

Abstract
Hereditary tyrosinemia type I (HTI) is a metabolic genetic disorder caused by mutation of fumarylacetoacetate hydrolase (FAH). Because of the accumulation of toxic metabolites, HTI causes severe liver cirrhosis, liver failure, and even hepatocellular carcinoma. HTI is an ideal model for gene therapy, and several strategies have been shown to ameliorate HTI symptoms in animal models. Although CRISPR/Cas9-mediated genome editing is able to correct the Fah mutation in mouse models, WT Cas9 induces numerous undesired mutations that have raised safety concerns for clinical applications. To develop a new method for gene correction with high fidelity, we generated a Fah mutant rat model to investigate whether Cas9 nickase (Cas9n)-mediated genome editing can efficiently correct the Fah. First, we confirmed that Cas9n rarely induces indels in both on-target and off-target sites in cell lines. Using WT Cas9 as a positive control, we delivered Cas9n and the repair donor template/single guide (sg)RNA through adenoviral vectors into HTI rats. Analyses of the initial genome editing efficiency indicated that only WT Cas9 but not Cas9n causes indels at the on-target site in the liver tissue. After receiving either Cas9n or WT Cas9-mediated gene correction therapy, HTI rats gained weight steadily and survived. Fah-expressing hepatocytes occupied over 95% of the liver tissue 9 months after the treatment. Moreover, CRISPR/Cas9-mediated gene therapy prevented the progression of liver cirrhosis, a phenotype that could not be recapitulated in the HTI mouse model. These results strongly suggest that Cas9n-mediated genome editing is a valuable and safe gene therapy strategy for this genetic disease.
Keywords: CRISPR/Cas, gene therapy, liver, fibrosis, genetic disease, Cas9 nickase, gene editing, genome editing, hereditary tyrosinemia
Introduction
Hereditary tyrosinemia type I (HTI)4 is a rare autosomal recessive liver disease caused by mutations in the enzyme fumarylacetoacetate hydrolase (FAH) (1). Defects in the FAH gene will cause the accumulation of hepatorenal toxic metabolites, including fumarylacetoacetate and maleylacetoacetate (2). These metabolites lead to severe liver and kidney damage, for example, liver failure, cirrhosis, hepatocellular carcinoma, and Fanconi syndrome (3). Current primary treatment for HTI is 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), a potent inhibitor of an enzyme upstream of FAH to prevent the generation of toxic metabolic intermediate products (2). However, a small proportion of the HTI patients respond poorly to the NTBC treatment, calling for alternative strategies that can fix the mutations in the Fah gene, for instance, gene therapies (4).
The revolutionary CRISPR/Cas9 technology is one of the most promising tools for genetic modification and therapy of genetic diseases. The programmable nuclease Cas9 is directed by a single guide RNA (sgRNA) to efficiently generate a double strand break (DSB) at a DNA target site (5). Site-specific DSBs are repaired simultaneously in the cells by two major mechanisms, the error-prone nonhomologous end-joining (NHEJ) pathway or the high fidelity homology-directed repair (HDR) pathway (6). Both of the above DNA repair pathways are exploited for gene therapy studies that use NHEJ to delete dominant active mutations or HDR to repair loss-of-function mutations (7, 8). Through HDR-mediated gene correction, several groups, including ours, have shown the feasibility of CRISPR/Cas9-mediated gene therapy in adult animal disease models, including mouse HTI, hyperammonemia, hemophilia, and other models (8–11). However, because HDR efficiency is usually much lower than NHEJ efficiency in Cas9-transduced cells in vivo, most of the DSBs are repaired through the NHEJ pathway to generate numerous undesired indels in the on-target site. It is well-known that a portion of genetic disorders are caused not by null mutations of genes but by point mutations leading to reduction of mRNA expression or protein activity. Large amounts of undesired on-target mutations could ablate the residual function of the original protein, threatening the safety of patients. Additionally, a recent study reported that Cas9-induced on-target indels had unexpected counteractive effects, which made the phenotype of the spfash mice more severe (11). This observation raises a critical issue of how to reduce the undesired indels in genome editing-mediated gene therapy.
Cas9 functions through two catalytic domains, a HNH domain and a RuvC-like domain. Introduction of a single amino acid substitution (usually D10A or H840A) in either of the domains generates the Cas9 nickase (Cas9n), which only creates a single strand break in targeted sites (12). Because a single strand DNA nick is predominantly repaired by the precise base excision repair pathway or HDR pathway (13–15), Cas9n is able to reduce both off-target and on-target indels in cells and embryos (12, 15). However, current usage of Cas9n is mainly focused on reducing the frequency of off-target events through a double-nicking strategy (16, 17). Hence, whether Cas9n-induced high fidelity DNA repair is feasible to correct genetic mutations for in vivo gene therapy is an area of active investigation. To test the therapeutic effects of Cas9n, we generated a rat HTI model with a 10-bp deletion in exon 2 of the Fah gene through the Cas9 system as previously described (18). As HTI rats are the only model to produce liver fibrosis and cirrhosis, we also evaluated whether gene therapy could prevent or reduce liver damage after long-term observation. A two-vector system was generated to deliver sgRNA/donor template and WT Cas9 or Cas9n through the tail vein into adult HTI rats. Through parallel comparisons, we demonstrated both Cas9 and Cas9n-mediated genome editing could correct the 10-bp Fah deletion in hepatocytes, which expanded in the absence of NTBC treatment and occupied 95% of the liver tissue after 9 months of the treatment. More importantly, unlike WT Cas9, which induced numerous on-target indels, Cas9n did not generate undesired mutations in the sites examined suggesting that Cas9n-mediated genome editing is a valuable and high fidelity gene therapy strategy for some genetic diseases.
