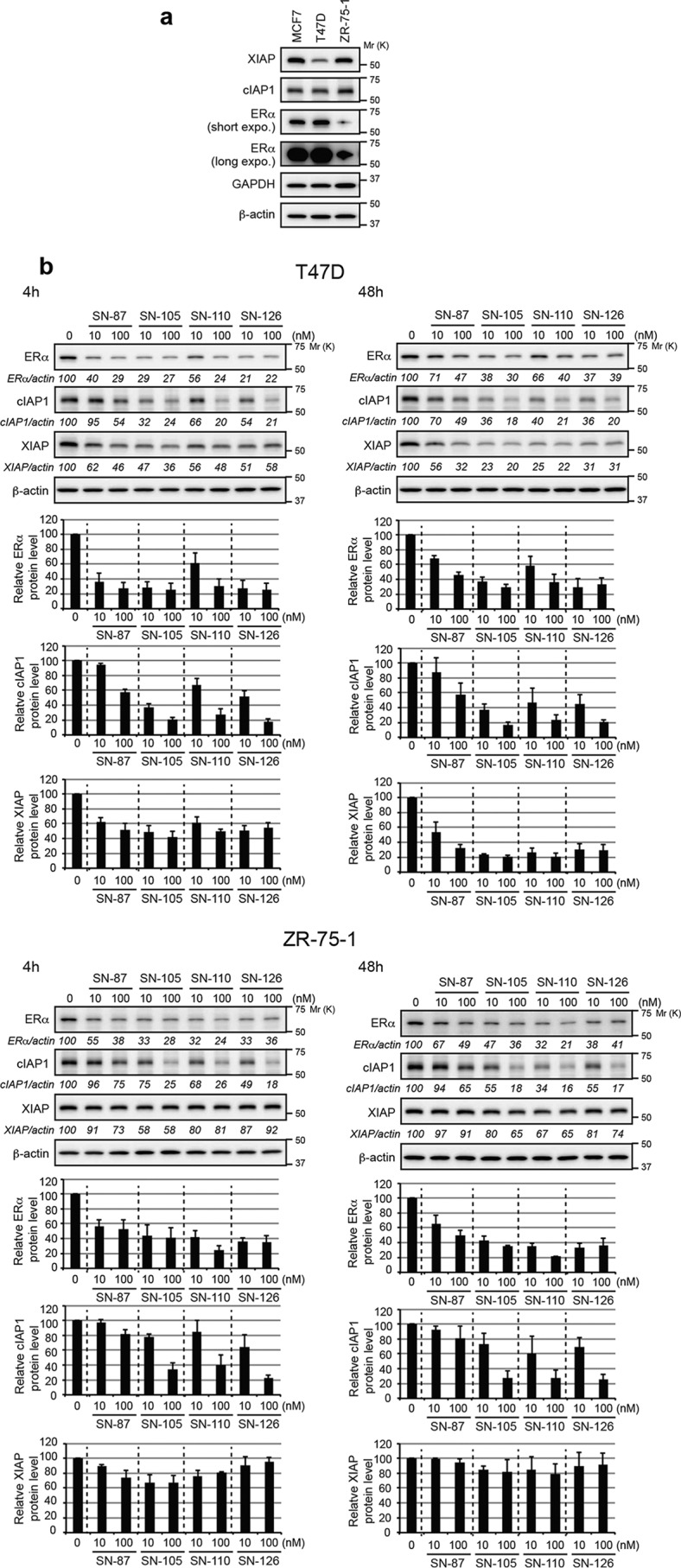
Protein-knockdown activities of SNIPER(ER)s in T47D and ZR-75-1 breast tumor cells. a, protein levels of IAPs and ERα in human ERα-positive breast tumor cells. b, knockdown activities of SNIPER(ER)s in T47D and ZR-75-1 cells. Cells were treated with the indicated concentrations of SNIPER(ER)s for the indicated periods. Whole-cell lysates were analyzed by Western blotting with the indicated antibodies. Numbers below the ERα, cIAP1, or XIAP panels represent ERα/actin, cIAP1/actin, or XIAP/actin ratios, normalized by designating the expression from the vehicle control condition as 100%. Data in the bar graph are the mean ± S.D. (error bars) of three independent experiments.
Degradation of cIAP1 and XIAP by SNIPER(ER)s
As observed in studies of many IAP antagonists, SNIPERs in our study rapidly induced proteasomal degradation of cIAP1, along with degradation of their target proteins (12–18). SNIPER(ER)-105, -110, -113, -119, and -126 each reduced the cIAP1 level more potently than SNIPER(ER)-87 (Figs. 2a and 3b), consistent with their increased binding affinities for cIAP1 (Table 1). Further, these SNIPER(ER)s reduced XIAP expression in MCF-7 cells after 48 h in a more potent fashion than SNIPER(ER)-87 (Fig. 2a). The reduction of XIAP by SNIPER(ER)s was prominent in T47D cells after 48 h but was weak after 4 h of exposure (Fig. 3b). The difference in the degradation pattern between cIAP1 and XIAP suggests that these IAPs are degraded through a different mechanism, as discussed below.
XIAP is required for the ERα degradation by SNIPER(ER)s
We previously reported that XIAP is preferentially recruited to ERα upon treatment with SNIPER(ER)-87 (13). To examine which IAP is recruited by the novel SNIPER(ER)s (SNIPER(ER)-105, -110, and -126) to induce ERα degradation, MCF-7 and T47D cells were treated with or without the SNIPER(ER)s in the presence of MG132, a proteasome inhibitor; cell lysates were analyzed by co-immunoprecipitation. Fig. 4a shows that, when cells were treated with SNIPER(ER)s, the anti-ERα precipitates contained both XIAP and cIAP1. We did not examine cIAP2, because it is not expressed in MCF-7 and T47D cells under normal cell culture and cIAP1-down-regulated conditions (Fig. S1, a–c). Upon comparison of IAP protein levels in total cellular lysates, we found that SNIPER(ER)s more effectively recruited XIAP to ERα, compared with cIAP1. Consistent with this, depletion of XIAP by siRNA substantially suppressed the ERα degradation induced in MCF-7 and T47D cells by our SNIPER(ER) molecules, whereas depletion of cIAP1 marginally suppressed the degradation (Fig. 4b). These results indicate that SNIPER(ER)-105, -110, and -126 preferentially recruit XIAP to degrade ERα in target cells, similar to the mechanism initiated by SNIPER(ER)-87.
Figure 4.

Role of XIAP in ERα degradation by SNIPER(ER)s. a, SNIPER(ER)s preferentially recruit XIAP to ERα. MCF-7 or T47D cells were treated with 10 nm SNIPER(ER)s in the presence of 10 μm MG132 for 3 h. Immunoprecipitates of anti-ERα (IP) and whole-cell lysates (total lysates) were analyzed by Western blotting (IB). b, depletion of XIAP suppresses the SNIPER(ER)-induced degradation of ERα. MCF-7 and T47D cells were transfected with the indicated siRNA for 42 h and treated with 10 nm SNIPER(ER)s for 3 h. Whole-cell lysates were analyzed by Western blotting with the indicated antibodies. Numbers below the ERα panel represent the ERα/actin ratio, normalized by designating the expression from the vehicle control condition as 100%. A mixture of three different siRNAs against each target was used to suppress expression.
