

Abstract
The human T-cell leukemia virus-1 (HTLV-1) oncoprotein Tax drives cell proliferation and resistance to apoptosis early in the pathogenesis of adult T-cell leukemia (ATL). Subsequently, probably as a result of specific immunoediting, Tax expression is down-regulated and functionally replaced by somatic driver mutations of the host genome. Both amplification and point mutations of interferon regulatory factor 4 (IRF4) have been previously detected in ATL., K59R is the most common single-nucleotide variation of IRF4 and is found exclusively in ATL. High-throughput whole-exome sequencing revealed recurrent activating genetic alterations in the T-cell receptor, CD28, and NF-κB pathways. We found that IRF4, which is transcriptionally activated downstream of these pathways, is frequently mutated in ATL. IRF4 RNA, protein, and IRF4 transcriptional targets are uniformly elevated in HTLV-1–transformed cells and ATL cell lines, and IRF4 was bound to genomic regulatory DNA of many of these transcriptional targets in HTLV-1–transformed cell lines. We further noted that the K59R IRF4 mutant is expressed at higher levels in the nucleus than WT IRF4 and is transcriptionally more active. Expression of both WT and the K59R mutant of IRF4 from a constitutive promoter in retrovirally transduced murine bone marrow cells increased the abundance of T lymphocytes but not myeloid cells or B lymphocytes in mice. IRF4 may represent a therapeutic target in ATL because ATL cells select for a mutant of IRF4 with higher nuclear expression and transcriptional activity, and overexpression of IRF4 induces the expansion of T lymphocytes in vivo.
Keywords: interferon regulatory factor (IRF), retrovirus, chromatin immunoprecipitation (ChiP), NF-kappaB transcription factor, cancer biology, ATL, HTLV, driver mutation
Introduction
Adult T-cell leukemia (ATL),4 a neoplasm of mature CD4+ T cells, was first described in 1977 by Uchiyama et al. (1) in a case series of 16 patients from southern Japan. The acute (60%) and lymphomatous (20%) types, which account for the majority of cases, are aggressive lymphomas characterized by rapid progression. In the absence of therapy, survival is measured in days. ATL patients are acutely ill with rapidly rising peripheral blood cell counts and leukemic infiltration of the liver, spleen, lungs, bone marrow, central nervous system, and gastrointestinal tract (1–6). ATL is also associated with severe hypercalcemia, which results from the expression of parathyroid hormone-related protein (PTHrP) and receptor activator of NF-κB ligand (RANKL) by leukemic cells (7, 8).
Treatment consists of combinations of cytotoxic chemotherapy, which include an alkylating agent (cyclophosphamide), microtubule poison (vincristine), anthracycline (hydroxydaunorubicin), and topoisomerase II inhibitor (etoposide); more aggressive regimens improve response rates at the cost of increased toxicity (9, 10). Lenalidomide, a thalidomide analog approved to treat multiple myeloma, has been shown to have activity in relapsed and refractory ATL. However, 70% of patients develop progressive disease within 5 months of treatment with this agent (11). The introduction in Japan of the novel anti-CCR4–directed mAb, mogamulizumab, improved response rates at the cost of increased risk of graft versus host disease in patients who subsequently receive an allogeneic stem cell transplant (12, 13). Allogeneic stem cell transplant remains the only known curative therapy, but it is associated with considerable short-term morbidity and mortality (14). Despite aggressive treatment, the majority of patients relapse, and the 4-year survival is <10%, with a median survival of 10 months.
Human T-cell leukemia virus type 1 (HTLV-1), the first retrovirus known to infect humans, was discovered by Gallo and colleagues in 1981 and is the etiological agent of ATL (15). ATL cells carry clonally integrated copies of the HTLV-1 genome, most commonly introduced through vertical transmission in infancy, followed by decades of asymptomatic latency. Expression of Tax, a potent oncogene and viral trans-activator, from the positive strand of the integrated HTLV-1 genome is necessary for leukemia initiation. Tax-induced activation of the NF-κB pathway plays a critical role in leukemia initiation (16). Activation of other pathways, such as the PI3K-Akt-mTOR by Tax, may also play a role in transformation (17). In addition, HTLV-1 up-regulates its own transcription via Tax-induced activation of the viral 5′-long terminal repeat. Progression to aggressive forms of ATL is marked by decreased Tax expression, decreased transcription from the positive strand of the virus, and proviral DNA methylation or mutation, as a result of Tax-specific immunoediting (18–21). At this stage, proliferation is sustained by cellular genes, mutated or epigenetically altered by events during leukemia initiation and progression. The identification and characterization of these cellular oncogenes and confirmation of their role as drivers of ATL will be critical for the design of new therapies.
The alterations that sustain the leukemic state recapitulate the effects of Tax, including activation of the NF-κB pathway (22). Targeting NF-κB is, therefore, a logical approach. However, a trial of bortezomib in ATL failed to demonstrate benefit over standard therapy (10). High-throughput whole-genome, exome, and RNA-Seq data revealed consistent overexpression and recurrent genetic alterations of the cellular proto-oncogene interferon regulatory factor 4 (IRF4/MUM1) in ATL (22). IRF4 expression is also associated with resistance to antiviral therapy (23, 24). Moreover, IRF4 is expressed in ATL but not in other nonneoplastic HTLV-1–associated pathologies, such as tropical spastic paraparesis, suggesting that its role is unique to the process of malignant transformation initiated by the virus (25).
IRF4 is an established oncogene in other lymphoid malignancies, such as activated B-cell diffuse large B-cell lymphoma (ABC-DLBCL), subtypes of pediatric germinal center DLBCL (GC-DLBCL) and follicular lymphoma, Hodgkin lymphoma, and multiple myeloma. IRF4 represses key genes involved in the mitotic checkpoint, DNA repair, and apoptotic pathways (26–31). In addition, IRF4 transforms fibroblasts in culture and is essential for Epstein-Barr virus–induced transformation of B cells in vitro, a phenomenon with many parallels to HTLV-1–induced T-cell transformation (32). Therefore, IRF4 may be relevant to the design of novel therapeutic strategies for these therapy-resistant malignancies.
IRF1 to -9 constitute a family of transcription factors that play key roles in innate and acquired immunity. IRF4, a lymphoid-specific IRF, was initially identified as a transcription factor that binds the 3′-enhancer of the κ immunoglobulin light chain gene as a heterodimer with the E-twenty-six (ETS) transcription factor PU.1 (Spi-1) (33, 34). IRF4 has two key domains, an N-terminal DNA-binding domain and a C-terminal regulatory domain, which contains an IRF autoinhibitory domain that hydrophobically interacts with and masks the DNA-binding domain (35). Although IRF4, by itself, weakly binds DNA, binding to specific heterodimerization partners relieves autoinhibition, leading to high-affinity DNA binding (36, 37). IRF4 binds as a homodimer to direct repeats of the IRF-binding element or interferon-stimulated response element (ISRE) (38).
In B cells, IRF4-PU.1 or IRF4-Spi-B heterodimers bind to ETS-IRF composite elements (EICEs) to activate transcription of B cell–specific IRF4 targets (35, 39). In T cells, IRF4 complexes with BATF and JunD to bind to AP1-IRF4 composite elements (AICEs) and activate an IRF4 transcriptional program unique to T cells (40). At the intersection of IRF4 transcriptional targets and ATL–up-regulated genes lies a set of genes that are potential transcriptional targets of IRF4 in ATL and may be critical for maintaining the transformed phenotype downstream of IRF4; these genes may represent additional targets for therapy of IRF4-driven malignancies.
Results
IRF4 is recurrently mutated in ATL
Previous studies suggested that IRF4 is recurrently mutated at K59R in ATL (Fig. 1, A and B) (22). We interrogated the COSMIC database to identify mutations of IRF4 previously detected in lymphoid malignancies. Of all single-nucleotide variations (SNVs) of IRF4 in the COSMIC database, K59R is the most common point mutation of IRF4 in lymphoid malignancies and is exclusive to ATL. We noted that other mutations of IRF4 also concentrate in the DNA-binding domain of IRF4 (Fig. 1B). We performed whole-exome sequencing on peripheral blood mononuclear cells (PBMCs) of nine patients with acute ATL. Five of nine patients (56%) had detectable somatic mutations of IRF4. Three of nine (33%) carried a K59R mutation, one patient had an L70V mutation, and one patient carried an S149N mutation. One patient, who initially presented with unfavorable chronic ATL with a white blood cell count of >50,000/μl, did not carry a K59R mutation at presentation (0 of 188 reads). This patient achieved a complete remission following treatment with interferon α and zidovudine but, 5 years later, relapsed with lymphoma subtype of ATL with bulky lymphadenopathy. Repeat sequencing at that time revealed the IRF4 K59R mutation in 30 of 156 reads, suggesting that the emergence of this mutation correlated with progression of disease.
Figure 1.

IRF4 alterations in ATL. A, pie chart depicting types of genetic alterations of IRF4 in ATL. B, distribution of SNVs across the IRF4 protein sequence in lymphoid malignancies, including ATL, obtained from the COSMIC database shows a predominance of SNVs in the DNA-binding domain. K59R and L70V, the most common recurrent mutations in ATL, are located in the DNA-binding domain. C, the Lys-59 region of IRF4 is conserved across species. D, sequences of other IRFs incorporate a lysine or arginine at the position corresponding to IRF4 Lys-59.
Lysine 59 of IRF4 lies within the DNA-binding domain of IRF4 (Fig. 1B), which has a helix-loop-helix tertiary structure with five conserved tryptophans, which are characteristic of the DNA-binding domains of interferon regulatory factors (41). Comparison of the amino acid sequence adjacent to the mutated lysine in IRF4 reveals that this region is highly conserved across evolutionarily diverse species (Fig. 1C), suggesting that this is a functionally important region of the protein. Moreover, the Lys-59 position in IRF4 corresponds to either a lysine or arginine in all other IRFs (Fig. 1D), suggesting that the K59R mutation is unlikely to result in loss of function.
IRF4 lies downstream of mutations in the TCR, CD28, and NF-κB signaling pathways, suggesting that it is a rational target for further study and for the design of future ATL therapies. Kataoka et al. (22) demonstrated that these pathways are frequently activated in ATL, as a result of gene amplification or gain-of-function mutations in Vav1, phospholipase Cγ1, protein kinases Cβ and Cθ, caspase-recruitment domain 11, tumor necrosis factor receptor–associated factor 3, and CD28 (Fig. 2).
Figure 2.
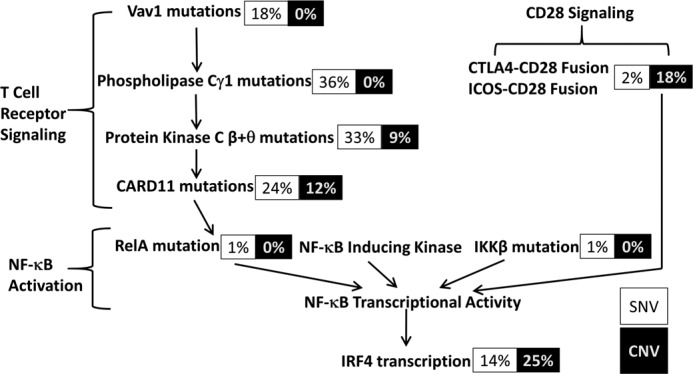
IRF4 lies downstream of constitutively activated T-cell receptor and CD28 pathways in ATL cells. Shown are the percentage of cases in which each of the proteins upstream of IRF4 in the T-cell receptor, CD28, and NF-κB pathways is mutated in Kataoka et al. (22). SNVs are depicted in black type and copy number variations (CNV) in white type. Note that in ATL, protein kinase Cβ is exclusively altered by point mutation and protein kinase Cθ exclusively by amplification.