Results
Cas9n induced less indels than WT Cas9 in cultured cells
To confirm that Cas9n-induced SSBs are precisely repaired in cells with greatly reduced on-target and off-target indels, we compared the indel rates induced by Cas9 and Cas9n (the D10A mutant of SpyCas9 used in this study) in the EMX1 and VEGFA loci by two sgRNAs, which had been reported to frequently induce off-target mutations (16) (Fig. 1A; Table S2). 48 h after transfection, targeted genomic fragments were PCR-amplified and cloned into TA vectors for sequencing. For each targeting site, 90 clones were sequenced. In accordance with previous publications (15, 17), WT Cas9 generated significantly higher indel rates in both on-target sites (41.75 and 42.16%, respectively) (Fig. 1B). Moreover, the indel sizes created by WT Cas9 ranged from −100 to +108 bp, showing an unpredictable pattern of NHEJ-induced indels (Fig. S1, A and C). On the other hand, Cas9n only created nucleotide substitutions at a low rate in on-target sites of the EMX1 locus (0.74%) but not in the VEGFA locus (Fig. 1B; Fig. S1B). Consistently, Cas9n induced rare off-target mutations in only one of the VEGF off-target sites (0.43% in OT-3-1), whereas off-target events caused by WT Cas9 were detected in each site tested, ranging from 0.81% for the lowest (OT) to 41.84% for the highest (OT-3-1), with the largest indel up to 57 bp (Fig. 1C; Fig. S2, A–C). Our results suggest that Cas9n induces indels extremely rarely.
Figure 1.

Comparison of indel frequencies between Cas9 and Cas9n in HEK293T cells. A, on-target and off-target sites in the EXM1 and VEGFA loci. Target sequences are labeled in red and protospacer adjacent motifs (PAMs) are in blue. Mismatches in the off-target sites are labeled in green. B, indel frequencies of Cas9 and Cas9n in EMX1 and VEGFA on-target sites. C, indel rate of Cas9 and Cas9n in the off-target sites of EMX1 and VEGFA. OT, off-target.
Generation and characterization of the rat HTI model
Earlier studies demonstrated that the rat HTI model recapitulates the key chronic symptoms of human HTI patient's liver fibrosis/cirrhosis (18, 19). In recent reports, the majority of the studies used point mutation animal models to test the feasibility of Cas9-mediated gene correction to treat genetic diseases (8, 10, 11, 20). In this study, we sought to generate a disease model with more nucleotide mutations to investigate the efficiency and efficacy of Cas9-mediated gene therapy. We generated a novel Fah mutant (FahΔ10/Δ10) rat strain in the Sprague-Dawley genetic background with a 10-bp deletion in exon 2 from a different founder than previously reported (18) (Fig. 2, A and B; Table S2). We confirmed that Fah expression is absent in the liver of homozygous mutants but not in WT or heterozygous rats by immunohistochemistry (Fig. 2C). After withdrawal of NTBC for 4 weeks the severe liver cirrhosis resulting from HTI was visualized by Masson's trichrome staining (Fig. 2D). Also, numerous activated hepatic stellate cells were detected by immunohistochemical staining of α-smooth muscle actin (α-SMA) (Fig. 2E). Serum biomarkers including aspartate aminotransferase (AST), alanine aminotransferase (ALT), and total bilirubin (TBIL), rose significantly, indicating severe liver damage in the FahΔ10/Δ10 rat (Fig. 2F). Thus, we successfully generated a Fah knockout rat that mimicked human HTI.
Figure 2.

Generation of the FahΔ10/Δ10 at and phenotype identification. A, the FahΔ10/Δ10 rat was generated by the CRISPR/Cas9 technology. sgRNA targeting the wildtype (WT) Fah sequence is indicated by the red line and the PAM sequence is labeled in blue. The reading frames of the WT and mutant Fah sequence are listed below. The 10-bp deletion in Fah results in an early STOP codon. B, sequencing results of the targeted Fah sites of WT and FahΔ10/Δ10 rats. The red box indicates the 10 missing bp in the FahΔ10/Δ10 rat. C, IHC staining to check Fah expression in WT, FahΔ10/+ and FahΔ10/Δ10 rat liver tissues. I, 100 μm. D and E, knockout of the Fah gene in rat caused severe liver damage including liver fibrosis/cirrhosis indicated by Masson's trichrome staining (D) and α-SMA (α-smooth muscle actin) (E) staining. F, WT and Fah rat serum AST, ALT, ALB, and TBIL levels were tested in WT and FahΔ10/Δ10 rats. *, p < 0.01 (n = 3) using two tailed unpaired Student's t test. Data are presented as mean ± S.D.