SNIPER(ER)s induce apoptosis in tumor cells requiring IAPs for survival
Because ERα plays an important role in the proliferation of most primary breast tumors (56, 57), we examined the effect of SNIPER(ER)s on the growth of breast tumor cells. SNIPER(ER)s and fulvestrant, a clinically approved inducer of ERα degradation, both efficiently suppressed the growth of ERα-positive breast tumor cells (MCF-7 and T47D) but not of ERα-negative breast tumor cells (MDA-MB-231) (Fig. 5a and Fig. S2). Notably, SNIPER(ER)-105, -110, and -126 exhibited superior antiproliferative effects on MCF-7 cells at concentrations above 50 nm, compared with SNIPER(ER)-87 (Fig. 5a and Fig. S2). In a microscopic analysis, we observed that substantial number of MCF-7 cells, but not T47D cells, underwent cell death upon treatment with SNIPER(ER)-105, -110, and -126 (Fig. 5b). Induction of apoptotic cell death in MCF-7 cells by SNIPER(ER)-105, -110, and -126 was confirmed by flow cytometric analysis and annexin V/propidium iodide (PI) staining (Fig. 5 (c and d) and Fig. S3). Consistent with this, a 100 nm concentration of these SNIPER(ER)s activated caspase-7 and caspase-6 (detected by reduction of their precursor forms), which resulted in the cleavage of two caspase substrates, PARP and lamin B1, in MCF-7 cells (Fig. 5e). These results indicate that SNIPER(ER)-105, -110, and -126 each induced apoptosis in MCF-7 cells more effectively than SNIPER(ER)-87; apoptosis was not observed in T47D cells.
Figure 5.

Induction of apoptosis by SNIPER(ER)s in MCF-7 breast tumor cells. a, antiproliferative effects on ERα-positive human breast tumor cells by SNIPER(ER)s. Cells were treated with the indicated concentrations of SNIPER(ER)s or fulvestrant for 72 h; the remaining living cells were stained with crystal violet. b–e, SNIPER(ER)s induce apoptosis in MCF-7 cells. MCF-7 or T47D cells were treated with 100 nm SNIPER(ER)s or fulvestrant for 48 h. b, phase-contrast images were obtained. Scale bars, 50 μm. c, cell cycle distribution was quantified by MultiCycle software. d, cell death was determined by Annexin V and PI staining. e, activation of caspases was analyzed by Western blotting.
To understand why these new SNIPER(ER)s effectively induce apoptosis in MCF-7 cells, but not in T47D cells, and because SNIPER(ER)-105, -110, and -126 each induced the degradation of cIAP1 and XIAP more potently than SNIPER(ER)-87 (Figs. 2a and 3b), we investigated the effects of IAP depletion in MCF-7 and T47D cells. In MCF-7 cells, individual silencing of cIAP1 and XIAP by siRNA slightly reduced the number of cells; combined silencing of cIAP1 and XIAP reduced the number of cells by ∼50%, compared with untreated cells (Fig. 6a), and was accompanied by caspase activation, as demonstrated by cleavage of lamin B1 and PARP (Fig. 6b). In contrast, similar silencing of IAPs in T47D cells neither reduced cell number nor activated caspases. When ERα was silenced in MCF-7 and T47D cells, the numbers of cells in both cell lines were effectively reduced without a requirement for caspase activation. However, triple silencing of ERα, XIAP, and cIAP1 efficiently reduced MCF-7 cell number, compared with T47D cells; further, caspase activation was observed only in MCF-7 cells. Additionally, an IAP antagonist, LCL161, sensitized tumor necrosis factor α (TNFα)-dependent apoptosis in MCF-7 cells, but not T47D cells (Fig. S4). These results suggest that MCF-7 cells, but not T47D cells, require IAPs for survival; thus, IAPs could be involved in the selective induction of apoptosis in MCF-7 cells that have been exposed to SNIPER(ER)-105, -110, and -126.
Figure 6.

Depletion of IAPs induces apoptosis in MCF-7 but not T47D cells. MCF-7 or T47D cells were transfected with the indicated siRNA for 72 h. a, cell proliferation was evaluated by a cell viability assay. Data in the bar graph are the mean ± S.D. (error bars) of a experiment performed in triplicate. b, activation of caspases was analyzed by Western blotting. A mixture of three different siRNAs against each target was used to suppress expression.
In vivo protein-knockdown activities and antitumor activities of SNIPER(ER)s
We measured the metabolic stabilities of SNIPER(ER)-87, -105, -110, -113, -119, and -126 in liver microsomes (Table S2) and carefully selected SNIPER(ER)-110 and -126 as test compounds for in vivo comparison with SNIPER(ER)-87. When female BALB/c mice were intraperitoneally injected with SNIPER(ER)-87, -110, and -126 (30 mg/kg body weight), ERα protein levels in ovarian tissue were effectively reduced by all SNIPERs (Fig. 7a). To evaluate SNIPER(ER)-knockdown activities in a tumor model, we next applied MCF-7 breast tumor xenografts to nude mice. The ERα protein levels in orthotopic tumors were reduced by 40–50% in SNIPER(ER)-treated mice, compared with those in vehicle-treated mice (Fig. 7, b and c); SNIPER(ER)-110 significantly showed a significant increase in protein-knockdown activity at 48 h, compared with SNIPER(ER)-87. Treatment with SNIPER(ER)-87, -110, and -126 each reduced cIAP1 and XIAP protein levels in tumor xenografts (Fig. 7, b and c), consistent with the results of our in vitro analyses (Fig. 2a). Notably, SNIPER(ER)-110 again exhibited a significant increase in the ability to reduce cIAP1 and XIAP at 24 and 48 h, compared with SNIPER(ER)-87.
Figure 7.

In vivo protein knockdown by SNIPER(ER)s in mice. a, in vivo protein knockdown in ovarian tissue. Female BALB/c mice were injected with vehicle or 30 mg/kg SNIPER(ER)s. After 24 or 48 h, the mice were sacrificed, and their ovaries were collected and analyzed by Western blotting with the indicated antibodies. Numbers below the ERα panel represent the ERα/actin ratio, normalized by designating the expression from the vehicle control condition as 100% (average of each group). b and c, MCF-7 human breast tumor cells were inoculated into mammary fat pads of 6-week-old female BALB/c nude mice. The tumor-bearing mice were intraperitoneally injected with SNIPER(ER)s. After 24 or 48 h, mice were sacrificed, and ERα protein levels in tumor xenografts were analyzed by Western blotting. Bar graphs represent the mean ± S.D. (error bars) of each group. *, p < 0.05 in two-tailed Student's t test compared with vehicle control. #, p < 0.05 in two-tailed Student's t test compared with SNIPER(ER)-87.
To demonstrate the therapeutic significance of these findings, we also evaluated the in vivo antitumor activity of SNIPER(ER)-110, compared with SNIPER(ER)-87, in an MCF-7 tumor xenograft model. When tumor-bearing mice received daily intraperitoneal injections of SNIPER(ER)-87 or SNIPER(ER)-110 (30 mg/kg body weight) for 16 days, SNIPER(ER)-110 attenuated MCF-7 tumor progression more effectively than SNIPER(ER)-87, as shown in measurements of tumor volume and tumor weight (Fig. 8, a–c). Notably, no obvious toxicities, including body weight changes, were observed during treatment (Fig. 8d). Thus, compared with SNIPER(ER)-87, SNIPER(ER)-110 showed superior antitumor activity against ERα-positive breast tumors that require IAPs for cellular survival.