IRF4 is highly expressed in HTLV-1–transformed and ATL cell lines
To determine the level of expression of IRF4 protein in HTLV-1–transformed and ATL cell lines (Fig. 3A), we performed Western blotting of whole-cell lysates of activated PBMCs (lane 1); HTLV-1–negative Jurkat cells (lane 2); HTLV-1–positive, Tax-positive cell lines MT2 (lane 3) and MT4 (lane 4); and HTLV-1–positive, Tax-negative cell lines TL-OM1 (lane 5), MT1 (lane 6), and ED40515 (lane 7). Densitometry was normalized to the levels observed in CD3/CD28-activated PBMCs. Ratios of IRF4 to actin measured by densitometry, normalized to that of CD3/CD28-activated PBMCs, demonstrated extremely low levels of IRF4 expression in Jurkat cells. The ratio of IRF4 to actin was higher in MT2 cells (1.36-fold), MT4 cells (2.89-fold), TL-OM1 (1.98-fold), MT1 (3.88-fold), and ED40515 (1.53-fold) than in activated PBMCs. Compared with Jurkat cells, MT2 cells showed 177-fold, MT4 cells 376-fold, TL-OM1 cells 258-fold, MT1 cells 504-fold, and ED40515 cells 198-fold elevation of IRF4 protein expression as determined by semiquantitative densitometry. A Tax blot demonstrates a specific band between 50 and 75 kDa corresponding to the Envelope-Tax fusion protein, which is expressed in MT2 cells (lane 3, middle blot, top arrow). Bands corresponding to Tax were seen at 40 kDa in MT2 cells and MT4 cells (bottom arrow). Tax expression was not seen in PBMCs or Jurkat cells and was undetectable in the Tax-negative ATL cell lines TL-OM1, MT1, and ED40515.
Figure 3.

IRF4 is overexpressed in ATL and bound to transcriptional target genes. A, Western blotting demonstrating IRF4 expression in CD3/CD28-activated PBMCs and Jurkat, MT2, MT4, TL-OM1, MT1, and ED40515 cell lines. An immunoblot shows Tax protein expression. Env-Tax fusion protein in lane 3 of the Tax blot (middle) is seen between 50 and 75 kDa and indicated by the top arrow. Tax-specific bands are seen in lanes 3 and 4 of the Tax blot at 40 kDa, as indicated by the bottom arrow. Immunoblot for actin (bottom) shows actin expression as loading control. B–F, binding of IRF4 to HELIOS 3′-regulatory region (B), CTLA4 gene (C), IFNβ gene (D), cFLIP gene (E), and a control genomic region 10 kilobases upstream of the IRF4 start site (F) by IRF4, as demonstrated by a ChIP assay in CD3/CD28-activated PBMCs and Jurkat, MT2, MT4, TL-OM1, and MT1 cells. B–F, results of four replicates. Error bars, S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
IRF4 is bound to genomic regulatory DNA of transcriptional targets
To determine whether IRF4 is bound to its transcriptional targets in HTLV-1–transformed and ATL cell lines, we performed ChIP assays on CD3/CD28-activated PBMCs and Jurkat, MT2, MT4, TL-OM1, and MT1 cells with an IRF4-specific goat polyclonal antibody and control goat immunoglobulin (Fig. 3B). We measured binding of IRF4 to the 3′-regulatory region of the HELIOS gene. Levels of IRF4 bound to this gene in PBMCs were 0.47% of input compared with 0.01% background levels as measured by qPCR. Jurkat cells showed no significant binding of IRF4. In contrast, IRF4 binding to the HELIOS gene was 2.2-, 9.7-, 330-, and 15.5-fold greater than background levels in MT2, MT4, TL-OM1, and MT-1 cells, respectively. There was a statistically significant difference of HELIOS DNA content between control and IRF4 immunoprecipitates from PBMCs and MT4, TL-OM1, and MT1 cells.
CTLA4 is an immune checkpoint receptor and transcriptional target of IRF4 that is overexpressed in ATL (40). We assayed for the presence of CTLA4 DNA in control IgG and IRF4 immunoprecipitates (Fig. 3C). Activated PBMC control immunoprecipitates and IRF4 immunoprecipitates yielded 0.03 and 0.42% of input DNA, respectively, as measured by qPCR. Jurkat cells showed no significant binding of IRF4 to the CTLA4 gene. In contrast, levels of IRF4 binding to the CTLA4 gene were 1.7-, 5.1-, 9.6-, and 7.8-fold above background in MT2, MT4, TL-OM1, and MT1 cells, respectively. A statistically significant difference of CTLA4 DNA content between control and IRF4 immunoprecipitates was detected in PBMCs, MT4, and MT1 cells.
IRF4 has been shown to activate an interferon β promoter–driven luciferase construct (42). We therefore analyzed IRF4 chromatin immunoprecipitates for enrichment of interferon β (IFNβ) promoter DNA (Fig. 3D). Activated PBMC IRF4-specific immunoprecipitates yielded no more IFNβ genomic DNA than control immunoglobulin immunoprecipitates. Levels of IRF4 binding to the IFNβ gene were 8.8-, 0.1, 2.7-, 15.3-, and 11.5-fold above background in Jurkat, MT2, MT4, TL-OM1, and MT1 cells, respectively. These results were statistically significant in PBMCs, Jurkat cells, MT4 cells, TL-OM1 cells, and MT1 cells.
cFLIP/cFLAR is a critical anti-apoptotic gene, which is a transcriptional target of IRF4 and overexpressed in ATL (43–47). We therefore analyzed control and IRF4 chromatin immunoprecipitates for cFLIP/cFLAR DNA (Fig. 3E). Activated PBMC control immunoprecipitates and IRF4-specific immunoprecipitates yielded 0.01 and 0.33% of input DNA. Levels of IRF4 binding to the cFLIP/cFLAR gene were 1.7-, 2.0-, >1,000-, 15-, and 2.6-fold above background in Jurkat, MT2, MT4, TL-OM1, and MT1 cells, respectively.
As a control, we assayed for a control region 10 kilobases upstream of the interferon β start site in control and IRF4 chromatin immunoprecipitates by qPCR. There was no significant difference in levels of this DNA region between control and IRF4 chromatin immunoprecipitates of any of the assayed T-cell lines.
IRF4 transcript is highly expressed in HTLV-transformed and ATL cell lines
We measured IRF4 transcript in a control HTLV-1–negative T-ALL cell line (Jurkat) and HTLV-1–positive cell lines MT2, MT4, TL-OM1, and MT1 (Fig. 4A). As compared with Jurkat cells, 37-, 121-, 10-, and 25-fold levels of expression of IRF4 transcript were observed in MT2, MT4, TL-OM1, and MT1 cells, respectively. These results were all statistically significant. We sequenced the entire DNA-binding domain coding sequence of IRF4 in MT2, MT4, TL-OM1, ED40515, and MT1 cells; we did not detect any mutations in the DNA-binding domain coding sequence of any of these cell lines.
Figure 4.

Expression of IRF4 and IRF4-target genes in HTLV-1–transformed and ATL cell lines. A–K, RNA expression of IRF4 and its transcriptional targets by qRT-PCR: IRF4 (A), BATF (B), CTLA4 (C), CCR4 (D), c-FLIP (E), IL-2 (F), IL-9 (G), IL-10 (H), IFN-β (I), HELIOS exon 7 (J), and exon 3 (K) in Jurkat, MT2, MT4, TL-OM1, and MT1 cells. Error bars (representing S.E.) are indicated as well as p values for comparisons with Jurkat; *, p < 0.05; **, p < 0.01; ***, p < 0.001. The values represent average of 3 replicates.
IRF4 transcriptional targets are highly expressed in HTLV-1–transformed and ATL cell lines
Previous investigations to identify an IRF4 transcriptional signature have identified a number of downstream targets (23). To identify genes that are relevant to ATL, we examined downstream targets of IRF4, which were previously identified to be highly expressed in ATL. BATF (43, 48), CTLA4 (40, 49), CCR4 (50, 51), c-FLIP (43–47), Helios (IKZF2) short isoform (40, 52), interleukin-2 (IL-2) (53, 54), IL-9 (55–58), and IL-10 (59–61) have been shown to be transcriptionally regulated by IRF4 and expressed in ATL. RNA levels of these genes in HTLV-1–transformed and ATL cell lines were measured by qRT-PCR and normalized to those of hypoxanthine guanine phosphoribosyl transferase (HGPRT) mRNA (Fig. 4). BATF transcript levels (Fig. 4B) were 41-, 124-, 3-, and 2-fold higher in MT2, MT4, TL-OM1, and MT1 cells, compared with transcript levels in Jurkat cells. CTLA4 transcript levels were 27-, 15-, 1-, and 2-fold higher in MT2, MT4, TL-OM1, and MT1 cells compared with Jurkat cells (Fig. 4C). Chemokine receptor CCR4 transcript levels were 41-, 26-, 2.0-, and 19-fold higher in MT2, MT4, TL-OM1, and MT1 cells compared with Jurkat cells (Fig. 4D). The cFLIP transcript levels were 18-, 2.0-, 1.0-, and 4.0-fold higher in MT2, MT4, TL-OM1, and MT1 cells compared with Jurkat cells (Fig. 4E). IL-2 transcript levels were 2.3-, 1.6-, 0.9-, and 2.4-fold greater in MT2, MT4, TL-OM1, and MT1 cells compared with Jurkat cells (Fig. 4F). IL-9 transcripts were 462-fold higher in MT2 cells than that in Jurkat cells, whereas no significant differences were observed in MT4, TL-OM1, and MT1 cells (1.0-, 0.9-, and 2.5-fold, respectively) (Fig. 4G). IL-10 transcript levels were 5.2-, 0.9-, 1.2-, and 2.3-fold higher in MT2, MT4, TL-OM1, and MT1 cells compared with transcript levels in Jurkat cells (Fig. 4H). IFNβ transcript levels were 1.4-, 1.0-, 0.6-, and 1.5-fold higher in MT2, MT4, TL-OM1, and MT1 cells compared with Jurkat cells (Fig. 4I).
Helios is a tumor suppressor that induces differentiation of Treg cells (62, 63). We assayed for transcripts containing HELIOS exon 7–coding sequence, which is usually present in all HELIOS transcripts, including the abbreviated transcripts previously detected in ATL. We detected 0.3-, 0.1-, 0.1-, and 1.1-fold higher transcript levels in MT2, MT4, TL-OM1, and MT1 cells compared with Jurkat cells (Fig. 4J). Truncated forms of Helios, lacking exon 2– or exon 3–coding sequences, are expressed in ATL. These forms of Helios are believed to act as dominant negative proteins of Ikaros (IKZF1) (22, 64). Therefore, we also assayed for HELIOS transcripts with intact exon 3 using primers complementary to the junctions of exons 2 and 3 and exons 3 and 4, and we observed that these transcripts were virtually undetectable in MT2, MT4, TL-OM1, and MT1 cells, consistent with previous literature, whereas robust signals were detected in Jurkat cells (Fig. 4K). Given binding of IRF4 to the HELIOS 3′ enhancer noted in Fig. 2B, these data would suggest that IRF4 may transcriptionally repress Helios.
Potential functional consequences of the K59R mutation
Whereas mutation of Lys-59 of IRF4 is unique to ATL, the corresponding residue in other IRF family members is frequently altered in malignant cells. Mutations in IRF2 Arg-43 and IRF6 Arg-45 have been described in cervical and ovarian cancers. Interestingly, IRF8 K45R is found in aggressive forms of DLBCL (65). The mutation K59R is a conservative change that replaces a positively charged lysine with arginine, which possesses an isoelectric pH that is higher than that of lysine (10.7 versus 9.7), and therefore it is likely to increase the net positive charge in this region. We therefore hypothesized that this amino acid change might preserve DNA binding while preventing ubiquitination, thus leading to increased protein levels or enhanced DNA binding and increased transcriptional activity. It is interesting to note that a K123R mutation of IRF4 detected in 2% of patients with chronic lymphocytic leukemia and multiple myeloma results in increased protein expression of the mutant protein as compared with the WT protein (66, 67). Alternatively, given the concentration of positively charged residues in this region of IRF4, K59R could lead to changes in subcellular localization, although we were unable to identify a canonical nuclear localization or nuclear export signal in this region.