Cas9n corrected the Fah gene in the rat HTI model with no on-target indels in vivo
Next, we investigated the ability of Cas9n to genetically correct the rat HTI model in vivo using WT Cas9 as a control. As we previously described (9), recombinant adenovirus was used for packaging Cas9, Cas9n, and the sgRNA/donor template, named AdV–Cas9, AdV–Cas9n, and AdV–HDR, respectively. Cas9 or Cas9n was linked with mCherry through a T2A self-cleaving peptide. The sgRNA and the corrective donor template with 800-bp homology arms on each side of the 10-bp deletion site were inserted into a GFP-expressing vector. We also introduced several silent mutations into the HDR template to prevent re-digestion of the repaired genome (Fig. 3A, left). Immunogenicity is the major side effect of AdV-mediated gene therapy (9). To avoid the curative effect being masked by an AdV-induced immune response, a low AdV dose was used to treat the HTI rats. Specifically, 1 × 1010 vector genomes (vgs) of AdV-HDR and 1 × 1010 vgs of AdV-Cas9 or AdV-Cas9n were injected into 4-week-old rats through the tail vein on day 0 (Fig. 3A, right). The control groups received only 1 × 1010 vgs of AdV-HDR or PBS (Table S1). On day 7, partial hepatectomy was performed and the tissues were collected for initial infection and genome editing efficiency analyses. Both GFP- and mCherry-positive hepatocytes were detected in the treated groups and the initial infectious rate was estimated to be 20–30% (Fig. 3B). Genomic fragments containing the targeted site were amplified and cloned into TA vectors. Sequencing of 98 clones from each group detected a 10.2% indel rate in AdV–Cas9-infected livers, with random indel sizes from −50 to +36 bp (Fig. 3C; Fig. S3). More importantly, no indels were identified in the AdV–Cas9n-infected group, which is consistent with our in vitro findings that Cas9n induces extremely few indels at the on-target site compared with WT Cas9. However, we did not detect any clone containing the perfectly fixed Fah sequence among the 98 clones sequenced in each group, suggesting very low repair efficiency. Alternatively, we employed immunohistochemistry to detect Fah-positive cells to examine the initial correction rate. Indeed, sporadic Fah-positive cells were discovered in liver tissues from both Cas9- and Cas9n-treated groups with a similar low efficiency, estimated at about 0.1% (Fig. 3D). These data suggested that AdV-delivered Cas9n did induce HDR and correct the genetic mutations in vivo. Moreover, Cas9n largely reduced the introduction of uncontrollable on-target indels, minimizing the risks from the NHEJ-mediated frame shifts and large deletions.
Figure 3.

In vivo delivery of the CRISPR/Cas9 system into the rat HTI model via recombinant adenoviral vectors. A, left, schematic views of the recombinant adenoviral vector design and the strategy to repair the Fah gene in FahΔ10/Δ10 rats. The sgRNA target site is listed under the FahΔ10/Δ10 allele. The dashed line in the target site sequence represents the 10-bp deletion in the FahΔ10/Δ10 rat. The theoretically repaired sequence is listed under the HDnR allele. The red-dashed rectangle indicates the fully repaired Fah sequence. The PAM sequence is labeled in blue. Silent mutations are labeled in green. Right, an overview of the NTBC withdrawal process; the black lines indicate the days under NTBC treatment and the gray lines indicate NTBC withdrawal periods. PHT, partial hepatectomy. B, co-infection of the AdV–Cas9/AdV–Cas9n (red) and the AdV-HDR (GFP). Scale bar, 200 μm. C, indel frequencies caused by AdV–Cas9 or AdV–Cas9n in the rat liver on day 8. Data were obtained after sequencing 98 clones. D, immunohistochemistry staining of rat Fah protein in liver tissues from AdV–Cas9 and AdV–Cas9n groups on day 8. FahΔ10/Δ10 and WT rats served as negative and positive controls, respectively.
Repopulation of the initially repaired hepatocytes in vivo
It has been demonstrated that Fah knockout rats can be rescued by WT hepatocyte transplantation and the transplanted cells were able to repopulate the whole liver (18). To check whether those initially repaired cells in our experiment can expand normally and take over the mutant cells, all rats were subjected to one cycle of NTBC withdrawal and re-feeding for 8 days and then left untreated for body weight monitoring. In the PBS and the AdV–HDR only control groups, the body weight of the rats sharply decreased 4 weeks after injection and none of them survived through day 40 of the experiment (Fig. 4A; Table S1). In contrast, similar to the AdV–Cas9/AdV–HDR-treated group, rats (3 of 4) that received AdV–Cas9n/AdV–HDR gained weight steadily 5 weeks after the treatment and the body weight ratio almost doubled on day 96 (Fig. 4A). In the meantime, liver damage conditions of both treated groups were monitored by periodic assessment of AST, ALT, and TBIL. As expected, all three markers declined gradually from high serum levels, indicating the acute liver damage after NTBC withdrawal and the gradual expansion of the repaired cells (Fig. 4B). Three months after the treatment, rat liver tissues were obtained via partial hepatectomy for further evaluation. Through IHC analyses, we discovered Fah+-cell-formed patches occupying over 60% of the livers in the AdV–Cas9n-treated group, which was comparable with the AdV–Cas9 group, suggesting successful restoration of repaired hepatocytes via expansion of those initially repaired cells (Fig. 4C). At this stage, liver damage markers (AST, ALT) were substantially reduced to normal levels compared with the NTBC off control group (Fig. 5A). Furthermore, DNA sequencing of 150 clones containing the mutated Fah site suggested that both Cas9 and Cas9n-mediated genome editing precisely repaired the 10-bp deletion in the Fah gene (Fig. 5B). These results above indicated that the repaired hepatocytes gradually expanded and rescued the liver from hepatic damage
Figure 4.