Figure 8.
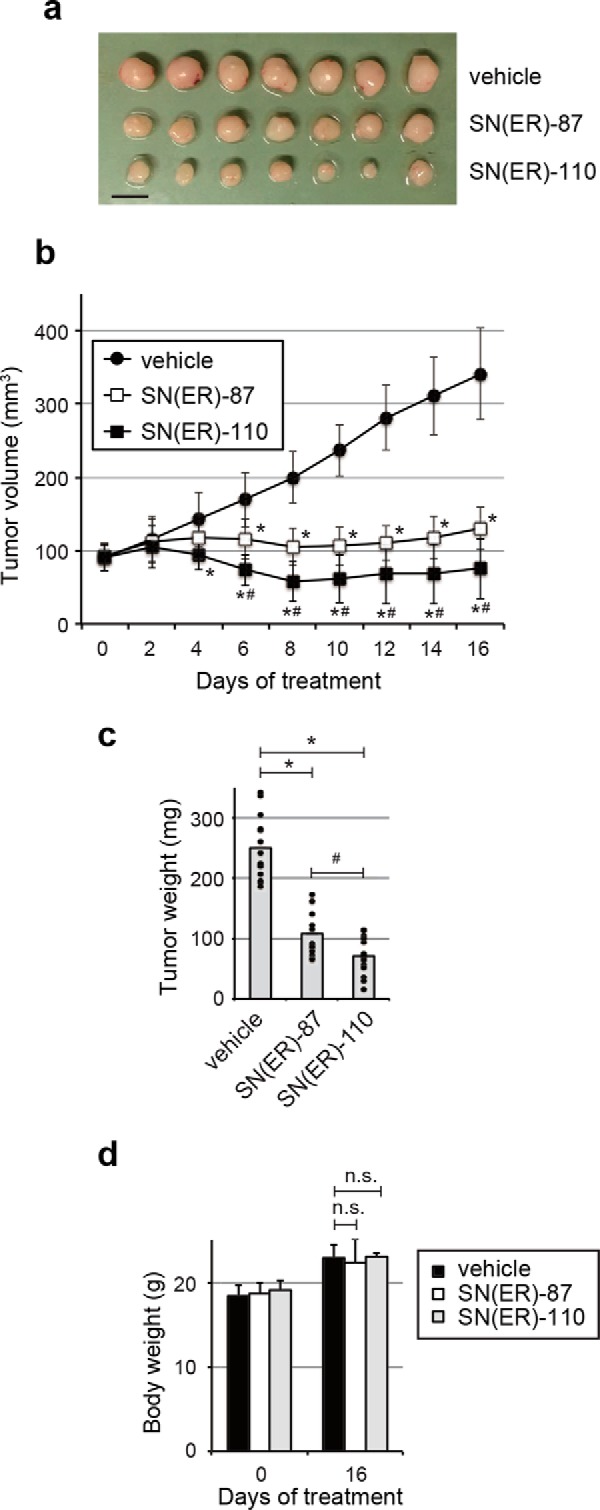
Antitumor activity of SNIPER(ER)s. Growth inhibition by SNIPER(ER)s of MCF-7 orthotopic breast tumor xenografts in nude mice. a, representative tumors. Scale bar, 10 mm. The tumor volume (b) or weight (c) represents the mean ± S.D. (error bars) of each group (mice, n = 7 each; tumors, n = 14 each; *, p < 0.05 in two-tailed Student's t test compared with vehicle; #, p < 0.05 in two-tailed Student's t test compared with SNIPER(ER)-87). Mice were administered vehicle or SNIPER(ER)s (30 mg/kg) intraperitoneally, every 24 h. d, treatment with SNIPER(ER)s did not induce significant body weight loss in mice after 16 days.
Discussion
Chemical protein knockdown technologies, utilizing chimeric molecules, such as PROTACs and SNIPERs, have been recently developed to induce degradation of target proteins in cells (5–7). Our SNIPER compounds degrade cIAP1 and XIAP along with their initial target proteins, which could be advantageous in attempts to kill cancer cells because of the frequent overexpression of IAPs to suppress apoptosis in cancer cells. In this study, we developed novel SNIPER(ER)s by incorporating various IAP ligands with higher binding affinities for IAP proteins. The resulting SNIPER(ER)s (SNIPER(ER)-105, -110, and -126) each induce degradation of ERα, cIAP1, and XIAP more potently than SNIPER(ER)-87, resulting in the induction of apoptosis in MCF-7 breast tumor cells. In an in vivo tumor xenograft model, the novel SNIPER(ER)s exhibited activity to reduce ERα and IAPs that was respectively comparable with, and better than, that of SNIPER(ER)-87; further, the novel SNIPER(ER)-110 showed superior antitumor activity against MCF-7 tumor xenograft, compared with SNIPER(ER)-87. These results suggest that incorporating IAP ligands with higher binding affinities may be a promising strategy to develop SNIPERs with better protein knockdown and antitumor activities.
The novel SNIPER(ER)s also exhibited better protein-knockdown activity in other ERα-positive breast tumor cells, compared with SNIPER(ER)-87 (Fig. 3). However, the novel SNIPER(ER)s did not exhibit improved inhibition of growth or induction of apoptosis in T47D cells (Fig. 5), which contrasts with their activities in MCF-7 cells. We speculate that the differences in sensitivity between the two cell lines derive from differing need for IAPs, because depletion of IAPs induces apoptosis in MCF-7 cells, but not in T47D cells (Fig. 6). The role of IAPs in cancer cell survival has been suggested in many papers (32, 42–45, 58), and IAP antagonists have shown antitumor activity against many tumor cells, including breast tumor (46, 47, 59). Therefore, we hypothesize that the ability of the SNIPERs to degrade IAPs could be advantageous in treatment of cancer cells, especially those dependent on the activity of IAPs. Recently, it has been reported that the different molecular signature other than IAP frequency also specifies a sensitivity to IAP antagonists in cancer cells; the mechanism depends on autocrine release of TNFα (60–64), constitutive ubiquitylation of receptor-interacting protein kinase 1 (RIP1) (61), and expression of cellular FLICE-inhibitory protein (c-FLIP) (60) and leucine-rich repeats and immunoglobulin-like domains protein 1 (LRIG1) (65). Thus, these signatures could be used to predict sensitivity to SNIPERs as well.