IRF4 is predominantly nuclear in HTLV-1–transformed cells
We examined the subcellular localization of IRF4 in control CD3/CD28-activated PBMCs as well as HTLV-1–transformed MT2, MT4, TL-OM1, and MT1 cells (Fig. 5, A and B). We detected predominantly nuclear localization of IRF4 in MT2 cells (Fig. 5A, lane 3 versus lane 4), MT4 cells (Fig. 5A, lane 5 versus lane 6), TL-OM1 cells (Fig. 5B, lane 3 versus lane 4), and MT1 cells (Fig. 5B, lane 5 versus lane 6). Histone H3 and HSP90 blots demonstrate purity of the nuclear and cytoplasmic fractions, respectively (middle and lower blots of Fig. 5, A and B).
Figure 5.

Preferential nuclear localization of WT and K59R IRF4. A, Western blotting of nuclear and cytoplasmic fractions of IRF4 in CD3/CD28 activated PBMCs and MT2 and MT4 cells showing predominantly nuclear localization of IRF4. B, Western blotting of nuclear and cytoplasmic fractions of IRF4 in CD3/CD28-activated PBMCs and TL-OM1 and MT1 cells, demonstrating predominantly nuclear localization of IRF4. C, Western blotting depicting expression of IRF4 in nuclear and cytoplasmic fractions of 293T cells transiently transfected with equal mass of MSCV-Empty-IRES-GFP, MSCV-IRF4 WT-IRES-GFP, and MSCV-IRF4 K59R-IRES-GFP. Lanes 1–3 show nuclear fractions. Lanes 4–6 show cytosolic fractions. The IRF4 immunoblot shows increased nuclear levels of the K59R mutant of IRF4 (top blot, lane 3) as compared with WT IRF4 (lane 2). Lanes 5 and 6 show equal IRF4 expression in the cytosolic fraction. The GFP blot shows equal expression of IRF4 in the cytosolic fraction from the IRES sequence of the vectors, indicating equal transfection efficiency. The HSP90 blot shows equal loading of cytosolic fractions, and lack of signal in the nuclear fractions confirms the purity of the subcellular fractions. The histone H3 blot shows equal loading of nuclear fractions, and lack of signal in the cytosolic fraction further confirms the purity of the subcellular fractions. A bar graph depicts results from three replicates of the experiment depicted in C. *, p < 0.05; **, p < 0.01; ***, p < 0.001. D, Western blotting depicting expression of IRF4 in nuclear and cytoplasmic fractions of Jurkat T cells transiently transfected with equal mass of MSCV-Empty, MSCV-IRF4 WT, and MSCV-IRF4 K59R. Lanes 1–3, nuclear fractions; lanes 4–6, cytosolic fractions; lanes 7–9, whole-cell lysates. The IRF4 immunoblot shows increased nuclear levels of the K59R mutant of IRF4 (top blot, lane 3) as compared with WT IRF4 (lane 2). Lanes 5 and 6 show equal IRF4 expression in the cytosolic fraction. The HSP90 blot shows equal loading of cytosolic fractions, and lack of signal in the nuclear fractions confirms the purity of the subcellular fractions. The histone H3 blot shows equal loading of the nuclear fractions, and lack of signal in the cytosolic fraction further confirms the purity of the subcellular fractions.
K59R mutation enhances nuclear expression levels of IRF4 protein without affecting cytosolic levels
To assess the effects of the K59R mutation on subcellular localization of IRF4, 293T cells were transfected with equal amounts of either empty murine stem cell virus (MSCV)-IRES-GFP vector or corresponding WT or K59R IRF4 expression vectors. Forty-eight hours after transfection, subcellular fractionation was performed, and nuclear and cytosolic fractions were separated by SDS-PAGE and then analyzed by immunoblot (Fig. 5C). Lanes 1, 2, and 3 show nuclear expression of IRF4 and histone H3 (nuclear loading control) and lack of nuclear expression of HSP90, which is a cytosolic protein. IRF4 was detected with an antibody to the C terminus of IRF4, a region that is least conserved among different IRFs, which is distal to the Lys-59 site and unlikely to affect antibody binding. Lane 1 of the IRF4 blot indicates lack of expression of endogenous IRF4 in 293T cells. Comparing lane 3 with lane 2 of the IRF4 blot, there is a 2.8-fold increase in nuclear levels of IRF4 K59R compared with WT IRF4. The histone H3 immunoblot shows equal loading of nuclear protein in lanes 1, 2, and 3. The HSP90 immunoblot shows lack of expression of HSP90 in nuclear fractions. Lanes 4–6 show expression of cytosolic IRF4 proteins, with equal expression of WT and K59R mutant IRF4. The histone H3 blot shows a lack of histone H3 in cytosolic fractions (lanes 4–6), confirming the purity of the subcellular fractions. The HSP90 immunoblot shows equal loading of cytosolic protein in lanes 4–6. Each of the transfected vectors expressed GFP from an internal ribosomal entry site. The GFP immunoblot shows equal expression of GFP in the cytosolic fractions (lanes 4–6). This experiment was confirmed with three biological replicates and with two different antibodies to distinct epitopes at the C terminus of IRF4. Densitometry of IRF4 blots revealed a statistically significant 2.8-fold increase in nuclear levels of K59R mutant IRF4 as compared with WT IRF4 (Fig. 5C, p = 0.03).
To assess whether similar findings are found in T-cell lines, we transfected, by electroporation, Jurkat T cells with 50 μg of either MSCV-Empty vector, MSCV-IRF4 WT, or MSCV-IRF4 K59R mutant plasmid and analyzed the localization of IRF4 by subcellular fractionation of cells at 24 h post-transfection (Fig. 5D). The IRF4 blot (first blot from the top) shows 6.2-fold increased nuclear levels of IRF4 in K59R mutant–transfected Jurkat cells as compared with WT IRF4–transfected cells. Cytoplasmic levels of IRF4 were not different. Note that there is a nonspecific band in the empty vector–transfected lane of the cytoplasmic fraction (lane 4, first blot from the top). Whole-cell extracts showed 2.7-fold increased levels of K59R mutant IRF4 as compared with WT IRF4, which is accounted for entirely by the increased nuclear levels of the K59R mutant. A histone H3 blot (second from the top) and HSP90 blot (third from the top) indicate the purity of the subcellular fractions.
K59R mutant IRF4 possesses increased transcriptional activity as compared with WT IRF4
To assess the transcriptional activity of the mutant compared with WT IRF4, we used luciferase reporter plasmids. We co-transfected 293T cells with interferon β firefly luciferase (interferon β Luc) plasmid and TK-Renilla (Renilla luciferase with the thymidine kinase promoter) and either MSCV-IRES-GFP empty vector or increasing quantities of vectors expressing WT or K59R IRF4 (Fig. 6A). Empty vector and IRF4 WT plasmids were transfected in a 3:1 ratio to K59R IRF4 plasmids to equalize nuclear levels based on our prior ∼3:1 K59R/WT IRF4 nuclear expression ratios. The difference in mass of transfected DNA was compensated by pcDNA3 plasmid, so that total transfected DNA remained constant. We observed 3-, 4-, and 5-fold induction of luciferase with MSCV-IRES-IRF4 WT relative to empty vector with 0.75, 1.5, and 3 μg of plasmid, respectively. With one-third the mass of K59R IRF4 expression plasmid (0.25, 0.5, and 1 μg) we observed 3.7-, 8.4-, and 11.6-fold induction of luciferase, respectively. The differences in results between 1.5 μg of WT IRF4 plasmid and 0.5 μg of K59R mutant IRF4 expression plasmid and between 3 μg of WT IRF4 expression plasmid and 1 μg of K59R expression plasmid were statistically significant (five replicates). We ensured that the nuclear expression level of K59R did not exceed that of WT IRF4 by separating nuclear extracts by SDS-PAGE and immunoblotting for IRF4 (Fig. 6B). The IRF4 immunoblot shows the amount of nuclear IRF4 protein increasing with increasing amounts of transfected plasmid for WT IRF4 (lanes 2–4) and for K59R mutant (lanes 5–7). The largest difference in luciferase activation was between 3 μg of transfected WT IRF4 plasmid and 1 μg of K59R mutant plasmid (bar 4 versus bar 7; 5- versus 11.6-fold induction); the corresponding lanes in the IRF4 immunoblot showed increased expression of IRF4 WT (lane 4) compared with K59R mutant IRF4 (lane 7), ruling out increased nuclear levels as the cause of this difference.
Figure 6.

Effects of WT and K59R IRF4 on interferon β promoter. A, luciferase expression from PGL2 vector with proximal promoter region of interferon β expressed as a ratio to Renilla Luc expressed from the constitutive thymidine kinase promoter in 293T cells, co-transfected with empty vector or increasing mass of MSCV-IRF4 WT-IRES-GFP plasmid or MSCV-IRF4 K59R-IRES-GFP plasmid; MSCV-IRF4 WT plasmid was transfected at a 3:1 ratio to MSCV-IRF4 K59R-IRES-GFP plasmid to equalize nuclear expression levels while keeping total DNA constant. 3 μg of total DNA was transfected (3 μg of empty vector DNA in bar 1 and the amount of empty vector decreased as IRF4 vector was increased to keep a total of 3 μg. The luciferase graph represents results of five replicates. Error bars, S.E. Significance was calculated using unpaired Student's t test. B, Western blotting showing expression of IRF4 in nuclear lysates corresponding to samples for the experiment shown in A.
Transcriptional activity of WT and K59R mutant IRF4 at AICEs, EICEs, and ISREs
IRF4 heterodimerizes with BATF and JunD to activate transcription from AICEs (40) and with Ets family transcription factors PU.1 and SpiB to activate transcription from EICEs (68). IRF4 also homodimerizes to activate direct repeats of the IRF binding sequence (GAAA), also known as an ISRE (34). We tested the capacity of WT IRF4 and K59R mutant IRF4 to induce transcription from these binding elements. For these experiments, we utilized S-tagged IRF4 constructs cloned into the TriExNeo backbone because these plasmids expressed protein at significantly higher levels than the MSCV vectors. We found consistent induction of the interferon β Luc reporter with S-tagged WT IRF4 (10-fold) and K59R IRF4 (23-fold), demonstrating that the transcriptional potential of the S-tagged constructs is similar to that of the untagged protein (Fig. 7A). AICE-Luc, incorporating three repeats of the AICE sequence, was co-transfected with TK-Renilla and either empty S-tagged vector, S-tagged IRF4 WT, or S-tagged K59R mutant IRF4 expression plasmids (Fig. 7B). We found 1.3-fold activation with WT IRF4 and 1.6-fold activation with K59R IRF4 mutant (four replicates). There was a statistically significant difference between induction with K59R mutant and WT IRF4; however, induction of the promoter by IRF4 was weak.
Figure 7.

Effects of WT and K59R IRF4 on transcriptional reporter elements in 293T cells. A, -fold induction of firefly luciferase expressed from the interferon β proximal promoter by WT S-tagged IRF4 and S-tagged K59R mutant IRF4 over empty vector expressed as a ratio to Renilla luciferase expressed from thymidine kinase promoter. B, induction of firefly luciferase expressed from an AP1-IRF composite element by S-tagged WT IRF4 and S-tagged K59R mutant IRF4 expressed as a ratio to Renilla luciferase expressed from thymidine kinase promoter. C, induction of firefly luciferase expressed from an AP1-IRF composite element by S-tagged WT IRF4 and S-tagged K59R mutant IRF4 transfected in combination with BATF expressed as a ratio to Renilla luciferase expressed from thymidine kinase promoter. D, induction of firefly luciferase expressed from an Ets-IRF composite element by S-tagged WT IRF4 and S-tagged K59R mutant IRF4 expressed as a ratio to Renilla luciferase expressed from thymidine kinase promoter. E, induction of firefly luciferase expressed from an Ets-IRF composite element by S-tagged WT IRF4 and S-tagged K59R mutant IRF4 in combination with PU.1 expressed as a ratio to Renilla luciferase expressed from thymidine kinase promoter. F, induction of firefly luciferase expressed from an ISRE element by WT IRF4 and K59R mutant IRF4 expressed as a ratio to Renilla luciferase expressed from thymidine kinase promoter. G, sequence from the IFN-β Luc plasmid corresponding to nucleotides −259 to +43 relative to the transcription start site was sequenced from the interferon β Luc plasmid used in experiments depicted in Figs. 6A and 7A. A mirror repeat element in that sequence is highlighted. H, induction of firefly luciferase expressed from a mirror repeat element by S-tagged WT IRF4 and S-tagged K59R mutant IRF4 expressed as a ratio to Renilla luciferase expressed from thymidine kinase promoter. A–G, results of four replicates; H, results of three replicates.