Delivery of AdV–Cas9/AdV–Cas9n with AdV–HDR cures HTI in FahΔ10/Δ10 rats. A, delivery of AdV–Cas9/AdV–Cas9n with AdV–HDR rescued the body weight loss following NTBC withdraw. Arrow indicates the partial hepatectomy (PHT) on day 8. B, liver damage marker levels (AST, ALT, and TBIL) of the AdV–Cas9- or AdV–Cas9n-treated groups decreased steadily over time. C, IHC staining of liver tissues to detect Fah expression 3 and 9 months after treatment. FahΔ10/Δ10 rat liver slide served as a negative control. Scale bar, 100 μm. PHT, partial hepatectomy.
Figure 5.

Detection of homology directed repair in rat Fah locus. A, rats treated with Cas9/Cas9n had comparable levels of serum AST, ALT, and TBIL (liver damage markers) to WT healthy rat, 3 months after treatment. *, p < 0.05; **, p < 0.01 (n = 3) using the two-tailed unpaired Student's t test. Data are presented as mean ± S.D. B, precise repair of the Fah locus was detected 3 months after treatment. The 10-bp lost in the FahΔ10/Δ10 HTI model were precisely repaired after the treatment as indicated by the red-dashed rectangle. C, detection of collagen deposition and α-SMA expression in rat liver tissues, 3 and 9 months after the treatment. HTI rats with NTBC on and with NTBC off for 1 month served negative and positive controls, respectively.
Progression to liver cirrhosis is rescued by repopulation with repaired hepatocytes
The key chronic manifestation of human HTI is severe liver fibrosis/cirrhosis. Nevertheless, due to the characteristics of the mouse HTI models, previous reports were not able to reveal whether CRISPR/Cas9 rescue in mouse HTI models also rescued mice from progression to liver cirrhosis (8, 10). Taking advantage of the rat HTI model, we examined liver fibrosis and cirrhosis in rat HTI liver tissues. Interestingly, although we did discover fibrous septa with strong collagen deposition in both groups by Sirius Red staining 3 months after the injection, we did not detect any activated hepatic stellate cells (HSCs) in the Cas9- or NTBC-treated liver slides as indicated by α-SMA staining (Fig. 5C). HSCs are the inducer of fibrosis and the main source of collagen production (21). The mismatch between Sirius Red and α-SMA staining may be explained if collagen was produced earlier when repaired hepatocytes were still in the minority. To confirm whether the HSCs were truly reversed to a quiescent state, AdV–Cas9n-treated HTI rats were analyzed at 9 months after injection. We found that Fah-positive cells expanded to ∼95% of the liver tissue (Fig. 4C) and the collagen deposition was significantly narrowed compared with rats treated for 3 months (Fig. 5C). This result suggested that liver fibrosis had already ceased 3 months after treatment.
Off-target and immune response analyses
Finally, we checked the two major side effects of the adenovirus-delivered CRISPR/Cas9 system: immune response and off-target events. We examined the mRNA levels of interleukin 6, interferon-β1, and interleukin 10 in the treated groups and the untreated WT group via RT-PCR. The low dose of AdV vectors only elicited a mild immune response as indicated by their relative liver cytokine mRNA levels (Fig. 6). Next, the off-target sites were predicted using the algorithm from the web-based tool (http://crispr.mit.edu).5 The top 10 predicted off-target sites were PCR-amplified and deep sequenced to analyze the indel rate (Table S2). As a result we did not find a significantly higher indel rate in either Adv–Cas9 or the Adv–Cas9n-treated groups than in the control group that only received PBS. Our results are consistent with previous discoveries that Cas9 induced off-target effects less frequently in vivo than in cells (22, 23).
Figure 6.