Although SNIPER(ER)s induce degradation of ERα, cIAP1, and XIAP, the degradation mechanism appears different for each protein. Degradation of ERα appears to require formation of a ternary complex that is composed of ERα, SNIPER, and XIAP; we suspect this because a treatment with the individual ligand or with an structural SNIPER(ER) analog that cannot bind to IAPs does not induce degradation of ERα (13) (Fig. S5). The pharmacological hook effect that is observed during SNIPER(ER)-induced ERα degradation further supports the hypothesis that ternary complex formation is required (13). In contrast, the degradation of cIAP1 is triggered by binding of the IAP antagonist module to the BIR3 domain of cIAP1, thus inducing autoubiquitylation. In this case, the ternary complex is not required, and no hook effect is observed (13). Notably, the degradation mechanism of XIAP remains ambiguous. Degradation of XIAP was induced by SNIPER(ER)s but not by IAP antagonists nor by the mixture of IAP antagonist and 4-hydroxytamoxifen (Fig. S5), suggesting that the ternary complex is required for XIAP degradation. Consistent with this hypothesis, XIAP degradation by SNIPER(ER)-87 was not observed in the ERα-negative MDA-MB-231 breast cancer cells (Fig. S5). Thus, XIAP degradation seems to require the same ternary complex formation that is required for ERα degradation. However, XIAP degradation requires more time and is inefficient, compared with degradation of the ERα target protein (Figs. 2a and 3). It is likely that part of the population of XIAP in the ternary complex is subjected to ubiquitylation and proteasomal degradation. Because XIAP degradation was more prominently observed in T47D cells than in MCF-7 and ZR-75-1 cells, we measured the expression of ERα, cIAP1, and XIAP in these cells (Fig. S6). We found that the ratio of XIAP to ERα is one-third and one-thirteenth less in T47D cells than in MCF-7 cells and in XR-75-1 cells, respectively, suggesting that the lower expression of XIAP may increase the significance of its degradation in T47D cells. Thus, although the degradation mechanisms differ, SNIPER(ER)s induce degradation of ERα, cIAP1, and XIAP, culminating in an anti-tumor effect that is directed against breast cancer cells.
It has been reported that ERα translocates from the cytosol to the nucleus upon binding with tamoxifen (66, 67). This is also observed after treatment with SNIPER(ER)s, because the relative amount of ERα increased in the nucleus, compared with the cytosol, upon treatment with SNIPER(ER)s; this was more clearly observed in the presence of MG132 (Fig. S7, a and b). Interestingly, the amount of XIAP increased in the nucleus upon treatment with SNIPER(ER)s; this was also more clearly observed in the presence of MG132. These results suggest that when a SNIPER(ER) penetrates a cell, it induces formation of the ternary complex composed of ERα, SNIPER(ER), and XIAP in the cytosol; ERα then translocates to the nucleus together with the bound XIAP. Thus, in the presence of SNIPER(ER), ERα interacts with XIAP sufficiently to tether XIAP in the nucleus, consistent with the preferential recruitment of XIAP, but not cIAP1, to ERα.
In summary, we have developed potent SNIPER(ER)s by incorporation of high-affinity IAP ligands. These SNIPER(ER)s induce degradation of ERα and IAPs and exhibit antitumor activity against breast tumors that require IAPs for survival. These IAP ligand modules could be utilized to develop novel SNIPERs with activity against various oncogenic proteins and enhanced antitumor capabilities.
Experimental procedures
Chemistry
Chemical synthesis and physicochemical data for SNIPER compounds are provided in the supporting information.
Cell culture
Human breast carcinoma MCF-7, T47D, and ZR-75-1 cells were maintained in RPMI 1640 medium containing 10% fetal bovine serum (FBS) and 100 μg/ml kanamycin. Human breast carcinoma MDA-MB-231 cells were maintained in Dulbecco's modified Eagle's medium containing 10% FBS and 100 μg/ml kanamycin. All of the cell lines were purchased from ATCC (Manassas, VA), and their cell authentications were confirmed by morphology, karyotyping, and PCR-based approaches in ATCC. Cells were treated with various concentrations of SNIPER compounds for the indicated times.
Western blotting
Cells were lysed with SDS lysis buffer (0.1 m Tris-HCl, pH 8.0, 10% glycerol, 1% SDS) and immediately boiled for 10 min to obtain clear lysates. Nuclear and cytoplasmic extracts were prepared with NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific). Protein concentration was measured by the BCA method (Pierce); lysates containing equal amounts of proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Darmstadt, Germany) for Western blot analysis using the appropriate antibodies. Immunoreactive proteins were visualized using the Immobilon Western chemiluminescent HRP substrate (Millipore) or Clarity Western ECL substrate (Bio-Rad); light emission intensity was quantified using an LAS-3000 lumino-image analyzer equipped with Image Gauge version 2.3 software (Fuji, Tokyo, Japan). The antibodies used in this study were as follows: anti-ERα rabbit monoclonal antibody (mAb) (Cell Signaling Technology (Danvers, MA), 8644), anti-ERα rabbit polyclonal antibody (pAb) (Santa Cruz Biotechnology, Inc. (Dallas, TX), sc-542 and sc-543), anti-cIAP1 goat pAb (R&D Systems (Minneapolis, MN), AF8181), anti-cIAP1 rat mAb (Enzo Life Sciences (Farmingdale, NY), 1E1-1-10), anti-β-actin mouse mAb (Sigma, A5316), anti-XIAP rabbit pAb (Cell Signaling Technology, 2042), anti-GAPDH rabbit pAb (Santa Cruz Biotechnology, sc-25778), anti-lamin B1 goat pAb (Santa Cruz Biotechnology, sc-6216), anti-histone H3 goat pAb (Santa Cruz Biotechnology, sc-8654), anti-PARP rabbit pAb (Cell Signaling Technology, 9542), anti-caspase-6 rabbit pAb (Cell Signaling Technology, 9762), anti-caspase-7 rabbit mAb (Cell Signaling Technology, 12827).
Measurement of binding affinities of ERα and IAPs
The bindings between test compounds and cIAP1, cIAP2, or XIAP were determined by a time-resolved FRET assay using His-IAP proteins (XIAP, cIAP1, or cIAP2), FITC-Smac, Tb-SA, and biotinylated anti-His antibody, as described previously (14).
The binding between test compounds and the human ERα protein was determined using the PolarScreenTM Estrogen Receptor-α Competitor Assay Green (Thermo Fisher Scientific, A15882) containing recombinant ERα full-length protein, Fluormone ES2 Green2, and ES2 screening buffer. Purified ERα and Fluormone ES2 were diluted with assay buffer to final concentrations of 25 and 4.5 nm, respectively, and 4 μl of each dilution was added to each well of a 384-well black low-volume assay plate (Greiner, 784076). Then 2 μl of the ES2 screening buffer, containing test compounds or DMSO, was added to the well. The plate was subjected to centrifugation and incubated at room temperature for 1 h; then the intensity of the fluorescence polarization signal was measured by a plate reader (Envision, PerkinElmer Life Sciences). Wells containing ERα and Fluormone ES2 were used as a positive control, and wells containing only Fluormone ES2 were used as a negative control. IC50 values and 95% confidence interval were calculated by XLfit (ID Business Solutions, fit model 204) from the data expressed as percentage of control inhibition.