We next assayed induction of the AICE Luc construct with WT and K59R mutant IRF4 in combination with BATF (Fig. 7C). We found 1.4- and 1.6-fold induction of AICE Luc with WT and K59R mutant IRF4, respectively (four replicates). The difference between empty vector and K59R IRF4 was statistically significant, although there was no statistically significant difference between the induction of WT and K59R IRF4.
We then assayed the induction of EICE-Luc by WT IRF4 and K59R IRF4. EICE-Luc incorporating three repeats of the EICE sequence was co-transfected with TK-Renilla and either empty vector or S-tagged IRF4 WT or S-tagged K59R mutant IRF4 expression plasmids (Fig. 7D). We observed 1.0- and 1.2-fold induction of EICE-Luc with WT IRF4 and K59R IRF4, respectively (four replicates). The differences between K59R and empty vector and K59R and WT IRF4 were statistically significant.
We assayed induction of EICE Luc by WT IRF4 and K59R mutant IRF4 in combination with the prototypical Ets family transcription factor PU.1 (Fig. 7E). We observed 0.9- and 1.2-fold induction with WT IRF4 and K59R IRF4, respectively (four replicates). These values were not statistically significant.
We next assayed the induction of ISRE Luc by WT IRF4 and K59R IRF4. ISRE-Luc incorporating three repeats of ISRE sequence was co-transfected with TK-Renilla and either empty vector or S-tagged IRF4 WT or S-tagged K59R mutant IRF4 expression plasmids (Fig. 7F). We observed 2.4- and 2.9-fold induction of ISRE Luc with co-transfection WT IRF4 and K59R mutant IRF4, respectively (four replicates). There was a statistically significant difference in luciferase values between empty vector and WT IRF4 and between empty vector and K59R mutant IRF4, but not between WT IRF4 and K59R IRF4.
Given that we observed only weak induction of AICE Luc and EICE Luc even with co-transcriptional activators of these elements, and weak induction of ISRE Luc by IRF4, we further examined the interferon β promoter, which was strongly induced by IRF4. Examination of this sequence revealed mirror repeats of the IRF-binding element (TTTCCTTT/AAAGGAAA), which is distinct from previously described AICEs, EICEs, and ISREs. We hypothesized that this sequence might account for the strong induction of the interferon β promoter by IRF4. We cloned three repeats of this sequence upstream of the luciferase gene and tested this luciferase reporter construct for induction by IRF4 (Fig. 7H). We observed 39- and 85-fold induction of luciferase with IRF4 WT and K59R IRF4, respectively. The differences between WT IRF4 and empty vector, between K59R IRF4 and empty vector, and between WT IRF4 and K59R IRF4 were statistically significant. This would suggest that this mirror repeat sequence, which allows for strong induction by IRF4, is a binding site for IRF4 in the interferon β promoter.
IRF4 knockdown increases apoptosis of an ATL cell line
Given the finding of recurrent mutations of IRF4 in ATL, the increased nuclear localization of the mutant, and increased transcriptional activity of the mutant, we further examined the effects of IRF4 on the viability of an ATL cell line. HuT102 cells were electroporated with control siRNA or an IRF4-targeted pool of siRNAs; cells were lysed at 72 h, and lysates were subjected to analysis by immunoblot (Fig. 8A). Cleaved caspase-3 was increased 7.5-fold in cells treated with IRF4-specific siRNAs as compared with control siRNA-treated cells (bottom blot, lane 2 versus lane 1). A blot for total caspase-3 shows a corresponding decrease in total caspase in IRF4 siRNA-treated cells as compared with control siRNA-treated cells (second blot from bottom, lane 2 versus lane 1). IRF4 was knocked down by 81% in cells treated with IRF4 siRNA compared with control siRNA-treated cells (top blot, lane 2 versus lane 1). Tax expression was not affected by IRF4 knockdown (second blot from top). An actin blot shows equal loading of protein in both lanes (third blot from top).
Figure 8.

Effect of WT and K59R IRF4 on T-lymphocyte abundance in mice. A, Western blotting depicting expression of cleaved caspase-3 (as a measure of apoptosis), total caspase-3, IRF4, Tax, and actin in lysates of HuT102 cells treated with control siRNA or a pool of siRNAs to IRF4. B and C, murine hematopoietic stem cells (CD45.1, c-kit+, Sca-1+ bone marrow cells) were transduced with MSCV retroviral vectors, which express IRF4, IRF4 (K59R), or vector alone, as well as GFP from an IRES and engrafted into irradiated CD45.2 recipients (n = 15, 10, and 10 for control, WT IRF4–overexpressing, and K59R-overexpressing mice). Four weeks after engraftment, peripheral blood was collected, and FACS was used to determine the abundance of CD45.1, GFP+ myeloid cells (Gr-1+, Mac-1+), B cells (B220+), and T cells (CD4+, CD8+). Whereas no effect was observed on B cells or myeloid cells, the T-cell populations in IRF4-overexpressing mice were significantly greater than controls, demonstrating that overexpression of IRF4 or IRF4 (K59R) results in T-cell proliferation in vivo. D, mechanisms for increased oncogenic activity of the ATL-specific IRF4 K59R mutation.
IRF4 increases abundance of murine T cells
In light of the finding of increased apoptosis in IRF4 knockdown cells in vitro, we further examined the effects of IRF4 on T-cell abundance in vivo. C57/B6 WT mice were transplanted with bone marrow-derived CD45.1+ Sca-1+ cells, which were transduced MSCV-IRES-GFP empty retroviral vectors or IRF4 WT– or IRF4 K59R–expressing vectors (Fig. 8, B and C). Four weeks after engraftment, peripheral blood was used for FACS analysis of donor cells. GFP+ CD45.1+ cells (donor cells successfully transduced with the MSCV-IRES-GFP retroviral vectors) were classified as myeloid based on reactivity for Gr-1 and Mac-1; B lymphocytes based on reactivity for B220; and T lymphocytes based on reactivity for CD4 or CD8. Fifteen replicates were performed with control mice and 10 replicates each of WT IRF4– and K59R–expressing mice (Fig. 8, A and B). We observed statistically and biologically significant increases in GFP+ donor T cells, but not for B lymphocytes or myeloid cells in mice that were transplanted with Sca-1+ bone marrow stem cells transduced with WT IRF4 and K59R mutant IRF4. This demonstrates that IRF4 selectively increased T-lymphocyte abundance in vivo.
Discussion
IRF4 is a key oncogenic transcription factor in common lymphoid malignancies, such as ABC-DLBCL and multiple myeloma, which are characterized by the activation of NF-κB (27, 28, 67, 68). HTLV-1-associated malignancies are initially sustained by the potent viral oncogene Tax. Subsequently, Tax expression is down-regulated to evade Tax-specific immunoediting, and somatically mutated host proteins take over as drivers of proliferation and confer resistance to apoptosis and therapy. ATL is characterized by mutations in the TCR, CD28, and NF-κB signaling pathways. IRF4 is transcriptionally up-regulated by all of these pathways. Moreover, IRF4 is genetically altered by amplification in 25% of ATL patients and point mutation in 14% of patients (22). Mutations at K59R and L70V are recurring mutations in ATL and represent the most common mutations of IRF4 identified in the COSMIC database of cancer-associated mutations (22). Our exome sequencing data revealed that 33% of acute ATL patients had a K59R mutation, and 11% of patients had an L70V mutation. In one patient, the K59R mutation emerged at progression from chronic ATL to acute ATL, suggesting that this mutation arose with disease progression. Therefore, there was a strong rationale for further characterization of the role of IRF4 and the functional consequences of IRF4 mutations in lymphoid malignancies. We did not detect mutations of the IRF4 DNA-binding domain in the HTLV-1–transformed cell lines we studied. It is possible that selection of this mutation only occurs in vivo; however, the small number of available cell lines precludes this conclusion.
We observed significantly increased IRF4 transcript and protein expression in HTLV-1–positive cell lines as compared with HTLV-1–negative cell lines and activated PBMCs, consistent with a role for IRF4 in their continued viability. We also observed greater binding of IRF4 to regulatory genomic DNA of its transcriptional targets in HTLV-1–positive cell lines and CD3/CD28 activated PBMCs as compared with control Jurkat cells.
We measured levels of transcriptional targets of IRF4, including BATF, CTLA4, CCR4, cFLIP, IL-2, IL-9, and IL-10, which were transcriptionally up-regulated, and also Helios transcripts, which were down-regulated by IRF4. We did not see a perfect correlation between IRF4 protein levels and expression levels of its transcriptional targets in HTLV-transformed and ATL cell lines, suggesting that other transcription factors may also be involved in their regulation.
We noted that the K59R mutation of IRF4 is in a highly conserved region of the DNA-binding domain. We hypothesized that this might affect protein levels, subcellular localization, or transcriptional activation potency. We noted that IRF4 is predominantly nuclear in HTLV-1–transformed and ATL cell lines. We observed increased nuclear expression of K59R mutant IRF4 as compared with WT protein, without changes in cytosolic levels in transiently transfected 293T cells and Jurkat T cells. This suggests that the difference in nuclear levels may be due to differences in import or to sequestration by other nuclear localized molecules.
When we tested the capacity of K59R mutant IRF4 to activate the interferon β promoter, we observed that at equal nuclear expression levels, the K59R mutant possessed more than twice the transcriptional activation potency as compared with WT IRF4. Surprisingly, when we tested the induction of more conventional IRF4-responsive promoter elements (i.e. AP1-IRF and Ets-IRF composite elements and interferon-stimulated response elements), we saw less increase over background. Although these promoters were also activated more potently by K59R mutant IRF4 than WT IRF4, the differences between signal and background were smaller.
Given the strong induction of the interferon β promoter by IRF4, we examined the promoter sequence for IRF-binding sequences. We observed a mirror repeat of the IRF response element in the proximal promoter. When we tested the effect of IRF4 on this promoter element, we saw strong induction, which would fully account for the high level of induction of the interferon β promoter by IRF4. When we measured binding of IRF4 to the interferon β promoter in HTLV-positive cell lines, CD3/CD28-activated PBMCs, and Jurkat cells by a ChIP assay, we observed binding in TL-OM1 cells and MT1 cells, both of which are ATL-derived cell lines. Although interferon β activates apoptosis of naive T cells, it allows for clonal expansion of TCR-activated T cells in a cell-intrinsic manner and can inhibit activation-induced cell death (69–73). However, it is unclear whether interferon β plays a role in survival of ATL cells. It is also possible that other genes with the same mirror repeat IRF-binding element may be the key targets of IRF4 required for viability of ATL cells.
When we knocked down IRF4 expression in HuT102 ATL cells in vitro, we found increased apoptosis, suggesting that ATL cell lines depend on IRF4 expression for viability. When we tested the capacity of hematopoietic cells transduced with IRF4 to expand in vivo, we noted a selective effect of IRF4 on T cells, further supporting a role for IRF4 in T-cell malignancies. We did not see a statistically significant difference in T-cell numbers between mice that received WT IRF4–transduced syngeneic bone marrow cells and those that received K59R mutant IRF4–transduced cells. It is unclear why a difference was not identified, but it is possible that the increased activity of K59R mutant IRF4 compared with WT IRF4 is only manifested in human cells due to interactions with co-transcriptional activators.