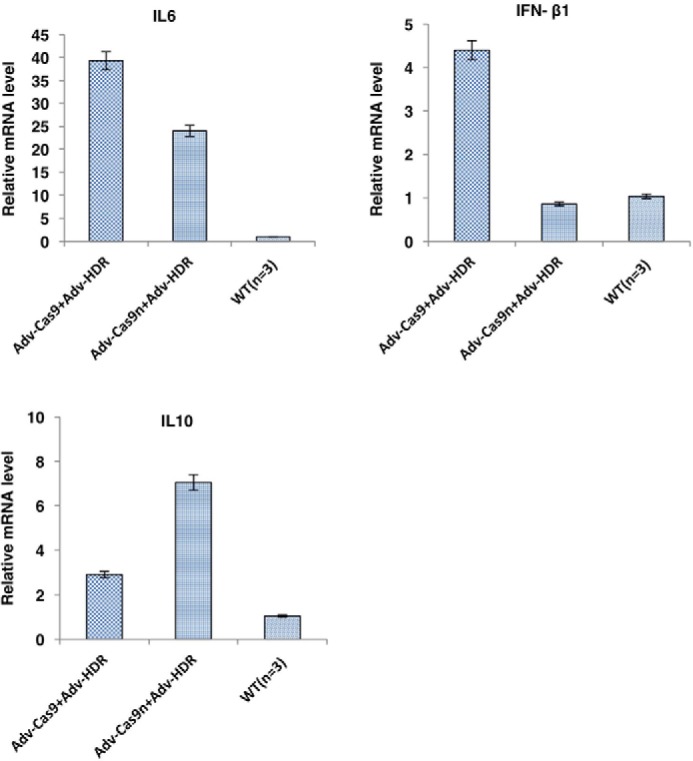
Low dose adenovirus treatment elicited a mild immune response in rats. On day 96, rat liver mRNA levels of inflammatory cytokines from each group were determined by real-time PCR. Data presented as mean ± S.D.
Discussion
Human HTI patients have mutation(s) in the FAH gene leading to accumulation of toxic intermediates in the tyrosine metabolic pathway. NTBC efficiently prevents liver and kidney damage in the majority of HTI patients by blocking the upstream tyrosine metabolic enzymes, however, some patients do not respond to NTBC treatment for unknown reasons (24). In addition, NTBC treatment with a low tyrosine/phenylalanine diet only ameliorates the symptoms but does not completely cure the disease (25). A current clinical cure of HTI is liver transplantation. However, the shortage of donor organs and the risk of graft versus host disease after the surgery are major obstacles confronting the patients. In this study, we cured HTI in a rat model by fixing the mutant Fah gene bearing a 10-bp deletion via delivery of two adenovirus-packaged elements, Cas9/Cas9n and a sgRNA-donor template, demonstrating the feasibility and advantages of nicking nuclease-facilitated in vivo gene therapy for genetic diseases.
HTI is an ideal model for gene therapy studies, because the hepatocytes, which obtain Fah expression have a repopulation advantage. However, all previous HTI gene therapy studies have used the mouse model, which does not develop liver fibrosis and cirrhosis. As liver cirrhosis is the key chronic manifestation of hereditary tyrosinemia type I in patients, it is critical to evaluate the symptoms after gene therapy treatment. The characteristics of the rat HTI model enabled us to monitor the effect of gene therapy on liver cirrhosis. Three months after treatment, activated HSCs were undetectable suggesting greatly reduced fibrogenic activity. More importantly, the deposition of collagen decreased gradually and Sirius Red staining positive signals almost disappeared at 9 months after treatment. This is similar to our previously reported findings that progression to liver cirrhosis was largely prevented after transplantation of WT hepatocytes into HTI rats (18). Our study demonstrates the potential effects of gene therapy to ameliorate HTI-induced liver fibrosis and cirrhosis.
Different strategies of gene therapy have been employed to treat the HTI model. For example, Paulk and others (26–28) showed that delivery of AAV donor templates spontaneously induced HDR in HTI mouse hepatocytes to ameliorate the disease. However, the autonomous HDR efficiency is extremely low and required a high virus titer (up to 10E11) and long homology arms. Numerous studies have shown that programmable nucleases induce DSBs that greatly stimulated HDR and they have been actively studied for gene therapy research. It is known that a DNA single strand nick could also induce HDR and avoid indels (14, 15), but it remains to be tested whether the customized nickases are applicable for high fidelity in vivo gene therapy. To avoid single-stranded DNA template-induced spontaneous HDR as observed in AAV vectors, we used an AdV vector, which is a double-stranded DNA vector and usually does not induce HDR with short homology arms. It was also confirmed in our experiments that HTI rats solely injected with sgRNA-donor templates could not survive in the absence of NTBC treatment. Delivery of Cas9n together with sgRNA-donor templates repaired the Fah mutation and ameliorated HTI liver damage in rats, suggesting that Cas9n induced site-specific DNA nicks that stimulated HDR efficiently. However, the initial correction rate in total hepatocytes was about 0.1%, which is very low due to many reasons. The main reason is that single-stranded DNA nicks are quickly repaired by the base excision repair pathway and only a small proportion of nicks are repaired through the HDR pathway. Second, the sgRNA activity was not high because the initial indel rate induced by WT Cas9 was only about 10% and the HDR rate was similar to Cas9n. The reason why we used this sgRNA was that it spans the 10-bp deletion region and would not re-digest the repaired alleles because it only recognizes the mutant Fah allele. Through screening for higher efficiency sgRNA targets, it is highly possible to increase the HDR rate for in vivo gene therapy. Third, to decrease AdV-induced hepatic toxicity, we used a relatively low viral titer and the infection efficiency was about 30% for each virus. Because we used a two-vector system to deliver both Cas9/Cas9n and sgRNA/donor templates, the HDR efficiency is greatly affected due to the low ratio of double-infected cells. As AdV has a maximum capacity of packaging 30-kb DNA fragments, a single vector AdV system should have a much higher repair efficiency for future applications.