Immunoprecipitation
MCF-7 or T47D cells were treated with the indicated concentrations of the indicated compounds, in combination with 10 μm MG132, for 3 h. Cells were lysed using immunoprecipitation lysis buffer (10 mm Hepes, pH 7.4, 142.5 mm KCl, 5 mm MgCl2, 1 mm EGTA, and 0.1% Triton X-100), containing protease inhibitor mixtures, rotated for 15 min at 4 °C, and centrifuged at 15,000 rpm for 10 min at 4 °C to obtain the supernatants. Lysates that had been precleared with naked protein G-Sepharose were immunoprecipitated with protein G-Sepharose beads that were preincubated with anti-ERα rabbit pAb (Santa Cruz Biotechnology, sc-543) for 2 h at 4 °C. The precipitates were washed with immunoprecipitation lysis buffer four times and analyzed by Western blotting.
siRNA transfection
MCF-7 or T47D cells were transiently transfected with a gene-specific siRNA or a negative control siRNA (Qiagen, Valencia, CA) using Lipofectamine RNAi MAX reagent (Life Technologies, Inc.). The siRNA sequences used in this study were as follows: human ERα-1 (5′-CGACAUGCUGCUGGCUACAUCAUCU-3′), ERα-2 (5′-UCACAGACACUUUGAUCCACCUGAU-3′), ERα-3 (5′-GACCGAAGAGGAGGGAGAAUGUUGA-3′), cIAP1–1 (5′-UCUAGAGCAGUUGAAGACAUCUCUU-3′), cIAP1-2 (5′-GCUGUAGCUUUAUUCAGAAUCUGGU-3′), cIAP1–3 (5′-GGAAAUGCUGCGGCCAACAUCUUCA-3′), XIAP-1 (5′-ACACUGGCACGAGCAGGGUUUCUUU-3′), XIAP-2 (5′-GAAGGAGAUACCGUGCGGUGCUUUA-3′), and XIAP-3 (5′-CCAGAAUGGUCAGUACAAAGUUGAA-3′).
Cell viability assay
Cell viability was evaluated by crystal violet staining. Cells were treated with graded concentrations of the SNIPER compounds for 72 h and then stained with 0.1% crystal violet (Wako, Osaka, Japan) in 1% ethanol for 15 min at room temperature. Cells were rinsed thoroughly with distilled water and then lysed in 1% SDS. The absorbance of the cell lysate at 600 nm was measured using an EnVision multilabel plate reader (PerkinElmer Life Sciences).
In vivo protein knockdown
All procedures of the animal experiments were reviewed and approved by the Institutional Animal Experiment Review Committee of the National Institute of Health Sciences (NIHS), and those experiments were conducted in accordance with the guidelines approved by the Care and Use of Animals published by the Institutional Animal Ethical Committee. Mice were housed in pathogen-free animal facilities at the NIHS with 12-h light/dark cycles and were fed rodent chow and water ad libitum. For Fig. 7a, female 6-week-old BALB/c mice (Clea Japan, Tokyo, Japan) were randomized and divided into eight treatment groups (n = 3) and then treated with vehicle or 30 mg/kg SNIPER(ER)s for 24 h or 48 h. For Fig. 7 (b and c), each suspension of 1 × 107 MCF-7 cells was mixed with an equal volume of Matrigel (Corning Inc.) and injected (100 μl total) into the left and right mammary fat pads of 6-week-old female BALB/c nude mice (Clea Japan). After cell inoculation, β-estradiol solution was subcutaneously injected into the neck twice at intervals of 6 days. Fourteen days after the last β-estradiol injection, tumor-bearing mice were randomized and divided into two or three treatment groups as follows: for Fig. 7b: 1) vehicle treatment for 24 h (n = 3), 2) 30 mg/kg SNIPER(ER)-87 treatment for 24 h (n = 2), 3) 30 mg/kg SNIPER(ER)-110 treatment for 24 h (n = 2), and 4) 30 mg/kg SNIPER(ER)-126 treatment for 24 h (n = 2); for Fig. 7c: 1) vehicle treatment for 48 h (n = 3), 2) 30 mg/kg SNIPER(ER)-87 treatment for 48 h (n = 3), 3) 30 mg/kg SNIPER(ER)-110 treatment for 48 h (n = 3), and 4) 30 mg/kg SNIPER(ER)-126 treatment for 48 h (n = 3). Compounds were administered via intraperitoneal injection. After the indicated treatment periods, the mice were sacrificed, and tissues were excised. Total lysates from the ovaries and tumors were analyzed by Western blotting.
In vivo tumor growth inhibition
Each suspension of 1 × 107 MCF-7 cells was mixed with an equal volume of Matrigel (BD Biosciences) and inoculated (100 μl total) into the left and right mammary fat pads of 6-week-old female BALB/c nude mice (Clea Japan) that had received an β-estradiol pellet (6 μg/day) (Innovative Research of America, Sarasota, FL) under the neck skin. After 4 days, mice bearing tumors of ∼100 mm3 in size were randomized and divided into two groups (n = 7). One group served as a control for the dosing vehicle; the other groups were administered SNIPER(ER)-87 or SNIPER(ER)-110 (30 mg/kg, intraperitoneally, every 24 h), respectively. Tumor volumes were measured every 2 days using a caliper and calculated according to the standard formula: (length × width2)/2. At 16 days, mice were sacrificed, and the tumors were weighed and excised.
Cell cycle analysis
After treatment, cells were gently trypsinized and washed with serum-containing medium. Cells were collected by centrifugation and then washed with PBS and fixed in 70% ice-cold ethanol for 1 h on ice. The cells were then washed, treated with 1 mg/ml RNase A for 1 h at 37 °C, and stained in PI solution (50 μg/ml in 0.1% sodium citrate, 0.1% NP-40). The stained cells were analyzed in a FACScan flow cytometer (BD Biosciences).
Measurement of apoptosis by flow cytometer
Apoptosis was analyzed with an annexin V-FITC apoptosis detection kit (BioVision). After treatment, cells were gently trypsinized and washed with serum-containing medium. Cells were collected by centrifugation and then washed with PBS and resuspended in binding buffer. The cells were stained with annexin V-FITC and PI at room temperature for 5 min in the dark, according to the manufacturer's instructions, and analyzed in a FACScan flow cytometer (BD Biosciences).
Statistical analysis
Student's t test was used to evaluate differences among the experimental groups. Values of p < 0.05 were considered significant.
Author contributions
N. O. and M. N. designed the experiments; Y. M., K. N., K. S., I. F., M. I., Y. H., Y. I., H. N., and N. C. designed and synthesized the compounds; N. O., K. O., N. S., T. H., T. S., O. S., and R. K. performed the experiments and analyzed the data; N. O., K. S., O. U., H. N., and M. N. wrote the manuscript; and M. N. supervised all research. All authors discussed and checked the manuscript.
Supplementary Material
Acknowledgments
We thank Mariko Seki for measurement of the protein-knockdown activities and Ryan Chastain-Gross, Ph.D. (Edanz Group) for editing a draft of the manuscript.
This work was supported in part by grants from the Japan Society for the Promotion of Science (KAKENHI Grants 26860049 (to N. O.) and 16H05090 and 16K15121 (to M. N.)), the Japan Agency for Medical Research and Development (AMED Grants JP 17ak0101073 (to M. N.) and JP17cm0106124 and JP17ak0101073 (to N. O.)), and Takeda Science Foundation (to N. O.). Y. M., K. N., K. S., O. U., I. F., M. I., Y. H., T. S., O. S., R. K., Y. I., H. N., and N. C. are employees of Takeda Pharmaceutical Co. Ltd.
This article contains Tables S1 and S2 and Figs. S1–S7.