These data suggest that novel IRF4-targeted agents may have efficacy in ATL and other IRF4-driven T-cell malignancies. The immunomodulatory agent lenalidomide had activity in a phase II trial in relapsed/refractory ATL (11). Lenalidomide has been shown to depress IRF4 levels in multiple myeloma cell lines, and sensitivity to lenalidomide correlates with IRF4 levels (74). We did not see inhibition of ATL cell lines by lenalidomide in vitro (data not shown), suggesting that, at least in ATL, responses to lenalidomide may not be due to cell-intrinsic effects. IRF4 is critical for the development of conventional type 2 dendritic cells, which drive Th2, Th9, and Th17 responses, as opposed to type 1 conventional dendritic cells, which drive Th1 responses (75). It is possible that in ATL, lenalidomide may skew immune responses toward a Th1-biased response, at the cost of a Th2 response to tumor-associated antigens, as demonstrated in multiple myeloma (76, 77).
In summary, IRF4 is highly expressed in ATL cell lines and HTLV-1–transformed cell lines and stimulates the proliferation of T lymphocytes. Moreover, the K59R mutant form of IRF4 is expressed at higher levels in the nucleus and possesses increased transcriptional activity compared with WT IRF4, strongly suggesting that this is an activating mutation in ATL.
Experimental procedures
Cell culture
TL-OM1, MT1, and ED40515 Tax-negative ATL cell lines were obtained from Edward Harhaj (Johns Hopkins University). The HuT102 ATL cell line was a gift from Patrick Green (Ohio State University). Jurkat, 293T, MT2, and MT4 cells were obtained from the National Institutes of Health AIDS Repository.
Cell lines were maintained at 37 °C and 5% CO2 in complete medium supplemented with 10% fetal bovine serum, 4 mm l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B. PBMCs and T-cell lines, including Jurkat, MT2, MT4, TL-OM1, MT1, and ED40515 cells, were maintained in complete RPMI medium. 293T cells were maintained in complete Dulbecco's modified Eagle's medium with 1 mm sodium pyruvate. For CD3/CD28 stimulation of PBMCs, preservative-free anti-CD3 (clone OKT3) and anti-CD28 (clone ANC28) at 1 μg/ml in PBS were bound to tissue culture–treated polystyrene plates by incubation overnight. Plates were then blocked for 30 min with complete RPMI medium before application of cells.
Transfections
293T cells were transiently transfected with Transit (Mirus Bio) as per the manufacturer's recommended protocol. Jurkat cells were transfected with 50 μg of MSCV-Empty vector, MSCV-IRF4 WT, or MSCV-IRF4 K59R by electroporation at 340 V with 400-microfarad capacitance and parallel resistance of 124 ohms in a volume of 150 μl of electroporation buffer consisting of RPMI without antibiotics, bicarbonate, or glutamine with 10 mm glycylglycine, pH 8.3, in a 4-mm gap cuvette (Bulldog Bio). HuT102 was transfected with either 75 pmol of control siRNAs or an siRNA pool targeting the IRF4 3′-UTR by electroporation at 325 V with 400-microfarad capacitance and parallel resistance of 124 ohms in a volume of 150 μl.
Plasmids
Plasmids expressing WT IRF4 and K59R mutant IRF4 were constructed by recombinational cloning of the respective cDNAs using LR clonase into MSCV gateway vector. S-tagged IRF4 and K59R mutant IRF4 constructs were made by amplifying the IRF4 coding sequences from the MSCV vectors described above using the left primer CGCGGATCCGATGAACCTGGAGGGCGG and right primer CGCGAATTCTCATTCTTGAATAGAGGAA, which introduced BamHI and EcoRI sites in termini of the amplified sequences that were ligated into the corresponding restriction sites in the Tri Ex Neo backbone vector, so that WT IRF4 and K59R mutant IRF4 were expressed with a hexahistidine tag and an S tag at the N terminus.
Interferon β Luc plasmid incorporating two repeats of nucleotides −259 to +43 of the interferon β proximal promoter was a gift from the late Dr. Paula Marie Pitha-Rowe (Johns Hopkins University). For the construction of AICE-Luc, EICE-Luc, ISRE-Luc, and (TTTCCTTT)3 firefly luciferase reporter plasmids, pairs of partially complementary 5′-phosphorylated oligonucleotides incorporating three repeats of AICE, EICE, ISRE, and TTTCCTTT mirror repeat sequences were annealed and ligated with 5′ NheI and 3′ HindIII sites in PGL4.25Luc2CPminP plasmid, obtained from Promega Corp. The sequences for the sense (S) and antisense (AS) oligonucleotides were as follows: AICE Luc (S), CTAGCGAAATGAGTCAGAAATGAGTCAGAAATGAGTCAA; AICE Luc (AS), AGCTTTGACTCATTTCTGACTCATTTCTGACTCATTTCG; EICE Luc (S), CTAGCGGAAGTGAAAGGAAGTGAAAGGAAGTGAAAA; EICE Luc (AS), AGCTTTTTCACTTCCTTTCACTTCCTTTCACTTCCG; ISRE Luc (S), CTAGC GAAAGGGAAAGTGAAAGGGAAAGTGAAAGGGAAAA; ISRE Luc (AS), AGCTTTTTCCCTTTCACTTTCCCTTTCACTTTCCCTTTCG; (TTTCCTTT)3 Luc (S), CTGAGCTTTCCTTTTTTCCTTTTTTCCTTTA; (TTTCCTTT)3 Luc (AS), AGCTTAAAGGAAAAAAGGAAAAAAGGAAAG. TK-R, in which Renilla luciferase is driven by the thymidine kinase promoter, obtained from Promega, was used as transfection control for all luciferase experiments. BATF cDNA cloned into pCMV SPORT6 backbone was obtained from the Harvard-DFCC DNA Repository (HsCD00327682, originally provided by the Mammalian Gene Corp.).
PU.1/Spi1 cDNA, cloned into pENTR223.1 backbone, was obtained from the Harvard-DFCC DNA Repository (HsCD00082621; originally provided by the ORFeome Collaboration).
siRNAs to IRF4
Sequences for IRF4 siRNA pool are CCACAGAUCUAUCCGCCAU, UGUCAGAGCUGCAAGCGUU, and GAAAAUGGUUGCCAGGUGA. siRNAs were purchased from Dharmacon/GE Life sciences.
Subcellular fractionation
Cells were washed twice with ice-cold PBS and resuspended in hypotonic buffer (20 mm Tris-HCl, pH 7.4, 10 mm NaCl, and 3 mm MgCl2). One-twentieth volume of 10% Igepal CA630 was added to each sample, and samples were vortexed vigorously before spinning down nuclei at 8,000 rpm at 4 °C in a refrigerated microcentrifuge. Supernatants were used as cytosolic fractions. Nuclear pellets were lysed in Tris-HCl, pH 6.8, with 2% Igepal CA630, 2% SDS, 10 mm sodium β-glycerophosphate, 10 mm sodium fluoride, 2.5 mm sodium pyrophosphate, 1 mm sodium orthovanadate, and EDTA-free protease inhibitor mixture (Roche Applied Science) and sonicated on ice for 20 s before use for analysis (modified from Ref. 78).
PAGE and Western blotting
Cells were lysed in Tris-HCl, pH 6.8, with 2% Igepal CA630, 2% SDS, 10 mm sodium β-glycerophosphate, 10 mm sodium fluoride, 2.5 mm sodium pyrophosphate, 1 mm sodium orthovanadate, and EDTA-free protease inhibitor mixture (Roche Applied Science/Sigma). Lysates were sonicated on ice for 20 s. Protein concentration was determined using the bicinchoninic acid assay (79). Equal amounts of protein (40 μg) were loaded onto polyacrylamide gels, and electrophoresis was performed according to methods described by Ornstein (80) and modified by Laemmli (81). Primary antibodies were diluted in 5% protease-free BSA, and secondary antibodies were diluted in 10% skim milk. Blots were imaged by enhanced chemiluminescence using a Chemidoc imager (Bio-Rad) and quantified using the Bio-Rad proprietary software.
The following antibodies were used for Western blots. Rabbit primary polyclonal antibody to HSP90 (SPA46) was obtained from Stressgen Corp.; rabbit primary mAb to histone H3 clone D1H2 was obtained from Cell Signaling Technology (catalog no. 4499); D175 rabbit primary polyclonal antibody to cleaved caspase-3 was obtained from Cell Signaling Technology (catalog no. 9661); mouse mAb to caspase-3 clone 3G2 was obtained from Cell Signaling Technology (catalog no. 9668); goat polyclonal antibody to IRF4 (M17; sc6059) was obtained from Santa Cruz Biotechnology, Inc.; goat polyclonal antibody to IRF4 was obtained from Sigma-Aldrich (SAB2502031); rabbit polyclonal antibody to full-length Aequorea coerulescens GFP was obtained from Clontech (catalog no. 632459); hybridoma 1316 for Tax1 was obtained from the NIH AIDS Repository.
ChIP
T-cell lines and CD3/CD28-activated (48 h) PBMCs were washed once in ice-cold PBS and resuspended in 2% paraformaldehyde in PBS for 5 min for cross-linking at room temperature. The reaction was quenched by resuspension in 200 mm glycine for 10 min. Cells were washed in PBS and then lysed in Tris-HCl, pH 7.4, 150 milliequivalents/liter NaCl, 0.1% sodium deoxycholate, 200 μm putrescine, and 10% glycerol. The lysate was sonicated in a bath sonicator with controlled temperature <10 °C for 20 min. The lysates were cleared by a brief centrifugation at 15,000 rpm for 10 min in a refrigerated centrifuge. Supernatants were used for ChIP analysis. DNA in each sample was quantified using Hoechst 33342 fluorescence, and equal mass of DNA from each sample was included in the immunoprecipitation reaction. Goat anti-IRF4 polyclonal antibody (M-17 from Santa Cruz Biotechnology) and magnetic protein G beads (Thermo Fisher Scientific) were added to the immunoprecipitation reaction overnight. After five washes with Tris-HCl, pH 7.4, 150 milliequivalents/liter NaCl, and 200 μm putrescine, protein and DNA were eluted from the beads by incubation at 65 °C overnight, which also served to reverse DNA–DNA and DNA–protein cross-links. Samples were simultaneously treated with RNase A overnight. Samples were used for qPCR to quantify genomic areas of interest in the IRF4 immunoprecipitates. Immunoprecipitates with equal amounts of DNA with IgG control were performed in parallel to determine the background signal by qPCR.
Genomic DNA was used as template for amplification in a three-step amplification protocol using iTaq Universal SYBR Green one-step kit on a Bio-Rad CFX Connect real-time system with an extension temperature of 65 °C. Primers were designed to flank IRF4-binding sites in the genomic neighborhood of genes of interest, as determined using ENCODE data viewed with the Integrated Genome Viewer (Broad Institute).
The following primers were used for qPCR: Helios-ChIP (forward), AAATGAAAGAATTTGGCAAGAAAAA; Helios-ChIP (reverse), CAAATATGAAAATTCAGGGTGAAAC; CTLA4-ChIP (forward), GAGGCAATAAATGAAGAGGAAGGAC; CTLA4-ChIP (reverse), TACTCAAATTGAAACCCTGCTCAGAA; IFNβ-ChIP (forward), TGCTTTCCTTTGCTTTCTCCCAAGT; IFNβ-ChIP (reverse), CCCACTTTCACTTCTCCCTTTCAGT; cFLIP-ChIP (forward), ATGTCTTGTGTGGTAACATTTCAGC; cFLIP-ChIP (reverse), CAGAAAGTCAAAAGCATCAACAGGT; IFNβ upstream-ChIP (forward),GCTTTCAATGCCTATAAAATTTGC; IFNβ upstream-ChIP (reverse), AATCAACATAAAAAGTCTGGTTCTTG.