Although recombinant AdVs have many advantages over other viral gene delivery systems, such as extremely efficient transduction of most tissues, a large packaging capacity and lack of integration into the host genome, its higher immunogenicity and toxicity add safety concerns to its clinical implementation. We previous showed that AdV-induced toxicity negatively affected Cas9-mediated gene correction therapy in a hemophilia model (9), but in this study HTI was completely cured, suggesting AdV as a gene delivery system has potential applications for certain diseases. Patients will greatly benefit from gene correction therapy through the AdV delivery system if further engineering of the AdV succeeds in reducing adenoviral immunogenicity and the innate immune response to the vector can be safely managed. Cas9n-induced HDR is sufficient to achieve precise gene correction and cure the genetic disease, whereas inducing minimal, if any, unpredictable indels that would ablate residual gene expression or create new dominant-negative proteins. Although Cas9n-induced initial HDR efficiency is not high, we believe that with the combination of other methods, as demonstrated by Nygaar and colleagues (30), Cas9n-mediated precise genome editing is a promising strategy for therapeutic applications in a range of genetic diseases.
Experimental procedures
Animal experiments
All rats (SLAC, Shanghai) were supplied with sufficient food (irradiated) and water (autoclaved) and maintained under a 12-h light cycle. The FahΔ10/Δ10 rat was created via the CRISPR/Cas9 technology as previously reported using the sgRNA targeting rat Fah gene (Table S2) (31). Primers for genotyping are listed in supporting Table S2. When homozygous FahΔ10/Δ10 rats were obtained, they were kept under NTBC treatment until they reached 6 weeks of age, at which point they were subjected to the experiment. All animal experiments conformed to the regulations drafted by the Association for Assessment and Accreditation of Laboratory Animal Care in Shanghai and were approved by the East China Normal University Center for Animal Research.
Construction of the adenovirus vectors and virus production
AdV–Cas9/AdV–Cas9n and AdV–HDR were produced by Obio Technology (Shanghai) using the AdMax packaging system. Adenovirus serotype 5 was used in this study because it has been widely used in clinical research (32, 33).
Cell culture and transfection
HEK293T cells were cultured in Dulbecco's modified Eagle's medium supplied with 10% FBS. Cells were seeded into a 12-well plate at 3 × 105 cells per well and cultured overnight. The next day, 1.2 μg of pX300 plasmid and 400 ng of U6-sgRNA amplicon were transfected into each well. 48 h after transfection, cells were harvested to extract the genomic DNA.
Indel rate assessment
For indel rate assessments by TA cloning, HEK293 or rat liver genomic DNA was extracted by phenol/chloroform and used as template to amplify the fragments containing the corresponding on-target or off-target sites. PCR products were then TA cloned into the pMD18-T (Takara, catalog number 6011) vector for Sanger sequencing. For indel rate assessments by deep sequencing, off-target sites corresponding to the targeting sgRNA were predicted by the web-based tool (crispr.mit.edu).5 For the top 10 predicted sites, DNA surrounding the potential cleavage sites were amplified by primers with unique barcodes. The amplicons were purified and sent for deep sequencing analysis by GENEWIZ (China). Any indel within 20 bp up- or downstream of the predicted cleavage sites is considered as a potential off-target event. The NGS data of this study have been submitted to the Sequence Read Archive database (accession number PRJNA433082). All target sites and primers in this study are listed in Table S2.
Tail vein injection of adenoviral particles
All adenoviral particles were diluted with PBS (pH 7.5) to adjust the final injection volume to be 800 μl. For each group, four FahΔ10/Δ10 rats (2 male, 2 female) were used. The treated group received 1 × 1010 vgs of Adv–HDR and 1 × 1010 vgs of Adv–Cas9n or Adv–Cas9. Control groups were injected with 1 × 1010 Adv–HDR in 800 μl of PBS or 800 μl of PBS only.
Partial hepatectomy, immunohistochemistry, and immunofluorescence
Partial hepatectomy was carried out 1 week after the adenoviral treatment. Rats were anesthetized by isoflurane and placed on a 37 °C warming pad. Thereafter, surgery was performed according to the previous report (29). About 30% of the liver was sectioned. For immunohistochemistry, liver tissues were freshly fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 4 μm. Liver slides were stained with anti-Fah (AbboMax, San Jose, CA) antibody for Fah expression or anti-SMA antibody (Abcam) for fibrosis measurement. For immunofluorescence, liver tissues were fixed in 4% paraformaldehyde, dehydrated in increasing concentrations of sucrose, and embedded in OCT. 4-μm liver sections were rinsed in PBS, stained with 4′,6-diamidino-2-phenylindole, sealed, and observed under the microscope. The infection efficiency was determined by manually counting the infected cells (green or red) using the ImageJ software.
Measurement of serum markers and cytokine mRNA levels
Rat blood was collected via retro-orbital bleeding and kept at 4 °C for 30 min. The supernatants were then transferred into a new tube followed by centrifuging at 900 × g for 10 min at 4 °C. The collected plasma was sent to Adicon Clinical Laboratories, Inc. (Shanghai, China) to measure liver damage markers. To check the cytokine levels, total mRNA from liver tissue was isolated with RNAiso Plus (TaKaRa) and subjected to RT-PCR. PCR primers are listed in supporting Table 2.