- PROTAC
- proteolysis targeting chimera
- BIR
- baculoviral IAP repeat
- BRD4
- bromodomain-containing protein 4
- cIAP1
- cellular inhibitor of apoptosis protein 1
- CRBN
- cereblon
- DIABLO
- direct IAP-binding protein with low pI
- FBS
- fetal bovine serum
- mAb
- monoclonal antibody
- pAb
- polyclonal antibody
- PI
- propidium iodide
- SMAC
- second mitochondria-derived activator of caspases
- SNIPER
- specific and non-genetic IAP-dependent protein eraser
- VHL
- von Hippel–Lindau
- XIAP
- X-linked inhibitor of apoptosis protein
- ERα
- estrogen receptor α
- PARP
- poly(ADP-ribose) polymerase
- TNF
- tumor necrosis factor.
References
- 1. Wells J. A., and McClendon C. L. (2007) Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450, 1001–1009 10.1038/nature06526 [DOI] [PubMed] [Google Scholar]
- 2. Arkin M. R., and Wells J. A. (2004) Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat. Rev. Drug Discov. 3, 301–317 10.1038/nrd1343 [DOI] [PubMed] [Google Scholar]
- 3. Mansoori B., Sandoghchian Shotorbani S., and Baradaran B. (2014) RNA interference and its role in cancer therapy. Adv. Pharm. Bull. 4, 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ozcan G., Ozpolat B., Coleman R. L., Sood A. K., and Lopez-Berestein G. (2015) Preclinical and clinical development of siRNA-based therapeutics. Adv. Drug Deliv. Rev. 87, 108–119 10.1016/j.addr.2015.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ohoka N., Shibata N., Hattori T., and Naito M. (2016) Protein knockdown technology: application of ubiquitin ligase to cancer therapy. Curr. Cancer Drug Targets 16, 136–146 10.2174/1568009616666151112122502 [DOI] [PubMed] [Google Scholar]
- 6. Toure M., and Crews C. M. (2016) Small-molecule PROTACS: new approaches to protein degradation. Angew. Chem. Int. Ed. Engl. 55, 1966–1973 10.1002/anie.201507978 [DOI] [PubMed] [Google Scholar]
- 7. Lai A. C., and Crews C. M. (2017) Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 16, 101–114 10.1038/nrd.2016.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bondeson D. P., Mares A., Smith I. E., Ko E., Campos S., Miah A. H., Mulholland K. E., Routly N., Buckley D. L., Gustafson J. L., Zinn N., Grandi P., Shimamura S., Bergamini G., Faelth-Savitski M., et al. (2015) Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11, 611–617 10.1038/nchembio.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu J., Qian Y., Altieri M., Dong H., Wang J., Raina K., Hines J., Winkler J. D., Crew A. P., Coleman K., and Crews C. M. (2015) Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 22, 755–763 10.1016/j.chembiol.2015.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Winter G. E., Buckley D. L., Paulk J., Roberts J. M., Souza A., Dhe-Paganon S., and Bradner J. E. (2015) Drug Development: phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 10.1126/science.aab1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soares P., Gadd M. S., Frost J., Galdeano C., Ellis L., Epemolu O., Rocha S., Read K. D., and Ciulli A. (2018) Group-based optimization of potent and cell-active inhibitors of the von Hippel-Lindau (VHL) E3 ubiquitin ligase: structure-activity relationships leading to the chemical probe (2S,4R)-1-((S)-2-(1-cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298). J. Med. Chem. 61, 599–618 10.1021/acs.jmedchem.7b00675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Itoh Y., Ishikawa M., Naito M., and Hashimoto Y. (2010) Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 132, 5820–5826 10.1021/ja100691p [DOI] [PubMed] [Google Scholar]
- 13. Ohoka N., Okuhira K., Ito M., Nagai K., Shibata N., Hattori T., Ujikawa O., Shimokawa K., Sano O., Koyama R., Fujita H., Teratani M., Matsumoto H., Imaeda Y., Nara H., et al. (2017) In vivo knockdown of pathogenic proteins via specific and nongenetic inhibitor of apoptosis protein (IAP)-dependent protein erasers (SNIPERs). J. Biol. Chem. 292, 4556–4570 10.1074/jbc.M116.768853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shibata N., Miyamoto N., Nagai K., Shimokawa K., Sameshima T., Ohoka N., Hattori T., Imaeda Y., Nara H., Cho N., and Naito M. (2017) Development of protein degradation inducers of oncogenic BCR-ABL protein by conjugation of ABL kinase inhibitors and IAP ligands. Cancer Sci. 108, 1657–1666 10.1111/cas.13284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimokawa K., Shibata N., Sameshima T., Miyamoto N., Ujikawa O., Nara H., Ohoka N., Hattori T., Cho N., and Naito M. (2017) Targeting the allosteric site of oncoprotein BCR-ABL as an alternative strategy for effective target protein degradation. ACS Med. Chem. Lett. 8, 1042–1047 10.1021/acsmedchemlett.7b00247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ohoka N., Nagai K., Shibata N., Hattori T., Nara H., Cho N., and Naito M. (2017) SNIPER(TACC3) induces cytoplasmic vacuolization and sensitizes cancer cells to Bortezomib. Cancer Sci. 108, 1032–1041 10.1111/cas.13198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Okuhira K., Ohoka N., Sai K., Nishimaki-Mogami T., Itoh Y., Ishikawa M., Hashimoto Y., and Naito M. (2011) Specific degradation of CRABP-II via cIAP1-mediated ubiquitylation induced by hybrid molecules that crosslink cIAP1 and the target protein. FEBS Lett. 585, 1147–1152 10.1016/j.febslet.2011.03.019 [DOI] [PubMed] [Google Scholar]
- 18. Okuhira K., Demizu Y., Hattori T., Ohoka N., Shibata N., Nishimaki-Mogami T., Okuda H., Kurihara M., and Naito M. (2013) Development of hybrid small molecules that induce degradation of estrogen receptor-α and necrotic cell death in breast cancer cells. Cancer Sci. 104, 1492–1498 10.1111/cas.12272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Demizu Y., Ohoka N., Nagakubo T., Yamashita H., Misawa T., Okuhira K., Naito M., and Kurihara M. (2016) Development of a peptide-based inducer of nuclear receptors degradation. Bioorg. Med. Chem. Lett. 26, 2655–2658 10.1016/j.bmcl.2016.04.013 [DOI] [PubMed] [Google Scholar]
- 20. Ohoka N., Misawa T., Kurihara M., Demizu Y., and Naito M. (2017) Development of a peptide-based inducer of protein degradation targeting NOTCH1. Bioorg. Med. Chem. Lett. 27, 4985–4988 10.1016/j.bmcl.2017.10.011 [DOI] [PubMed] [Google Scholar]
- 21. Sun B., Fiskus W., Qian Y., Rajapakshe K., Raina K., Coleman K. G., Crew A. P., Shen A., Saenz D. T., Mill C. P., Nowak A. J., Jain N., Zhang L., Wang M., Khoury J. D., et al. (2018) BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 32, 343–352 10.1038/leu.2017.207 [DOI] [PubMed] [Google Scholar]