Input DNA was diluted 100-fold before qPCR. Cycle number (Ct) for immunoprecipitated DNA was converted to percentage of input by dividing antilog to the base 2 of (−1 × Ct) for immunoprecipitates by antilog to the base 2 of (−1 × Ct) for input to give a percentage of input (accounting for the dilution factor of 100 for input DNA).
Genomic DNA sequencing
Genomic DNA from HTLV-1–transformed cell lines was used as template to amplify sequence corresponding to exons 2 and 3 of the IRF4 gene. Forward and reverse primers used for PCR were used for sequencing reactions to obtain forward and reverse sequences. Sequences for forward and reverse primers for amplification of exon 2 with the 3′ position from the IRF4 transcription start site indicated are as follows: IRF4–1351 (forward), CCCCGCAGTGCAGAGCAGA; IRF4–1677 (reverse), GGGGACCCCGGGCTCTGTCT.
Sequences for forward and reverse primers for amplification of exon 3 with 3′-position from the IRF4 transcription start site indicated are as follows: IRF4–3063 (forward), GACATGTATTTTGACTTTTCGTTCT; IRF4–3293 (reverse), GCTGCCTCTGTTAGGTGA.
Whole-exome sequencing
Genomic DNA was extracted from PBMCs of nine ATL patients and sonicated to generate 200–300-base pair fragments. Exonic DNA fragments were captured with biotin-labeled probes covering the entire human exome. Pulldown was performed with streptavidin beads, and eluted DNA was used to make libraries with Illumina linkers and amplified using specific index primers for each patient's DNA sample. For the sequencing process, the ultra-high-throughput sequencing system, HiSeq 2500, was used, generating paired-end reads of 250 nucleotides each. Alignment and variant calling was performed using Basespace.
qRT-PCR
RNA was extracted using TRIzol reagent. Total RNA was used as template for reverse transcription and amplification of cDNAs in a three-step amplification protocol using the iTaq Universal SYBR Green one-step kit (Bio-Rad) on a Bio-Rad CFX Connect real-time system with an extension temperature of 65 °C. Exon-exon junction–spanning primers were designed to reduce background from genomic DNA (except for interferon β, which has no introns). The following primers were used for qRT-PCR: HGPRT1 (forward), GCCCTGGCGTCGTGATTAGT; HGPRT1 (reverse), GTTGACTGGTCATTACAATA; IRF4 (forward), TTGGCGTTCTCAGACTGCCG; IRF4 (reverse), AACGCTTGCAGCTCTGACAA; BATF-F (forward), CTCTCCTCCCCCTGGCAAACAGGACTC; BATF (reverse), TCTGTTTCTCCAGGTCTTCGCTCTCCAGGT; CTLA4 (forward), CATCCCTGTCTTCTGCAAAGCAATG; CTLA4 (reverse), GTTGTAAGAGGGCTTCTTTTCTTTAGCATT; CCR4 (forward), CGGATATAGCAGACACCACCCTCGA; CCR4 (reverse), AACCCACTGGTCTGCTGCATAGTAG; cFLIP (forward), ATCCAGAAGTACAAGCAGTCTGTTCAA; cFLIP (reverse), GAAGGTGTCTCGAAGAAGCTCTGTC; IFNβ (forward), GCAGCAGTTCCAGAAGGAGGAC; IFNβ (reverse), ACGAGTACTCAAAAGGGGACCA; Helios-exon7 (forward), TCTGATGCAGCACCCGCCAA; Helios-exon7 (reverse), GGCCTCTCTTTCCTGGGGTCG; Helios exon2–3 (forward), GCCTCACCAAGTCACATGACAAGCACAAAT; Helios exon3–4 (reverse), CAGTGGAAGGGGCGTTCACCAGTG; IL2 (forward), GGATTTACAGATGATTTTGAATGGAATTAATAATTA; IL2 (reverse), ATGAATGTTGTTTCAGATCCCTTTA; IL9 (forward), TTCCTCATCAACAAGATGCAGGAAGA; IL9 (reverse), ATGGCTGTTCACAGGAAAAATATGGAC; IL10 (forward), CTTCAGCAGAGTGAAGACTTTCTTTCAAAT; IL10 (reverse), TTCACAGGGAAGAAATCGATGACAGC.
Cycle number (Ct value) for each measured transcript was converted to ratios of transcript abundance by taking the antilog to the base 2 of (−1 × Ct) value for each transcript and dividing that by the antilog to the base 2 of (−1 × Ct) value for hypoxanthine guanine phosphoribosyl transferase based on the assumption of a 100% amplification efficiency.
Transduction of murine hematopoietic stem cells and transplantation into recipient mice
Donor B6 CD45.1 mice were treated with 150 mg/kg 5-fluorouracil 6 days before marrow harvest. Harvested bone marrow was enriched for Sca-1+ cells by autoMACS (Miltenyi). Cell concentrations were adjusted to 5 × 105 cells/ml in medium containing Stempro34 (Gibco) serum-free medium supplement, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mm glutamine, 10 ng/ml stem cell factor (R&D Systems), 100 ng/ml mouse thrombopoietin, and 4 μg/ml Polybrene. Virus-like particles were generated in 293T cells by calcium phosphate–mediated co-transfection of pCL-ECO packaging plasmid with either the empty MSCV-IRES-GFP retroviral vector or vector with coding sequence for WT IRF4 or K59R mutant IRF4 inserted by LR clonase recombinational cloning. Virus-like particles were harvested from the medium by ultracentrifugation. Sca-1+ bone marrow cells were transduced with MSCV retroviral vectors by spin-infection at 250 × g at room temperature for 2 h, followed by incubation for 1 h at 37 °C, and then transplanted by retro-orbital injection into lethally irradiated (9.5 grays) CD45.2 (C57/B6 WT) recipients (n = 15 for empty vector and 10 each for WT IRF4 and mutant IRF4). Four weeks after engraftment, peripheral blood was collected, and FACS was used to determine the abundance of CD45.1+, GFP+ myeloid cells (Gr-1+, Mac-1+), B cells (B220+), and T cells (CD4+, CD8+). This was an unblinded experiment. Mouse strains used for this study were obtained from the Jackson Laboratory (Bar Harbor, ME).
Statistics
Data were analyzed by two-tailed unpaired Student's t test for samples of equal variance for statistical significance. Error bars specify S.E.
All animal studies were performed as per animal protocols, which were approved by the division of comparative medicine at Washington University in Saint Louis. All human studies were approved by the institutional review board of Washington University in Saint Louis and abide by the principles of the Declaration of Helsinki.
Author contributions
M. A. C., D. R., G. A. C., and L. R. conceived of the experiments; M. A. C., S. O., H. S., K. C., X. C., M. T., J. H., and A. M. performed the experiments. All authors revised and approved the manuscript.
Acknowledgments
We thank Juan Carlos Ramos (University of Miami), Stefan Barta (Montefiore Hospital), and Ariela Noy (Memorial Sloan Kettering Hospital) for providing samples from ATL patients. We thank the McDonnell Genome Institute and the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis. The center is partially supported by NCI, National Institutes of Health (NIH), Cancer Center Support Grant P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant UL1 TR000448 from the National Center for Research Resources (NCRR), a component of NIH and NIH Roadmap for Medical Research.
This work was supported by National Institutes of Health (NIH) Grants CA063417, CA94056, CA100730, and CA63413; Lymphoma and Leukemia Foundation Grant LLS6067-10 (to L. R.); and NIH Grant K12 CA167540-06 (to M. A. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ATL
- adult T-cell leukemia
- BATF
- basic leucine zipper transcription factor ATF-like
- CTLA4
- cytotoxic T lymphocyte–associated antigen 4
- CCR4
- C-C chemokine receptor 4
- cFLIP
- cellular FLICE (Fas-associated death domain-like interleukin 1β-converting enzyme-like inhibitory protein)
- HTLV
- human T-cell leukemia virus
- IL
- interleukin
- IRF
- interferon regulatory factor
- NF-κB
- nuclear factor of the κ light chain enhancer of activated B cells
- OKT3
- Muromonab-CD3
- PBMC
- peripheral blood mononuclear cell
- RPMI medium
- Roswell Park Memorial Institute medium
- Tax
- HTLV viral transactivator
- TCR
- T-cell receptor
- SNV
- single-nucleotide variation
- ISRE
- interferon-stimulated response element
- EICE
- ETS-IRF composite element
- AICE
- AP1-IRF4 composite element
- qPCR and qRT-PCR
- quantitative PCR and RT-PCR, respectively
- HGPRT
- hypoxanthine guanine phosphoribosyl transferase
- ABC
- activated B-cell
- DLBCL
- diffuse large B-cell lymphoma
- MSCV
- murine stem cell virus
- IRES
- internal ribosome entry site
- GFP
- green fluorescent protein
- TK
- thymidine kinase.