Author contributions
Y. S., L. W., N. G., S. W., L. Y., Y. L., M. W., and S. Y. data curation; Y. S., L. W., N. G., S. W., and L. Y. formal analysis; Y. S., L. W., N. G., M. L., and D. L. investigation; Y. S., M. L., and D. L. methodology; Y. S., L. W., and D. L. writing-original draft; Y. S., M. L., and D. L. project administration; Y. S., L. W., and D. L. writing-review and editing; L. W., N. G., L. Zhang, and J. Z. validation; H. H., L. Zeng, L. Zhang, L. H., Q. D., H. G., M. L., and D. L. resources; M. L. and D. L. supervision; M. L. and D. L. funding acquisition; D. L. conceptualization.
Supplementary Material
Acknowledgment
We thank Dr. Stefan Siwko for scientific editing and comments.
This work was supported by National Natural Science Foundation of China Grants 81670470 and 81600149 and Shanghai Municipal Commission for Science and Technology Grants 14140901600 and 15JC1400201. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Tables S1–S3 and Figs. S1–S3.
The NGS data of this study have been submitted to the Sequence Read Archive database with accession number PRJNA433082.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- HTI
- hereditary tyrosinemia type I
- FAH
- fumarylacetoacetate hydrolase
- NTBC
- 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3– cyclohexanedione
- sgRNA
- single guide RNA
- DSB
- double strand break
- NHEJ
- nonhomologous end-joining
- HDR
- homology-directed repair
- Cas9n
- Cas9 nickase
- VEGFA
- vascular endothelia growth factor A
- α-SMA
- α-smooth muscle actin
- AST
- aspartate aminotransferase
- ALT
- alanine aminotransferase
- TBIL
- total bilirubin
- GFP
- green fluorescent protein
- AdV
- adenovirus
- vgs
- vector genomes
- HSC
- hepatic stellate cell
- IHC
- immunohistochemistry
- AAV
- adeno-associated virus
- PAM
- protospacer adjacent motif.
References
- 1. Grompe M., al-Dhalimy M., Finegold M., Ou C. N., Burlingame T., Kennaway N. G., and Soriano P. (1993) Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes Dev. 7, 2298–2307 10.1101/gad.7.12a.2298 [DOI] [PubMed] [Google Scholar]
- 2. de Laet C., Dionisi-Vici C., Leonard J. V., McKiernan P., Mitchell G., Monti L., de Baulny H. O., Pintos-Morell G., and Spiekerkötter U. (2013) Recommendations for the management of tyrosinaemia type 1. Orphanet. J. Rare Dis. 8, 8 10.1186/1750-1172-8-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grompe M. (2001) The pathophysiology and treatment of hereditary tyrosinemia type 1. Semin. Liver Dis. 21, 563–571 10.1055/s-2001-19035 [DOI] [PubMed] [Google Scholar]
- 4. Masurel-Paulet A., Poggi-Bach J., Rolland M.-O., Bernard O., Guffon N., Dobbelaere D., Sarles J., de Baulny H. O., and Touati G. (2008) NTBC treatment in tyrosinaemia type I: long-term outcome in French patients. J. Inherit. Metab. Dis. 31, 81–87 10.1007/s10545-008-0793-1 [DOI] [PubMed] [Google Scholar]
- 5. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., and Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Doudna J. A., and Charpentier E. (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096–1258096 10.1126/science.1258096 [DOI] [PubMed] [Google Scholar]
- 7. Ding Q., Strong A., Patel K. M., Ng S. L., Gosis B. S., Regan S. N., Cowan C. A., Rader D. J., and Musunuru K. (2014) Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 115, 488–492 10.1161/CIRCRESAHA.115.304351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yin H., Xue W., Chen S., Bogorad R. L., Benedetti E., Grompe M., Koteliansky V., Sharp P. A., Jacks T., and Anderson D. G. (2014) Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 32, 551–553 10.1038/nbt.2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guan Y., Ma Y., Li Q., Sun Z., Ma L., Wu L., Wang L., Zeng L., Shao Y., Chen Y., Ma N., Lu W., Hu K., Han H., Yu Y., et al. (2016) CRISPR/Cas9-mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse. EMBO Mol. Med. 8, 477–488 10.15252/emmm.201506039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yin H., Song C.-Q., Dorkin J. R., Zhu L. J., Li Y., Wu Q., Park A., Yang J., Suresh S., Bizhanova A., Gupta A., Bolukbasi M. F., Walsh S., Bogorad R. L., Gao G., et al. (2016) Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 34, 328–333 10.1038/nbt.3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang Y., Wang L., Bell P., McMenamin D., He Z., White J., Yu H., Xu C., Morizono H., Musunuru K., Batshaw M. L., and Wilson J. M. (2016) A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 34, 334–338 10.1038/nbt.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., and Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hegde M. L., Hazra T. K., and Mitra S. (2008) Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 18, 27–47 10.1038/cr.2008.