- 22. Remillard D., Buckley D. L., Paulk J., Brien G. L., Sonnett M., Seo H. S., Dastjerdi S., Wühr M., Dhe-Paganon S., Armstrong S. A., and Bradner J. E. (2017) Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angew. Chem. Int. Ed. Engl. 56, 5738–5743 10.1002/anie.201611281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Winter G. E., Mayer A., Buckley D. L., Erb M. A., Roderick J. E., Vittori S., Reyes J. M., di Iulio J., Souza A., Ott C. J., Roberts J. M., Zeid R., Scott T. G., Paulk J., Lachance K., et al. (2017) BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell 67, 5–18.e19 10.1016/j.molcel.2017.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schiedel M., Herp D., Hammelmann S., Swyter S., Lehotzky A., Robaa D., Oláh J., Ovádi J., Sippl W., and Jung M. (2018) Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). J. Med. Chem. 61, 482–491 10.1021/acs.jmedchem.6b01872 [DOI] [PubMed] [Google Scholar]
- 25. Zhou B., Hu J., Xu F., Chen Z., Bai L., Fernandez-Salas E., Lin M., Liu L., Yang C. Y., Zhao Y., McEachern D., Przybranowski S., Wen B., Sun D., and Wang S. (2018) Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J. Med. Chem. 61, 462–481 10.1021/acs.jmedchem.6b01816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crew A. P., Raina K., Dong H., Qian Y., Wang J., Vigil D., Serebrenik Y. V., Hamman B. D., Morgan A., Ferraro C., Siu K., Neklesa T. K., Winkler J. D., Coleman K. G., and Crews C. M. (2018) Identification and characterization of Von Hippel-Lindau-recruiting proteolysis targeting chimeras (PROTACs) of TANK-binding kinase. J. Med. Chem. 61, 583–598 10.1021/acs.jmedchem.7b00635 [DOI] [PubMed] [Google Scholar]
- 27. Ohoka N., Nagai K., Hattori T., Okuhira K., Shibata N., Cho N., and Naito M. (2014) Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin-proteasome pathway. Cell Death Dis. 5, e1513 10.1038/cddis.2014.471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shibata N., Nagai K., Morita Y., Ujikawa O., Ohoka N., Hattori T., Koyama R., Sano O., Imaeda Y., Nara H., Cho N., and Naito M. (2018) Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands. J. Med. Chem. 61, 543–575 10.1021/acs.jmedchem.7b00168 [DOI] [PubMed] [Google Scholar]
- 29. Chan K. H., Zengerle M., Testa A., and Ciulli A. (2018) Impact of target warhead and linkage vector on inducing protein degradation: comparison of bromodomain and extra-terminal (BET) degraders derived from triazolodiazepine (JQ1) and tetrahydroquinoline (I-BET726) BET inhibitor scaffolds. J. Med. Chem. 61, 504–513 10.1021/acs.jmedchem.6b01912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lai A. C., Toure M., Hellerschmied D., Salami J., Jaime-Figueroa S., Ko E., Hines J., and Crews C. M. (2016) Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 55, 807–810 10.1002/anie.201507634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Raina K., Lu J., Qian Y., Altieri M., Gordon D., Rossi A. M., Wang J., Chen X., Dong H., Siu K., Winkler J. D., Crew A. P., Crews C. M., and Coleman K. G. (2016) PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 113, 7124–7129 10.1073/pnas.1521738113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fulda S., and Vucic D. (2012) Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 11, 109–124 10.1038/nrd3627 [DOI] [PubMed] [Google Scholar]
- 33. Deveraux Q. L., and Reed J. C. (1999) IAP family proteins: suppressors of apoptosis. Genes Dev. 13, 239–252 10.1101/gad.13.3.239 [DOI] [PubMed] [Google Scholar]
- 34. Salvesen G. S., and Duckett C. S. (2002) IAP proteins: blocking the road to death's door. Nat. Rev. Mol. Cell Biol. 3, 401–410 10.1038/nrm830 [DOI] [PubMed] [Google Scholar]
- 35. Deveraux Q. L., Takahashi R., Salvesen G. S., and Reed J. C. (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388, 300–304 10.1038/40901 [DOI] [PubMed] [Google Scholar]
- 36. Hao Y., Sekine K., Kawabata A., Nakamura H., Ishioka T., Ohata H., Katayama R., Hashimoto C., Zhang X., Noda T., Tsuruo T., and Naito M. (2004) Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat. Cell Biol. 6, 849–860 10.1038/ncb1159 [DOI] [PubMed] [Google Scholar]
- 37. Suzuki Y., Nakabayashi Y., and Takahashi R. (2001) Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc. Natl. Acad. Sci. U.S.A. 98, 8662–8667 10.1073/pnas.161506698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kikuchi R., Ohata H., Ohoka N., Kawabata A., and Naito M. (2014) APOLLON protein promotes early mitotic CYCLIN A degradation independent of the spindle assembly checkpoint. J. Biol. Chem. 289, 3457–3467 10.1074/jbc.M113.514430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Imoto I., Tsuda H., Hirasawa A., Miura M., Sakamoto M., Hirohashi S., and Inazawa J. (2002) Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Res. 62, 4860–4866 [PubMed] [Google Scholar]
- 40. Imoto I., Yang Z. Q., Pimkhaokham A., Tsuda H., Shimada Y., Imamura M., Ohki M., and Inazawa J. (2001) Identification of cIAP1 as a candidate target gene within an amplicon at 11q22 in esophageal squamous cell carcinomas. Cancer Res. 61, 6629–6634 [PubMed] [Google Scholar]
- 41. Tamm I., Kornblau S. M., Segall H., Krajewski S., Welsh K., Kitada S., Scudiero D. A., Tudor G., Qui Y. H., Monks A., Andreeff M., and Reed J. C. (2000) Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin. Cancer Res. 6, 1796–1803 [PubMed] [Google Scholar]
- 42. Jaffer S., Orta L., Sunkara S., Sabo E., and Burstein D. E. (2007) Immunohistochemical detection of antiapoptotic protein X-linked inhibitor of apoptosis in mammary carcinoma. Hum. Pathol. 38, 864–870 10.1016/j.humpath.2006.11.016 [DOI] [PubMed] [Google Scholar]
- 43. Wang J., Liu Y., Ji R., Gu Q., Zhao X., Liu Y., and Sun B. (2010) Prognostic value of the X-linked inhibitor of apoptosis protein for invasive ductal breast cancer with triple-negative phenotype. Hum. Pathol. 41, 1186–1195 10.1016/j.humpath.2010.01.013 [DOI] [PubMed] [Google Scholar]
- 44. Foster F. M., Owens T. W., Tanianis-Hughes J., Clarke R. B., Brennan K., Bundred N. J., and Streuli C. H. (2009) Targeting inhibitor of apoptosis proteins in combination with ErbB antagonists in breast cancer. Breast Cancer Res. 11, R41 10.1186/bcr2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang L., Cao Z., Yan H., and Wood W. C. (2003) Coexistence of high levels of apoptotic signaling and inhibitor of apoptosis proteins in human tumor cells: implication for cancer specific therapy. Cancer Res. 63, 6815–6824 [PubMed] [Google Scholar]