References
- 1. Uchiyama T., Yodoi J., Sagawa K., Takatsuki K., and Uchino H. (1977) Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood 50, 481–492 [PubMed] [Google Scholar]
- 2. Tamura K. (1996) [Clinical classification of adult T-cell leukemia and its complications]. Rinsho Byori 44, 19–23 [PubMed] [Google Scholar]
- 3. Shimoyama M. (1991) Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma: a report from the Lymphoma Study Group (1984–87). Br. J. Haematol. 79, 428–437 10.1111/j.1365-2141.1991.tb08051.x [DOI] [PubMed] [Google Scholar]
- 4. Teshima T., Akashi K., Shibuya T., Taniguchi S., Okamura T., Harada M., Sumida I., Hanada M., and Niho Y. (1990) Central nervous system involvement in adult T-cell leukemia/lymphoma. Cancer 65, 327–332 10.1002/1097-0142(19900115)65:2%3C327::AID-CNCR2820650224%3E3.0.CO%3B2-W [DOI] [PubMed] [Google Scholar]
- 5. Ma W. L., Li C. C., Yu S. C., and Tien H. F. (2014) Adult T-cell lymphoma/leukemia presenting as isolated central nervous system T-cell lymphoma. Case Rep. Hematol. 2014, 917369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Isomoto H., Ohnita K., Mizuta Y., Maeda T., Onizuka Y., Miyazaki M., Omagari K., Takeshima F., Murase K., Haraguchi M., Murata I., and Kohno S. (2001) Clinical and endoscopic features of adult T-cell leukemia/lymphoma with duodenal involvement. J. Clin. Gastroenterol. 33, 241–246 10.1097/00004836-200109000-00017 [DOI] [PubMed] [Google Scholar]
- 7. Richard V., Nadella M. V., Green P. L., Lairmore M. D., Feuer G., Foley J. G., and Rosol T. J. (2005) Transcriptional regulation of parathyroid hormone-related protein promoter P3 by ETS-1 in adult T-cell leukemia/lymphoma. Leukemia 19, 1175–1183 10.1038/sj.leu.2403787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nosaka K., Miyamoto T., Sakai T., Mitsuya H., Suda T., and Matsuoka M. (2002) Mechanism of hypercalcemia in adult T-cell leukemia: overexpression of receptor activator of nuclear factor κB ligand on adult T-cell leukemia cells. Blood 99, 634–640 10.1182/blood.V99.2.634 [DOI] [PubMed] [Google Scholar]
- 9. Tsukasaki K., Utsunomiya A., Fukuda H., Shibata T., Fukushima T., Takatsuka Y., Ikeda S., Masuda M., Nagoshi H., Ueda R., Tamura K., Sano M., Momita S., Yamaguchi K., Kawano F., et al. (2007) VCAP-AMP-VECP compared with biweekly CHOP for adult T-cell leukemia-lymphoma: Japan Clinical Oncology Group Study JCOG9801. J. Clin. Oncol. 25, 5458–5464 10.1200/JCO.2007.11.9958 [DOI] [PubMed] [Google Scholar]
- 10. Ratner L., Rauch D., Abel H., Caruso B., Noy A., Barta S. K., Parekh S., Ramos J. C., Ambinder R., Phillips A., Harding J., Baydoun H. H., Cheng X., and Jacobson S. (2016) Dose-adjusted EPOCH chemotherapy with bortezomib and raltegravir for human T-cell leukemia virus-associated adult T-cell leukemia lymphoma. Blood Cancer J. 6, e408 10.1038/bcj.2016.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ishida T., Fujiwara H., Nosaka K., Taira N., Abe Y., Imaizumi Y., Moriuchi Y., Jo T., Ishizawa K., Tobinai K., Tsukasaki K., Ito S., Yoshimitsu M., Otsuka M., Ogura M., et al. (2016) Multicenter phase II study of lenalidomide in relapsed or recurrent adult T-cell leukemia/lymphoma: ATLL-002. J. Clin. Oncol. 34, 4086–4093 10.1200/JCO.2016.67.7732 [DOI] [PubMed] [Google Scholar]
- 12. Ishida T., Jo T., Takemoto S., Suzushima H., Uozumi K., Yamamoto K., Uike N., Saburi Y., Nosaka K., Utsunomiya A., Tobinai K., Fujiwara H., Ishitsuka K., Yoshida S., Taira N., et al. (2015) Dose-intensified chemotherapy alone or in combination with mogamulizumab in newly diagnosed aggressive adult T-cell leukaemia-lymphoma: a randomized phase II study. Br. J. Haematol. 169, 672–682 10.1111/bjh.13338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sugio T., Kato K., Aoki T., Ohta T., Saito N., Yoshida S., Kawano I., Henzan H., Kadowaki M., Takase K., Muta T., Miyawaki K., Yamauchi T., Shima T., Takashima S., et al. (2016) Mogamulizumab treatment prior to allogeneic hematopoietic stem cell transplantation induces severe acute graft-versus-host disease. Biol. Blood Marrow Transplant. 22, 1608–1614 10.1016/j.bbmt.2016.05.017 [DOI] [PubMed] [Google Scholar]
- 14. Ishida T., Hishizawa M., Kato K., Tanosaki R., Fukuda T., Taniguchi S., Eto T., Takatsuka Y., Miyazaki Y., Moriuchi Y., Hidaka M., Akashi K., Uike N., Sakamaki H., Morishima Y., et al. (2012) Allogeneic hematopoietic stem cell transplantation for adult T-cell leukemia-lymphoma with special emphasis on preconditioning regimen: a nationwide retrospective study. Blood 120, 1734–1741 10.1182/blood-2012-03-414490 [DOI] [PubMed] [Google Scholar]
- 15. Poiesz B. J., Ruscetti F. W., Reitz M. S., Kalyanaraman V. S., and Gallo R. C. (1981) Isolation of a new type C retrovirus (HTLV) in primary uncultured cells of a patient with Sezary T-cell leukaemia. Nature 294, 268–271 10.1038/294268a0 [DOI] [PubMed] [Google Scholar]
- 16. Robek M. D., and Ratner L. (2000) Immortalization of T lymphocytes by human T-cell leukemia virus type 1 is independent of the tax-CBP/p300 interaction. J. Virol. 74, 11988–11992 10.1128/JVI.74.24.11988-11992.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cherian M. A., Baydoun H. H., Al-Saleem J., Shkriabai N., Kvaratskhelia M., Green P., and Ratner L. (2015) Akt pathway activation by human T-cell leukemia virus type 1 Tax oncoprotein. J. Biol. Chem. 290, 26270–26281 10.1074/jbc.M115.684746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taniguchi Y., Nosaka K., Yasunaga J., Maeda M., Mueller N., Okayama A., and Matsuoka M. (2005) Silencing of human T-cell leukemia virus type I gene transcription by epigenetic mechanisms. Retrovirology 2, 64 10.1186/1742-4690-2-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyazaki M., Yasunaga J., Taniguchi Y., Tamiya S., Nakahata T., and Matsuoka M. (2007) Preferential selection of human T-cell leukemia virus type 1 provirus lacking the 5′ long terminal repeat during oncogenesis. J. Virol. 81, 5714–5723 10.1128/JVI.02511-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tamiya S., Matsuoka M., Etoh K., Watanabe T., Kamihira S., Yamaguchi K., and Takatsuki K. (1996) Two types of defective human T-lymphotropic virus type I provirus in adult T-cell leukemia. Blood 88, 3065–3073 [PubMed] [Google Scholar]
- 21. Takeda S., Maeda M., Morikawa S., Taniguchi Y., Yasunaga J., Nosaka K., Tanaka Y., and Matsuoka M. (2004) Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int. J. Cancer 109, 559–567 10.1002/ijc.20007 [DOI] [PubMed] [Google Scholar]
- 22. Kataoka K., Nagata Y., Kitanaka A., Shiraishi Y., Shimamura T., Yasunaga J. I., Totoki Y., Chiba K., Sato-Otsubo A., Nagae G., Ishii R., Muto S., Kotani S., Watatani Y., Takeda J., et al. (2015) Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 47, 1304–1315 10.1038/ng.3415 [DOI] [PubMed] [Google Scholar]
- 23. Mamane Y., Grandvaux N., Hernandez E., Sharma S., Innocente S. A., Lee J. M., Azimi N., Lin R., and Hiscott J. (2002) Repression of IRF-4 target genes in human T cell leukemia virus-1 infection. Oncogene 21, 6751–6765 10.1038/sj.onc.1205843 [DOI] [PubMed] [Google Scholar]
- 24. Ramos J. C., Ruiz P. Jr., Ratner L., Reis I. M., Brites C., Pedroso C., Byrne G. E. Jr., Toomey N. L., Andela V., Harhaj E. W., Lossos I. S., and Harrington W. J. Jr. (2007) IRF-4 and c-Rel expression in antiviral-resistant adult T-cell leukemia/lymphoma. Blood 109, 3060–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mamane Y., Sharma S., Grandvaux N., Hernandez E., and Hiscott J. (2002) IRF-4 activities in HTLV-I-induced T cell leukemogenesis. J. Interferon Cytokine Res. 22, 135–143 10.1089/107999002753452746 [DOI] [PubMed] [Google Scholar]
- 26. Mamane Y., Loignon M., Palmer J., Hernandez E., Césaire R., Alaoui-Jamali M., and Hiscott J. (2005) Repression of DNA repair mechanisms in IRF-4-expressing and HTLV-I-infected T lymphocytes. J. Interferon Cytokine Res. 25, 43–51 10.1089/jir.2005.25.43 [DOI] [PubMed] [Google Scholar]
- 27. Zhang L. H., Kosek J., Wang M., Heise C., Schafer P. H., and Chopra R. (2013) Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. Br. J. Haematol. 160, 487–502 10.1111/bjh.12172 [DOI] [PubMed] [Google Scholar]
- 28. Shaffer A. L., Emre N. C., Lamy L., Ngo V. N., Wright G., Xiao W., Powell J., Dave S., Yu X., Zhao H., Zeng Y., Chen B., Epstein J., and Staudt L. M. (2008) IRF4 addiction in multiple myeloma. Nature 454, 226–231 10.1038/nature07064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Salaverria I., Philipp C., Oschlies I., Kohler C. W., Kreuz M., Szczepanowski M., Burkhardt B., Trautmann H., Gesk S., Andrusiewicz M., Berger H., Fey M., Harder L., Hasenclever D., Hummel M., et al. (2011) Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood 118, 139–147 10.1182/blood-2011-01-330795 [DOI] [PubMed] [Google Scholar]
- 30. Aldinucci D., Celegato M., Borghese C., Colombatti A., and Carbone A. (2011) IRF4 silencing inhibits Hodgkin lymphoma cell proliferation, survival and CCL5 secretion. Br. J. Haematol. 152, 182–190 10.1111/j.1365-2141.2010.08497.x [DOI] [PubMed] [Google Scholar]
- 31. Iida S., Rao P. H., Butler M., Corradini P., Boccadoro M., Klein B., Chaganti R. S., and Dalla-Favera R. (1997) Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat. Genet. 17, 226–230 10.1038/ng1097-226 [DOI] [PubMed] [Google Scholar]
- 32. Xu D., Meyer F., Ehlers E., Blasnitz L., and Zhang L. (2011) Interferon regulatory factor 4 (IRF-4) targets IRF-5 to regulate Epstein-Barr virus transformation. J. Biol. Chem. 286, 18261–18267 10.1074/jbc.M110.210542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pongubala J. M., Nagulapalli S., Klemsz M. J., McKercher S. R., Maki R. A., and Atchison M. L. (1992) PU.1 recruits a second nuclear factor to a site important for immunoglobulin κ 3′ enhancer activity. Mol. Cell. Biol. 12, 368–378 10.1128/MCB.12.1.368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Matsuyama T., Grossman A., Mittrücker H. W., Siderovski D. P., Kiefer F., Kawakami T., Richardson C. D., Taniguchi T., Yoshinaga S. K., and Mak T. W. (1995) Molecular cloning of LSIRF, a lymphoid-specific member of the interferon regulatory factor family that binds the interferon-stimulated response element (ISRE). Nucleic Acids Res. 23, 2127–2136 10.1093/nar/23.12.2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brass A. L., Zhu A. Q., and Singh H. (1999) Assembly requirements of PU.1-Pip (IRF-4) activator complexes: inhibiting function in vivo using fused dimers. EMBO J. 18, 977–991 10.1093/emboj/18.4.977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. De Silva N. S., Simonetti G., Heise N., and Klein U. (2012) The diverse roles of IRF4 in late germinal center B-cell differentiation. Immunol. Rev. 247, 73–92 10.1111/j.1600-065X.2012.01113.x [DOI] [PubMed] [Google Scholar]
- 37. Tamura T., Yanai H., Savitsky D., and Taniguchi T. (2008) The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26, 535–584 10.1146/annurev.immunol.26.021607.090400 [DOI] [PubMed] [Google Scholar]
- 38. Forero A., Moore P. S., and Sarkar S. N. (2013) Role of IRF4 in IFN-stimulated gene induction and maintenance of Kaposi sarcoma-associated herpesvirus latency in primary effusion lymphoma cells. J. Immunol. 191, 1476–1485 10.4049/jimmunol.1202514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Escalante C. R., Brass A. L., Pongubala J. M., Shatova E., Shen L., Singh H., and Aggarwal A. K. (2002) Crystal structure of PU.1/IRF-4/DNA ternary complex. Mol. Cell 10, 1097–1105 10.1016/S1097-2765(02)00703-7 [DOI] [PubMed] [Google Scholar]