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramirez C. L., Certo M. T., Mussolino C., Goodwin M. J., Cradick T. J., McCaffrey A. P., Cathomen T., Scharenberg A. M., and Joung J. K. (2012) Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res. 40, 5560–5568 10.1093/nar/gks179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gao Y., Wu H., Wang Y., Liu X., Chen L., Li Q., Cui C., Liu X., Zhang J., and Zhang Y. (2017) Single Cas9 nickase induced generation of NRAMP1 knockin cattle with reduced off-target effects. Genome Biol. 18, 13 10.1186/s13059-016-1144-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ran F. A., Hsu P. D., Lin C.-Y., Gootenberg J. S., Konermann S., Trevino A. E., Scott D. A., Inoue A., Matoba S., Zhang Y., and Zhang F. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 10.1016/j.cell.2013.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shen B., Zhang W., Zhang J., Zhou J., Wang J., Chen L., Wang L., Hodgkins A., Iyer V., Huang X., and Skarnes W. C. (2014) Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Meth. 11, 399–402 10.1038/nmeth.2857 [DOI] [PubMed] [Google Scholar]
- 18. Zhang L., Shao Y., Li L., Tian F., Cen J., Chen X., Hu D., Zhou Y., Xie W., Zheng Y., Ji Y., Liu M., Li D., and Hui L. (2016) Efficient liver repopulation of transplanted hepatocyte prevents cirrhosis in a rat model of hereditary tyrosinemia type I. Sci. Rep. 6, 31460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hickey R. D., Lillegard J. B., Fisher J. E., McKenzie T. J., Hofherr S. E., Finegold M. J., Nyberg S. L., and Grompe M. (2011) Efficient production of Fah-null heterozygote pigs by chimeric adeno-associated virus-mediated gene knockout and somatic cell nuclear transfer. Hepatology 54, 1351–1359 10.1002/hep.24490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bengtsson N. E., Hall J. K., Odom G. L., Phelps M. P., Andrus C. R., Hawkins R. D., Hauschka S. D., Chamberlain J. R., and Chamberlain J. S. (2017) Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 8, 14454 10.1038/ncomms14454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ismail M. H., and Pinzani M. (2009) Reversal of liver fibrosis. Saudi J. Gastroenterol. 15, 72–79 10.4103/1319-3767.45072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iyer V., Shen B., Zhang W., Hodgkins A., Keane T., Huang X., and Skarnes W. C. (2015) Off-target mutations are rare in Cas9-modified mice. Nat. Meth. 12, 479–479 10.1038/nmeth.3408 [DOI] [PubMed] [Google Scholar]
- 23. Kim D., Kim S., Kim S., Park J., and Kim J.-S. (2016) Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res. 26, 406–415 10.1101/gr.199588.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crone J., Möslinger D., Bodamer O. A., Schima W., Huber W. D., Holme E., and Stöckler Ipsiroglu S. (2003) Reversibility of cirrhotic regenerative liver nodules upon NTBC treatment in a child with tyrosinaemia type I. Acta Paediatr. 92, 625–628 10.1111/j.1651-2227.2003.tb02518.x [DOI] [PubMed] [Google Scholar]
- 25. Malik S., NiMhurchadha S., Jackson C., Eliasson L., Weinman J., Roche S., and Walter J. (2015) Treatment adherence in type 1 hereditary tyrosinaemia (HT1): a mixed-method investigation into the beliefs, attitudes and behaviour of adolescent patients, their families and their health-care team. JIMD Rep. 18, 13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paulk N. K., Wursthorn K., Wang Z., Finegold M. J., Kay M. A., and Grompe M. (2010) Adeno-associated virus gene repair corrects a mouse model of hereditary tyrosinemia in vivo. Hepatology 51, 1200–1208 10.1002/hep.23481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paulk N. K., Loza L. M., Finegold M. J., and Grompe M. (2012) AAV-mediated gene targeting is significantly enhanced by transient inhibition of nonhomologous end joining or the proteasome in vivo. Hum. Gene Ther. 23, 658–665 10.1089/hum.2012.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Z., Lisowski L., Finegold M. J., Nakai H., Kay M. A., and Grompe M. (2012) AAV vectors containing rDNA homology display increased chromosomal integration and transgene persistence. Mol. Ther. 20, 1902–1911 10.1038/mt.2012.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mitchell C., and Willenbring H. (2008) A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat. Protoc. 3, 1167–1170 10.1038/nprot.2008.80 [DOI] [PubMed] [Google Scholar]
- 30. Nygaard S., Barzel A., Haft A., Major A., Finegold M., Kay M. A., and Grompe M. (2016) A universal system to select gene-modified hepatocytes in vivo. Sci. Transl. Med. 8, 342ra79–342ra79 10.1126/scitranslmed.aad8166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shao Y., Guan Y., Wang L., Qiu Z., Liu M., Chen Y., Wu L., Li Y., Ma X., Liu M., and Li D. (2014) CRISPR/Cas-mediated genome editing in the rat via direct injection of one-cell embryos. Nat. Protoc. 9, 2493–2512 10.1038/nprot.2014.171 [DOI] [PubMed] [Google Scholar]
- 32. Lusky M. (2005) Good manufacturing practice production of adenoviral vectors for clinical trials. Hum. Gene Ther. 16, 281–291 10.1089/hum.2005.16.281 [DOI] [PubMed] [Google Scholar]
- 33. Wold W. S., and Toth K. (2013) Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 13, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.