- 46. Cohen P., and Tcherpakov M. (2010) Will the ubiquitin system furnish as many drug targets as protein kinases? Cell 143, 686–693 10.1016/j.cell.2010.11.016 [DOI] [PubMed] [Google Scholar]
- 47. Wang S., Bai L., Lu J., Liu L., Yang C. Y., and Sun H. (2012) Targeting inhibitors of apoptosis proteins (IAPs) for new breast cancer therapeutics. J. Mammary Gland Biol. Neoplasia 17, 217–228 10.1007/s10911-012-9265-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Varfolomeev E., Blankenship J. W., Wayson S. M., Fedorova A. V., Kayagaki N., Garg P., Zobel K., Dynek J. N., Elliott L. O., Wallweber H. J., Flygare J. A., Fairbrother W. J., Deshayes K., Dixit V. M., and Vucic D. (2007) IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 131, 669–681 10.1016/j.cell.2007.10.030 [DOI] [PubMed] [Google Scholar]
- 49. Vince J. E., Wong W. W., Khan N., Feltham R., Chau D., Ahmed A. U., Benetatos C. A., Chunduru S. K., Condon S. M., McKinlay M., Brink R., Leverkus M., Tergaonkar V., Schneider P., Callus B. A., et al. (2007) IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 131, 682–693 10.1016/j.cell.2007.10.037 [DOI] [PubMed] [Google Scholar]
- 50. Bertrand M. J., Milutinovic S., Dickson K. M., Ho W. C., Boudreault A., Durkin J., Gillard J. W., Jaquith J. B., Morris S. J., and Barker P. A. (2008) cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 10.1016/j.molcel.2008.05.014 [DOI] [PubMed] [Google Scholar]
- 51. Sekine K., Takubo K., Kikuchi R., Nishimoto M., Kitagawa M., Abe F., Nishikawa K., Tsuruo T., and Naito M. (2008) Small molecules destabilize cIAP1 by activating auto-ubiquitylation. J. Biol. Chem. 283, 8961–8968 10.1074/jbc.M709525200 [DOI] [PubMed] [Google Scholar]
- 52. Oost T. K., Sun C., Armstrong R. C., Al-Assaad A. S., Betz S. F., Deckwerth T. L., Ding H., Elmore S. W., Meadows R. P., Olejniczak E. T., Oleksijew A., Oltersdorf T., Rosenberg S. H., Shoemaker A. R., Tomaselli K. J., Zou H., and Fesik S. W. (2004) Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J. Med. Chem. 47, 4417–4426 10.1021/jm040037k [DOI] [PubMed] [Google Scholar]
- 53. Hennessy E. J., Saeh J. C., Sha L., MacIntyre T., Wang H., Larsen N. A., Aquila B. M., Ferguson A. D., Laing N. M., and Omer C. A. (2012) Discovery of aminopiperidine-based Smac mimetics as IAP antagonists. Bioorg. Med. Chem. Lett. 22, 1690–1694 10.1016/j.bmcl.2011.12.109 [DOI] [PubMed] [Google Scholar]
- 54. Cohen F., Koehler M. F., Bergeron P., Elliott L. O., Flygare J. A., Franklin M. C., Gazzard L., Keteltas S. F., Lau K., Ly C. Q., Tsui V., and Fairbrother W. J. (2010) Antagonists of inhibitor of apoptosis proteins based on thiazole amide isosteres. Bioorg. Med. Chem. Lett. 20, 2229–2233 10.1016/j.bmcl.2010.02.021 [DOI] [PubMed] [Google Scholar]
- 55. Flygare J. A., Beresini M., Budha N., Chan H., Chan I. T., Cheeti S., Cohen F., Deshayes K., Doerner K., Eckhardt S. G., Elliott L. O., Feng B., Franklin M. C., Reisner S. F., Gazzard L., et al. (2012) Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152). J. Med. Chem. 55, 4101–4113 10.1021/jm300060k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hall J. M., and McDonnell D. P. (2005) Coregulators in nuclear estrogen receptor action: from concept to therapeutic targeting. Mol. Interv. 5, 343–357 10.1124/mi.5.6.7 [DOI] [PubMed] [Google Scholar]
- 57. Nadji M., Gomez-Fernandez C., Ganjei-Azar P., and Morales A. R. (2005) Immunohistochemistry of estrogen and progesterone receptors reconsidered: experience with 5,993 breast cancers. Am. J. Clin. Pathol. 123, 21–27 10.1309/4WV79N2GHJ3X1841 [DOI] [PubMed] [Google Scholar]
- 58. LaCasse E. C., Mahoney D. J., Cheung H. H., Plenchette S., Baird S., and Korneluk R. G. (2008) IAP-targeted therapies for cancer. Oncogene 27, 6252–6275 10.1038/onc.2008.302 [DOI] [PubMed] [Google Scholar]
- 59. Fulda S. (2015) Promises and challenges of Smac mimetics as cancer therapeutics. Clin. Cancer Res. 21, 5030–5036 10.1158/1078-0432.CCR-15-0365 [DOI] [PubMed] [Google Scholar]
- 60. Cheung H. H., Mahoney D. J., Lacasse E. C., and Korneluk R. G. (2009) Down-regulation of c-FLIP enhances death of cancer cells by smac mimetic compound. Cancer Res. 69, 7729–7738 10.1158/0008-5472.CAN-09-1794 [DOI] [PubMed] [Google Scholar]
- 61. Varfolomeev E., Izrael-Tomasevic A., Yu K., Bustos D., Goncharov T., Belmont L. D., Masselot A., Bakalarski C. E., Kirkpatrick D. S., and Vucic D. (2015) Ubiquitination profiling identifies sensitivity factors for IAP antagonist treatment. Biochem. J. 466, 45–54 10.1042/BJ20141195 [DOI] [PubMed] [Google Scholar]
- 62. Pilling A. B., Hwang O., Boudreault A., Laurent A., and Hwang C. (2017) IAP antagonists enhance apoptotic response to enzalutamide in castration-resistant prostate cancer cells via autocrine TNF-α signaling. Prostate 77, 866–877 10.1002/pros.23327 [DOI] [PubMed] [Google Scholar]
- 63. Sumi H., Inazuka M., Hashimoto K., Ishikawa T., Yoshida S., and Yabuki M. (2016) T-3256336, a novel and orally available small molecule IAP antagonist, induced tumor cell death via induction of systemic TNFα production. Biochem. Biophys. Res. Commun. 479, 179–185 10.1016/j.bbrc.2016.09.019 [DOI] [PubMed] [Google Scholar]
- 64. Petersen S. L., Wang L., Yalcin-Chin A., Li L., Peyton M., Minna J., Harran P., and Wang X. (2007) Autocrine TNFα signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 12, 445–456 10.1016/j.ccr.2007.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bai L., McEachern D., Yang C. Y., Lu J., Sun H., and Wang S. (2012) LRIG1 modulates cancer cell sensitivity to Smac mimetics by regulating TNFα expression and receptor tyrosine kinase signaling. Cancer Res. 72, 1229–1238 10.1158/0008-5472.CAN-11-2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Raam S., Richardson G. S., Bradley F., MacLaughlin D., Sun L., Frankel F., and Cohen J. L. (1983) Translocation of cytoplasmic estrogen receptors to the nucleus: immunohistochemical demonstration utilizing rabbit antibodies to estrogen receptors of mammary carcinomas. Breast Cancer Res. Treatment 3, 179–199 10.1007/BF01803561 [DOI] [PubMed] [Google Scholar]
- 67. Maruvada P., Baumann C. T., Hager G. L., and Yen P. M. (2003) Dynamic shuttling and intranuclear mobility of nuclear hormone receptors. J. Biol. Chem. 278, 12425–12432 10.1074/jbc.M202752200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.