- 40. Li P., Spolski R., Liao W., Wang L., Murphy T. L., Murphy K. M., and Leonard W. J. (2012) BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 490, 543–546 10.1038/nature11530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Remesh S. G., Santosh V., and Escalante C. R. (2015) Structural studies of IRF4 reveal a flexible autoinhibitory region and a compact linker domain. J. Biol. Chem. 290, 27779–27790 10.1074/jbc.M115.678789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang L., and Ning S. (2013) Interferon regulatory factor 4 is activated through c-Src-mediated tyrosine phosphorylation in virus-transformed cells. J. Virol. 87, 9672–9679 10.1128/JVI.01435-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang L., Yao Z. Q., Moorman J. P., Xu Y., and Ning S. (2014) Gene expression profiling identifies IRF4-associated molecular signatures in hematological malignancies. PLoS One 9, e106788 10.1371/journal.pone.0106788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lamy L., Ngo V. N., Emre N. C., Shaffer A. L. 3rd, Yang Y., Tian E., Nair V., Kruhlak M. J., Zingone A., Landgren O., and Staudt L. M. (2013) Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 23, 435–449 10.1016/j.ccr.2013.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morelli E., Leone E., Cantafio M. E., Di Martino M. T., Amodio N., Biamonte L., Gullà A., Foresta U., Pitari M. R., Botta C., Rossi M., Neri A., Munshi N. C., Anderson K. C., Tagliaferri P., and Tassone P. (2015) Selective targeting of IRF4 by synthetic microRNA-125b-5p mimics induces anti-multiple myeloma activity in vitro and in vivo. Leukemia 29, 2173–2183 10.1038/leu.2015.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krueger A., Fas S. C., Giaisi M., Bleumink M., Merling A., Stumpf C., Baumann S., Holtkotte D., Bosch V., Krammer P. H., and Li-Weber M. (2006) HTLV-1 Tax protects against CD95-mediated apoptosis by induction of the cellular FLICE-inhibitory protein (c-FLIP). Blood 107, 3933–3939 10.1182/blood-2005-06-2567 [DOI] [PubMed] [Google Scholar]
- 47. Okamoto K., Fujisawa J., Reth M., and Yonehara S. (2006) Human T-cell leukemia virus type-I oncoprotein Tax inhibits Fas-mediated apoptosis by inducing cellular FLIP through activation of NF-κB. Genes Cells 11, 177–191 10.1111/j.1365-2443.2006.00927.x [DOI] [PubMed] [Google Scholar]
- 48. Hasegawa H., Utsunomiya Y., Kishimoto K., Tange Y., Yasukawa M., and Fujita S. (1996) SFA-2, a novel bZIP transcription factor induced by human T-cell leukemia virus type I, is highly expressed in mature lymphocytes. Biochem. Biophys. Res. Commun. 222, 164–170 10.1006/bbrc.1996.0700 [DOI] [PubMed] [Google Scholar]
- 49. Alizadeh A. A., Bohen S. P., Lossos C., Martinez-Climent J. A., Ramos J. C., Cubedo-Gil E., Harrington W. J. Jr., and Lossos I. S. (2010) Expression profiles of adult T-cell leukemia-lymphoma and associations with clinical responses to zidovudine and interferon α. Leukemia Lymphoma 51, 1200–1216 10.3109/10428191003728628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jabeen R., Goswami R., Awe O., Kulkarni A., Nguyen E. T., Attenasio A., Walsh D., Olson M. R., Kim M. H., Tepper R. S., Sun J., Kim C. H., Taparowsky E. J., Zhou B., and Kaplan M. H. (2013) Th9 cell development requires a BATF-regulated transcriptional network. J. Clin. Invest. 123, 4641–4653 10.1172/JCI69489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nakagawa M., Schmitz R., Xiao W., Goldman C. K., Xu W., Yang Y., Yu X., Waldmann T. A., and Staudt L. M. (2014) Gain-of-function CCR4 mutations in adult T cell leukemia/lymphoma. J. Exp. Med. 211, 2497–2505 10.1084/jem.20140987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fujii K., Ishimaru F., Nakase K., Tabayashi T., Kozuka T., Naoki K., Miyahara M., Toki H., Kitajima K., Harada M., and Tanimoto M. (2003) Over-expression of short isoforms of Helios in patients with adult T-cell leukaemia/lymphoma. Br. J. Haematol. 120, 986–989 10.1046/j.1365-2141.2003.04216.x [DOI] [PubMed] [Google Scholar]
- 53. Shindo H., Yasui K., Yamamoto K., Honma K., Yui K., Kohno T., Ma Y., Chua K. J., Kubo Y., Aihara H., Ito T., Nagayasu T., Matsuyama T., and Hayashi H. (2011) Interferon regulatory factor-4 activates IL-2 and IL-4 promoters in cooperation with c-Rel. Cytokine 56, 564–572 10.1016/j.cyto.2011.08.014 [DOI] [PubMed] [Google Scholar]
- 54. Farcet J. P., Lebargy F., Lavignac C., Gaulard P., Dautry A., Gazzolo L., Roméo P. H., and Vainchenker W. (1991) Constitutive IL-2 expression in HTLV-I-infected leukaemic T cell lines. Clin. Exp. Immunol. 84, 415–421 [PMC free article] [PubMed] [Google Scholar]
- 55. Staudt V., Bothur E., Klein M., Lingnau K., Reuter S., Grebe N., Gerlitzki B., Hoffmann M., Ulges A., Taube C., Dehzad N., Becker M., Stassen M., Steinborn A., Lohoff M., et al. (2010) Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity 33, 192–202 10.1016/j.immuni.2010.07.014 [DOI] [PubMed] [Google Scholar]
- 56. Tamiya T., Ichiyama K., Kotani H., Fukaya T., Sekiya T., Shichita T., Honma K., Yui K., Matsuyama T., Nakao T., Fukuyama S., Inoue H., Nomura M., and Yoshimura A. (2013) Smad2/3 and IRF4 play a cooperative role in IL-9-producing T cell induction. J. Immunol. 191, 2360–2371 10.4049/jimmunol.1301276 [DOI] [PubMed] [Google Scholar]
- 57. Chen J., Petrus M., Bryant B. R., Phuc Nguyen V., Stamer M., Goldman C. K., Bamford R., Morris J. C., Janik J. E., and Waldmann T. A. (2008) Induction of the IL-9 gene by HTLV-I Tax stimulates the spontaneous proliferation of primary adult T-cell leukemia cells by a paracrine mechanism. Blood 111, 5163–5172 10.1182/blood-2007-09-113654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lavorgna A., Matsuoka M., and Harhaj E. W. (2014) A critical role for IL-17RB signaling in HTLV-1 tax-induced NF-kappaB activation and T-cell transformation. PLoS Pathog. 10, e1004418 10.1371/journal.ppat.1004418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee C. G., Hwang W., Maeng K. E., Kwon H. K., So J. S., Sahoo A., Lee S. H., Park Z. Y., and Im S. H. (2011) IRF4 regulates IL-10 gene expression in CD4(+) T cells through differential nuclear translocation. Cell. Immunol. 268, 97–104 10.1016/j.cellimm.2011.02.008 [DOI] [PubMed] [Google Scholar]
- 60. Ahyi A. N., Chang H. C., Dent A. L., Nutt S. L., and Kaplan M. H. (2009) IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J. Immunol. 183, 1598–1606 10.4049/jimmunol.0803302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mori N., Gill P. S., Mougdil T., Murakami S., Eto S., and Prager D. (1996) Interleukin-10 gene expression in adult T-cell leukemia. Blood 88, 1035–1045 [PubMed] [Google Scholar]
- 62. Daley S. R., Hu D. Y., and Goodnow C. C. (2013) Helios marks strongly autoreactive CD4+ T cells in two major waves of thymic deletion distinguished by induction of PD-1 or NF-κB. J. Exp. Med. 210, 269–285 10.1084/jem.20121458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hu D. Y., Wirasinha R. C., Goodnow C. C., and Daley S. R. (2017) IL-2 prevents deletion of developing T-regulatory cells in the thymus. Cell Death Differ. 24, 1007–1016 10.1038/cdd.2017.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Asanuma S., Yamagishi M., Kawanami K., Nakano K., Sato-Otsubo A., Muto S., Sanada M., Yamochi T., Kobayashi S., Utsunomiya A., Iwanaga M., Yamaguchi K., Uchimaru K., Ogawa S., and Watanabe T. (2013) Adult T-cell leukemia cells are characterized by abnormalities of Helios expression that promote T cell growth. Cancer Sci. 104, 1097–1106 10.1111/cas.12181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fabbri G., Khiabanian H., Holmes A. B., Wang J., Messina M., Mullighan C. G., Pasqualucci L., Rabadan R., and Dalla-Favera R. (2013) Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J. Exp. Med. 210, 2273–2288 10.1084/jem.20131448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Havelange V., Pekarsky Y., Nakamura T., Palamarchuk A., Alder H., Rassenti L., Kipps T., and Croce C. M. (2011) IRF4 mutations in chronic lymphocytic leukemia. Blood 118, 2827–2829 10.1182/blood-2011-04-350579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Melchor L., Brioli A., Wardell C. P., Murison A., Potter N. E., Kaiser M. F., Fryer R. A., Johnson D. C., Begum D. B., Hulkki Wilson S., Vijayaraghavan G., Titley I., Cavo M., Davies F. E., Walker B. A., and Morgan G. J. (2014) Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia 28, 1705–1715 10.1038/leu.2014.13 [DOI] [PubMed] [Google Scholar]
- 68. Care M. A., Cocco M., Laye J. P., Barnes N., Huang Y., Wang M., Barrans S., Du M., Jack A., Westhead D. R., Doody G. M., and Tooze R. M. (2014) SPIB and BATF provide alternate determinants of IRF4 occupancy in diffuse large B-cell lymphoma linked to disease heterogeneity. Nucleic Acids Res. 42, 7591–7610 10.1093/nar/gku451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Marrack P., Kappler J., and Mitchell T. (1999) Type I interferons keep activated T cells alive. J. Exp. Med. 189, 521–530 10.1084/jem.189.3.521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Davis A. M., Ramos H. J., Davis L. S., and Farrar J. D. (2008) Cutting edge: a T-bet-independent role for IFN-α/β in regulating IL-2 secretion in human CD4+ central memory T cells. J. Immunol. 181, 8204–8208 10.4049/jimmunol.181.12.8204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Havenar-Daughton C., Kolumam G. A., and Murali-Krishna K. (2006) Cutting edge: the direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J. Immunol. 176, 3315–3319 10.4049/jimmunol.176.6.3315 [DOI] [PubMed] [Google Scholar]
- 72. Kolumam G. A., Thomas S., Thompson L. J., Sprent J., and Murali-Krishna K. (2005) Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202, 637–650 10.1084/jem.20050821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Aichele P., Unsoeld H., Koschella M., Schweier O., Kalinke U., and Vucikuja S. (2006) CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J. Immunol. 176, 4525–4529 10.4049/jimmunol.176.8.4525 [DOI] [PubMed] [Google Scholar]
- 74. Lopez-Girona A., Heintel D., Zhang L. H., Mendy D., Gaidarova S., Brady H., Bartlett J. B., Schafer P. H., Schreder M., Bolomsky A., Hilgarth B., Zojer N., Gisslinger H., Ludwig H., Daniel T., et al. (2011) Lenalidomide downregulates the cell survival factor, interferon regulatory factor-4, providing a potential mechanistic link for predicting response. Br. J. Haematol. 154, 325–336 10.1111/j.1365-2141.2011.08689.x [DOI] [PubMed] [Google Scholar]
- 75. Bajaña S., Turner S., Paul J., Ainsua-Enrich E., and Kovats S. (2016) IRF4 and IRF8 act in CD11c+ cells to regulate terminal differentiation of lung tissue dendritic cells. J. Immunol. 196, 1666–1677 10.4049/jimmunol.1501870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bae J., Keskin D. B., Cowens K., Lee A. H., Dranoff G., Munshi N. C., and Anderson K. C. (2015) Lenalidomide polarizes Th1-specific anti-tumor immune response and expands XBP1 antigen-specific central memory CD3+CD8+ T cells against various solid tumors. J. Leuk. (Los Angel.) 3, 178 10.4172/2329-6917.1000178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Luptakova K., Rosenblatt J., Glotzbecker B., Mills H., Stroopinsky D., Kufe T., Vasir B., Arnason J., Tzachanis D., Zwicker J. I., Joyce R. M., Levine J. D., Anderson K. C., Kufe D., and Avigan D. (2013) Lenalidomide enhances anti-myeloma cellular immunity. Cancer Immunol. Immunother. 62, 39–49 10.1007/s00262-012-1308-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gagnon K. T., Li L., Chu Y., Janowski B. A., and Corey D. R. (2014) RNAi factors are present and active in human cell nuclei. Cell Rep. 6, 211–221 10.1016/j.celrep.2013.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., and Klenk D. C. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85 10.1016/0003-2697(85)90442-7 [DOI] [PubMed] [Google Scholar]
- 80. Ornstein L. (1964) Disc electrophoresis. I. Background and theory. Ann. N.Y. Acad. Sci. 121, 321–349 [DOI] [PubMed] [Google Scholar]
- 81. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]